

ABSTRACT
Our recent study revealed that non-neuroendocrine small cell lung cancer (SCLC) is sensitive to the induction of ferroptosis due to upregulation of ether lipid synthesis. While neuroendocrine SCLC is ferroptosis resistant, it acquires addiction to the thioredoxin pathway. Combined redox pathway targeting therefore achieves efficient anti-tumor activity in heterogenous SCLC.
KEYWORDS: Ferroptosis, cell death, SCLC, neuroendocrine cancer, lipid peroxidation, ether lipids, PUFA, GPX4, GSH, thioredoxin
Cell death is mainly caused by two different types of cell death, accidental cell death (ACD) and programmed cell death (PCD), an important regulated mechanism controlling essential physiological processes such as development, immunity and tissue homeostasis. PCD can be triggered by intracellular or extracellular perturbations overcoming stress response mechanisms in cells which in turn activate a “programmed” molecular cascade ultimately resulting in cell death. The most in-depth researched PCD, to date, is apoptosis which can be induced by intracellular triggers such as DNA damage or stress signals resulting in an activation of pro-apoptotic over anti-apoptotic factors leading to mitochondria outer membrane permeabilization (MOMP). Cytochrome C released from mitochondria then activates the caspase (CASP) cascade consisting of initiator CASP8/9/10 and effector CASP3/6/7 preventing uncontrolled membrane rupture and cell content release. Similarly, extrinsic apoptosis is induced upon binding of death ligands belonging to the tumor necrosis factor (TNF) superfamily to their cognate receptors on the cell surface resulting in either direct recruitment of Fas-associated protein with death domain (FADD) to membrane complexes or intracellular secondary complexes followed by activation of CASP cascade. Contrarily, necrotic PCDs such as necroptosis result in membrane rupture accompanied by a release of cellular content. Upon extra- or intracellular stimulation, the necroptotic machinery depends on the sequential activation of receptor interacting serine/threonine kinase 1 (RIPK1) activating RIPK3 and mixed lineage kinase domain like pseudokinase (MLKL) forming the necrosome triggering membrane permeabilization.
These tightly regulated processes are often inactivated in prevalent human pathologies such as cancer. Fortunately, PCD can often be re-instated through genetic or pharmacological interventions enabling therapy options for these cancers. Moreover, the recent advent of immunotherapy has exploited the fact that also immune effector cells can be re-activated to effectively trigger PCD in tumor cells. Herein, binding of death ligands expressed by immune effector cells to their cognate receptor on tumor cells induces extrinsic apoptosis and/or necroptosis. Interestingly, ferroptosis – another type of regulated necrosis - was recently described to be amplified by interferon gamma (IFNγ) released from CD8+ T cell activated by immunotherapy1.
So far, execution of ferroptosis has been described as a collapse of the cellular antioxidant defense machinery rather than as an agonist-driven molecular cascade actively inducing cell death. An important hallmark of ferroptosis is the characteristic accumulation of peroxidized membrane lipids, allowing for membrane pore formation and destabilization of the lipid bilayer2. Within the plasma membrane, polyunsaturated fatty acids (PUFAs) within phospholipids harboring methylene bridges with highly reactive hydrogen atoms are especially sensitive to lipid peroxidation which are by orders of magnitude less peroxidized during other types of PCD3. Furthermore, ether-linked phospholipids synthesized in peroxisomes were recently identified to substantially contribute to the lipid peroxide pool and thereby drive ferroptosis susceptibility4.
Lipid peroxidation can take place spontaneously in the presence of free redox active divalent iron (Fe2+) to promote a Fenton reaction generating hydroxyl radicals (HO∙) or Fe2+ serves as a cofactor for lipoxygenase (LOX) to catalyze peroxidation enzymatically.
In order to protect cells from spontaneous or enzymatically catalyzed lipid peroxidation, glutathione peroxidase 4 (GPX4) constitutively hydrolyzes lipid hydroperoxides (L-OOH) in a glutathione (GSH)-dependent manner and thereby prevents ferroptosis5. Recently, ferroptosis suppressor protein 1 (FSP1) was shown to be recruited to the plasma membrane where it acts as an oxidoreductase reducing ubiquinone (=Coenzyme Q10) to the lipophilic radical scavenger ubiquinol preventing lipid peroxidation in the absence of GPX46,7.
