

ABSTRACT
Genome instability is a hallmark of cancer. ATP-dependent chromatin remodelers are frequently altered in cancer. We have recently reported that the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex protects the genome by limiting R-loop-mediated genome instability, mainly that caused by transcription-replication conflicts. Here we discuss the significance and biomedical applications of this finding.
KEYWORDS: Cancer, SWI/SNF, cBAF, transcription-replication conflicts, R-loops, genome instability, chromatin, epigenetics
In addition to the transcription machinery, non-B DNA structures generated by transcription may pose an obstacle to replication fork (RF) progression. Co-transcriptional R-loops, three-stranded nucleic acid structures composed of a DNA-RNA hybrid plus a displaced single strand DNA (ssDNA), may occur naturally to serve a physiological role but its unscheduled formation have been shown to menace genome integrity.1 Avoidance of such situations is achieved thru multiple mechanisms, also involving the chromatin network.
Genome instability is a hallmark of cancer cells. Well-known cancer-associated factors such as Breast cancer type 1/2 susceptibility proteins (BRCA1/2) have been demonstrated to impact R-loop homeostasis.1 ATP-dependent chromatin remodelers are frequently altered in cancer, with the SWItch/Sucrose Non-Fermentable (SWI/SNF) complex genes accumulating mutation at frequencies close to those of the Tumor Protein P53 (TP53) gene.2 SWI/SNF’s contribution to transcription had been extensively studied, but little was known about its impact on genome integrity. Our recently published work3 unveils a novel role for SWI/SNF in preventing DNA replication stress by helping solve transcription-replication conflicts (TRCs), which may be an important source of DNA damage and genome instability. In particular, SWI/SNF mutations effect in genome instability was found epistatic to Fanconi Anemia Complementation Group D2 (FANCD2), a member of the Fanconi Anemia pathway of repair accumulating at sites of RF stalling, directly linking SWI/SNF to TRC resolution rather than prevention.
A major contribution of SWI/SNF-related Matrix-associated Actin-dependent Regulator of Chromatin subfamily A (SMARCA), member 4 (SMARCA4), best known as Brahma-related gene 1 (BRG1), but not SMARCA member 2 (SMARCA2), also named Brahma (BRM), the two ATPase subunits of the human SWI/SNF, was shown to play a central role in TRC resolution. Consistently, BRG1 generally associates with proliferative tissues with high DNA replication rates, whereas BRM is mostly linked to differentiated tissues with low replication.4,5 SWI/SNF may exist in several complex subtypes including canonical BRG1-associated factor (cBAF), non-canonical BAF (ncBAF) and polybromo BRG1-associated factor (PBAF) with subunits specific to each subtype. Interestingly, a major contribution of cBAF complex subtype to TRC outcome was evidenced by the observation that AT-Rich Interaction Domain 1A (ARID1A) also accumulates at TRC and its depletion results in R-loop-dependent DNA damage. Polybromo 1 (PBRM1), a member of the PBAF complex subtype, also influences R-loop homeostasis, but to a lesser extent. Consistently, ARID1A and PBRM1 inactivation also induces R-loops.6,7 Interestingly, BRG1, ARID1A and PBRM1 are among the ATP-dependent chromatin remodeling subunit genes most frequently altered in cancer,2 suggesting a direct connection between their recurrent mutation in malignant cells and their function preventing TRC-derived genome instability, and highlighting their importance as putative tumor suppressors. BRM, which does not impact TRCs, present much lower mutation rates in transformed cells.
Defining the origin and cause of malignancies is crucial to understand the disease and to generate new therapeutic approaches. According to the Integrative Onco Genomics (Intogen) database, BRG1, ARID1A and PBRM1 act as cancer-drivers in several cancer types.8 In these cases, malignancy could be mediated by a sustained R-loop-induced genome instability eventually leading to cell transformation. Nevertheless, this process would imply that pre-malignant and/or malignant cells develop additional mechanisms to deal with DNA replication stress and genome instability to proliferate (Figure 1a). Indeed, sustained genome instability prior to malignant transformation might promote mutations that could increase cell fitness and malignant cell proliferation in contrast to other cells presenting genome instability. The same rational would apply when malignancy is acquired, since cancer cells able to deal with its inherent genome instability would proliferate better.
Figure 1.
