

ABSTRACT
Cisplatin is an effective chemotherapeutic agent in facilitating the inhibition of proliferation, migration, and invasion in cancerous cells. However, the detailed mechanism of the regulation by cisplatin of human breast cancer cells is still unclear. This study aimed to investigate the mechanism of kruppel-like factor 2 (KLF2) transcription factor in cisplatin therapy for breast cancer. RT-qPCR was performed to quantify the expression of KLF2 and WEE1 in clinical tissue samples from breast cancer patients and in MDA-MB-231 cells. ChIP assay and dual-luciferase reporter assay were used to analyze the potential-binding sites of KLF2 and WEE1 promoter. Gain- or loss-of-function approaches were used to manipulate KLF2 and WEE1 in cisplatin-treated MDA-MB-231 cells, and the mechanism of KLF2 in breast cancer was evaluated both via CCK-8 assay, flow cytometry, Transwell assay, and Western blot. Further validation of the KLF2 was performed on nude mouse models. Breast cancer tissues and cells showed a relative decline of KLF2 expression and abundant WEE1 expression. Cisplatin inhibited the proliferation, migration, and invasion of MDA-MB-231 cells. Overexpression of KLF2 enhanced the inhibitory effect of cisplatin on the malignant characteristics of MDA-MB-231 cells in vitro. KLF2 targeted WEE1 and negatively regulated its expression, thus enhancing the sensitivity to cisplatin of breast cancer cells as well as tumor-bearing mice. Overall, these results suggest that KLF2 can potentially inhibit WEE1 expression and sensitize breast cancer cells to cisplatin, thus presenting a promising adjunct treatment.
KEYWORDS: KLF2, wee1, cisplatin, breast cancer, proliferation, invasion, migration
Introduction
Breast cancer is a malignant tumor occurring in the breast gland epithelium tissues, which has surpassed cervical cancer and lung cancer to become the most frequently diagnosed and lethal tumor in women in most countries in the world, including China.1,2 In 2018, breast cancer contributed to 11.6% of newly diagnosed cancer cases in both genders and 24.2% in females, while the mortality rates of breast cancer were 6.6% and 15.0%, respectively.1 The rapid increasing incidence of breast cancer continues to threaten female health, while placing greater demands on medical care. Patients diagnosed with breast cancer at an early stage have a high survival rate through improvements of various interventions, especially traditional local surgery and chemotherapy, the molecular heterogeneity and drug resistance characteristics diminish the effectiveness of various potential options in systemic chemotherapies in metastatic breast cancer.3,4 Therefore, identification of potential molecular characteristics through next-generation sequencing and bioinformatics analysis for subtype markers, and the development of new therapeutic targets is essential for innovative progress in identifying combinational therapeutics in breast cancer treatment.
The most important clinically relevant and validated therapeutic targets of breast cancer are human epidermal growth factor receptor 2 (c-erb-B2), estrogen receptor (ER) and progesterone receptor (PgR).3 However, resistance to endocrine therapy targeting the ER and PgR almost inevitably occurs in advanced and metastatic breast cancer. Combination therapy against emerging potential targets like phosphoinositide-3-kinase, mammalian target of rapamycin, breast cancer 1 (BRCA1), or BRCA2 in conjunction with existing chemotherapies have so far brought modest improvements in oncologic outcome.5,6 Efforts in molecular portrait analysis conducted by The Cancer Genome Atlas,7 as well as ongoing initiatives and trials including PRAEGNANT and AURORA,8,9 have provided invaluable experience and opportunities for identification and assessment of novel prognostic and predictive biomarkers. This innovation enables an integration of theory- and evidence-based therapies and drugs in multistep treatment clinical trials against cases with acquired resistance to monotherapy.
Cis-Dichlorodiammine platinum, usually referred as cisplatin, possesses a broad anticancer spectrum, and is widely applied in solid tumor treatment, including but, not restricted to ovarian, testicular, and breast cancers.10 Cisplatin is also one of the most commonly used combination chemotherapeutic drugs due to its good synergistic effect with a variety of other anti-tumor drugs. In multiple clinical trials of metastatic breast cancer, cisplatin showed good effects both in monotherapy and in combination therapy with drugs like gemcitabine.11,12 However, although cisplatin consistently shows a good initial response, further treatment commonly results in the development of cancer cell chemoresistance, which may limit the treatment efficacy because of the systemic toxicity of high-dose cisplatin.13 Thus, it would be of great clinical value to find effective approaches to increase the sensitivity to cisplatin in the treatment of breast cancer.
A recent phase II clinical trial demonstrated that the WEE1 inhibitor adavosertib combined with cisplatin-improved clinical outcomes for patients with metastatic triple-negative breast cancer (mTNBC).14 WEE1, a tyrosine kinase that arrests the cell cycle at the G2 checkpoint.14 Kruppel-like factor 2 (KLF2), which is a typical member of the KLF transcription factor subfamily, plays an important role in many cell differentiation and maturation processes, including vasculogenesis and embryonic erythropoiesis.15–17 KLF2 was found to negatively regulate the expression of the WEE1.18,19 KLF2 was also reported to be a tumor suppressor gene, as well as a potential therapeutic target for modulating cancer cell growth, proliferation, and apoptosis.20,21 KLF2 expression is significantly downregulated in many cancers, such as liver cancer, breast cancer, and non-small cell lung cancer,21–23 the negating effect of recovery of KLF2 expression on the efficacy of anticancer drugs has not been examined. Here, we report that restoration of KLF2 expression enhanced the antitumor effect of cisplatin in breast cancer cells by reducing their proliferation, invasion, and migration capacity.
Materials and methods
Ethics statement
The study was approved by the Ethics Committee and Animal Ethics Committee of The Second Affiliated Hospital of Soochow University and conducted in strict accordance with the Declaration of Helsinki. Informed consent documentation was obtained from each participant prior to sample collection. Animal experiments were approved by the Animal Care and Use Committee of our hospital and complied with the National Institutes of Health guidelines. Extensive efforts were made to ensure minimal suffering of the included animals.
Sample collection
A total of 53 female patients with breast cancer at the Department of Thyroid and Breast Surgery of The Second Affiliated Hospital of Soochow University from January 2010 to January 2014 were enrolled in the current study. The baseline characteristics of these enrolled patients are shown in Supplementary Table S1. All patients were pathologically diagnosed with primary breast cancer, and surgically obtained cancer tissues and adjacent normal tissues were immediately frozen in liquid nitrogen and stored at −80°C for later use.
Cell culture and treatment
Human normal breast cell line MCF10A (AE-H620, American Type Culture Collection, Manassas, VA, USA) and human triple-negative breast cancer (TNBC) cell line MDA-MB-231 (C0001 CCC000013, Cell Resource Center of the Institute of Basic Medicine of the Chinese Academy of Medical Sciences, Shanghai, China) were utilized in this study. The MCF10A cell line was cultured in Dulbecco’s modified Eagle’s medium (DMEM, 12800017, Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS). MDA-MB-231 cell line was cultured using Roswell Park Memorial Institute (RPMI) 1640 medium (12633012, Haoran Biotech., Shanghai, China) containing 10% FBS in a 5% CO2 humidified incubator at 37°C. The cells were seeded into 96-well plates at a density of 5 × 103 cells/well.
The MDA-MB-231 cells were cultured in a medium containing 1 μg/L of cisplatin. MDA-MB-231 cells without any transfection served as control material, while cisplatin-treated MDA-MB-231 cells were transfected with sequences of overexpression-negative control (oe-NC), oe-KLF2, short hairpin RNA (sh)-NC, sh-WEE1, oe-KLF2 + oe-NC and oe-KLF2 + oe-WEE1.
The WEE1 gene was searched using the GenBank database and shRNA sequences were then designed. Two shRNAs (Table 1) were obtained by BLAST screening with the exclusion of nonspecific binding and were then constructed into pshRNA-neo plasmids. Enzyme digestion and sequencing confirmed the successful construction of the WEE1 gene silencing plasmids, which were designated as shWEE1-1 and shWEE1-2. WEE1-shRNA vectors and the NC vectors were transfected into MDA-MB-231 cells, and WEE1 expression was detected by Western blot to screen the most effective of the two shRNAs.
Table 1.