Amongst lung cancer entities, small cell lung cancer (SCLC) is the most aggressive form, defined by highly proliferative, small cells mostly of neuroendocrine origin with a high tumor mutational burden (TMB) closely associated with tobacco smoking. SCLC often presents with heterogenous tumors consisting of neuroendocrine (NE) cells and non-neuroendocrine (non-NE) cells, a fact complicating therapy. Despite initial response of most SCLC patients to chemotherapy, they almost invariably develop relapse.
In our recent study8, we surprisingly identified that in treatment-naïve SCLC patient tumor tissue expression of many PCD pathway components, including those driving apoptosis and necroptosis, was already lost at diagnosis. These data strongly suggest that SCLC likely undergoes stringent selection against PCD pathways prior to therapy. As SCLC is known to present with high TMB, this prior selection pressure may stem from immune effector cells trying to kill tumor cells with high TMB. Interestingly, neuroendocrine status of SCLC has recently been linked to immune gene signature expression9. NE cells repress expression of immune signature genes especially major histocompatibility complex (MHC) I, while MHC I proteins were described to be re-expressed on non-NE chemoresistant relapsed cells in some cases.
Despite this broad inactivation of PCD, we identified that non-NE mouse and human SCLC cells show a strong response to ferroptosis inducing agents, while NE SCLC cells are instead highly vulnerable to thioredoxin (TRX) pathway inhibition. Importantly, we found that non-NE differentiation, an SCLC subtype associated with chemotherapy resistance upon relapse, renders SCLC exquisitely ferroptosis sensitive by upregulating ether-linked PUFA synthesis resulting in a ferroptosis-prone lipidome (Figure 1).
Figure 1.
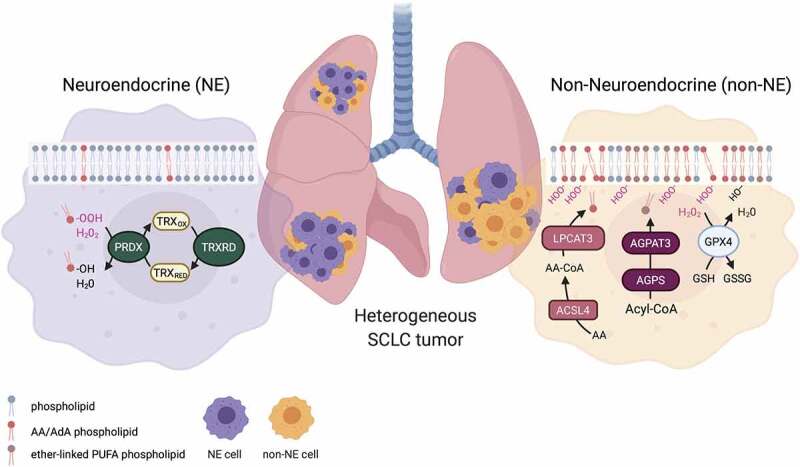
Phenotypic heterogeneity of small cell lung cancer (SCLC): Heterogenous SCLC tumors contain non-neuroendocrine (non-NE) and neuroendocrine (NE) cells. Non-NE cell membranes are characterized by a high arachidonic acid (AA) and adrenic acid (AdA) phospholipid and ether-linked polyunsaturated fatty acid (PUFA) phospholipid content prone to be peroxidized during ferroptosis. Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are crucial for the synthesis of AA and AdA containing phospholipids and alkylglycerone phosphate synthase (AGPS) and 1-acylglycerol-3-phosphate O-acyltransferase 3 (AGPAT3) catalyze ether phospholipid synthesis from Acyl-Coenzyme A (Acyl-CoA) and are highly expressed in non-NE SCLC cells. Peroxidation of membrane lipids is counteracted by glutathione peroxidase 4 (GPX4) reducing lipid peroxides (L-OOH) to their respective lipid alcohol (L-OH) while oxidizing glutathione (GSH) to glutathione disulfide (GSSG) thereby preventing ferroptosis. NE SCLC cells present with a lipidome less prone to ferroptosis-induced lipid peroxidation and are highly dependent on the thioredoxin (TRX) redox pathway. Here, reactive oxygen (ROS) species such as hydrogen peroxide (H2O2) as well as lipid peroxides are reduced respectively by peroxiredoxins (PRDX) using TRX as a redox equivalent, which is restored by thioredoxin reductases (TRXRD). Inhibition of the thioredoxin pathways results in ROS-dependent cell death in NE SCLC. The scheme was drawn using fully licensed Biorender.com
Metabolic remodeling is one of the hallmarks of cancer and changes in lipid metabolism have been described to fuel cancer progression and proliferation. Interestingly, Acyl-CoA synthetase long-chain family member 4 (ACSL4), important for PUFA phospholipid synthesis, has been reported to increase chemoresistance in breast cancer by regulating ATP binding cassette (ABC) transporter expression10 hinting at the possibility that acquired metabolic changes in NE to non-NE plasticity may further fuel chemotherapy resistance of non-NE SCLC. Yet, the exact mechanism by which increased lipid metabolism might support chemoresistance in non-NE SCLC remains to be further examined. Based on our finding that non-NE SCLC is specifically ferroptosis sensitive due to elevated ether lipid synthesis, ferroptosis induction could serve as alternative treatment strategy specifically targeting non-NE chemoresistant tumors upon relapse.
