

Abstract
Hereditary colorectal cancer (CRC) syndromes attributable to high penetrance mutations represent 9–26% of young-onset CRC cases. The clinical significance of many of these mutations is understood well enough to be used in diagnostics and as an aid in patient care. However, despite the advances made in the field, a significant proportion of familial and early-onset cases remains molecularly uncharacterized and extensive work is still needed to fully understand the genetic nature of CRC susceptibility. With the emergence of next-generation sequencing and associated methods, several predisposition loci have been unraveled, but validation is incomplete. Individuals with cancer-predisposing mutations are currently enrolled in life-long surveillance, but with the development of new treatments, such as cancer vaccinations, this might change in the not so distant future for at least some individuals. For individuals without a known cause for their disease susceptibility, prevention and therapy options are less precise. Herein, we review the progress achieved in the last three decades with a focus on how CRC predisposition genes were discovered. Furthermore, we discuss the clinical implications of these discoveries and anticipate what to expect in the next decade.
Introduction
Familial clustering of colorectal cancer (CRC) and gastrointestinal polyposis has been recognized for well over a century (1,2), but little was known about the heritable changes in this heterogeneous group of diseases until the last three decades. Familial clustering is observed in up to 25% of CRC cases (3) and twin studies have yielded heritability estimates of ~ 35% (4).
Inherited predisposition to CRC because of high penetrance germline mutations (Table 1) has been implicated in 9–26% of patients diagnosed before the age of 50 years (5–9). Hereditary CRC syndromes caused by these mutations are phenotypically divided into polyposis and non-polyposis syndromes (Fig. 1). The more common non-polyposis syndromes include Lynch syndrome (OMIM#120435; LS), caused by DNA mismatch repair (MMR) gene mutations and resulting in a hypermutated tumor phenotype, and familial colorectal cancer type X (FCCTX) describing the remaining MMR-proficient cases with a heterogeneous genetic background. Polyposis syndromes are distinguished by adenomatous or non-adenomatous (hamartomatous/serrated) polyp histology (10). Of these, the causative gene for familial adenomatous polyposis (OMIM#175100; FAP), APC, was discovered first (11) (Fig. 2). CRC syndromes also involve predisposition to other tumors, including breast and gastrointestinal cancers in hamartomatous polyposes (12–14), endometrial cancer in LS (15,16), and fundic gland polyps (17) and desmoid tumors (18) in FAP.
Table 1.
Established (in bold) and putative high penetrance genes associated with hereditary CRC and polyposis
| Gene symbol | Gene name | Gene function | Cancer syndrome(s) | Mode of inheritance | Method of discovery | Type of genetic instability | PubMed ID | Ref. no. |
|---|---|---|---|---|---|---|---|---|
| APC | Adenomatous polyposis coli | Inhibitor of Wnt signaling pathway | FAP | Autosomal dominant | Chromosomal deletion | CIN | 3 789 010 1 651 174 1 651 562 1 651 563 1 678 319 |
(21) (11) (26) (221) (222) |
| AXIN2 | Axis inhibition protein 2 | Inhibitor of Wnt signaling pathway | Familial tooth agenesis and attenuated familial polyposis | Autosomal dominant | Linkage and candidate gene analysis | CIN? | 15 042 511 | (62) |
| BMPR1A | BMP receptor, type IA | Activates SMAD transcriptional regulators | JPS FCCTX |
Autosomal dominant | Linkage analysis | CIN (low) | 11 381 269 (JPS) 21 640 116 (FCCTX) |
(73) (JPS) (223) (FCCTX) |
| BRF1 | BRF1 RNA polymerase III transcription initiation factor subunit | Activator of RNA polymerase | FCCTX Cerebellofaciodental syndrome (CFDS) |
Autosomal dominant | Exome sequencing | CIN | 28 912 018 (FCCTX) 25 561 519 (CFDS) |
(224) (FCCTX) (225) (CFDS) |
| EPCAM | Epithelial cell adhesion molecule | Cell adhesion | LS (MSH2 hypermethylation from transcriptional read-through of EPCAM) Congenital tufting enteropathy (CTE) |
Autosomal dominant (LS) Autosomal recessive (CTE) |
Tumor phenotype | MSI (LS) | 16 951 683 (LS) 19 098 912 (LS) 18 572 020 (CTE) |
(226) (LS) (227) (LS) (228) (CTE) |
| ERCC6 | ERCC excision repair 6, chromatin remodeling factor | Transcription-coupled nucleotide excision repair | FCCTX Cockayne’s syndrome (CKS) |
Autosomal dominant/codominant with WRN? (FCCTX) Autosomal recessive (CKS) |
Exome sequencing | CIN | 26 344 056 (FCCTX) 1 339 317 (CKS) 9 443 879 (CKS) |
(83) (FCCTX) (229) (CKS) (230) (CKS) |
| FAF1 | Fas-associated factor 1 | DNA replication | FCCTX | Autosomal dominant | Exome sequencing | CIN | 32 179 092 | (231) |
| FAN1 | FANCD2- and FANCI-associated nuclease 1 | DNA cross-link repair | FCCTX Karyomegalic interstitial nephritis (KMIN) |
Autosomal dominant (FCCTX) Autosomal recessive (KMIN) |
Exome sequencing | CIN | 26 052 075 (FCCTX) 22 772 369 (KMIN) |
(232) (FCCTX) (233) (KMIN) |
| GALNT12 | Polypeptide N-acetylgalactosaminyl-transferase 12 | Oligosaccharide biosynthesis | FCCTX | Autosomal dominant (possibly dominant negative) | Candidate gene approach | ? | 19 617 566 | (234) |
| GREM1 | Gremlin 1, DAN family BMP antagonist | BMP antagonist | Mixed polyposis syndrome | Autosomal dominant | Candidate gene approach | CIN (low?) | 22 561 515 | (235) |
| MBD4 | Methyl-CpG binding domain 4, DNA glycosylase | BER | MBD4-associated neoplasia syndrome (MANS) | Autosomal recessive | Whole-genome sequencing | CpG > TpG | 30 049 810 | (236) (237) |
| MLH1 | MutL homolog 1 | MMR | LS (monoallelic) Constitutional MMR deficiency syndrome (CMMRD; biallelic) | Autosomal dominant (LS) Autosomal recessive (CMMRD) |
Linkage analysis | MSI-H Hypermutated |
7 903 889 8 128 251 8 145 827 |
(53) (51) (52) |
| MLH3 | MutL homolog 3 | MMR recombination |
MLH3-associated adenomatous polyposis (biallelic) Low-penetrance CRC (monoallelic) |
Autosomal recessive (polyposis) | Exome sequencing (polyposis) Candidate gene approach (CRC) |
CIN (polyposis) MSI-low (CRC) |
30 573 798 (polyposis) 11 586 295 (CRC) |
(91) (polyposis) (238) CRC |
| MRE11 | MRE11 homolog, DSB nuclease | Telomere maintenance | FCCTX Ataxia-telangiectasia-like disorder (ATLD) |
Autosomal dominant? Autosomal recessive (ATLD) |
Exome sequencing | CIN? | 27 329 137 (FCCTX) 10 612 394 (ATLD) |
(239) (FCCTX) (240) (ATLD) |
| MSH2 | MutS homolog 2, colon cancer, nonpolyposis type 1 (E. coli) | MMR | LS (monoallelic) CMMRD; biallelic | Autosomal dominant (LS) Autosomal recessive (CMMRD) |
Linkage analysis and tumor phenotype | MSI-H Hypermutated |
8 484 120 8 252 616 8 261 515 8 484 121 |
(46) (47) (45) (57) |
| MSH3 | MutS homolog 3 | MMR | MSH3-associated adenomatous polyposis (biallelic) | Autosomal recessive (polyposis) | Exome sequencing | MSI (EMAST) | 27 476 653 | (90) |
| MSH6 | MutS homolog 6 | MMR | LS (monoallelic) CMMRD; biallelic | Autosomal dominant (LS) Autosomal recessive (CMMRD) |
Candidate gene approach | MSI-H | 9 354 786 | (81) |
| MUTYH | MutY DNA glycosylase | BER | MUTYH-associated adenomatous polyposis | Autosomal recessive | Tumor phenotype | G:C > T:A | 11 818 965 | (38) |
| NTHL1 | Nth-like DNA glycosylase 1 | BER | NTHL1-associated adenomatous polyposis | Autosomal recessive | Exome sequencing | C:G > T:A Hypermutated |
25 938 944 | (89) |
| OGG1 | 8-Oxoguanine DNA glycosylase | BER | FCCTX | Autosomal dominant/codominant | Candidate gene approach | G:C > T:A? | 21 195 604 | (114) |
| PMS2 | PMS2 postmeiotic segregation increased 2 | MMR | LS (monoallelic) CMMRD; biallelic | Autosomal dominant (LS) Autosomal recessive (CMMRD) |
Candidate gene approach | MSI | 8 072 530 | (241) |
| POLD1 | DNA polymerase delta 1, catalytic subunit | DNA replication | PPAP | Autosomal dominant | Linkage analysis and genome sequencing | Ultrahypermutated Possible MSI |
23 263 490 | (87) |
| POLE | DNA polymerase epsilon, catalytic subunit | DNA replication | PPAP | Autosomal dominant | Linkage analysis and genome sequencing | Ultrahypermutated Possible MSI |
23 263 490 | (87) |
| POLE2 | DNA polymerase epsilon 2, catalytic subunit | DNA repair and replication | FCCTX Adenomatous polyposis |
Autosomal dominant? (27329137) Autosomal recessive? (25529843) |
Exome sequencing | Hypermutant? | 25 529 843 27 329 137 |
(242) (239) |
| POT1 | Protection of telomeres 1 | Telomere maintenance | FCCTX Familial melanoma (FM) Familial glioma (FG) Li-Fraumeni-like syndrome (LFL) |
Autosomal dominant? | Exome sequencing | CIN? | 27 329 137 (FCCTX) 24 686 849 (FM) 24 686 846 (FM) 25 482 530 (FG) 26 403 419 (LFL) |
(239) (FCCTX) (243) (FM) (244) (FM) (245) (FG) (246) (LFL) |
| PTEN | Phosphatase and tensin homolog | Phospatase | CS PTEN hamartoma tumor syndrome |
Autosomal dominant | Linkage and candidate gene approach | CIN (low?) | 8 673 088 9 072 974 9 140 396 |
(66) (247) (67) |
| RNF43 | Ring finger protein 43 | DNA damage response | Serrated polyposis syndrome | Autosomal dominant | Exome sequencing | CIN | 24 512 911 | (248) |
| RPS20 | Ribosomal protein 20 | Ribosome biogenesis | FCCTX DBA |
Autosomal dominant | Exome sequencing | CIN? | 24 941 021 (FCCTX) 32 790 018 (DBA) |
(88) (FCCTX) (98) (DBA) |
| SEMA4A | Semaphorin 4A | Semaphorin | FCCTX Retinis pigmentosa (RP) |
Autosomal dominant (FCCTX) Autosomal recessive (RP) |
Linkage analysis and exome sequencing | CIN | 25 307 848 (FCCTX) 16 199 541 (RP) |
(249) (FCCTX) (250) (RP) |
| SMAD4 | SMAD family member 4 | Cytoplasmic mediators of TGF-beta signaling | Juvenile polyposis syndrome | Autosomal dominant | Candidate gene approach | CIN (low?) | 9 582 123 | (71) |
| STK11 | Serine/threonine kinase 11 | AMPK activity regulator | PJS | Autosomal dominant | Chromosomal deletion | CIN (low) | 8 988 175 9 428 765 |
(68) (69) |
| WRN | WRN RecQ like helicase | DNA DSB repair | FCCTX Werner syndrome |
Autosomal dominant/co-dominant with ERCC6 (FCCTX) Autosomal recessive (Werner) |
Exome sequencing (FCCTX) Candidate gene approach (Werner) |
CIN | 26 344 056 (FCCTX) 8 602 509 (Werner) |
(83) (FCCTX) (251) (Werner) |
Figure 1.
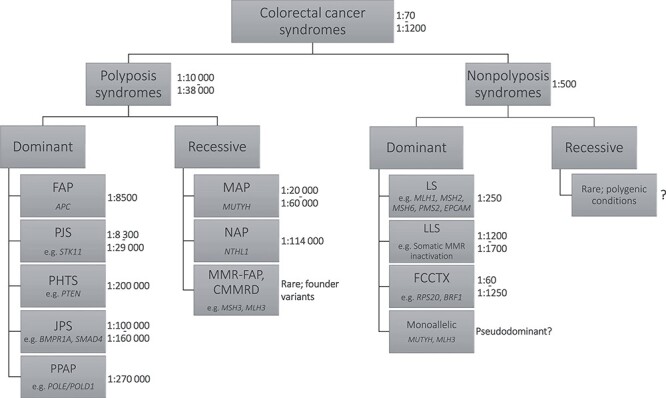
CRC syndromes. Division between syndromes is traditionally based on the number of and histopathology of intestinal polyps and mode of inheritance. The most common germline mutant genes for each syndrome are given. Ratios indicate estimates of prevalence of each syndrome based on literature (201–213).