Sequences for shRNAs and the negative control
| Gene | Sequences (5ʹ-3ʹ) |
|---|---|
| sh-WEE1-1 | 5ʹ-CATCGATATGCACCTTGAGTCTCAA-3’ |
| sh-WEE1-2 | 5ʹ-CAGCCCATGTCCTTTGACACCATAA-3’ |
| sh-negative control | 5ʹ-TTCTCCGAACGTGTCACGTTT-3’ |
Immunohistochemistry
Breast cancer tissues were fixed in 4% paraformaldehyde for 12 h, paraffin embedded, dewaxed with xylene, and rehydrated using gradient alcohol (100, 95, and 75%). The tissues were then immersed in 0.01 M citrate buffer solution for 15–20 min, and after cooling to room temperature were washed with phosphate-buffered saline (PBS). Next, the tissues were blocked with goat serum and allowed to stand for 20 min at room temperature, followed by the removal of excess liquid. The tissues were then immunostained with primary antibodies against KLF2 (30 μL, ab244507, 1:50, Abcam Inc., Cambridge, UK) and WEE1 (rabbit, ab137337, 1:100, Abcam) at room temperature for 1 h. Following PBS washing, 30 μL of secondary goat anti-rabbit IgG (ab6721, 1:2000, Abcam) was added to the tissues and reacted for 1 h at room temperature, followed by addition of streptavidin-peroxidase (SP) for 30 min of treatment at 37°C. Next, the tissues were developed with 3,3ʹ-diaminobenzidine tetrahydrochloride (DAB) for 5–10 min, counterstained with hematoxylin for 2 min, differentiated by hydrochloric acid alcohol, and washed with tap water for 10 min. The tissues were dehydrated, cleared, sealed, and observed under a microscope. The percentage of positive cells was rated as 0 (0%), 1 (<10%), 2 (<50%), 3 (<75%), and 4 (≥75%). The staining intensity of cells was graded as follows: 0 for no staining, 1 of light yellow, 2 for moderate, yellowish brown, and 3 for dark brown staining. The staining index was the calculated using the formula: the proportion of positive cells × staining intensity, which yielded grades of 0–9 and 12.
Chromatin Immunoprecipitation (ChIP) assay
ChIP was performed as previously described.24 In brief, cells were fixed in paraformaldehyde for 10 min, and sheared into fragments using an ultrasound disruptor. The supernatant was collected by centrifugation (4°C, 12000 × g, 10 min) and divided into two tubes. To one tube was added normal nude mouse antibody to IgG (ab172730, 1:1000, Abcam) as the NC antibody. To the other, anti-KLF2 (ab17008, 1:32000; ab32381, 1:1000, Abcam) was applied. After mixing, the specimens were incubated overnight at 4°C. The DNA-protein complex was then precipitated by Protein A-Agarose/Salmon Sperm DNA, and the pellet was then centrifuged at 12000 × g for 5 min. The supernatant was discarded and the nonspecific complex was washed, followed by incubation at 65°C overnight to undo the crosslinking, whereupon the DNA fragments were purified and recovered by phenol/chloroform extraction.
Reverse Transcription quantitative Polymerase Chain Reaction (RT-qPCR)
Total mRNA samples were loaded using the SYBR Premix EX Taq kit (RR420A, Takara, Japan) and subjected to analysis on real-time fluorescence quantitative PCR (ABI 7500, Applied Biosystems, Foster City, CA, USA). The reaction system consisted of SYBR Mix 9 μL, positive primer 0.5 μL and negative primer 0.5 μL, cDNA 2 μL, and RNase-free ddH2O 8 μL. Each sample was set with three replicates. The primers were synthesized by Shanghai Sangon Biotechnology Co., Ltd. (Shanghai, China) and the primer sequences are listed in Supplementary Table S2. The relative expression of the mRNA was calculated by the 2−ΔΔCt method.
Western blot analysis
Total protein was extracted from cells in each group and the concentration was then determined using a bicinchoninic acid (BCA) kit (Thermo Fisher, Waltham, MA, USA). Next, 30 μg samples of protein were separated using polyacrylamide gel electrophoresis (PAGE) and transferred onto the polyvinylidene fluoride (PVDF) membrane (Amersham, GE Healthcare, Chicago, IL, USA). The membrane was probed overnight at 4°C with rabbit antibodies against KLF2 (1:1000, ab17008, Abcam), WEE1 (1:1000, ab137337, Abcam), matrix metalloproteinase (MMP)-2 (1:1000, ab37150, Abcam), MMP-9 (1:1000, ab73734, Abcam), Bcl-2-associated X protein (Bax, 1:1000, ab32503, Abcam), B-cell lymphoma 2 (Bcl-2, 1:2000, ab182858, Abcam), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:10000, ab181602, Abcam). The next day, horseradish peroxidase-labeled goat anti-rabbit IgG (1:10000, ab6721, Abcam) was added to the membrane, which was then incubated at room temperature for 1 h. After being developed with enhanced chemiluminescence reagents and imaged using the optical luminescence instrument (GE Healthcare, Chicago, IL, USA). the relative expression of protein was analyzed by Image-Pro Plus 6.0 (Medical Cybernetics, Los Angeles, CA, USA). All experiments were conducted independently in triplicate.
Dual-luciferase reporter assay
The binding site of KLF2 and WEE1 promoter was predicted using the JASPAR website (http://jaspar.genereg.net/). The recombinant luciferase reporter gene vector with truncated or mutated binding site was co-transfected with the KLF2 expression vector into MDA-MB-231 cells. After 48 h of transfection, the cells were collected and lysed. Then luciferase activity was measured with the dual-luciferase assay system (Promega, Madison, WI, USA) using the luciferase assay kit (K801-200, Biovision, San Francisco, CA, USA). The relative luciferase (RLU) activity was calculated as the RLU activity of firefly luciferase/RLU activity of renilla luciferase.
Cell viability assay using cell counting kit-8 (CCK-8) assay
Cells were seeded into a 96-well plate at a density of 2 × 103 cells/well. The blank control group was set with no cells in the medium for zeroing of the assay. After transfection for 24 h, the cells were sampled at 0, 24, 48, 72, and 96 h, respectively, whereupon 10 μL of CCK-8 reagent was added to each well and incubated for 14 h at 37°C. The absorbance at 450 nm was measured using a microplate reader (Bio-Rad, Hercules, CA, USA).
Cell apoptosis assay using flow cytometry
Apoptosis was analyzed by Annexin V-fluorescein isothiocyanate/propidium iodide (FITC/PI) (556547, Shujia Biotech., Shanghai, China). The cell samples were digested with for 24 h 0.25% trypsin after transfection, and were then fixed by addition of 70% pre-cooled ethanol at 4°C. Next, the cells were incubated with FITC and PI in a 4°C freezer for 15–30 min, and read by flow cytometry with excitation at 480 nm, and absorbance at 530 nm for FITC and 575 nm for PI (Beckman Coulter, Brea, CA, USA). Each experiment was repeated three times independently.
Migration and invasion assay
The transfected cells were diluted with 100 μL serum-free medium and inoculated into the upper chamber of a Transwell chamber at a concentration of 1 × 106 cells/mL. DMEM medium containing 20% FBS (500 μL) was added to the lower chamber to the Transwell. Each sample was analyzed in triplicate. After culturing for 24 h at 37°C, the cells were fixed with 5% glutaraldehyde and stained with 0.1% crystal violet for 5 min. Finally, five random fields were chosen for imaging (400 ×) by an inverted fluorescence microscope (TE2000, Nikon, Tokyo, Japan) and the average number of cells passing through the chamber was counted. The experiment was repeated three times independently.
After 48 h of transfection, the Matrigel (Ybio, Shanghai, China) gel kept at −80°C was conditioned and melted 4°C overnight. To 200 μL serum-free medium, 200 μL liquid Matrigel was added and mixed well and to give a diluted matrix. For the cell invasion experiment, 50 μL diluted matrix gel mixture was added in the upper chamber of the Transwell plate, incubated for 2–3 h for solidification while the cells were digested, and then counted and resuspended in serum-free medium. The suspension (200 μL) was added to the upper chamber, while 600 μL medium containing 20% FBS was placed in the lower chamber. After the cells were cultured at 37°C for 20–24 h, they were fixed with formaldehyde for 10 min and stained with 0.1% crystal violet. Cells on the surface were wiped off with a cotton ball. After drying, cells in five random visual fields were counted using an inverted microscope (Leica, Wetzlar, Germany), and the average value was recorded. The experiment was repeated three times independently.
Tumor formation experiment in nude mice
Female 5–7-week-old BALB/c nude mice purchased from the Experimental Animal Center of the Chinese Academy of Sciences (Shanghai, China) were fed with pelleted diet, and given ad libitum access to water in a room with a 12-h light/dark cycle. Mice were randomly selected and then inoculated with MDA-MB-231 cells transfected with oe-KLF2 and oe-WEE1 at a density of 1 × 106 cells/mouse. The mice were grouped as cisplatin + oe-NC group, cisplatin + oe-KLF2 + oe-NC group, and cisplatin + oe-KLF2 + oe-WEE1 group (with six mice per group). From day 5 after modeling, cisplatin was intraperitoneally injected to the mice for 21 days at a dose of 3 mg/kg per day delivered in 0.15 mL. Twenty-six days after modeling, the mice were euthanized and the tumor was removed. Tumor volume was then measured using a Vernier caliper and calculated using the formula: volume = W2 × L × 0.52, where W represents the length and L represents the width. At the same time, the lung tissues of mice were collected to observe the lung metastasis and the number of tumor cells was counted under a wide-field microscope. The body weight and overall health status of the nude mice were recorded every three days, and findings are listed in Table 2.