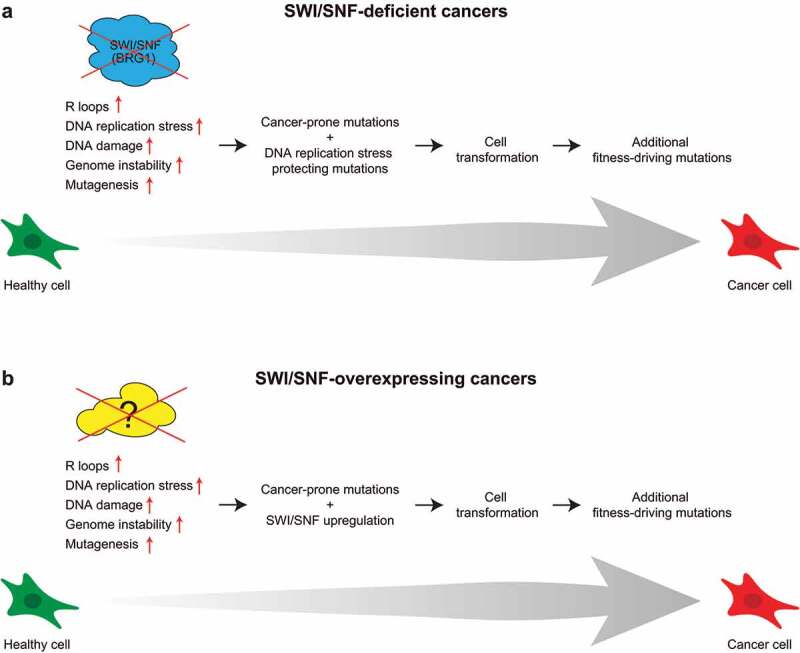
Potential impact of SWItch/Sucrose Non-Fermentable (SWI/SNF) complexes on DNA replication stress-driven oncogenic processes. (a), In SWI/SNF-deficient cells, high unscheduled R-loop formation causes DNA replication stress, DNA damage and genome instability. Under these conditions, mutagenesis increases and results in accumulation of cancer-prone alterations. Its co-occurrence with alterations favoring proliferation under DNA replication stress may permit cell survival and eventually lead to cell transformation. (b), Deficiencies in molecular activities leading to high R-loop-mediated genome instability scenarios increase mutagenesis and promote cancer-prone alterations. SWI/SNF upregulation might help these cells to deal with its inherent genome instability, given its role at transcription-replication conflicts (TRCs), and contribute to cell transformation. Additional mutations after transformation may further increase cell fitness. Grey arrow indicates direction and timing of events leading to cell transformation. Healthy (green) and malignant (red) cell fates are also shown. Events occurring during transformation process are described over the gray arrow
BRG1 is overexpressed in a wide range of cancers.9 It is possible that under DNA replication stress higher levels of BRG1 confer a cell proliferation advantage given its role in TRC resolution and genome integrity (Figure 1b). If so, the use of BRG1-targeting drugs would be a plausible approach to limit cancer cell proliferation, by restoring the high genome instability, thus affecting cell fitness. A similar mechanism has been suggested for Inositol requiring 80 (INO80) complex, which also impacts on R-loop homeostasis.10 INO80 depletion inhibits cell growth in PC3 (prostate cancer), MCF7 (breast cancer) and WM1361 (melanoma) cancer cells, but co-depletion of INO80 and ribonuclease H1 (RNase H1), which degrades the RNA moiety from DNA-RNA hybrids, restores cell proliferation.
Therefore, even though dysfunction of SWI/SNF triggers genome instability and might promote cell transformation, different survival mechanisms can be used in some cancer types with high genome instability. Unveiling the contribution of the chromatin-remodeling network is vital to understand how cells proliferate under DNA replication stress. Identifying the chromatin activities used by SWI/SNF-deficient cancer cells to grow despite its high R-loop-dependent genome instability could serve to design new drugs targeting specifically such activities. The combined use of such drugs with DNA replication stress-inducing drugs could block cell proliferation in malignancies with up-regulated SWI/SNF levels. In support of this possibility, PBRM1 deficiency was found to be synthetic lethal with Poly(ADP-Ribose) Polymerase (PARP) and Ataxia Telangiectasia And Rad3-Related (ATR) inhibitors.7
In conclusion, identification of the cause-effect relationship of SWI/SNF alterations in several cancer types could permit designing new strategies to specifically target malignant cells. The identification of R-loop-mediated replication stress may suppose a step forward in this direction.
Funding Statement
A.B-F. is supported by a Juan de la Cierva postdoctoral contract from the Spanish Ministry of Science and Innovation. Research in A.A.’s lab is funded by the European Research Council, the Spanish Ministries of Science and Innovation, the European Union (FEDER) and Foundation “Vencer el Cancer”.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Crossley MP, Bocek M, Cimprich KA.. R-loops as cellular regulators and genomic threats. Mol Cell. 2019;73(3):1–3. doi: 10.1016/j.molcel.2019.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR.. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bayona-Feliu A, Barroso S, Muñoz S, Aguilera A. The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription–replication conflicts. Nat Genet. 2021;53(7):1050–1063. doi: 10.1038/s41588-021-00867-2. [DOI] [PubMed] [Google Scholar]
- 4.Muchardt C, Bourachot B, Reyes JC, Yaniv M. ras transformation is associated with decreased expression of the brm/SNF2α ATPase from the mammalian SWI-SNF complex. EMBO J. 1998;17(1):223–231. doi: 10.1093/emboj/17.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2α). EMBO J. 1998;17(23):6979–6991. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai S, Fournier LA, Chang EYC, Wells JP, Minaker SW, Zhu YD, Wang AYH, Wang Y, Huntsman DG, Stirling PC, et al. ARID1A regulates R-loop associated DNA replication stress. PLoS Genet. 2021;17(4):e1009238. doi: 10.1371/journal.pgen.1009238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chabanon RM, Morel D, Eychenne T, Colmet-Daage L, Bajrami I, Dorvault N, Garrido M, Meisenberg C, Lamb A, Ngo C, et al. PBRM1 deficiency confers synthetic lethality to DNA repair inhibitors in cancer. Cancer Res. 2021;81(11):2888–2902. doi: 10.1158/0008-5472.CAN-21-0628. [DOI] [PubMed] [Google Scholar]
- 8.Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, Mularoni L, Pich O, Bonet J, Kranas H, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer. 2020;20(10):555–572. doi: 10.1038/s41568-020-0290-x. [DOI] [PubMed] [Google Scholar]
- 9.Guerrero-Martínez JA, Reyes JC. High expression of SMARCA4 or SMARCA2 is frequently associated with an opposite prognosis in cancer. Sci Rep. 2018;8(1). doi: 10.1038/s41598-018-20217-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prendergast L, McClurg UL, Hristova R, Berlinguer-Palmini R, Greener S, Veitch K, Hernandez I, Pasero P, Rico D, Higgins JMG, et al. Resolution of R-loops by INO80 promotes DNA replication and maintains cancer cell proliferation and viability. Nat Commun. 2020;11(1). doi: 10.1038/s41467-020-18306-x. [DOI] [PMC free article] [PubMed] [Google Scholar]