Figure 2.
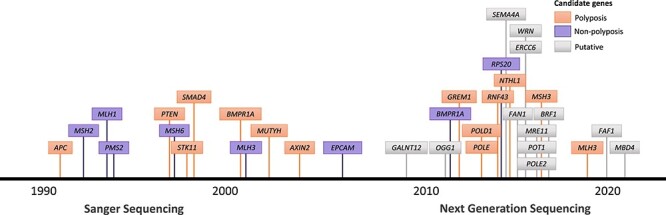
Timeline of CRC and polyposis susceptibility gene discoveries in the Sanger sequencing and NGS eras. Timeline assignment is based on the discovery of the gene as a CRC or polyposis susceptibility gene (the gene itself may have been identified before). Established susceptibility genes are colored and putative are indicated with a gray box.
In this anniversary review, we give an overview on genetics of hereditary CRC syndromes with a focus on Mendelian conditions and developments in genetic analysis techniques over the past three decades.
Clues From Molecular Tumor Phenotype
Chromosomal abnormalities have been attributed to the genesis of cancer for over a century (19), and deletions of the long arm of chromosome 5 (5q) have been recognized as the most frequent chromosome anomaly in secondary leukemia and in some solid cancers for some 40 years (20). As 5q was already a prominent factor in cancer development and familial clustering of colon polyposis was acknowledged, a clinical report of a Gardner syndrome patient with a de novo interstitial deletion within the 5q arm (21) provided the ‘missing link’ connecting 5q to polyposis predisposition. Soon, several research groups confirmed frequent allelic loss in 5q in CRC (22) and linked the adenomatous polyposis phenotype to the locus (23–25), leading to the detailed characterization of the APC gene (26) and initiating the hunt for gene defects responsible for inherited CRC and polyposes. Today, germline APC mutations are recognized as the underlying cause of FAP (Table 1); of these, 10–25% arise de novo. Typical of disorders with high de novo mutation rates (27), somatic APC mutations are sometimes restricted to certain tissues or organs, reflecting the timing of the mutation occurrence in embryogenesis. In these cases multiple genotypes are present in the patient, collectively referred to as genetic mosaicism. Patients with somatic APC mosaicism generally manifest as sporadic cases, as a disease phenotype only occurs when a substantial number of cells with the mutation are present in the target tissue. The detection of somatic APC mosaicism with conventional methods often proves difficult; the mutations might not be observable in samples derived from peripheral blood (28–30). Somatic APC mutations are commonplace in colorectal tumors and key players initiating colonic tumorigenesis (31–33).
Sometimes the landscape of somatic mutations can provide direct clues to the underlying germline mutation. In a British polyposis family negative for APC germline mutations, 11 tumors revealed somatic inactivation of APC because of G:C → T:A transversions (34). Previous efforts had identified spontaneous G:C → T:A transversions in OGG1 (35), MUTYH (36,37), and MTH mutant Escherichia coli and Saccharomyces cerevisiae, which gave researchers an incentive to test the aforementioned genes for mutations. This resulted in the discovery of two MUTYH missense mutations in the family, p.Tyr165Cys and p.Gly382Asp, segregating with the disease in an autosomal recessive mode. Nowadays both variants are recognized as common causes for MUTYH-associated polyposis (OMIM#608456) when biallelic (34,38,39). Later mutational signature analyses have attributed the G:C → T:A transversions in MUTYH-deficient tumors to the mutational signatures 18 (40) and 36 (41). Tumors deficient of the base-excision repair protein NTHL1 also have a conserved mutational signature characterized by C:G → T:A transitions (mutational signature 30) (42,43). Similarly, small insertions and deletions at repetitive sequences were consistently observed in tumors belonging to the spectrum of hereditary non-polyposis CRC (44), simultaneously attributed to genes in charge of MMR (45–47). This tumor phenotype is widely in use for diagnostic purposes as we will later discuss.
Gene-Hunting With Linkage Analysis—Narrowing It Down
Genetic linkage analysis remains a powerful tool for rare variant detection when combined with whole-genome sequencing (48,49) and has played an important role in gene discoveries made in CRC syndromes. LS is caused by germline mutations in MMR genes MLH1 (42% of unique variants), MSH2 (33%), MSH6 (18%), and PMS2 (7.5%) (50) (Table 1, Fig. 3). MSH2 and MLH1 were the first LS genes discovered in 1993–1994 (45,47,51,52), fueled by mapping of the respective predisposing loci by genetic linkage analysis (46,53,54). For MSH2, two large LS kindreds were studied using 345 microsatellite markers across the genome, revealing highly significant linkage with marker D2S123 at 2p15-16 (46). For MLH1, restriction fragment length polymorphisms and microsatellite markers studied in Swedish LS families revealed a predisposing locus at 3p21.3-23 (53,54). Subsequent gene identification by positional cloning was aided by the evolving understanding of MMR proteins in bacteria and yeast, providing a connection with the instability of DNA microsatellite markers observed in LS and other human tumors (55–61).
Figure 3.
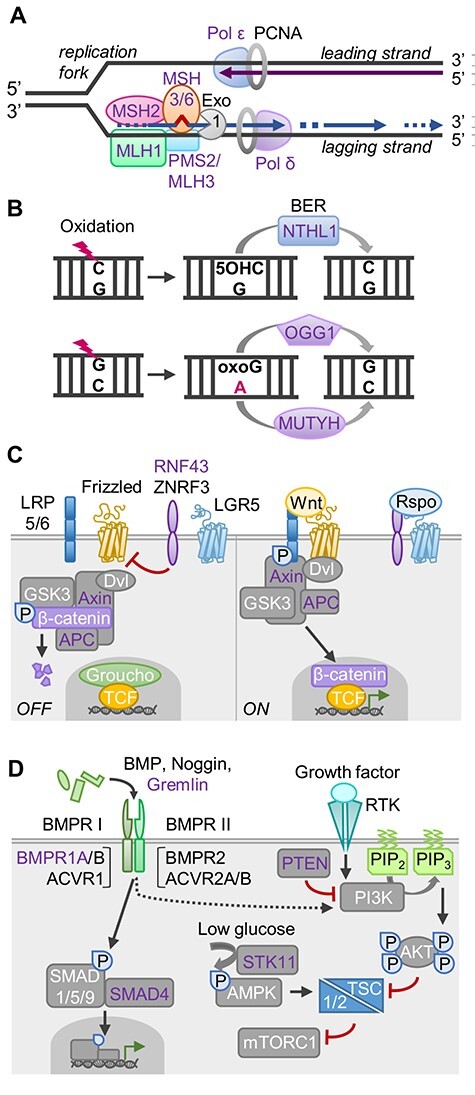
Key biological pathways associated with hereditary CRC syndromes. Genes with known germline mutations are shown in purple font. (A) Fidelity of DNA replication depends primarily on DNA polymerases ε and δ for correct base pairing and proofreading exonuclease activity. Any persisting base–base mispairs or insertion–deletion loops both on the leading and lagging strands are subsequently targeted by MMR proteins. MSH2:MSH3/6 dimers recognize the error. Interaction with MLH1:PMS2 triggers downstream repair events initiated by removal of the erroneous DNA by exonuclease 1 (Exo1). (B) Oxidation, alkylation, and deamination of DNA bases are repaired by base excision repair (BER) pathway. Eleven DNA glycosylases recognize and remove the damaged/mispaired bases; these are divided into monofunctional (e.g. MUTYH) and bifunctional (e.g. NTHL1, OGG1) glycosylases based on the presence of additional endonuclease activity. Their function is exemplified by repair of oxidative 8-oxoguanine (oxoG) and 5-hydroxycytosine (5-OHC) lesions. (C) Wnt signaling regulates cell development and stemness and is commonly hyperactivated in cancer. In the absence of secreted Wnt ligands, a cytoplasmic destruction complex directs β-catenin for proteasomal degradation and RNF43/ZNRF3 ubiquitin ligases downregulate Frizzled receptors. Upon receptor binding of Wnts, β-catenin is released and translocates to the nucleus, where it binds TCF/LEF transcription factors and displaces the Groucho repressor to activate target gene transcription. Binding of R-spondins (Rspo) to LGR5 inhibits RNF43/ZNRF3, enhancing Wnt signaling. (D) TGF-β signaling restricts proliferation of colonic epithelial cells. Bone morphogenetic proteins (BMPs) are TGF-β superfamily ligands that trigger receptor dimerization and activation via trans-phosphorylation, resulting in SMAD-dependent target gene transcription. Also, non-SMAD signaling pathways, including the PI3K–AKT–mTOR pathway, can be activated. Growth factor signaling via receptor tyrosine kinases (RTK) activates PI3K to generate phosphoinositide-3,4,5-triphosphate (PIP3), resulting in AKT-mediated derepression of mTOR complex 1 (mTORC1). mTOR integrates nutrient and growth factor signals, promoting cell growth upon activation. PTEN lipid phosphatase antagonizes this pathway by converting PIP3 back to PIP2. Conversely, low glucose conditions trigger the STK11-dependent activation of AMPK, suppressing mTORC1. See references (214–220) for further details.
AXIN2 locus underlying oligodontia-CRC syndrome (OMIM#608615) (Table 1) was discovered in a genome-wide search utilizing microsatellite markers in an affected Finnish family (62). Linkage was excluded for APC and candidate loci associated with tooth development, pointing to a region in chromosome 17. AXIN2 (17q24.1) became the strongest candidate owing to its role in WNT signaling (Fig. 3) and somatic mutations in CRC (63,64). Sequencing revealed an AXIN2 nonsense mutation segregating with the phenotype (62) as later confirmed in unrelated cases (65).
The genes for the main hamartomatous polyposis syndromes—Cowden syndrome (OMIM#158350; CS), Peutz-Jeghers syndrome (OMIM#175200; PJS), and juvenile polyposis syndrome (OMIM#174900; JPS)—were all identified through linkage studies. For CS, a traditional linkage and candidate gene analysis identified PTEN as the culprit gene (66,67) (Table 1, Fig. 3). In case of PJS, a targeted linkage analysis based on allelic loss detected in PJS polyps by comparative genomic hybridization revealed the disease locus, followed by fine mapping and systematic sequencing of transcripts in the region to identify the gene, LKB1/STK11 (68,69). For JPS, targeted linkage analysis excluded loci previously associated with CRC and closely related hamartomatous polyposis syndromes, revealing linkage to 18q21.1 (70). This led to the discovery of SMAD4 (18q21.2) mutations (71), but these were found to explain only a minority of the cases (72). Genome-wide linkage analysis in four JPS families without SMAD4 or PTEN mutations resulted in the discovery of germline BMPR1A (10q23.2) nonsense mutations in all families (73). Together, mutations in SMAD4 and BMPR1A, both involved in transforming growth factor beta (TGF-β) signaling, account for 50–60% of JPS cases (74) (Table 1, Fig. 3).
Candidate Genes Pinpointed by Biological Function
Existing knowledge of biological functions of candidate genes can greatly facilitate predisposition gene discoveries. Human MSH6 was identified as a 160 kDa protein dimerizing with MSH2 in a mismatch-binding factor purified from HeLa cells (75–77) (Fig. 3). Both proteins were required for restoring MMR in extracts of hypermutable CRC cell lines (76–78). Sequence analysis of proteolytic peptides from p160 allowed cDNA identification, revealing homology to human and yeast MSH2 and E. coli MutS (75–77). An Msh6-deficient mouse model gave the first evidence for a role in cancer susceptibility (79). While initial attempts to find germline mutations of MSH6 in 20 LS kindreds failed (78), mutations were soon found in atypical LS cases with weak family history (80,81). In contrast to MLH1 or MSH2 mutant tumors, microsatellite instability (MSI) is not always detected in MSH6 mutant tumors (79,82), in agreement with its preferential role in the repair of single-nucleotide mismatches (76–78).