Table 2.
The body weight and overall health status of nude mice during treatment
| Group | Hair | Activity | Intake | Faeces | Urine | Death | Body weight (g, Mean ± SD) | |
|---|---|---|---|---|---|---|---|---|
| Cisplatin + oe-NC | Day 0 ~ 6 | Bright and clean | Normal | Normal | Normal | Normal | NO | 20.53 ± 1.87 |
| Day 9 | Bright and clean | Normal | Normal | Normal | Normal | NO | 20.96 ± 2.13 | |
| Day 12 | Bright and clean | Normal | Normal | Normal | Normal | NO | 20.21 ± 1.71 | |
| Day 15 | Dull and messy | Reduce | Reduce | Reduce | Reduce | NO | 19.23 ± 2.25 | |
| Day 18 | Dull and messy | Reduce | Reduce | Reduce | Reduce | NO | 17.53 ± 2.34 | |
| Day 21 | Dull and messy | Reduce | Reduce | Reduce | Reduce | NO | 16.05 ± 1.96 | |
| Day 24 | messy and Dry | Reduce | Reduce | Reduce | Reduce | NO | 14.28 ± 2.73 | |
| Cisplatin + oe-KLF2 + oe-NC | Day 0 ~ 6 | Bright and clean | Normal | Normal | Normal | Normal | NO | 21.19 ± 2.36 |
| Day 9 | Bright and clean | Normal | Normal | Normal | Normal | NO | 20.87 ± 1.92 | |
| Day 12 | Bright and clean | Normal | Reduce | Reduce | Reduce | NO | 20.16 ± 2.54 | |
| Day 15 | Dull and messy | Reduce | Reduce | Reduce | Reduce | NO | 19.38 ± 1.78 | |
| Day 18 | Dull and messy | Reduce | Reduce | Reduce | Reduce | NO | 18.04 ± 2.21 | |
| Day 21 | Dull and messy | Reduce | Reduce | Reduce | Reduce | NO | 16.72 ± 2.45 | |
| Day 24 | Messy and Dry | Reduce | Reduce | Reduce | Reduce | NO | 15.14 ± 2.16 | |
| Cisplatin + oe-KLF2 + oe-WEE1 | Day 0 ~ 6 | Bright and clean | Normal | Normal | Normal | Normal | NO | 20.82 ± 1.65 |
| Day 9 | Bright and clean | Normal | Normal | Normal | Normal | NO | 20.95 ± 2.08 | |
| Day 12 | Bright and clean | Normal | Normal | Normal | Normal | NO | 20.63 ± 2.29 | |
| Day 15 | Bright and clean | Normal | Normal | Normal | Normal | NO | 19.92 ± 2.41 | |
| Day 18 | Dull and messy | Reduce | Reduce | Reduce | Reduce | NO | 19.18 ± 2.32 | |
| Day 21 | Dull and messy | Reduce | Reduce | Reduce | Reduce | NO | 18.59 ± 1.94 | |
| Day 24 | Dull and messy | Reduce | Reduce | Reduce | Reduce | NO | 17.84 ± 2.28 |
Hematoxylin and Eosin (H&E) staining
The tumor tissues of nude mice were fixed, paraffin-embedded, and sliced into 4-µm-thick sections. The sections were then dewaxed in xylene and rehydrated with gradient alcohol (100, 95, 80, and 75%), and washed with distilled water. Next, the sections were stained with hematoxylin for 5 min, and differentiated in hydrochloric acid ethanol for 30 s. After soaking in tap water for 15 min, sections were stained in eosin solution for 2 min, and then dehydrated, cleared, and sealed with neutral resin. Finally, the sections were observed under an inverted microscope (XSP-8CA, Shanghai Optical Instrument Factory, Shanghai, China).
Statistical analysis
Statistical analysis was performed using the SPSS versio 21.0 statistical software (IBM Corp. Armonk, NY, USA). Measurement data were expressed as mean ± standard deviation. Independent sample t-test was used for data comparison between two groups and one-way analysis of variance (ANOVA) was used for multi-group data comparison. p < .05 was considered to be statistically significantly different.
Results
Cisplatin inhibits breast cancer cell proliferation, invasion, and migration
It has been reported that cisplatin is a chemotherapeutic agent that effectively inhibits the proliferation and migration of various kinds of cancer cells;25 although the effect of cisplatin on breast cancer cells is not fully elucidated. To determine the effect of cisplatin on the TNBC cell line MDA-MB-231, we initially performed a CCK-8 assay, which showed a gradual downward progression in the viability of MDA-MB-231 cells with duration of cisplatin treatment (Figure 1a). Additionally, flow cytometric data revealed an enhancement of cell apoptosis following cisplatin treatment (Figure 1b). The results from the Transwell assay showed that cisplatin treatment decreased the migration and invasion ability of MDA-MB-231 cells (Figure 1c). Meanwhile, Western blot displayed that the protein expression of MMP-2, MMP-9 and Bcl-2 was lower in the cisplatin-treated MDA-MB-231 cells than in the control cells, while the protein expression of Bax was increased (Figure 1d). These results demonstrated that cisplatin could inhibit breast cancer cell proliferation, invasion, and migration.
Figure 1.
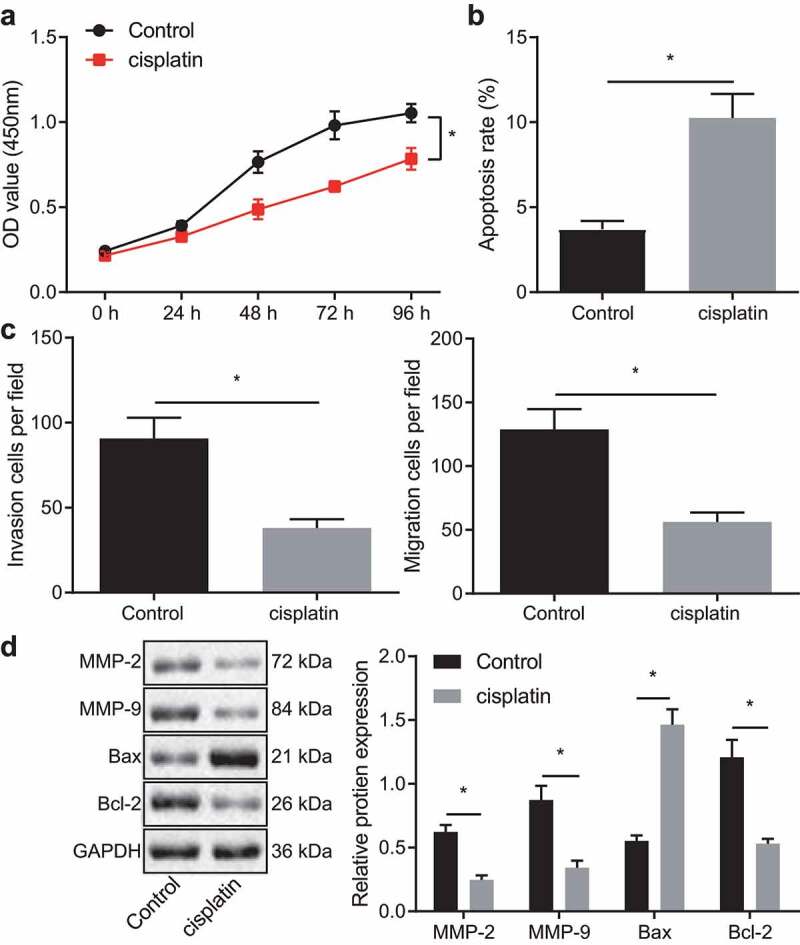
Cisplatin inhibits breast cancer cell proliferation, invasion, and migration. A, Viability of cisplatin-treated MDA-MB-231 cells measured using the CCK-8 assay. B, Apoptosis of cisplatin-treated MDA-MB-231 cells detected by flow cytometry. C, Migration and invasion of MDA-MB-231 cells exposed to cisplatin measured using Transwell assay. D, Expression of invasion and migration-related proteins MMP-2, MMP-9 and proliferation and apoptosis-related proteins Bax, Bcl-2 in cisplatin-treated MDA-MB-231 cells detected by Western blot. * p < .05 indicates the comparison between two groups. The cell experiment was repeated three times
Overexpression of KLF2 enhances the sensitivity of breast cancer cells to cisplatin
Existing literature reports that there is poor expression of KLF2 in breast cancer.22 The Immunohistochemistry employed in the current study also revealed a decline of KLF2 expression in clinical tissue samples collected from 53 breast cancer patients, relative to their adjacent healthy tissue (Figure 2a). RT-qPCR and Western blot further analyzed that KLF2 expression was significantly suppressed in the MDA-MB-231 cells compared with normal breast epithelial cell line MCF10A (Figure 2b, c). In addition, both KLF2 mRNA and protein expressions were upregulated in the cisplatin-treated MDA-MB-231 cells and even more remarkably so in the MDA-MB-231 cells overexpressing KLF2 (Figure 2d, e). Moreover, treatment with cisplatin was shown to reduce cell viability (figure 2f), migration, and invasion (Figure 2h) while the apoptosis rate (Figure 2g) was significantly increased. These trends were more pronounced upon dual treatment with cisplatin and oe-KLF2. As shown in Figure 2i, protein expression of MMP-2, MMP-9, and Bcl-2 were repressed, while that of Bax was increased in MDA-MB-231 cells treated with cisplatin, and that additional oe-KLF2 transfection caused a more significant effect. Thus, we find that KLF2 overexpression sensitized breast cancer cells to cisplatin.