Of note, our study also showed that NE SCLC did not present with elevated ether lipid synthesis and was resistant to ferroptosis. Instead, we identified that NE cells rely on the TRX pathway for antioxidant defense and Auranofin, a thioredoxin reductase (TRXRD) inhibitor, which is already clinically approved for the treatment of rheumatoid arthritis, was highly and selectively active against NE SCLC cells. Consequently, we also identified a novel Achilles heel of the NE SCLC subtype. Importantly, the combined therapy of ferroptosis induction and targeting the TRX pathway allows to target heterogenous SCLC tumors and subtype plasticity oppressing escape mechanisms.
Funding Statement
This work was supportedby the German Cancer Aid (Deutsche Krebshilfe) [Max-Eder-Project 701125509], a project grant [432038712], a collaborative research center grant on SCLC [SFB1399, A05], a collaborative research center grant on cell death [SFB1403, A05] all funded by the German Research Foundation [Deutsche Forschungsgemeinschaft, DFG], by an eMed consortium grant [01ZX1901] funded by the German state (BMBF) and a project grant [A06] funded by the center for molecular medicine (CMMC), Cologne, Germany.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Wang W, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019. doi: 10.1038/s41586-019-1170-y [DOI] [PMC free article] [PubMed]
- 2.Pedrera L, Espiritu RA, Ros U, Weber J, Schmitt A, Stroh J, Hailfinger S, Von Karstedt S, García-Sáez AJ.. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2020. doi: 10.1038/s41418-020-00691-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiernicki B, Dubois H, Tyurina YY, Hassannia B, Bayir H, Kagan VE, Vandenabeele P, Wullaert A, Vanden Berghe T. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 2020;11:1. doi: 10.1038/s41419-020-03118-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, Paradkar S, Boehnke N, Deik AA, Reinhardt F, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603–3. doi: 10.1038/s41586-020-2732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang WS, SriRamaratnam R, Welsch M, Shimada K, Skouta R, Viswanathan V, Cheah J, Clemons P, Shamji A, Clish C, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doll S, Freitas FP, Shah R, Aldrovandi M, Da Silva MC, Ingold I, Goya Grocin A, Xavier Da Silva TN, Panzilius E, Scheel CH, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 8.Bebber CM, Thomas ES, Stroh J, Chen Z, Androulidaki A, Schmitt A, Höhne MN, Stüker L, De Pádua Alves C, Khonsari A, et al. Ferroptosis response segregates small cell lung cancer (SCLC) neuroendocrine subtypes. Nat Commun. 2021;12:2048. doi: 10.1038/s41467-021-22336-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahadevan NR, et al. Intrinsic immunogenicity of small cell lung carcinoma revealed by its cellular plasticity. Cancer Discov. 2021. candisc.0913.2020. doi: 10.1158/2159-8290.CD-20-0913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orlando UD, Castillo AF, Medrano MAR, Solano AR, Maloberti PM, Podesta EJ. Acyl-CoA Synthetase-4 Is Implicated in Drug Resistance in Breast Cancer Cell Lines Involving the Regulation of Energy-Dependent Transporter Expression. Biochem. Pharmacol. 2019;159:52–63. 10.1016/j.bcp.2018.11.005 [DOI] [PubMed]