In a study of 25 patients with familial CRC of undefined genetic basis (83), mostly falling into the category of FCCTX (Fig. 1), the authors hypothesized to find mutations predisposing to DNA double-strand breaks (DSBs) based on earlier reports on replication stress as the underlying mechanism of chromosomal instability in CRC (84). Compared with controls, CRC patient T cells had increased susceptibility to DSBs under ultraviolet radiation or aphidicolin-induced replication stress, and exome sequencing showed significantly higher frequency of rare disruptive germline mutations in DSB repair-related genes, found in 25 genes in 17/20 patients (83). Heterozygous mutations in WRN (genome stability during DNA repair) and ERCC6 (transcription-coupled nucleotide excision repair) found in one of the patients were functionally validated, further supporting a role for constitutional defects in suppression of DSBs as a new molecular class of hereditary CRC. Homozygous mutations cause Werner (OMIM#277700) and Cockayne (OMIM#133540) syndromes, respectively, the former associated with various neoplasms and the latter with heightened sensitivity to ultraviolet. Interestingly, the WRN-encoded DNA helicase was suggested to have a cell/tissue type specific role in MMR (85), and a related RecQ family DNA helicase BLM directly interacts with MSH6 (86).
From Single Genes to the Exome and Genome
The emergence of new sequencing techniques has enabled scientists to make advances in oncogenomics, especially in the study of somatic changes in tumors. Nevertheless, success in research of CRC predisposition has been limited. In conjunction with family information, be it in the form of linkage or other methods, a handful of novel causative genes have been identified in the last decade (Fig. 2, Table 1) (87–91). One of the successful results is the characterization of polymerase proofreading-associated polyposis (PPAP) by discovery of the associated genes: POLE and POLD1 (OMIM#615083 and OMIM#612591, respectively). Whole exome sequencing (WES) analysis of 15 mutation negative index cases with polyposis and family history of CRC, as well as some of their family members, initially left the researchers empty-handed: after excluding common and benign variants across multiple families, no susceptibility genes were identified (87). However, previous linkage data from a few families in this study (92,93) pinpointed the analysis to POLE and POLD1 genes, resulting in the identification of three families wherein the mutations segregated with the disease. Subsequent research has further defined the roles of POLE and POLD1 in familial CRC and polyposis (94).
The genetic background of FCCTX has been a particularly difficult one to crack: the underlying genes have eluded geneticists worldwide despite being under investigation for almost 20 years (95). In a similar fashion to the discovery of PPAP, a combination of linkage analysis and WES identified a truncating variant of RPS20 in association with MMR-proficient CRC in a four-generation family (88,95). Subsequent studies have since identified three RPS20 mutations in familial CRC cases (96,97) and two in Diamond-Blackfan anemia (DBA) patients (98). Available experience suggests that only a small fraction of FCCTX susceptibility genes have been discovered so far, and that private genes and mutations are common, strengthening the idea of the heterogeneous genetic background of FCCTX (94,99).
WES has become the main approach to detect novel cancer susceptibility genes and has enabled much of the recent success in the discovery of novel CRC and polyposis susceptibility genes. In addition to POLE, POLD1, and RPS20, recent successful reports of novel CRC and polyposis susceptibility variant discoveries include NTHL1 (89), MSH3 (90), and MLH3 (91) (Table 1). While WES is able to detect (or exclude) mutations in exons of protein-coding genes, a disadvantage of the method is that only ~2% of the genome is covered and many of the regulatory elements controlling the expression of these genes remain unrevealed. For example, intronic variants resulting in a splicing acceptor or donor domains can create pseudoexons, intronic sequences inserted in the mature mRNA of a gene. Detection of such variants is impossible with just WES and requires simultaneous transcriptome and whole genome-based approaches. Few cases of APC pseudoexons have been reported (100,101).
Toward Understanding ‘the Missing Heritability’—Oligogenic and Polygenic Etiology
Though much has been learned from hereditary CRC, mutations in known susceptibility genes explain only a fraction of familial CRC risk, and multiple explanations for this ‘missing heritability’ have been proposed (102–105). Genome-wide association studies (GWAS) continue to uncover novel susceptibility loci and common low-risk variants associated with CRC at the population level, further shaping our view of the genetic landscape of CRC (105,106). A recent effort combining whole-genome sequencing and imputation into GWAS data almost doubled the number of known association signals for sporadic CRC into ~ 100 independent signals at > 80 CRC risk loci (107), implicating novel pathways and gene families in CRC risk. These GWAS findings may provide further clues into etiology of hereditary CRC syndromes, as genes previously linked with hereditary syndromes have been found to harbor also common CRC risk variants with weaker effects (105). Common variants may modify CRC risk in patients with hereditary CRC syndromes, possibly explaining some of the observed differences in the ages of cancer diagnosis and disease presentation among individuals carrying the same high-penetrance mutation (108–112). Moreover, combinations of multiple common low-penetrance variants may work synergistically to substantially contribute to overall disease heritability. A recent example is from serrated polyposis syndrome, a hereditary CRC predisposition with a poorly understood genetic basis, which was analyzed in a case–control study for contribution of 65 common low-penetrance CRC risk variants, reporting significant associations of the condition with seven of these (113). These findings, together with emerging reports on di- or oligogenic inheritance via rare moderately penetrant variants (114–117), highlight the complex genetic make-up of hereditary CRC.
Clinical Implications
Molecular diagnostics of hereditary CRC has advanced significantly in the past decades, enabling today efficient screening strategies to identify individuals at high risk for referral to genetic testing and specialized cancer surveillance programs for each hereditary CRC predisposition (118). The genetic testing of affected individuals and predictive testing of their at-risk relatives, combined with intensive cancer surveillance, has an enormous cancer-preventive potential in these families, while also reducing unnecessary procedures and anxiety in family members who are found mutation negative.
Compared with polyposis syndromes, LS lacks distinctive endoscopic features and presents a particular challenge for diagnostics. Diagnosis was initially based on so-called Amsterdam criteria formulated in 1991 (119,120); these focused on family history and age of onset and were highly useful for selection of families for scientific gene identification studies. More practical and less stringent Bethesda guidelines (121,122) followed, utilizing tumor MSI for the selection of patients for further genetic testing. Both criteria became suboptimal for identifying LS cases as tools for genetic diagnostics advanced, prompting recommendations to offer genetic testing to all individuals with newly diagnosed CRC (123,124). The discovery of a high frequency of DNA replication errors in LS tumors compared with normal tissue paved the way for universal screening based on MSI (57,125–127). MSI testing involves polymerase chain reaction (PCR) amplification of a set of short repetitive DNA sequences (microsatellites) to determine changes in their length in tumors (128). Apart from LS, MSI occurs in ~ 12% of sporadic CRCs because of defective MMR caused by MLH1 promoter methylation or, rarely, bi-allelic somatic mutations of MMR genes (129,130). The former can be excluded by testing the tumors for the BRAF-V600E mutation and/or MLH1 promoter hypermethylation (131). Immunohistochemistry for detecting loss of expression of MMR proteins in tumor tissue is another prevalent LS screening approach (132–134). MSI testing and immunohistochemistry identify LS cases with an estimated clinical sensitivity of 85/83% and specificity of 90/89%, respectively (124). Subsequent detection of germline mutation in an MMR gene by sequencing then establishes the diagnosis of LS.
In recent years, the sequencing of each established hereditary CRC gene individually has become increasingly replaced by next-generation sequencing (NGS)-based diagnostic gene panels. These allow simultaneous germline mutation testing of a wide selection of CRC predisposition genes in a cost-effective manner in suspected CRC/polyposis syndrome cases, supporting utility as a first line test (135). The overlapping clinical presentation and genetic heterogeneity in CRC syndromes may also be better addressed by panel testing. The diagnostic yield and clinical actionability of results from CRC panel testing vary depending on the cohort selected for testing, genes included in the panel, and variant interpretation (6–8,136–147). In cohorts of unselected CRC patients representing different populations and ethnicities, panel testing of 25, 18, 73, 83, and 27 cancer-predisposing genes identified pathogenic or likely pathogenic germline mutations in 9.9%, 2%, 18.1%, 15.5%, and 3.3% of the patients, respectively (136,137,143,144,147). A large variety of commercial CRC gene panels are available (148). These show inconsistent criteria for inclusion of genes, including both high and low penetrance genes with highly variable levels of supporting evidence (148). To address these and other concerns, the Collaborative Group of the Americas on Inherited Gastrointestinal Cancer recently published a position statement proposing a minimal set of 11 genes to be included on a multigene panel for evaluation of hereditary CRC/polyposis; MLH1, MSH2, MSH6, PMS2, EPCAM, APC, BMPR1A, MUTYH, PTEN, STK11, and SMAD4 (149). The statement also recommends an additional set of 16 genes to be considered for panel testing (149), including genes with low-to-moderate CRC risk, preliminary but limited data on CRC risk, or actionable mutations without proven causation in CRC. Notably, this set includes several genes with germline mutations not traditionally associated with CRC/polyposis, including TP53, CHEK2, and BRCA1/2, highlighting the utility of a broader analysis of hereditary cancer genes in diagnostic panels.
The hypermutable state in MSI tumors causes accumulation of frameshift mutations that generate neoantigens, immunogenic peptides recognized as non-self by the immune system (150–156). Neoantigen burden is significantly increased in MSI versus microsatellite stable (MSS) CRCs and in LS versus sporadic MSI CRCs (154,155). The neoantigens can be presented at the tumor cell surface in human leukocyte antigen type I (HLA I) molecules, eliciting antitumor cytotoxic T cell responses (150–152,154,157,158). These observations likely explain the high level of lymphocyte infiltration long known as a hallmark of MSI CRCs (159,160) and more recently shown to be a predictor of better prognosis (154,161) similar to MSI (162). The highly immunogenic tumor microenvironment in MSI CRCs is balanced by elevated expression of several immune checkpoint molecules that block antitumor T cell responses, making this subgroup an excellent candidate for treatment using immune checkpoint inhibitors (163,164). Clinical studies have consistently demonstrated efficacy of immune checkpoint inhibition with anti-PD-1 antibodies (pembrolizumab, nivolumab) in MSI tumors from colorectum and many other tissues (165–171). Both drugs have received US Food and Drug Administration approval for treatment of patients with MSI-high or MMR-deficient metastatic CRC, followed by approval of nivolumab combination immunotherapy with anti-CTLA-4 antibody (ipilimumab) for the same indication (169). Similar to MSI, also the presence of POLE and POLD1 mutations, associated with an ultramutator phenotype in tumors, may predict survival benefit from immune checkpoint therapy in CRC and other cancers (172,173).
Conclusions and Future Perspectives
Advances in the dissection of hereditary CRC syndromes offer a remarkable example of how the understanding of human disease has improved in the past 30 years. Interestingly, with the new knowledge, the borders between established CRC syndromes have become blurrier: for example, evidence shows that some LS patients can mimic attenuated FAP (91,174) and few families exhibiting digenic inheritance of CRC have been reported (114,117). Thus, genotype–phenotype diversity in inherited CRC will likely continue to be a source of difficulty for diagnostics (140) and treatment (175). Much further work will be required to better understand the key components of CRC susceptibility and optimal management strategies for most syndromes, though effective targeted treatments already exist for some of the affected signaling pathways (Fig. 3). For example, patients with APC germline mutations respond well to treatment with the nonsteroidal anti-inflammatory drug sulindac and COX inhibitor celecoxib. In these patients, adenoma burden was significantly reduced (176), likely because of the suppression of canonical WNT/β-catenin signaling (177,178). Very recent breakthroughs have been made in LS vaccination by exploiting the recurrent frameshift-induced neoantigens consequent of the defective MMR machinery (179). Dense lymphocyte infiltration accompanies the MSI tumors of LS patients (159,160,180) and specific T cell reactivity to frameshift peptides is detectable already in healthy LS family members without a history of cancer (158). These observations, combined with the fact that LS carcinomas often lose HLA I expression likely because of a selection pressure (181,182), suggest that the LS patient’s immune system effectively recognizes MMR-deficient cells and may control tumor outgrowth. Therefore, vaccination is an attractive option for cancer prevention in the future. Moreover, vaccination could direct the development of MSI tumors toward a surgically curable phenotype by increasing the immunoselective pressure and likelihood of HLA I loss via mutations in beta-2-microglobulin (182); these mutations have been associated with an excellent prognosis with a low risk of relapse (183) and metastases (184,185). The first clinical trials for frameshift peptide vaccines are promising, with low toxicity and high immune response (179).