Figure 2.
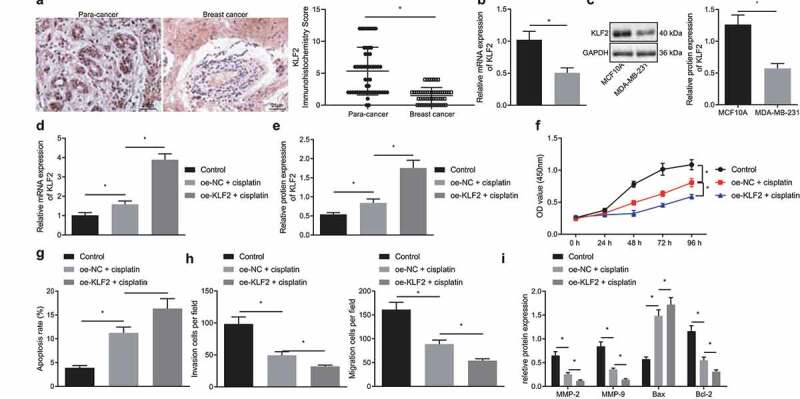
Overexpression of KLF2 sensitizes breast cancer cells to cisplatin. A, Immunohistochemistry of KLF2 protein in cancer tissues and adjacent normal tissues collected from 53 breast cancer patients. B, The expression of KLF2 in MDA-MB-231 and MCF10A cells determined by RT-qPCR. C, The protein expression of KLF2 in MDA-MB-231 and MCF10A cells determined by Western blot. D, RT-qPCR detection of KLF2 mRNA expression in MDA-MB-231 cells treated with cisplatin or in combination with oe-KLF2. E, The protein expression of KLF2 detected by Western blot in MDA-MB-231 cells treated with cisplatin or in combination with oe-KLF2. F, Viability of MDA-MB-231 cells treated with cisplatin or in combination with oe-KLF2. G, Apoptosis rate of MDA-MB-231 cells treated with cisplatin or in combination with oe-KLF2 detected by flow cytometry. H, Migration and invasion of MDA-MB-231 cells treated with cisplatin or in combination with oe-KLF2 detected by Transwell assay. I, The protein expression of MMP-2, MMP-9, Bax, and Bcl-2 detected by Western blot in MDA-MB-231 cells treated with cisplatin or in combination with oe-KLF2. * p < .05 indicates the comparison between two groups. All experiments were conducted independently in triplicate
WEE1 is a target gene of KLF2
We had previously confirmed that KLF2 could sensitize breast cancer cells to cisplatin, and KLF2 has meanwhile been reported to negatively regulate WEE1 expression at the transcriptional level.18 However, the regulatory relationship of KLF2 and WEE1 in breast cancer cells has been unclear. Experimental results from immunohistochemistry documented that WEE1 expression was robustly induced in cancer tissues compared to adjacent normal tissues (Figure 3a). Western blot disclosed that WEE1 was highly expressed in MDA-MB-231 cells, while it was decreased in cisplatin-treated MDA-MB-231 cells (Figure 3b). As Figure 3c shows, overexpression of KLF2 suppressed WEE1 expression in MDA-MB-231 cells. In addition, ChIP assay confirmed that the WEE1 enrichment in the anti-KLF2 group was boosted compared to the that in the IgG group (Figure 3d). JASPAR (http://jaspar.genereg.net/) predicted the presence of KLF2 binding sites in the WEE1 promoter region (Figure 3e). Three binding sites were selected to verify the binding of the WEE1 positive strand. Dual-luciferase reporter assay (figure 3f, g) further showed that luciferase activity was related to binding site 2 and independent of the other two WEE1 promoter-binding sites. Therefore, binding site 2 was determined to be the WEE1 promoter and KLF2 transcription factor. These results confirmed that KLF2 could target WEE1 and negatively regulate its expression in MDA-MB-231 cells treated with cisplatin.
Figure 3.
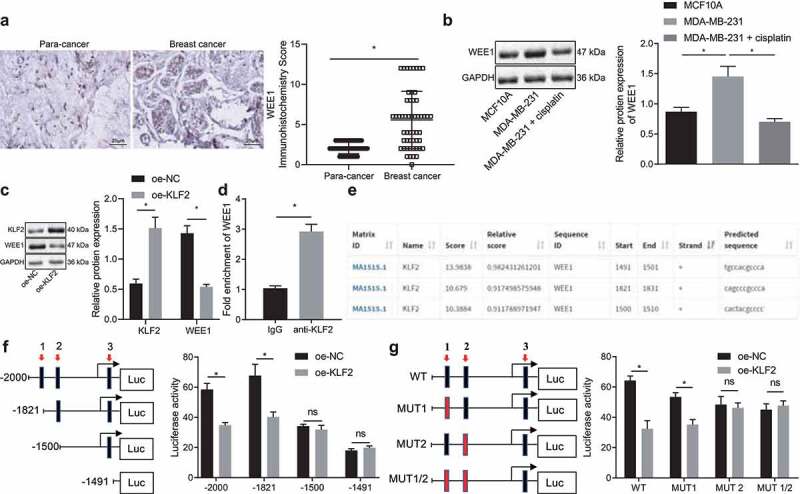
KLF2 negatively regulates WEE1 expression in cisplatin-treated MDA-MB-231 cells. A, Immunohistochemistry of WEE1 protein in cancer tissues and adjacent normal tissues collected from 53 breast cancer patients. B, Protein expression of WEE1 detected by Western blot in human normal breast cells MCF10A (control) and MDA-MB-231 breast cancer cells. C, The protein expression of KLF2 and WEE1 in KLF2-overexpressed MDA-MB-231 cells detected by Western blot. D, KLF2 binding to WEE1 promoter evaluated by ChIP assay. E, KLF2 protein binding site to the WEE1 promoter predicted by the JASPAR website. F, The luciferase activity of MDA-MB-231 cells by dual luciferase reporter assay; G, Effect of the mutation o of different WEE1 promoter binding sites on the luciferase activity in MDA-MB-231 cells detected by dual luciferase reporter assay. * p < .05 indicates the comparison between two groups. Cell experiment was repeated three times independently
WEE1 inhibition enhances the sensitivity of breast cancer cells to cisplatin
We then moved on to investigate the role of WEE1 in the biological functions of breast cancer cells. The silencing effect of two WEE1 shRNAs (sh-WEE1-1 and sh-WEE1-2) in the MDA-MB-231 cells was detected by Western blot, with the results showing that sh-WEE1-2 was of superior effect in repressing the target than sh-WEE1-1 (Figure 4a), and was therefore selected for subsequent experiments. The expression WEE1 detected by RT-qPCR and Western blot displayed that WEE1 was down-regulated in cells treated with cisplatin, and was further down-regulated in the presence of WEE1 silencing (Figure 4b, c). Moreover, the results of CCK-8 assay (Figure 4d) and flow cytometry (Figure 4e) showed that, after cisplatin treatment, the viability of MDA-MB-231 cells was decreased, while the apoptosis rate was increased. A more prominent effect was noted in response to sh-WEE1. Transwell assay (figure 4f) showed that the migration and invasion ability of MDA-MB-231 cells was reduced upon cisplatin treatment, with a more pronounced decline following WEE1 silencing. Meanwhile, MDA-MB-231 cells treated with cisplatin exhibited repressed MMP-2, MMP-9, Bcl-2 expression and elevated Bax expression, and furthermore, WEE1 silencing led to a more pronounced effect (Figure 4g). These results indicated that inhibition of WEE1 had the potential to enhance the inhibitory effect of cisplatin on the proliferation, invasion, and migration of breast cancer cells.
Figure 4.
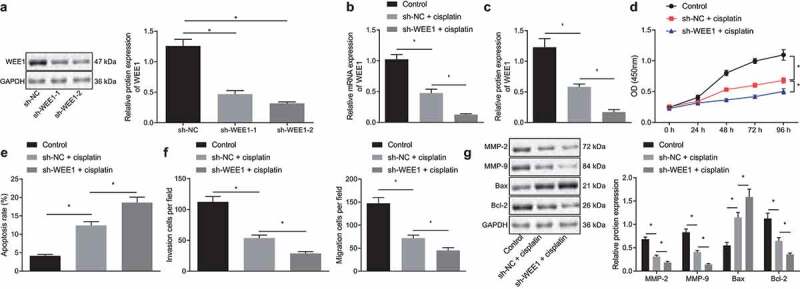
Inhibition of WEE1 advances the inhibitory effect of cisplatin on proliferation, invasion, and migration of breast cancer cell lines. A, Silencing efficiency of sh-WEE1-1 and sh-WEE1-2 in MDA-MB-231 cells detected by Western blot. B, mRNA expression of WEE1 detected by RT-qPCR in MDA-MB-231 cells treated with cisplatin or in combination with sh-WEE1. C, Protein expression of WEE1 detected by Western blot in MDA-MB-231 cells treated with cisplatin or in combination with sh-WEE1. D, Viability of MDA-MB-231 cells treated with cisplatin or in combination with sh-WEE1 detected by CCK-8. E, Apoptosis rate of MDA-MB-231 cells treated with cisplatin or in combination with sh-WEE1 detected by flow cytometry. F, Migration and invasion of MDA-MB-231 cells treated with cisplatin or in combination with sh-WEE1 measured by Transwell assay. G, The protein expression of MMP-2, MMP-9, Bax, and Bcl-2 in MDA-MB-231 cells treated with cisplatin or in combination with sh-WEE1 detected by Western blot. * p < .05, indicates the comparison between two groups. The cell experiment was repeated three times
KLF2 enhances the sensitivity of breast cancer cells to cisplatin by inhibiting WEE1
Based on the preceding results, we then attempted to explore whether KLF2 sensitized breast cancer cells to cisplatin by inhibiting WEE1. Concomitant treatment with oe-KLF2 and cisplatin markedly declined the expression of WEE1 in MDA-MB-231 cells as compared to cisplatin treatment alone, as revealed by RT-qPCR and Western blot. In addition, when WEE1 and KLF2 were overexpressed simultaneously in cisplatin-treated MDA-MB-231 cells, KLF2 expression was not affected, while WEE1 expression showed an increase (Figure 5a, b). The cell viability, migration, and invasion were found to be decreased upon overexpression of KLF2 in MDA-MB-231 cells treated with cisplatin, while concomitant overexpression of KLF2 and WEE1 brought about the opposite effects (Figure 5c, e). The flow cytometric analysis unveiled that KLF2 overexpression increased MDA-MB-231 cell apoptosis, which was repressed following KLF2 and WEE1 co-overexpression (Figure 5d). Furthermore, Western blot detected down-regulated expression of MMP-2, MMP-9, and Bcl-2, and up-regulated Bax expression in the KLF2-overexpressed MDA-MB-231 cells; however, opposite effects were seen following KLF2 and WEE1 co-overexpression (figure 5f). The above data indicated that KLF2 sensitized breast cancer cells to cisplatin by inhibiting WEE1.
Figure 5.
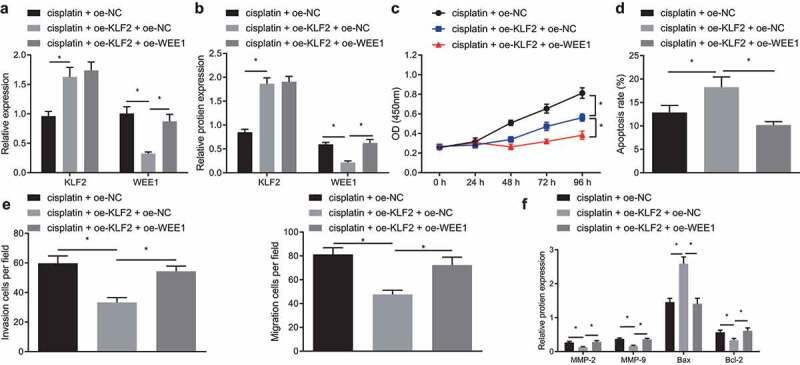
KLF2 increased the sensitivity of breast cancer cells to cisplatin by inhibiting WEE1. A, mRNA expression of KLF2 and WEE1 detected by RT-qPCR in cisplatin-treated MDA-MB-231 cells overexpressing KLF2 or combined with WEE1. B, Protein expression of KLF2 and WEE1 detected by Western blot in cisplatin-treated MDA-MB-231 cells overexpressing KLF2 or combined with WEE1. C, Viability of cisplatin-treated MDA-MB-231 cells overexpressing KLF2 or combined with WEE1 measured by CCK-8 assay. D, Apoptosis of cisplatin-treated MDA-MB-231 cells overexpressing KLF2 or combined with WEE1 detected by flow cytometry. E, Migration and invasion of cisplatin-treated MDA-MB-231 cells overexpressing KLF2 or combined with WEE1 measured by Transwell assay. F, The protein expression of MMP-2, MMP-9, Bax, and Bcl-2 detected by Western blot in cisplatin-treated MDA-MB-231 cells overexpressing KLF2 or combined with WEE1. * p < .05, indicates the comparison between two groups. The cell experiment was repeated three times
KLF2 facilitates the sensitivity of tumor-bearing mice to cisplatin by suppression WEE1 in vivo
Finally, we aimed to characterize the effect of KLF2 on the sensitivity of tumor-bearing mice by downregulating WEE1 in a mouse xenograft model. The expression of WEE1 examined by RT-qPCR and Western blot exhibited a reduction, while that of KLF2 presented with an increase in tumor tissues of cisplatin-treated mice overexpressing KLF2 alone. However, simultaneous overexpression of WEE1 and KLF2 did not affect KLF2 expression, but decreased WEE1 expression (Figure 6a, b). Besides, tumor weight was decreased when KLF2 was overexpressed in the xenografted nude mice. When KLF2 and WEE1 were co-overexpressed, the body weight was increased (Figure 6c), indicating better health. Analysis of lung tissues using H&E staining suggested that staining in the stroma was increased, and decreased in the parenchyma and lymph nodes in cisplatin-treated mice with KLF2 overexpression. This result was opposite to that following co-overexpression of KLF2 and WEE2 (Figure 6d). Furthermore, Western blot data (Figure 6e) indicated that expression of MMP-2, MMP-9, Bcl-2 was inhibited and Bax was up-regulated in tumor tissues of cisplatin-treated mice with KLF2 overexpression, while further overexpression of WEE1 reversed that result. These results confirmed that KLF2 sensitized tumor-bearing mice to cisplatin by downregulating WEE1 expression in vivo.
Figure 6.
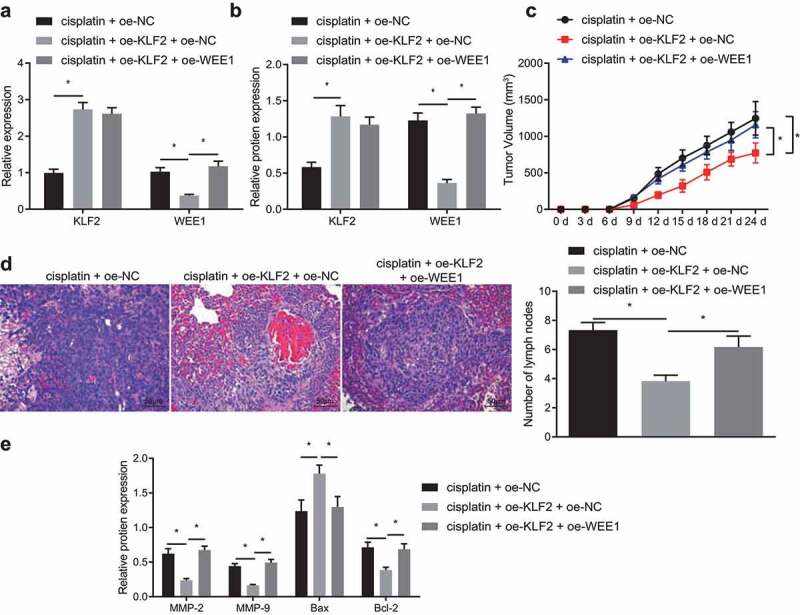
KLF2 increased the sensitivity of tumor-bearing mice to cisplatin by inhibiting WEE1 in vivo. Mice were injected with cisplatin-treated MDA-MB-231 cells that had been transfected with oe-KLF2 or in combination with oe-WEE1. A, The mRNA expression of KLF2 and WEE1 detected in tumor tissues of nude mice by RT-qPCR. B, The protein expression of KLF2 and WEE1 in tumor tissues of nude mice by Western blot. C, Tumor weight in nude mice. D, H&E staining of lung tissues of nude mice. E, The protein expression of MMP-2, MMP-9, Bax, and Bcl-2 in tumor tissues of nude mice detected by Western blot. * p < .05, indicates the comparison between two groups. n = 6
Cisplatin induces the increase of KLF2 expression in breast cancer cells
Flow cytometry was performed to detect cell apoptosis. The results showed that cisplatin induction significantly increased the apoptosis rate of the TNBC cell line BT549 and non-TNBC cell line MCF7 (Supplementary Figure 1a). Relative to the MCF10A cells, the mRNA expression of KLF2 was decreased in the MCF7 and BT549 cells (Supplementary Figure 1b). In addition, cisplatin treatment was observed to increase the protein level of KLF2 and decreased the protein level of WEE1 in the MCF7 and BT549 cells but not in the MCF10A cells (Supplementary Figure 1c).