Similar to the use of MSI-induced frameshift products in LS-diagnosis, tumor-specific mutation spectra or signatures have laid the groundwork for connections between the molecular profiles of cancers and the tumor development (186). One notable advance in the last decade is the detailed characterization of mutational signatures across different cancer types (187,188). As each signature is a reflection of the mutational processes in cancer, especially prominent in samples whose DNA repair machinery is defective (35–37,40,41,43,189), they can serve as a hint toward the underlying cancer-causing mechanisms and serve as potential diagnostic or prognostic biomarkers (190). Furthermore, mutational signatures may provide evidence for accurate classification of variants of unknown significance as pathogenic or benign (191). For example, owing to the discovery of the mutational signature 30 in a breast cancer sample, a discovery of a germline mutation of NTHL1 was made (42). Finding characteristic features, such as MSI or specific mutational signatures, in tumor samples could reflect the germline defect in molecularly unexplained familial CRC or polyposis cases, acting as a starting point for finding the underlying cause of disease. Unfortunately, availability of tumor DNA is often a limiting factor for NGS-based studies. Liquid biopsies could be a reliable source of tumor DNA for such purposes, as significant numbers of clinically actionable variants have been present in cell-free DNA samples when tumor samples were either unavailable or had not yielded any results (192). Similar conclusions can be drawn from studies utilizing mutation-specific PCR (193) and methylation analysis (194,195) of cell-free DNA from patients with metastatic disease. MSI status and mutational signatures observed in cell-free DNA seem to reflect those of the primary tumor as well (196,197).
Despite major advances in the field, CRC remains one of the leading causes of cancer-related death and a major economic burden (198,199) that personalized medicine is thought to alleviate (200). The decreasing costs of genomic research technologies bring the onset of precision medicine closer to reality as more research is carried out. Our current knowledge of germline and somatic changes in CRC predisposition has given us meaningful clinical predictors for the best-characterized syndromes; however, extending this to the rarer or lesser-known syndromes and understanding the genotype–phenotype correlations present in recognized syndromes is still awaiting further research. This includes the analysis of the noncoding genome and disease-modifying variants, which are anticipated to shed light onto the susceptibility to cancer as well as its progression and treatment. The work of cancer genomics projects, such as The Cancer Genome Atlas, The International Cancer Genome Consortium, or the Pan-Cancer Analysis of Whole Genomes Consortium, will likely deepen our understanding of CRC through the development of research tools and techniques able to identify generalized patterns and characteristics of cancer, such as mutational signatures, contributing to the missing factors prohibiting detailed molecular profiling of patient-matched cancer and interpretation of variants of unknown significance.
Currently, many of the syndromes are not surgically curable, and patients need life-long monitoring. With the rate of new technological advances, the next decade will be exciting and will likely yield a better understanding of the genetic and environmental determinants of CRC susceptibility, the phenotype–genotype correlations of the already established susceptibility genes, and optimal management of heritable CRC in its various forms.
Conflicts of Interest statement. None declared.
Funding
The Biomedicum Helsinki Foundation (to A.P.O.); The Finnish Center of Excellence in Tumor Genetics (to L.A.A.); the Jane and Aatos Erkko Foundation (to P.T.P.); the Academy of Finland (grant number 294643 to P.T.P.); the Academy of Finland’s professorship (L.A.A.); the Cancer Foundation Finland (to P.T.P. and L.A.A.); iCAN Digital Precision Cancer Medicine Flagship (to L.A.A.); the Sigrid Juselius Foundation (to P.T.P. and L.A.A.); The Doctoral Programme of Biomedicine of Helsinki University offered a paid doctoral student position to A.P.O.
Contributor Information
Alisa P Olkinuora, Department of Medical and Clinical Genetics, Medicum, University of Helsinki, 00014 Helsinki, Finland; iCAN Digital Precision Cancer Medicine Flagship, University of Helsinki, 00014 Helsinki, Finland.
Päivi T Peltomäki, Department of Medical and Clinical Genetics, Medicum, University of Helsinki, 00014 Helsinki, Finland; iCAN Digital Precision Cancer Medicine Flagship, University of Helsinki, 00014 Helsinki, Finland.
Lauri A Aaltonen, Department of Medical and Clinical Genetics, Medicum, University of Helsinki, 00014 Helsinki, Finland; iCAN Digital Precision Cancer Medicine Flagship, University of Helsinki, 00014 Helsinki, Finland; Applied Tumor Genomics Research Program, Research Programs Unit, University of Helsinki, 00014 Helsinki, Finland.
Kristiina Rajamäki, Department of Medical and Clinical Genetics, Medicum, University of Helsinki, 00014 Helsinki, Finland; Applied Tumor Genomics Research Program, Research Programs Unit, University of Helsinki, 00014 Helsinki, Finland.
References
- 1.Bülow, S., Berk, T. and Neale, K. (2006) The history of familial adenomatous polyposis. Fam. Cancer, 5, 213–220. [DOI] [PubMed] [Google Scholar]
- 2.Boland, C.R. and Lynch, H.T. (2013) The history of Lynch syndrome. Fam. Cancer, 12, 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kerber, R.A., Neklason, D.W., Samowitz, W.S. and Burt, R.W. (2005) Frequency of familial colon cancer and hereditary nonpolyposis colorectal cancer (Lynch syndrome) in a large population database. Fam. Cancer, 4, 239–244. [DOI] [PubMed] [Google Scholar]
- 4.Lichtenstein, P., Holm, N.V., Verkasalo, P.K., Iliadou, A., Kaprio, J., Koskenvuo, M., Pukkala, E., Skytthe, A. and Hemminki, K. (2000) Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med., 343, 78–85. [DOI] [PubMed] [Google Scholar]
- 5.Mork, M.E., Nancy You, Y., Ying, J., Bannon, S.A., Lynch, P.M., Rodriguez-Bigas, M.A. and Vilar, E. (2015) High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer. J. Clin. Oncol., 33, 3544–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeRycke, M.S., Gunawardena, S., Balcom, J.R., Pickart, A.M., Waltman, L.A., French, A.J., McDonnell, S., Riska, S.M., Fogarty, Z.C., Larson, M.C. et al. (2017) Targeted sequencing of 36 known or putative colorectal cancer susceptibility genes. Mol. Genet. Genomic Med., 5, 553–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearlman, R., Frankel, W.L., Swanson, B., Zhao, W., Yilmaz, A., Miller, K., Bacher, J., Bigley, C., Nelsen, L., Goodfellow, P.J. et al. (2017) Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol., 3, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoffel, E.M., Koeppe, E., Everett, J., Ulintz, P., Kiel, M., Osborne, J., Williams, L., Hanson, K., Gruber, S.B. and Rozek, L.S. (2018) Germline genetic features of young individuals with colorectal cancer. Gastroenterology, 154, 897–905.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daca Alvarez, M., Quintana, I., Terradas, M., Mur, P., Balaguer, F. and Valle, L. (2021) The inherited and familial component of early-onset colorectal cancer. Cell, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geboes, K., De Hertogh, G., Van Caillie, M.-A. and Van Eyken, P. (2007) Non-adenomatous colorectal polyposis syndromes. Curr. Diag. Pathol., 13, 479–489. [Google Scholar]
- 11.Groden, J., Thliveris, A., Samowitz, W., Carlson, M., Gelbert, L., Albertsen, H., Joslyn, G., Stevens, J., Spirio, L. and Robertson, M. (1991) Identification and characterization of the familial adenomatous polyposis coli gene. Cell, 66, 589–600. [DOI] [PubMed] [Google Scholar]
- 12.van Lier, M.G.F., Wagner, A., Mathus-Vliegen, E.M.H., Kuipers, E.J., Steyerberg, E.W. and van Leerdam, M.E. (2010) High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am. J. Gastroenterol., 105, 1258–1264 author reply 1265. [DOI] [PubMed] [Google Scholar]
- 13.Howe, J.R., Mitros, F.A. and Summers, R.W. (1998) The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann. Surg. Oncol., 5, 751–756. [DOI] [PubMed] [Google Scholar]
- 14.Pilarski, R. (2019) Hamartoma tumor syndrome: a clinical overview. Cancer, 11, 844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Møller, P., Seppälä, T., Bernstein, I., Holinski-Feder, E., Sala, P., Evans, D.G., Lindblom, A., Macrae, F., Blanco, I., Sijmons, R. et al. (2017) Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut, 66, 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Møller, P., Seppälä, T.T., Bernstein, I., Holinski-Feder, E., Sala, P., Gareth Evans, D., Lindblom, A., Macrae, F., Blanco, I., Sijmons, R.H. et al. (2018) Cancer risk and survival in carriers by gene and gender up to 75 years of age: a report from the prospective Lynch syndrome database. Gut, 67, 1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abraham, S.C., Nobukawa, B., Giardiello, F.M., Hamilton, S.R. and Wu, T.T. (2000) Fundic gland polyps in familial adenomatous polyposis: neoplasms with frequent somatic adenomatous polyposis coli gene alterations. Am. J. Pathol., 157, 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Righetti, A.E.M., Jacomini, C., Parra, R.S., de Almeida, A.L.N.R., Rocha, J.J.R. and Féres, O. (2011) Familial adenomatous polyposis and desmoid tumors. Clinics, 66, 1839–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boveri, T. (1914) Zur Frage der Entstehung maligner Tumoren. Naturwissenschaften, 2, 676–679. [Google Scholar]
- 20.Van den Berghe, H., Vermaelen, K., Mecucci, C., Barbieri, D. and Tricot, G. (1985) The 5q-anomaly. Cancer Genet. Cytogenet., 17, 189–255. [DOI] [PubMed] [Google Scholar]
- 21.Herrera, L., Kakati, S., Gibas, L., Pietrzak, E. and Sandberg, A.A. (1986) Gardner syndrome in a man with an interstitial deletion of 5q. Am. J. Med. Genet., 25, 473–476. [DOI] [PubMed] [Google Scholar]
- 22.Solomon, E., Voss, R., Hall, V., Bodmer, W.F., Jass, J.R., Jeffreys, A.J., Lucibello, F.C., Patel, I. and Rider, S.H. (1987) Chromosome 5 allele loss in human colorectal carcinomas. Nature, 328, 616–619. [DOI] [PubMed] [Google Scholar]
- 23.Leppert, M., Dobbs, M., Scambler, P., O’Connell, P., Nakamura, Y., Stauffer, D., Woodward, S., Burt, R., Hughes, J. and Gardner, E. (1987) The gene for familial polyposis coli maps to the long arm of chromosome 5. Science, 238, 1411–1413. [DOI] [PubMed] [Google Scholar]
- 24.Bodmer, W.F., Bailey, C.J., Bodmer, J., Bussey, H.J.R., Ellis, A., Gorman, P., Lucibello, F.C., Murday, V.A., Rider, S.H., Scambler, P. et al. (1987) Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature, 328, 614–616. [DOI] [PubMed] [Google Scholar]
- 25.Leppert, M., Burt, R., Hughes, J.P., Samowitz, W., Nakamura, Y., Woodward, S., Gardner, E., Lalouel, J.M. and White, R. (1990) Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N. Engl. J. Med., 322, 904–908. [DOI] [PubMed] [Google Scholar]
- 26.Kinzler, K.W., Nilbert, M.C., Su, L.K., Vogelstein, B., Bryan, T.M., Levy, D.B., Smith, K.J., Preisinger, A.C., Hedge, P. and McKechnie, D. (1991) Identification of FAP locus genes from chromosome 5q21. Science, 253, 661–665. [DOI] [PubMed] [Google Scholar]
- 27.Hall, J.G. (1988) Review and hypotheses: somatic mosaicism: observations related to clinical genetics. Am. J. Hum. Genet., 43, 355–363. [PMC free article] [PubMed] [Google Scholar]