KLF2 inhibits the occurrence and development of breast cancer by downregulating WEE1
RT qPCR was used to detect the knockdown efficiency of KLF2 in the MDA-MB-231 cells. Both siRNAs of KLF2 (sh-KLF2-1 and sh-KLF2-2) successfully knocked down the level of KLF2 expression (Supplementary Figure. S2A). In the MDA-MB-231 and MCF7 cell lines, KLF2 knockdown inhibited the cisplatin-induced cell apoptosis, but had no effect on the apoptosis of the MCF10A cell line (Supplementary Figure. S2B). Moreover, in the MDA-MB-231 cells overexpressing KLF2, KLF2 silencing significantly decreased the protein level of KLF2 and increased the protein level of WEE1 (Supplementary Figure. S2C). Next, we further verified whether WEE1 knockdown affects the expression of KLF2. We found that that, in the MDA-MB-231 cells, WEE1 knockdown markedly decreased the protein level of WEE1, but did not affect the KLF2 protein level (Supplementary Figure. S2D). In the MDA-MB-231 cells, there was no significant change in the protein levels of HIF-1α and Hedgehog (Hh) after KLF2 overexpression (Supplementary Figure. S2E). This suggested that KLF2 inhibits the occurrence and development of breast cancer via the downregulation of WEE1.
KLF2 overexpression or WEE1 silencing inhibits tumor growth in nude mice
MDA-MB-231 cells were used to construct a nude mouse model with xenografted breast cancer. Western blot experiments revealed that oe-KLF2 or sh-WEE1 treatments both increased the expression of KLF2 and decreased the expression of WEE1 in the cisplatin-treated mice (Supplementary Figure. S3A). Meanwhile, the tumor volume of nude mice was recorded, revealing that the KLF2 overexpression treatment and WEE1 silencing treatment further inhibited the tumor volume of cisplatin-treated nude mice (Supplementary Figure. S3B).
Discussion
Cisplatin achieves its antitumor effects by incorporating into the rapid-replicating genomic DNA of cancer cells and inducing double-strand breaks, thus preventing further replication and transcription of the genome.26 Also, increased DNA repair capacity has been demonstrated to contribute to the development of cisplatin resistance.27 Therefore, enhanced DNA damage response and inhibition of cell apoptosis may be the main reason for the development of cisplatin resistance in tumors, leading to the idenitification of many therapeutic targets to potentially overcome the resistance effect.28,29 In this study, we identified the transcription factor KLF2 as a potential target to enhance the sensitivity of breast cancer to cisplatin by directly binding to the promoter of WEE1 and downregulating its expression in vitro and in vivo.
While regulating multiple processes in development and immunity, KLF2 was also reported to inhibit DNA synthesis and sensitize DNA damage-induced apoptosis in tumors.18,30 KLF2 was poorly expressed in breast cancer tissues, and abnormal epigenetic silencing of KLF2 promoted the proliferation capacity in multiple breast cancer cell lines.22 However, the therapeutic effect of KLF2 restoration alone or in combination with other drugs has rarely been discussed in the literature. Our results further demonstrated more than 50% downregulation of KLF2 both on the mRNA and protein levels in MDA-MB-231 cells compared with MCF10A cells. Interestingly, cisplatin treatment alone showed partial recovery of KLF2 expression in MDA-MB-231 cells, which may contribute to the total therapeutic effect of cisplatin. Overexpression of KLF2 in MDA-MB-231 cells with cisplatin treatment recovered the expression level of KLF2 similar to that in normal MCF10A cells and enhanced the antitumor effect of cisplatin, indicating that restoration of KLF2 expression may be a feasible way to overcome cisplatin resistance and thus serve as a potential adjunct molecular therapy for breast cancer.
KLF2 was reported to negatively regulate WEE1 expression in ovarian cancer cells.18 The major function of the WEE1 kinase is to regulate cell mitosis through the G2/M checkpoint,31 cell size checkpoint,32 and DNA damage checkpoint.33 WEE1 has been detected to be upregulated in breast cancer, while knockdown of WEE1 significantly inhibited the proliferation of breast cancer cells,34 while also aggravating DNA damage and inducing apoptosis.35 Since WEE1 is closely related to cell cycle and DNA damage responses, WEE1 has been long considered as a potential anti-tumor target in many cancers.36 For example, MK-1775, the small molecule inhibitor of WEE1, could be applied clinically to treat multiple tumors alone or in combination with platinum-based drugs like cisplatin.37–39 In addition, WEE1 inhibitor AZD1775 combined with cisplatin has been also reported to increase DNA damage and cell apoptosis, thus potentiating the anticancer activity against gastric cancer.40 It was also demonstrated that WEE1 inhibitor analog of AZD1775 could maintain synergy with cisplatin, which resulted in reduced single-agent cytotoxicity in medulloblastoma cells.41 We thus analyzed WEE1 expression in KLF2 overexpressed MDA-MB-231 cells cultured under cisplatin treatment and found that the protein level of WEE1 was also decreased. We further confirmed the direct binding of KLF2 to the promoter region of WEE1 in breast cancer cells, as indicated in HEK293 cells.18 Transcriptional repression of WEE1 by KLF2 is involved in DNA damage-induced cell apoptosis in ovarian cancer.18 Knockdown of WEE1 in MDA-MB-231 cells enhanced the inhibitory effect of cisplatin on cell proliferation, invasion, and migration, which was consistent both with a previous basic study and a clinical trial.34,37 Besides, WEE1 inhibition targeted cell cycle checkpoints for TNBCs to overcome cisplatin resistance.42 These results together demonstrate that KLF2 may enhance the sensitivity of breast cancer cells to cisplatin through inhibition of WEE1 expression.
Because KLF2 transcriptionally regulates a large number of genes and pathways,16,21,43,44 we attempted to study whether the therapeutic effect of KLF2 results from inhibition specifically of WEE1. We thus overexpressed both KLF2 and WEE1 in MDA-MB-231 cells with cisplatin treatment. Our results showed that overexpression of WEE1 nearly counteracted the positive effect of KLF2 treatment. Considering that the expression level of WEE1 in double overexpression MDA-MB-231 cells treated with cisplatin was similar to that in cells treated only with cisplatin, we thus demonstrated that the effect of KLF2 in breast cancer was sufficiently explicable through downregulation of WEE1, without involving regulation of HIF-1α/Notch-1 or Hh pathway, as reported in colorectal cancer or liver cancer.21,43 Restoration of KLF2 expression also exhibited a significant biological effect in the in vivo mouse breast cancer model. The tumor volume of KLF2 overexpressed MDA-MB-231 cells was significantly smaller and had less metastasis than the tumor generated by normal MDA-MB-231 cells under cisplatin treatment, and WEE1 overexpression consistently counteracted that positive effect. Successful validation of the efficacy of KLF2 overexpression with cisplatin treatment in an animal model indicated that restoration of KLF2 expression has potential as a molecular therapy against breast cancer in combination with cisplatin treatment.
Prior to embarking on clinical trials of this principle, some questions still remain to be answered. First, abnormal downregulation of KLF2 expression in tumors mainly results from epigenetically silence mediated by histone methylation.45,46 Since no small molecule to activate the expression or activity of KLF2 is currently available, precise and targeted recovery of KLF2 expression with limited side effect in patients might only be achieved by delivery of exogenous KLF2,47,48 or targeted epigenetic modification on KLF2 promoter by CRISPR/Cas based epigenome editing approaches.49,50 Both potential methods are difficult to achieve in the clinic since they require exogenous delivery of mRNAs or proteins. Therefore, further studies are required to find a clinically applicable way to activate KLF2 as a molecular therapy combined with cisplatin. Second, as cisplatin treatment is mainly applied against triple negative breast cancers,11,12,51 validation of the efficacy of KLF2 overexpression combined with cisplatin in cell lines like triple negative MDA-MB-231 would be a more appropriate and valuable approach to provide functional evidence in support of broad clinical application. We note that small molecule inhibition of WEE1 activity alone has been demonstrated as a safe and efficient adjunct to platinum-based treatment against ovarian and breast cancer in clinical trials.37,52 Thus, the necessity and benefits of targeting upstream KLF2 should be further evaluated. Targeting KLF2 could be applied in combination of WEE1 inhibition, or in patients resistant or insensitive supplementary to MK-1775 treatment.
In summary, we showed in a series of experiments that restored expression of abnormally downregulated KLF2 could effectively enhance the sensitivity of breast cancer cells to cisplatin. The effect of restored KLF2 expression was mainly reflected in an inhibition of the abnormal WEE1 expression obtained by direct binding of KLF2 to the promoter region of WEE1. The resultant upregulation promoted cell proliferation and resisted DNA damage-induced apoptosis in breast cancer cells. Our results in cultured MDA-MB-231 cells, together with evidence in the xenograft mouse model, demonstrated the potential of KLF2 to be a sensitizer and enhancer of cisplatin treatment of breast cancer. We shall further validate the efficacy of KLF2 in this context, and also develop appropriate methods for activation of KLF2 expression in animal breast cancer models in vivo.
Supplementary Material
Acknowledgments
We would like to give our sincere appreciation to the reviewers for their helpful comments on this article.
Notes on contributions
Ruiqing Li, Jiejing Chen, Xiaokang Gao and Guoqin Jiang designed the study. Ruiqing Li collated the data, designed and developed the database, carried out data analyses and produced the initial draft of the manuscript. Jiejing Chen, Xiaokang Gao, and Guoqin Jiang contributed to drafting the manuscript. All authors have read and approved the final submitted manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A.. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. PMID: 30207593. [DOI] [PubMed] [Google Scholar]
- 2.Fan L, Strasser-Weippl K, Li JJ, St Louis J, Finkelstein DM, Yu KD, Chen WQ, Shao ZM, Goss PE. Breast cancer in China. Lancet Oncol. 2014;15(7):e279–289. doi: 10.1016/S1470-2045(13)70567-9. PMID: 24872111. [DOI] [PubMed] [Google Scholar]
- 3.Harbeck N, Gnant M. Breast cancer. Lancet. 2017;389(10074):1134–1150. doi: 10.1016/S0140-6736(16)31891-8. PMID: 27865536. [DOI] [PubMed] [Google Scholar]
- 4.Isakoff SJ. Triple-negative breast cancer: role of specific chemotherapy agents. Cancer J. 2010;16(1):53–61. doi: 10.1097/PPO.0b013e3181d24ff7. PMID: 20164691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner NC, Neven P, Loibl S, Andre F. Advances in the treatment of advanced oestrogen-receptor-positive breast cancer. Lancet. 2017;389(10087):2403–2414. doi: 10.1016/S0140-6736(16)32419-9. PMID: 27939057. [DOI] [PubMed] [Google Scholar]
- 6.Loibl S, Gianni L. HER2-positive breast cancer. Lancet. 2017;389(10087):2415–2429. doi: 10.1016/S0140-6736(16)32417-5. PMID: 27939064. [DOI] [PubMed] [Google Scholar]
- 7.Cancer Genome AN. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. PMID: 23000897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fasching PA, Brucker SY, Fehm TN, Overkamp F, Janni W, Wallwiener M, Hadji P, Belleville E, Haberle L, Taran FA, et al. Biomarkers in patients with metastatic breast cancer and the PRAEGNANT study network. Geburtshilfe Frauenheilkd. 2015;75(1):41–50. 10.1055/s-0034-1396215. PMID: 25684786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zardavas D, Maetens M, Irrthum A, Goulioti T, Engelen K, Fumagalli D, Salgado R, Aftimos P, Saini KS, Sotiriou C, et al. The AURORA initiative for metastatic breast cancer. Br J Cancer. 2014;111(10):1881–1887. 10.1038/bjc.2014.341. PMID: 25225904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickson MA, Carvajal RD, Merrill AH Jr., Gonen M, Cane LM, Schwartz GK. A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clin Cancer Res. 2011;17(8):2484–2492. doi: 10.1158/1078-0432.CCR-10-2323. PMID: 21257722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu XC, Zhang J, Xu BH, Cai L, Ragaz J, Wang ZH, Wang BY, Teng YE, Tong ZS, Pan YY, et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015;16(4):436–446. 10.1016/S1470-2045(15)70064-1. PMID: 25795409. [DOI] [PubMed] [Google Scholar]
- 12.Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, Rugo HS, Liu MC, Stearns V, Come SE, et al. TBCRC009: a multicenter Phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer. J Clin Oncol. 2015;33(17):1902–1909. 10.1200/JCO.2014.57.6660. PMID: 25847936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun CY, Zhang QY, Zheng GJ, Feng B. Phytochemicals: current strategy to sensitize cancer cells to cisplatin. Biomed Pharmacother. 2019;110:518–527. PMID: 30530287. doi: 10.1016/j.biopha.2018.12.010. [DOI] [PubMed] [Google Scholar]
- 14.Keenan TE, Li T, Vallius T, Guerriero JL, Tayob N, Kochupurakkal B, Davis J, Pastorello R, Tahara RK, Anderson L, et al. Clinical efficacy and molecular response correlates of the WEE1 inhibitor adavosertib combined with cisplatin in patients with metastatic triple-negative breast cancer. Clin Cancer Res. 2021;27(4):983–991. 10.1158/1078-0432.CCR-20-3089. PMID: 33257427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song Y, Li X, Wang D, Fu C, Zhu Z, Zou MH, Zhu Y. Transcription factor Kruppel-like factor 2 plays a vital role in endothelial colony forming cells differentiation. Cardiovasc Res. 2013;99(3):514–524. doi: 10.1093/cvr/cvt113. PMID: 23667185. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Boon RA, Leyen TA, Fontijn RD, Fledderus JO, Baggen JM, Volger OL, van Nieuw Amerongen GP, Horrevoets AJ. KLF2-induced actin shear fibers control both alignment to flow and JNK signaling in vascular endothelium. Blood. 2010;115(12):2533–2542. doi: 10.1182/blood-2009-06-228726. PMID: 20032497. [DOI] [PubMed] [Google Scholar]
- 17.Basu P, Lung TK, Lemsaddek W, Sargent TG, Williams DC Jr., Basu M, Redmond LC, Lingrel JB, Haar JL, Lloyd JA. EKLF and KLF2 have compensatory roles in embryonic beta-globin gene expression and primitive erythropoiesis. Blood. 2007;110(9):3417–3425. doi: 10.1182/blood-2006-11-057307. PMID: 17675555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang F, Zhu Y, Huang Y, McAvoy S, Johnson WB, Cheung TH, Chung TK, Lo KW, Yim SF, Yu MM, et al. Transcriptional repression of WEE1 by Kruppel-like factor 2 is involved in DNA damage-induced apoptosis. Oncogene. 2005;24(24):3875–3885. 10.1038/sj.onc.1208546. PMID: 15735666. [DOI] [PubMed] [Google Scholar]
- 19.Xie P, Tang Y, Shen S, Wang Y, Xing G, Yin Y, He F, Zhang L. Smurf1 ubiquitin ligase targets Kruppel-like factor KLF2 for ubiquitination and degradation in human lung cancer H1299 cells. Biochem Biophys Res Commun. 2011;407(1):254–259. doi: 10.1016/j.bbrc.2011.03.016. PMID: 21382345. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Tu S, Zeng Y, Zhang C, Deng T, Luo W, Lian L, Chen L, Xiong X, Yan X. KLF2 inhibits TGF-beta-mediated cancer cell motility in hepatocellular carcinoma. Acta Biochim Biophys Sin (Shanghai). 2020;52(5):485–494. doi: 10.1093/abbs/gmaa024. PMID: 32318691. [DOI] [PubMed] [Google Scholar]
- 21.Lin J, Tan H, Nie Y, Wu D, Zheng W, Lin W, Zhu Z, Yang B, Chen X, Chen T. Kruppel-like factor 2 inhibits hepatocarcinogenesis through negative regulation of the Hedgehog pathway. Cancer Sci. 2019;110(4):1220–1231. doi: 10.1111/cas.13961. PMID: 30719823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu CC, Liang YW, Hu JL, Liu LF, Liang JW, Wang R. LncRNA RUSC1-AS1 promotes the proliferation of breast cancer cells by epigenetic silence of KLF2 and CDKN1A. Eur Rev Med Pharmacol Sci. 2019;23(15):6602–6611. doi: 10.26355/eurrev_201908_18548. PMID: 31378902. [DOI] [PubMed] [Google Scholar]
- 23.Xiao S, Jin-Xiang Y, Long T, Xiu-Rong L, Hong G, Jie-Cheng Y, Fei Z. Kruppel-like factor 2 disturb non-small cell lung cancer energy metabolism by inhibited glutamine consumption. J Pharm Pharmacol. 2020;72(6):843–851. doi: 10.1111/jphp.13252. PMID: 32196690. [DOI] [PubMed] [Google Scholar]