- 28.Aretz, S., Stienen, D., Friedrichs, N., Stemmler, S., Uhlhaas, S., Rahner, N., Propping, P. and Friedl, W. (2007) Somatic APC mosaicism: a frequent cause of familial adenomatous polyposis (FAP). Hum. Mutat., 28, 985–992. [DOI] [PubMed] [Google Scholar]
- 29.Spier, I., Drichel, D., Kerick, M., Kirfel, J., Horpaopan, S., Laner, A., Holzapfel, S., Peters, S., Adam, R., Zhao, B. et al. (2016) Low-level APC mutational mosaicism is the underlying cause in a substantial fraction of unexplained colorectal adenomatous polyposis cases. J. Med. Genet., 53, 172–179. [DOI] [PubMed] [Google Scholar]
- 30.Hes, F.J., Nielsen, M., Bik, E.C., Konvalinka, D., Wijnen, J.T., Bakker, E., Vasen, H.F.A., Breuning, M.H. and Tops, C.M.J. (2008) Somatic APC mosaicism: an underestimated cause of polyposis coli. Gut, 57, 71–76. [DOI] [PubMed] [Google Scholar]
- 31.Rustin, R.B., Jagelman, D.G., McGannon, E., Fazio, V.W., Lavery, I.C. and Weakley, F.L. (1990) Spontaneous mutation in familial adenomatous polyposis. Dis Colon Rectum, 33, 52–55. [DOI] [PubMed] [Google Scholar]
- 32.Ripa, R., Bisgaard, M.L., Bülow, S. and Nielsen, F.C. (2002) De novo mutations in familial adenomatous polyposis (FAP). Eur. J. Hum. Genet., 10, 631–637. [DOI] [PubMed] [Google Scholar]
- 33.Aretz, S., Uhlhaas, S., Caspari, R., Mangold, E., Pagenstecher, C., Propping, P. and Friedl, W. (2004) Frequency and parental origin of de novo APC mutations in familial adenomatous polyposis. Eur. J. Hum. Genet., 12, 52–58. [DOI] [PubMed] [Google Scholar]
- 34.Cleary, S.P., Cotterchio, M., Jenkins, M.A., Kim, H., Bristow, R., Green, R., Haile, R., Hopper, J.L., LeMarchand, L., Lindor, N. et al. (2009) Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology, 136, 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas, D., Scot, A.D., Barbey, R., Padula, M. and Boiteux, S. (1997) Inactivation of OGG1 increases the incidence of G:C-->T:A transversions in Saccharomyces cerevisiae: evidence for endogenous oxidative damage to DNA in eukaryotic cells. Mol. Gen. Genet., 254, 171–178. [DOI] [PubMed] [Google Scholar]
- 36.Nghiem, Y., Cabrera, M., Cupples, C.G. and Miller, J.H. (1988) The mutY gene: a mutator locus in Escherichia coli that generates G.C----T.A transversions. Proc. Natl. Acad. Sci. U. S. A., 85, 2709–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moriya, M. and Grollman, A.P. (1993) Mutations in the mutY gene of Escherichia coli enhance the frequency of targeted G:C-->T:A transversions induced by a single 8-oxoguanine residue in single-stranded DNA. Mol. Gen. Genet., 239, 72–76. [DOI] [PubMed] [Google Scholar]
- 38.Al-Tassan, N., Chmiel, N.H., Maynard, J., Fleming, N., Livingston, A.L., Williams, G.T., Hodges, A.K., Davies, D.R., David, S.S., Sampson, J.R. et al. (2002) Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat. Genet., 30, 227–232. [DOI] [PubMed] [Google Scholar]
- 39.Nielsen, M., Joerink-van de Beld, M.C., Jones, N., Vogt, S., Tops, C.M., Vasen, H.F.A., Sampson, J.R., Aretz, S. and Hes, F.J. (2009) Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH-associated polyposis. Gastroenterology, 136, 471–476. [DOI] [PubMed] [Google Scholar]
- 40.Pilati, C., Shinde, J., Alexandrov, L.B., Assié, G., André, T., Hélias-Rodzewicz, Z., Ducoudray, R., Le Corre, D., Zucman-Rossi, J., Emile, J.-F. et al. (2017) Mutational signature analysis identifies MUTYH deficiency in colorectal cancers and adrenocortical carcinomas. J. Pathol., 242, 10–15. [DOI] [PubMed] [Google Scholar]
- 41.Viel, A., Bruselles, A., Meccia, E., Fornasarig, M., Quaia, M., Canzonieri, V., Policicchio, E., Urso, E.D., Agostini, M., Genuardi, M. et al. (2017) A specific mutational signature associated with DNA 8-oxoguanine persistence in MUTYH-defective colorectal cancer. EBioMedicine, 20, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drost, J., van Boxtel, R., Blokzijl, F., Mizutani, T., Sasaki, N., Sasselli, V., de Ligt, J., Behjati, S., Grolleman, J.E., van Wezel, T. et al. (2017) Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science, 358, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grolleman, J.E., de Voer, R.M., Elsayed, F.A., Nielsen, M., Weren, R.D.A., Palles, C., Ligtenberg, M.J.L., Vos, J.R., Ten Broeke, S.W., de Miranda, N.F.C.C. et al. (2019) Mutational signature analysis reveals NTHL1 deficiency to cause a multi-tumor phenotype. Cancer Cell, 35, 256–266.e5. [DOI] [PubMed] [Google Scholar]
- 44.Peltomäki, P., Lothe, R.A., Aaltonen, L.A., Pylkkänen, L., Nyström-Lahti, M., Seruca, R., David, L., Holm, R., Ryberg, D. and Haugen, A. (1993) Microsatellite instability is associated with tumors that characterize the hereditary non-polyposis colorectal carcinoma syndrome. Cancer Res., 53, 5853–5855. [PubMed] [Google Scholar]
- 45.Leach, F.S., Nicolaides, N.C., Papadopoulos, N., Liu, B., Jen, J., Parsons, R., Peltomäki, P., Sistonen, P., Aaltonen, L.A. and Nyström-Lahti, M. (1993) Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell, 75, 1215–1225. [DOI] [PubMed] [Google Scholar]
- 46.Peltomäki, P., Aaltonen, L.A., Sistonen, P., Pylkkänen, L., Mecklin, J.P., Järvinen, H., Green, J.S., Jass, J.R., Weber, J.L. and Leach, F.S. (1993) Genetic mapping of a locus predisposing to human colorectal cancer. Science, 260, 810–812. [DOI] [PubMed] [Google Scholar]
- 47.Fishel, R., Lescoe, M.K., Rao, M.R., Copeland, N.G., Jenkins, N.A., Garber, J., Kane, M. and Kolodner, R. (1993) The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell, 75, 1027–1038. [DOI] [PubMed] [Google Scholar]
- 48.Ott, J., Wang, J. and Leal, S.M. (2015) Genetic linkage analysis in the age of whole-genome sequencing. Nat. Rev. Genet., 16, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teare, M.D., Dawn Teare, M. and Barrett, J.H. (2005) Genetic linkage studies. Lancet, 366, 1036–1044. [DOI] [PubMed] [Google Scholar]
- 50.Plazzer, J.P., Sijmons, R.H., Woods, M.O., Peltomäki, P., Thompson, B., Den Dunnen, J.T. and Macrae, F. (2013) The InSiGHT database: utilizing 100 years of insights into Lynch syndrome. Fam. Cancer, 12, 175–180. [DOI] [PubMed] [Google Scholar]
- 51.Papadopoulos, N., Nicolaides, N.C., Wei, Y.F., Ruben, S.M., Carter, K.C., Rosen, C.A., Haseltine, W.A., Fleischmann, R.D., Fraser, C.M. and Adams, M.D. (1994) Mutation of a mutL homolog in hereditary colon cancer. Science, 263, 1625–1629. [DOI] [PubMed] [Google Scholar]
- 52.Bronner, C.E., Baker, S.M., Morrison, P.T., Warren, G., Smith, L.G., Lescoe, M.K., Kane, M., Earabino, C., Lipford, J. and Lindblom, A. (1994) Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature, 368, 258–261. [DOI] [PubMed] [Google Scholar]
- 53.Lindblom, A., Tannergård, P., Werelius, B. and Nordenskjöld, M. (1993) Genetic mapping of a second locus predisposing to hereditary non-polyposis colon cancer. Nat. Genet., 5, 279–282. [DOI] [PubMed] [Google Scholar]
- 54.Tannergård, P., Zabarovsky, E., Stanbridge, E., Nordenskjöld, M. and Lindblom, A. (1994) Sublocalization of a locus at 3p21.3-23 predisposing to hereditary nonpolyposis colon cancer. Hum. Genet., 94, 210–214. [DOI] [PubMed] [Google Scholar]
- 55.Levinson, G. and Gutman, G.A. (1987) High frequencies of short frameshifts in poly-CA/TG tandem repeats borne by bacteriophage M13 in Escherichia coli K-12. Nucleic Acids Res., 15, 5323–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strand, M., Prolla, T.A., Liskay, R.M. and Petes, T.D. (1993) Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature, 365, 274–276. [DOI] [PubMed] [Google Scholar]
- 57.Aaltonen, L.A., Peltomäki, P., Leach, F.S., Sistonen, P., Pylkkänen, L., Mecklin, J.P., Järvinen, H., Powell, S.M., Jen, J. and Hamilton, S.R. (1993) Clues to the pathogenesis of familial colorectal cancer. Science, 260, 812–816. [DOI] [PubMed] [Google Scholar]
- 58.Ionov, Y., Peinado, M.A., Malkhosyan, S., Shibata, D. and Perucho, M. (1993) Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature, 363, 558–561. [DOI] [PubMed] [Google Scholar]
- 59.Thibodeau, S.N., Bren, G. and Schaid, D. (1993) Microsatellite instability in cancer of the proximal colon. Science, 260, 816–819. [DOI] [PubMed] [Google Scholar]
- 60.Risinger, J.I., Berchuck, A., Kohler, M.F., Watson, P., Lynch, H.T. and Boyd, J. (1993) Genetic instability of microsatellites in endometrial carcinoma. Cancer Res., 53, 5100–5103. [PubMed] [Google Scholar]
- 61.Han, H.J., Yanagisawa, A., Kato, Y., Park, J.G. and Nakamura, Y. (1993) Genetic instability in pancreatic cancer and poorly differentiated type of gastric cancer. Cancer Res., 53, 5087–5089. [PubMed] [Google Scholar]
- 62.Lammi, L., Arte, S., Somer, M., Jarvinen, H., Lahermo, P., Thesleff, I., Pirinen, S. and Nieminen, P. (2004) Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am. J. Hum. Genet., 74, 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Behrens, J., Jerchow, B.A., Würtele, M., Grimm, J., Asbrand, C., Wirtz, R., Kühl, M., Wedlich, D. and Birchmeier, W. (1998) Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science, 280, 596–599. [DOI] [PubMed] [Google Scholar]
- 64.Liu, W., Dong, X., Mai, M., Seelan, R.S., Taniguchi, K., Krishnadath, K.K., Halling, K.C., Cunningham, J.M., Boardman, L.A., Qian, C. et al. (2000) Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat. Genet., 26, 146–147. [DOI] [PubMed] [Google Scholar]
- 65.Marvin, M.L., Mazzoni, S.M., Herron, C.M., Edwards, S., Gruber, S.B. and Petty, E.M. (2011) AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am. J. Med. Genet. A, 155A, 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nelen, M.R., Padberg, G.W., Peeters, E.A., Lin, A.Y., van den Helm, B., Frants, R.R., Coulon, V., Goldstein, A.M., van Reen, M.M., Easton, D.F. et al. (1996) Localization of the gene for Cowden disease to chromosome 10q22-23. Nat. Genet., 13, 114–116. [DOI] [PubMed] [Google Scholar]
- 67.Liaw, D., Marsh, D.J., Li, J., Dahia, P.L., Wang, S.I., Zheng, Z., Bose, S., Call, K.M., Tsou, H.C., Peacocke, M. et al. (1997) Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet., 16, 64–67. [DOI] [PubMed] [Google Scholar]
- 68.Hemminki, A., Tomlinson, I., Markie, D., Järvinen, H., Sistonen, P., Björkqvist, A.M., Knuutila, S., Salovaara, R., Bodmer, W., Shibata, D. et al. (1997) Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat. Genet., 15, 87–90. [DOI] [PubMed] [Google Scholar]
- 69.Hemminki, A., Markie, D., Tomlinson, I., Avizienyte, E., Roth, S., Loukola, A., Bignell, G., Warren, W., Aminoff, M., Höglund, P. et al. (1998) A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature, 391, 184–187. [DOI] [PubMed] [Google Scholar]
- 70.Howe, J.R., Ringold, J.C., Summers, R.W., Mitros, F.A., Nishimura, D.Y. and Stone, E.M. (1998) A gene for familial juvenile polyposis maps to chromosome 18q21.1. Am. J. Hum. Genet., 62, 1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Howe, J.R., Roth, S., Ringold, J.C., Summers, R.W., Järvinen, H.J., Sistonen, P., Tomlinson, I.P., Houlston, R.S., Bevan, S., Mitros, F.A. et al. (1998) Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science, 280, 1086–1088. [DOI] [PubMed] [Google Scholar]
- 72.Houlston, R., Bevan, S., Williams, A., Young, J., Dunlop, M., Rozen, P., Eng, C., Markie, D., Woodford-Richens, K., Rodriguez-Bigas, M.A. et al. (1998) Mutations in DPC4 (SMAD4) cause juvenile polyposis syndrome, but only account for a minority of cases. Hum. Mol. Genet., 7, 1907–1912. [DOI] [PubMed] [Google Scholar]
- 73.Howe, J.R., Bair, J.L., Sayed, M.G., Anderson, M.E., Mitros, F.A., Petersen, G.M., Velculescu, V.E., Traverso, G. and Vogelstein, B. (2001) Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet., 28, 184–187. [DOI] [PubMed] [Google Scholar]