- 24.Lorentzen A, Mitchelmore C. NDRG2 gene copy number is not altered in colorectal carcinoma. World J Clin Oncol. 2017;8(1):67–74. doi: 10.5306/wjco.v8.i1.67. PMID: 28246586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JO, Kang MJ, Byun WS, Kim SA, Seo IH, Han JA, Moon JW, Kim JH, Kim SJ, Lee EJ, et al. Metformin overcomes resistance to cisplatin in triple-negative breast cancer (TNBC) cells by targeting RAD51. Breast Cancer Res. 2019;21(1):115. 10.1186/s13058-019-1204-2. PMID: 31640742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zdraveski ZZ, Mello JA, Marinus MG, Essigmann JM. Multiple pathways of recombination define cellular responses to cisplatin. Chem Biol. 2000;7(1):39–50. doi: 10.1016/s1074-5521(00)00064-8. PMID: 10662689. [DOI] [PubMed] [Google Scholar]
- 27.Crul M, Schellens JH, Beijnen JH, Maliepaard M. Cisplatin resistance and DNA repair. Cancer Treat Rev. 1997;23(5–6):341–366. doi: 10.1016/s0305-7372(97)90032-3. PMID: 9465884. [DOI] [PubMed] [Google Scholar]
- 28.Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. PMID: 24874729. doi: 10.1038/cddis.2013.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016;106:27–36. PMID: 26804248. doi: 10.1016/j.phrs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 30.Wu J, Lingrel JB. KLF2 inhibits Jurkat T leukemia cell growth via upregulation of cyclin-dependent kinase inhibitor p21WAF1/CIP1. Oncogene. 2004;23(49):8088–8096. doi: 10.1038/sj.onc.1207996. PMID: 15361832. [DOI] [PubMed] [Google Scholar]
- 31.Coleman TR, Dunphy WG. Cdc2 regulatory factors. Curr Opin Cell Biol. 1994;6(6):877–882. doi: 10.1016/0955-0674(94)90060-4. PMID: 7880537. [DOI] [PubMed] [Google Scholar]
- 32.Kellogg DR. Wee1-dependent mechanisms required for coordination of cell growth and cell division. J Cell Sci. 2003;116(24):4883–4890. doi: 10.1242/jcs.00908. PMID: 14625382. [DOI] [PubMed] [Google Scholar]
- 33.Walworth NC. DNA damage: chk1 and Cdc25, more than meets the eye. Curr Opin Genet Dev. 2001;11(1):78–82. doi: 10.1016/s0959-437x(00)00160-x. PMID: 11163155. [DOI] [PubMed] [Google Scholar]
- 34.Iorns E, Lord CJ, Grigoriadis A, McDonald S, Fenwick K, Mackay A, Mein CA, Natrajan R, Savage K, Tamber N, et al. Integrated functional, gene expression and genomic analysis for the identification of cancer targets. PLoS One. 2009;4(4):e5120. 10.1371/journal.pone.0005120. PMID: 19357772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murrow LM, Garimella SV, Jones TL, Caplen NJ, Lipkowitz S. Identification of WEE1 as a potential molecular target in cancer cells by RNAi screening of the human tyrosine kinome. Breast Cancer Res Treat. 2010;122(2):347–357. doi: 10.1007/s10549-009-0571-2. PMID: 19821025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stathis A, Oza A. Targeting Wee1-like protein kinase to treat cancer. Drug News Perspect. 2010;23(7):425–429. doi: 10.1358/dnp.2010.23.7.1490760. PMID: 20862394. [DOI] [PubMed] [Google Scholar]
- 37.Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, Lam R, Demuth T, Rose S, Lee MA, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34(36):4371–4380. 10.1200/JCO.2016.67.5991. PMID: 27601554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendez E, Rodriguez CP, Kao MC, Raju S, Diab A, Harbison RA, Konnick EQ, Mugundu GM, Santana-Davila R, Martins R, et al. A Phase I Clinical Trial of AZD1775 in Combination with Neoadjuvant Weekly Docetaxel and Cisplatin before Definitive Therapy in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2018;24(12):2740–2748. 10.1158/1078-0432.CCR-17-3796. PMID: 29535125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cole KA, Pal S, Kudgus RA, Ijaz H, Liu X, Minard CG, Pawel BR, Maris JM, Haas-Kogan DA, Voss SD, et al. Phase I clinical trial of the Wee1 inhibitor adavosertib (AZD1775) with irinotecan in children with relapsed solid tumors: a COG Phase I consortium report (ADVL1312). Clin Cancer Res. 2020;26(6):1213–1219. 10.1158/1078-0432.CCR-19-3470. PMID: 31857431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen D, Lin X, Gao J, Shen L, Li Z, Dong B, Zhang C, Zhang X. Wee1 Inhibitor AZD1775 Combined with Cisplatin Potentiates Anticancer Activity against Gastric Cancer by Increasing DNA Damage and Cell Apoptosis. Biomed Res Int. 2018;2018:5813292. PMID: 29977914. doi: 10.1155/2018/5813292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matheson CJ, Venkataraman S, Amani V, Harris PS, Backos DS, Donson AM, Wempe MF, Foreman NK, Vibhakar R, Reigan P. A WEE1 Inhibitor Analog of AZD1775 Maintains Synergy with Cisplatin and Demonstrates Reduced Single-Agent Cytotoxicity in Medulloblastoma Cells. ACS Chem Biol. 2016;11(4):921–930. doi: 10.1021/acschembio.5b00725. PMID: 26745241. [DOI] [PubMed] [Google Scholar]
- 42.Zheng H, Shao F, Martin S, Xu X, Deng CX. WEE1 inhibition targets cell cycle checkpoints for triple negative breast cancers to overcome cisplatin resistance. Sci Rep. 2017;7(1):43517. doi: 10.1038/srep43517. PMID: 28262781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang HG, Cao B, Zhang LX, Song N, Li H, Zhao WZ, Li YS, Ma SM, Yin DJ. KLF2 inhibits cell growth via regulating HIF-1alpha/Notch-1 signal pathway in human colorectal cancer HCT116 cells. Oncol Rep. 2017;38(1):584–590. doi: 10.3892/or.2017.5708. PMID: 28628187. [DOI] [PubMed] [Google Scholar]
- 44.Zhuang T, Liu J, Chen X, Zhang L, Pi J, Sun H, Li L, Bauer R, Wang H, Yu Z, et al. Endothelial foxp1 suppresses atherosclerosis via modulation of Nlrp3 inflammasome activation. Circ Res. 2019;125(6):590–605. 10.1161/CIRCRESAHA.118.314402. PMID: 31318658. [DOI] [PubMed] [Google Scholar]
- 45.Xu M, Chen X, Lin K, Zeng K, Liu X, Pan B, Xu X, Xu T, Hu X, Sun L, et al. The long noncoding RNA SNHG1 regulates colorectal cancer cell growth through interactions with EZH2 and miR-154-5p. Mol Cancer. 2018;17(1):141. 10.1186/s12943-018-0894-x. PMID: 30266084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li X, Li Z, Liu Z, Xiao J, Yu S, Song Y. Long non-coding RNA DLEU1 predicts poor prognosis of gastric cancer and contributes to cell proliferation by epigenetically suppressing KLF2. Cancer Gene Ther. 2018;25(3–4):58–67. doi: 10.1038/s41417-017-0007-9. PMID: 29282356. [DOI] [PubMed] [Google Scholar]
- 47.Kowalski PS, Rudra A, Miao L, Anderson DG. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther. 2019;27(4):710–728. doi: 10.1016/j.ymthe.2019.02.012. PMID: 30846391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sikder S, Gote V, Alshamrani M, Sicotte J, Pal D. Long-term delivery of protein and peptide therapeutics for cancer therapies. Expert Opin Drug Deliv. 2019;16(10):1113–1131. doi: 10.1080/17425247.2019.1662785. PMID: 31498012. [DOI] [PubMed] [Google Scholar]
- 49.Pulecio J, Verma N, Mejia-Ramirez E, Huangfu D, Raya A. CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell. 2017;21(4):431–447. doi: 10.1016/j.stem.2017.09.006. PMID: 28985525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tadic V, Josipovic G, Zoldos V, Vojta A. CRISPR/Cas9-based epigenome editing: an overview of dCas9-based tools with special emphasis on off-target activity. Methods. 2019;164-165:109–119. PMID: 31071448. doi: 10.1016/j.ymeth.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 51.Koshy N, Quispe D, Shi R, Mansour R, Burton GV. Cisplatin-gemcitabine therapy in metastatic breast cancer: improved outcome in triple negative breast cancer patients compared to non-triple negative patients. Breast. 2010;19(3):246–248. doi: 10.1016/j.breast.2010.02.003. PMID: 20227277. [DOI] [PubMed] [Google Scholar]
- 52.Leijen S, van Geel RM, Sonke GS, de Jong D, Rosenberg EH, Marchetti S, Pluim D, van Werkhoven E, Rose S, Lee MA, et al. Phase II study of WEE1 Inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol. 2016;34(36):4354–4361. 10.1200/JCO.2016.67.5942. PMID: 27998224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.