- 74.Brosens, L.A., Langeveld, D., van Hattem, W.A., Giardiello, F.M. and Offerhaus, G.J.A. (2011) Juvenile polyposis syndrome. World J. Gastroenterol., 17, 4839–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hughes, M.J. and Jiricny, J. (1992) The purification of a human mismatch-binding protein and identification of its associated ATPase and helicase activities. J. Biol. Chem., 267, 23876–23882. [PubMed] [Google Scholar]
- 76.Palombo, F., Gallinari, P., Iaccarino, I., Lettieri, T., Hughes, M., D’Arrigo, A., Truong, O., Hsuan, J.J. and Jiricny, J. (1995) GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science, 268, 1912–1914. [DOI] [PubMed] [Google Scholar]
- 77.Drummond, J.T., Li, G.M., Longley, M.J. and Modrich, P. (1995) Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science, 268, 1909–1912. [DOI] [PubMed] [Google Scholar]
- 78.Papadopoulos, N., Nicolaides, N.C., Liu, B., Parsons, R., Lengauer, C., Palombo, F., D’Arrigo, A., Markowitz, S., Willson, J.K. and Kinzler, K.W. (1995) Mutations of GTBP in genetically unstable cells. Science, 268, 1915–1917. [DOI] [PubMed] [Google Scholar]
- 79.Edelmann, W., Yang, K., Umar, A., Heyer, J., Lau, K., Fan, K., Liedtke, W., Cohen, P.E., Kane, M.F., Lipford, J.R. et al. (1997) Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell, 91, 467–477. [DOI] [PubMed] [Google Scholar]
- 80.Akiyama, Y., Sato, H., Yamada, T., Nagasaki, H., Tsuchiya, A., Abe, R. and Yuasa, Y. (1997) Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res., 57, 3920–3923. [PubMed] [Google Scholar]
- 81.Miyaki, M., Konishi, M., Tanaka, K., Kikuchi-Yanoshita, R., Muraoka, M., Yasuno, M., Igari, T., Koike, M., Chiba, M. and Mori, T. (1997) Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat. Genet., 17, 271–272. [DOI] [PubMed] [Google Scholar]
- 82.Wu, Y., Berends, M.J., Mensink, R.G., Kempinga, C., Sijmons, R.H., van Der Zee, A.G., Hollema, H., Kleibeuker, J.H., Buys, C.H. and Hofstra, R.M. (1999) Association of hereditary nonpolyposis colorectal cancer-related tumors displaying low microsatellite instability with MSH6 germline mutations. Am. J. Hum. Genet., 65, 1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arora, S., Yan, H., Cho, I., Fan, H.-Y., Luo, B., Gai, X., Bodian, D.L., Vockley, J.G., Zhou, Y., Handorf, E.A. et al. (2015) Genetic variants that predispose to DNA double-strand breaks in lymphocytes from a subset of patients with familial colorectal carcinomas. Gastroenterology, 149, 1872–1883.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Burrell, R.A., McClelland, S.E., Endesfelder, D., Groth, P., Weller, M.-C., Shaikh, N., Domingo, E., Kanu, N., Dewhurst, S.M., Gronroos, E. et al. (2013) Replication stress links structural and numerical cancer chromosomal instability. Nature, 494, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bennett, S.E., Umar, A., Oshima, J., Monnat, R.J., Jr. and Kunkel, T.A. (1997) Mismatch repair in extracts of Werner syndrome cell lines. Cancer Res., 57, 2956–2960. [PubMed] [Google Scholar]
- 86.Pedrazzi, G., Bachrati, C.Z., Selak, N., Studer, I., Petkovic, M., Hickson, I.D., Jiricny, J. and Stagljar, I. (2003) The Bloom’s syndrome helicase interacts directly with the human DNA mismatch repair protein hMSH6. Biol. Chem., 384, 1155–1164. [DOI] [PubMed] [Google Scholar]
- 87.Palles, C., Cazier, J.-B., Howarth, K.M., Domingo, E., Jones, A.M., Broderick, P., Kemp, Z., Spain, S.L., Guarino, E., Salguero, I. et al. (2013) Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet., 45, 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nieminen, T.T., O’Donohue, M.-F., Wu, Y., Lohi, H., Scherer, S.W., Paterson, A.D., Ellonen, P., Abdel-Rahman, W.M., Valo, S., Mecklin, J.-P. et al. (2014) Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology, 147, 595–598.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weren, R.D.A., Ligtenberg, M.J.L., Kets, C.M., de Voer, R.M., Verwiel, E.T.P., Spruijt, L., van Zelst-Stams, W.A.G., Jongmans, M.C., Gilissen, C., Hehir-Kwa, J.Y. et al. (2015) A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet., 47, 668–671. [DOI] [PubMed] [Google Scholar]
- 90.Adam, R., Spier, I., Zhao, B., Kloth, M., Marquez, J., Hinrichsen, I., Kirfel, J., Tafazzoli, A., Horpaopan, S., Uhlhaas, S. et al. (2016) Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am. J. Hum. Genet., 99, 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Olkinuora, A., Nieminen, T.T., Mårtensson, E., Rohlin, A., Ristimäki, A., Koskenvuo, L., Lepistö, A., Swedish Extended Genetic Analysis of Colorectal Neoplasia (SWEN) Study Group, Gebre-Medhin, S., Nordling, M. et al. (2019) Biallelic germline nonsense variant of MLH3 underlies polyposis predisposition. Genet. Med., 21, 1868–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kemp, Z., Carvajal-Carmona, L., Spain, S., Barclay, E., Gorman, M., Martin, L., Jaeger, E., Brooks, N., Bishop, D.T., Thomas, H. et al. (2006) Evidence for a colorectal cancer susceptibility locus on chromosome 3q21-q24 from a high-density SNP genome-wide linkage scan. Hum. Mol. Genet., 15, 2903–2910. [DOI] [PubMed] [Google Scholar]
- 93.Papaemmanuil, E., Carvajal-Carmona, L., Sellick, G.S., Kemp, Z., Webb, E., Spain, S., Sullivan, K., Barclay, E., Lubbe, S., Jaeger, E. et al. (2008) Deciphering the genetics of hereditary non-syndromic colorectal cancer. Eur. J. Hum. Genet., 16, 1477–1486. [DOI] [PubMed] [Google Scholar]
- 94.Valle, L., de Voer, R.M., Goldberg, Y., Sjursen, W., Försti, A., Ruiz-Ponte, C., Caldés, T., Garré, P., Olsen, M.F., Nordling, M. et al. (2019) Update on genetic predisposition to colorectal cancer and polyposis. Mol. Asp. Med., 69, 10–26. [DOI] [PubMed] [Google Scholar]
- 95.Lindor, N.M., Rabe, K., Petersen, G.M., Haile, R., Casey, G., Baron, J., Gallinger, S., Bapat, B., Aronson, M., Hopper, J. et al. (2005) Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA, 293, 1979–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Broderick, P., Dobbins, S.E., Chubb, D., Kinnersley, B., Dunlop, M.G., Tomlinson, I. and Houlston, R.S. (2017) Validation of recently proposed colorectal cancer susceptibility gene variants in an analysis of families and patients-a systematic review. Gastroenterology, 152, 75–77.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thompson, B.A., Snow, A.K., Koptiuch, C., Kohlmann, W.K., Mooney, R., Johnson, S., Huff, C.D., Yu, Y., Teerlink, C.C., Feng, B.-J. et al. (2020) A novel ribosomal protein S20 variant in a family with unexplained colorectal cancer and polyposis. Clin. Genet., 97, 943–944. [DOI] [PubMed] [Google Scholar]
- 98.Bhar, S., Zhou, F., Reineke, L.C., Morris, D.K., Khincha, P.P., Giri, N., Mirabello, L., Bergstrom, K., Lemon, L.D., Williams, C.L. et al. (2020) Expansion of germline RPS20 mutation phenotype to include Diamond-Blackfan anemia. Hum. Mutat., 41, 1918–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peltomäki, P., Olkinuora, A. and Nieminen, T.T. (2020) Updates in the field of hereditary nonpolyposis colorectal cancer. Expert Rev. Gastroenterol. Hepatol., 14, 707–720. [DOI] [PubMed] [Google Scholar]
- 100.Nieminen, T.T., Pavicic, W., Porkka, N., Kankainen, M., Järvinen, H.J., Lepistö, A. and Peltomäki, P. (2016) Pseudoexons provide a mechanism for allele-specific expression of APC in familial adenomatous polyposis. Oncotarget, 7, 70685–70698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Spier, I., Horpaopan, S., Vogt, S., Uhlhaas, S., Morak, M., Stienen, D., Draaken, M., Ludwig, M., Holinski-Feder, E., Nöthen, M.M. et al. (2012) Deep intronic APC mutations explain a substantial proportion of patients with familial or early-onset adenomatous polyposis. Hum. Mutat., 33, 1045–1050. [DOI] [PubMed] [Google Scholar]
- 102.Aaltonen, L., Johns, L., Järvinen, H., Mecklin, J.-P. and Houlston, R. (2007) Explaining the familial colorectal cancer risk associated with mismatch repair (MMR)-deficient and MMR-stable tumors. Clin. Cancer Res., 13, 356–361. [DOI] [PubMed] [Google Scholar]
- 103.Win, A.K., Jenkins, M.A., Dowty, J.G., Antoniou, A.C., Lee, A., Giles, G.G., Buchanan, D.D., Clendenning, M., Rosty, C., Ahnen, D.J. et al. (2017) Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol. Biomark. Prev., 26, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schubert, S.A., Morreau, H., de Miranda, N.F.C.C. and van Wezel, T. (2020) The missing heritability of familial colorectal cancer. Mutagenesis, 35, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Peters, U., Bien, S. and Zubair, N. (2015) Genetic architecture of colorectal cancer. Gut, 64, 1623–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tenesa, A. and Dunlop, M.G. (2009) New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat. Rev. Genet., 10, 353–358. [DOI] [PubMed] [Google Scholar]
- 107.Huyghe, J.R., Bien, S.A., Harrison, T.A., Kang, H.M., Chen, S., Schmit, S.L., Conti, D.V., Qu, C., Jeon, J., Edlund, C.K. et al. (2019) Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet., 51, 76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Talseth-Palmer, B.A., Wijnen, J.T., Grice, D.M. and Scott, R.J. (2013) Genetic modifiers of cancer risk in Lynch syndrome: a review. Fam. Cancer, 12, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Donald, N., Malik, S., McGuire, J.L. and Monahan, K.J. (2018) The association of low penetrance genetic risk modifiers with colorectal cancer in lynch syndrome patients: a systematic review and meta-analysis. Fam. Cancer, 17, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fahed, A.C., Wang, M., Homburger, J.R., Patel, A.P., Bick, A.G., Neben, C.L., Lai, C., Brockman, D., Philippakis, A., Ellinor, P.T. et al. (2020) Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat. Commun., 11, 3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wiik, M.U., Evans, T.-J., Belhadj, S., Bolton, K.A., Dymerska, D., Jagmohan-Changur, S., Capellá, G., Kurzawski, G., Wijnen, J.T., Valle, L. et al. (2021) A genetic variant in telomerase reverse transcriptase (TERT) modifies cancer risk in Lynch syndrome patients harbouring pathogenic MSH2 variants. Sci. Rep., 11, 11401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jenkins, M.A., Buchanan, D.D., Lai, J., Makalic, E., Dite, G.S., Win, A.K., Clendenning, M., Winship, I.M., Hayes, R.B., Huyghe, J.R. et al. (2021) Assessment of a polygenic risk score for colorectal cancer to predict risk of Lynch syndrome colorectal cancer. JNCI Cancer Spectr, 5, kab022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Arnau-Collell, C., Soares de Lima, Y., Díaz-Gay, M., Muñoz, J., Carballal, S., Bonjoch, L., Moreira, L., Lozano, J.J., Ocaña, T., Cuatrecasas, M. et al. (2020) Colorectal cancer genetic variants are also associated with serrated polyposis syndrome susceptibility. J. Med. Genet., 57, 677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Morak, M., Massdorf, T., Sykora, H., Kerscher, M. and Holinski-Feder, E. (2011) First evidence for digenic inheritance in hereditary colorectal cancer by mutations in the base excision repair genes. Eur. J. Cancer, 47, 1046–1055. [DOI] [PubMed] [Google Scholar]
- 115.Ciavarella, M., Miccoli, S., Prossomariti, A., Pippucci, T., Bonora, E., Buscherini, F., Palombo, F., Zuntini, R., Balbi, T., Ceccarelli, C. et al. (2018) Somatic APC mosaicism and oligogenic inheritance in genetically unsolved colorectal adenomatous polyposis patients. Eur. J. Hum. Genet., 26, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fernández-Rozadilla, C., Álvarez-Barona, M., Quintana, I., López-Novo, A., Amigo, J., Cameselle-Teijeiro, J.M., Roman, E., Gonzalez, D., Llor, X., Bujanda, L. et al. (2021) Exome sequencing of early-onset patients supports genetic heterogeneity in colorectal cancer. Sci. Rep., 11, 11135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schubert, S.A., Ruano, D., Tiersma, Y., Drost, M., de Wind, N., Nielsen, M., van Hest, L.P., Morreau, H., de Miranda, N.F.C.C. and van Wezel, T. (2020) Digenic inheritance of MSH6 and MUTYH variants in familial colorectal cancer. Genes Chromosomes Cancer, 59, 697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stoffel, E.M. and Richard Boland, C. (2015) Genetics and genetic testing in hereditary colorectal cancer. Gastroenterology, 149, 1191–1203.e2. [DOI] [PubMed] [Google Scholar]
- 119.Vasen, H.F., Mecklin, J.P., Khan, P.M. and Lynch, H.T. (1991) The International Collaborative group on hereditary non-polyposis colorectal cancer (ICG-HNPCC). Dis Colon Rectum, 34, 424–425. [DOI] [PubMed] [Google Scholar]
- 120.Vasen, H.F., Watson, P., Mecklin, J.P. and Lynch, H.T. (1999) New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology, 116, 1453–1456. [DOI] [PubMed] [Google Scholar]
- 121.Rodriguez-Bigas, M.A., Boland, C.R., Hamilton, S.R., Henson, D.E., Jass, J.R., Khan, P.M., Lynch, H., Perucho, M., Smyrk, T., Sobin, L. et al. (1997) A National Cancer Institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and Bethesda guidelines. J. Natl. Cancer Inst., 89, 1758–1762. [DOI] [PubMed] [Google Scholar]
- 122.Umar, A., Boland, C.R., Terdiman, J.P., Syngal, S., de la Chapelle, A., Rüschoff, J., Fishel, R., Lindor, N.M., Burgart, L.J., Hamelin, R. et al. (2004) Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst., 96, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group (2009) Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet. Med., 11, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Palomaki, G.E., McClain, M.R., Melillo, S., Hampel, H.L. and Thibodeau, S.N. (2009) EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet. Med., 11, 42–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Aaltonen, L.A., Peltomäki, P., Mecklin, J.P., Järvinen, H., Jass, J.R., Green, J.S., Lynch, H.T., Watson, P., Tallqvist, G. and Juhola, M. (1994) Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Res., 54, 1645–1648. [PubMed] [Google Scholar]
- 126.Aaltonen, L.A., Salovaara, R., Kristo, P., Canzian, F., Hemminki, A., Peltomäki, P., Chadwick, R.B., Kääriäinen, H., Eskelinen, M., Järvinen, H. et al. (1998) Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N. Engl. J. Med., 338, 1481–1487. [DOI] [PubMed] [Google Scholar]
- 127.Salovaara, R., Loukola, A., Kristo, P., Kääriäinen, H., Ahtola, H., Eskelinen, M., Härkönen, N., Julkunen, R., Kangas, E., Ojala, S. et al. (2000) Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J. Clin. Oncol., 18, 2193–2200. [DOI] [PubMed] [Google Scholar]
- 128.Zhang, L. (2008) Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J. Mol. Diagn., 10, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Boland, C.R., Richard Boland, C. and Goel, A. (2010) Microsatellite instability in colorectal cancer. Gastroenterology, 138, 2073–2087.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carethers, J.M. (2014) Differentiating Lynch-like from Lynch syndrome. Gastroenterology, 146, 602–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Parsons, M.T., Buchanan, D.D., Thompson, B., Young, J.P. and Spurdle, A.B. (2012) Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J. Med. Genet., 49, 151–157. [DOI] [PubMed] [Google Scholar]
- 132.Hampel, H., Frankel, W.L., Martin, E., Arnold, M., Khanduja, K., Kuebler, P., Nakagawa, H., Sotamaa, K., Prior, T.W., Westman, J. et al. (2005) Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N. Engl. J. Med., 352, 1851–1860. [DOI] [PubMed] [Google Scholar]
- 133.Piñol, V., Castells, A., Andreu, M., Castellví-Bel, S., Alenda, C., Llor, X., Xicola, R.M., Rodríguez-Moranta, F., Payá, A., Jover, R. et al. (2005) Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA, 293, 1986–1994. [DOI] [PubMed] [Google Scholar]
- 134.Shia, J. (2008) Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. the utility of immunohistochemistry. J. Mol. Diagn., 10, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gallego, C.J., Shirts, B.H., Bennette, C.S., Guzauskas, G., Amendola, L.M., Horike-Pyne, M., Hisama, F.M., Pritchard, C.C., Grady, W.M., Burke, W. et al. (2015) Next-generation sequencing panels for the diagnosis of colorectal cancer and polyposis syndromes: a cost-effectiveness analysis. J. Clin. Oncol., 33, 2084–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yurgelun, M.B., Kulke, M.H., Fuchs, C.S., Allen, B.A., Uno, H., Hornick, J.L., Ukaegbu, C.I., Brais, L.K., McNamara, P.G., Mayer, R.J. et al. (2017) Cancer susceptibility gene mutations in individuals with colorectal cancer. J. Clin. Oncol., 35, 1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kraus, C., Rau, T.T., Lux, P., Erlenbach-Wünsch, K., Löhr, S., Krumbiegel, M., Thiel, C.T., Stöhr, R., Agaimy, A., Croner, R.S. et al. (2015) Comprehensive screening for mutations associated with colorectal cancer in unselected cases reveals penetrant and nonpenetrant mutations. Int. J. Cancer, 136, E559–E568. [DOI] [PubMed] [Google Scholar]
- 138.Cragun, D., Radford, C., Dolinsky, J.S., Caldwell, M., Chao, E. and Pal, T. (2014) Panel-based testing for inherited colorectal cancer: a descriptive study of clinical testing performed by a US laboratory. Clin. Genet., 86, 510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hansen, M.F., Johansen, J., Sylvander, A.E., Bjørnevoll, I., Talseth-Palmer, B.A., Lavik, L.A.S., Xavier, A., Engebretsen, L.F., Scott, R.J., Drabløs, F. et al. (2017) Use of multigene-panel identifies pathogenic variants in several CRC-predisposing genes in patients previously tested for Lynch syndrome. Clin. Genet., 92, 405–414. [DOI] [PubMed] [Google Scholar]
- 140.Rohlin, A., Rambech, E., Kvist, A., Törngren, T., Eiengård, F., Lundstam, U., Zagoras, T., Gebre-Medhin, S., Borg, Å., Björk, J. et al. (2017) Expanding the genotype-phenotype spectrum in hereditary colorectal cancer by gene panel testing. Fam. Cancer, 16, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Martin-Morales, L., Rofes, P., Diaz-Rubio, E., Llovet, P., Lorca, V., Bando, I., Perez-Segura, P., de la Hoya, M., Garre, P., Garcia-Barberan, V. et al. (2018) Novel genetic mutations detected by multigene panel are associated with hereditary colorectal cancer predisposition. PLoS One, 13, e0203885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.LaDuca, H., Stuenkel, A.J., Dolinsky, J.S., Keiles, S., Tandy, S., Pesaran, T., Chen, E., Gau, C.-L., Palmaer, E., Shoaepour, K. et al. (2014) Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet. Med., 16, 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gong, R., He, Y., Liu, X.-Y., Wang, H.-Y., Sun, L.-Y., Yang, X.-H., Li, B., Cao, X.-K., Ye, Z.-L., Kong, L.-H. et al. (2019) Mutation spectrum of germline cancer susceptibility genes among unselected Chinese colorectal cancer patients. Cancer Manag. Res., 11, 3721–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Uson, P.L.S., Jr., Riegert-Johnson, D., Boardman, L., Kisiel, J., Mountjoy, L., Patel, N., Lizaola-Mayo, B., Borad, M.J., Ahn, D., Sonbol, M.B. et al. (2021) Germline cancer susceptibility gene testing in unselected patients with colorectal adenocarcinoma: a multicenter prospective study. Clin. Gastroenterol. Hepatol., S1542-3565(21)00447-X. [DOI] [PubMed] [Google Scholar]
- 145.Yurgelun, M.B., Allen, B., Kaldate, R.R., Bowles, K.R., Judkins, T., Kaushik, P., Roa, B.B., Wenstrup, R.J., Hartman, A.-R. and Syngal, S. (2015) Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology, 149, 604–13.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Susswein, L.R., Marshall, M.L., Nusbaum, R., Vogel Postula, K.J., Weissman, S.M., Yackowski, L., Vaccari, E.M., Bissonnette, J., Booker, J.K., Cremona, M.L. et al. (2016) Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med., 18, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fujita, M., Liu, X., Iwasaki, Y., Terao, C., Mizukami, K., Kawakami, E., Takata, S., Inai, C., Aoi, T., Mizukoshi, M. et al. (2020) Population-based screening for hereditary colorectal cancer variants in Japan. Clin. Gastroenterol. Hepatol., S1542-3565(20)31664-5. [DOI] [PubMed] [Google Scholar]
- 148.Lorans, M., Dow, E., Macrae, F.A., Winship, I.M. and Buchanan, D.D. (2018) Update on hereditary colorectal cancer: improving the clinical utility of multigene panel testing. Clin. Colorectal Cancer, 17, e293–e305. [DOI] [PubMed] [Google Scholar]
- 149.Heald, B., the Collaborative Group of the Americas on Inherited Gastrointestinal Cancer, Hampel, H., Church, J., Dudley, B., Hall, M.J., Mork, M.E., Singh, A., Stoffel, E., Stoll, J. et al. (2020) Collaborative Group of the Americas on Inherited Gastrointestinal Cancer Position statement on multigene panel testing for patients with colorectal cancer and/or polyposis. Fam. Cancer, 19, 223–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Saeterdal, I., Bjørheim, J., Lislerud, K., Gjertsen, M.K., Bukholm, I.K., Olsen, O.C., Nesland, J.M., Eriksen, J.A., Møller, M., Lindblom, A. et al. (2001) Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. U. S. A., 98, 13255–13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Linnebacher, M., Gebert, J., Rudy, W., Woerner, S., Yuan, Y.P., Bork, P. and von Knebel Doeberitz, M. (2001) Frameshift peptide-derived T-cell epitopes: a source of novel tumor-specific antigens. Int. J. Cancer, 93, 6–11. [DOI] [PubMed] [Google Scholar]
- 152.Tougeron, D., Fauquembergue, E., Rouquette, A., Le Pessot, F., Sesboüé, R., Laurent, M., Berthet, P., Mauillon, J., Di Fiore, F., Sabourin, J.-C. et al. (2009) Tumor-infiltrating lymphocytes in colorectal cancers with microsatellite instability are correlated with the number and spectrum of frameshift mutations. Mod. Pathol., 22, 1186–1195. [DOI] [PubMed] [Google Scholar]
- 153.Williams, D.S., Bird, M.J., Jorissen, R.N., Yu, Y.L., Walker, F., Zhang, H.H., Nice, E.C. and Burgess, A.W. (2010) Nonsense mediated decay resistant mutations are a source of expressed mutant proteins in colon cancer cell lines with microsatellite instability. PLoS One, 5, e16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mlecnik, B., Bindea, G., Angell, H.K., Maby, P., Angelova, M., Tougeron, D., Church, S.E., Lafontaine, L., Fischer, M., Fredriksen, T. et al. (2016) Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity, 44, 698–711. [DOI] [PubMed] [Google Scholar]
- 155.Liu, G.-C., Liu, R.-Y., Yan, J.-P., An, X., Jiang, W., Ling, Y.-H., Chen, J.-W., Bei, J.-X., Zuo, X.-Y., Cai, M.-Y. et al. (2018) The heterogeneity between Lynch-associated and sporadic MMR deficiency in colorectal cancers. J. Natl. Cancer Inst., 110, 975–984. [DOI] [PubMed] [Google Scholar]
- 156.Ballhausen, A., Przybilla, M.J., Jendrusch, M., Haupt, S., Pfaffendorf, E., Seidler, F., Witt, J., Hernandez Sanchez, A., Urban, K., Draxlbauer, M. et al. (2020) The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat. Commun., 11, 4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Maby, P., Tougeron, D., Hamieh, M., Mlecnik, B., Kora, H., Bindea, G., Angell, H.K., Fredriksen, T., Elie, N., Fauquembergue, E. et al. (2015) Correlation between density of CD8 T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res., 75, 3446–3455. [DOI] [PubMed] [Google Scholar]
- 158.Schwitalle, Y., Kloor, M., Eiermann, S., Linnebacher, M., Kienle, P., Knaebel, H.P., Tariverdian, M., Benner, A. and von Knebel Doeberitz, M. (2008) Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology, 134, 988–997. [DOI] [PubMed] [Google Scholar]
- 159.Kim, H., Jen, J., Vogelstein, B. and Hamilton, S.R. (1994) Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am. J. Pathol., 145, 148–156. [PMC free article] [PubMed] [Google Scholar]
- 160.Smyrk, T.C., Watson, P., Kaul, K. and Lynch, H.T. (2001) Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer, 91, 2417–2422. [PubMed] [Google Scholar]
- 161.Galon, J., Costes, A., Sanchez-Cabo, F., Kirilovsky, A., Mlecnik, B., Lagorce-Pagès, C., Tosolini, M., Camus, M., Berger, A., Wind, P. et al. (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science, 313, 1960–1964. [DOI] [PubMed] [Google Scholar]
- 162.Popat, S., Hubner, R. and Houlston, R.S. (2005) Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol., 23, 609–618. [DOI] [PubMed] [Google Scholar]
- 163.Llosa, N.J., Cruise, M., Tam, A., Wicks, E.C., Hechenbleikner, E.M., Taube, J.M., Blosser, R.L., Fan, H., Wang, H., Luber, B.S. et al. (2015) The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov., 5, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Xiao, Y. and Freeman, G.J. (2015) The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov., 5, 16–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Brahmer, J.R., Drake, C.G., Wollner, I., Powderly, J.D., Picus, J., Sharfman, W.H., Stankevich, E., Pons, A., Salay, T.M., McMiller, T.L. et al. (2010) Phase I study of single-agent anti–programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol., 28, 3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Le, D.T., Uram, J.N., Wang, H., Bartlett, B.R., Kemberling, H., Eyring, A.D., Skora, A.D., Luber, B.S., Azad, N.S., Laheru, D. et al. (2015) PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med., 372, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Le, D.T., Durham, J.N., Smith, K.N., Wang, H., Bartlett, B.R., Aulakh, L.K., Lu, S., Kemberling, H., Wilt, C., Luber, B.S. et al. (2017) Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science, 357, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Overman, M.J., McDermott, R., Leach, J.L., Lonardi, S., Lenz, H.-J., Morse, M.A., Desai, J., Hill, A., Axelson, M., Moss, R.A. et al. (2017) Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol., 18, 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Overman, M.J., Lonardi, S., Wong, K.Y.M., Lenz, H.-J., Gelsomino, F., Aglietta, M., Morse, M.A., Van Cutsem, E., McDermott, R., Hill, A. et al. (2018) Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol., 36, 773–779. [DOI] [PubMed] [Google Scholar]
- 170.Marabelle, A., Le, D.T., Ascierto, P.A., Di Giacomo, A.M., De Jesus-Acosta, A., Delord, J.-P., Geva, R., Gottfried, M., Penel, N., Hansen, A.R. et al. (2020) Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J. Clin. Oncol., 38, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Le, D.T., Kim, T.W., Van Cutsem, E., Geva, R., Jäger, D., Hara, H., Burge, M., O’Neil, B., Kavan, P., Yoshino, T. et al. (2020) Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J. Clin. Oncol., 38, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang, F., Zhao, Q., Wang, Y.-N., Jin, Y., He, M.-M., Liu, Z.-X. and Xu, R.-H. (2019) Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol., 5, 1504–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mehnert, J.M., Panda, A., Zhong, H., Hirshfield, K., Damare, S., Lane, K., Sokol, L., Stein, M.N., Rodriguez-Rodriquez, L., Kaufman, H.L. et al. (2016) Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J. Clin. Invest., 126, 2334–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kalady, M.F., Kravochuck, S.E., Heald, B., Burke, C.A. and Church, J.M. (2015) Defining the adenoma burden in lynch syndrome. Dis Colon Rectum, 58, 388–392. [DOI] [PubMed] [Google Scholar]
- 175.Roukos, D.H., Katsios, C. and Liakakos, T. (2010) Genotype-phenotype map and molecular networks: a promising solution in overcoming colorectal cancer resistance to targeted treatment. Expert. Rev. Mol. Diagn., 10, 541–545. [DOI] [PubMed] [Google Scholar]
- 176.Kim, B. and Giardiello, F.M. (2011) Chemoprevention in familial adenomatous polyposis. Best Pract. Res. Clin. Gastroenterol., 25, 607–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lee, H.-J., Wang, N.X., Shi, D.-L. and Zheng, J.J. (2009) Sulindac inhibits canonical Wnt signaling by blocking the PDZ domain of the protein dishevelled. Angew. Chem. Int. Ed. Engl., 48, 6448–6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Egashira, I., Takahashi-Yanaga, F., Nishida, R., Arioka, M., Igawa, K., Tomooka, K., Nakatsu, Y., Tsuzuki, T., Nakabeppu, Y., Kitazono, T. et al. (2017) Celecoxib and 2,5-dimethylcelecoxib inhibit intestinal cancer growth by suppressing the Wnt/β-catenin signaling pathway. Cancer Sci., 108, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kloor, M., Reuschenbach, M., Pauligk, C., Karbach, J., Rafiyan, M.-R., Al-Batran, S.-E., Tariverdian, M., Jäger, E. and von Knebel Doeberitz, M. (2020) A frameshift peptide neoantigen-based vaccine for mismatch repair-deficient cancers: a phase I/IIa clinical trial. Clin. Cancer Res., 26, 4503–4510. [DOI] [PubMed] [Google Scholar]
- 180.Risio, M., Reato, G., di Celle, P.F., Fizzotti, M., Rossini, F.P. and Foà, R. (1996) Microsatellite instability is associated with the histological features of the tumor in nonfamilial colorectal cancer. Cancer Res., 56, 5470–5474. [PubMed] [Google Scholar]
- 181.Bernal, M., García-Alcalde, F., Concha, A., Cano, C., Blanco, A., Garrido, F. and Ruiz-Cabello, F. (2012) Genome-wide differential genetic profiling characterizes colorectal cancers with genetic instability and specific routes to HLA class I loss and immune escape. Cancer Immunol. Immunother., 61, 803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kloor, M., Becker, C., Benner, A., Woerner, S.M., Gebert, J., Ferrone, S. and von Knebel Doeberitz, M. (2005) Immunoselective pressure and human leukocyte antigen class I antigen machinery defects in microsatellite unstable colorectal cancers. Cancer Res., 65, 6418–6424. [DOI] [PubMed] [Google Scholar]
- 183.Tikidzhieva, A., Benner, A., Michel, S., Formentini, A., Link, K.-H., Dippold, W., von Knebel Doeberitz, M., Kornmann, M. and Kloor, M. (2012) Microsatellite instability and Beta2-microglobulin mutations as prognostic markers in colon cancer: results of the FOGT-4 trial. Br. J. Cancer, 106, 1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kloor, M., Michel, S., Buckowitz, B., Rüschoff, J., Büttner, R., Holinski-Feder, E., Dippold, W., Wagner, R., Tariverdian, M., Benner, A. et al. (2007) Beta2-microglobulin mutations in microsatellite unstable colorectal tumors. Int. J. Cancer, 121, 454–458. [DOI] [PubMed] [Google Scholar]
- 185.Koelzer, V.H., Baker, K., Kassahn, D., Baumhoer, D. and Zlobec, I. (2012) Prognostic impact of β-2-microglobulin expression in colorectal cancers stratified by mismatch repair status. J. Clin. Pathol., 65, 996–1002. [DOI] [PubMed] [Google Scholar]
- 186.Pfeifer, G.P. and Besaratinia, A. (2009) Mutational spectra of human cancer. Hum. Genet., 125, 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Alexandrov, L.B., Nik-Zainal, S., Wedge, D.C., Aparicio, S.A.J.R., Behjati, S., Biankin, A.V., Bignell, G.R., Bolli, N., Borg, A., Børresen-Dale, A.-L. et al. (2013) Signatures of mutational processes in human cancer. Nature, 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Alexandrov, L.B., Kim, J., Haradhvala, N.J., Huang, M.N., Tian Ng, A.W., Wu, Y., Boot, A., Covington, K.R., Gordenin, D.A., Bergstrom, E.N. et al. (2020) The repertoire of mutational signatures in human cancer. Nature, 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Haradhvala, N.J., Kim, J., Maruvka, Y.E., Polak, P., Rosebrock, D., Livitz, D., Hess, J.M., Leshchiner, I., Kamburov, A., Mouw, K.W. et al. (2018) Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun., 9, 1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium (2020) Pan-cancer analysis of whole genomes. Nature, 578, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Van Hoeck, A., Tjoonk, N.H., van Boxtel, R. and Cuppen, E. (2019) Portrait of a cancer: mutational signature analyses for cancer diagnostics. BMC Cancer, 19, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tsui, D.W.Y., Cheng, M.L., Shady, M., Yang, J.L., Stephens, D., Won, H., Srinivasan, P., Huberman, K., Meng, F., Jing, X. et al. (2021) Tumor fraction-guided cell-free DNA profiling in metastatic solid tumor patients. Genome Med., 13, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bettegowda, C., Sausen, M., Leary, R.J., Kinde, I., Wang, Y., Agrawal, N., Bartlett, B.R., Wang, H., Luber, B., Alani, R.M. et al. (2014) Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med., 6, 224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Shen, S.Y., Singhania, R., Fehringer, G., Chakravarthy, A., Roehrl, M.H.A., Chadwick, D., Zuzarte, P.C., Borgida, A., Wang, T.T., Li, T. et al. (2018) Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature, 563, 579–583. [DOI] [PubMed] [Google Scholar]
- 195.Liu, M.C., Oxnard, G.R., Klein, E.A., Swanton, C., Seiden, M.V. and CCGA Consortium (2020) Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol., 31, 745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Willis, J., Lefterova, M.I., Artyomenko, A., Kasi, P.M., Nakamura, Y., Mody, K., Catenacci, D.V.T., Fakih, M., Barbacioru, C., Zhao, J. et al. (2019) Validation of microsatellite instability detection using a comprehensive plasma-based genotyping panel. Clin. Cancer Res., 25, 7035–7045. [DOI] [PubMed] [Google Scholar]
- 197.Kong, S.L., Liu, X., Suhaimi, N.-A.M., Koh, K.J.H., Hu, M., Lee, D.Y.S., Cima, I., Phyo, W.M., Lee, E.X.W., Tai, J.A. et al. (2017) Molecular characterization of circulating colorectal tumor cells defines genetic signatures for individualized cancer care. Oncotarget, 8, 68026–68037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mariotto, A.B., Warren, J.L., Zeruto, C., Coughlan, D., Barrett, M.J., Zhao, L. and Yabroff, K.R. (2020) Cancer-attributable medical costs for colorectal cancer patients by phases of care: what is the effect of a prior cancer history? J. Natl. Cancer Inst. Monogr., 2020, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Paszat, L., Sutradhar, R., Luo, J., Rabeneck, L., Tinmouth, J. and Baxter, N.N. (2021) Overall health care cost during the year following diagnosis of colorectal cancer stratified by history of colorectal evaluative procedures. J.Can. Assoc. Gastroenterol, gwab001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Schilsky, R.L. (2010) Personalized medicine in oncology: the future is now. Nat. Rev. Drug Discov., 9, 363–366. [DOI] [PubMed] [Google Scholar]
- 201.Järvinen, H.J. (1992) Epidemiology of familial adenomatous polyposis in Finland: impact of family screening on the colorectal cancer rate and survival. Gut, 33, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Half, E., Bercovich, D. and Rozen, P. (2009) Familial adenomatous polyposis. Orphanet J. Rare Dis., 4, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.InSiGHT . (2016) InSiGHT. https://www.insight-group.org/ (accessed Jun 30, 2021).
- 204.Hamzaoui, N., Alarcon, F., Leulliot, N., Guimbaud, R., Buecher, B., Colas, C., Corsini, C., Nuel, G., Terris, B., Laurent-Puig, P. et al. (2020) Genetic, structural, and functional characterization of POLE polymerase proofreading variants allows cancer risk prediction. Genet. Med., 22, 1533–1541. [DOI] [PubMed] [Google Scholar]
- 205.Kuiper, R.P., Nielsen, M., De Voer, R.M. and Hoogerbrugge, N. (2020) NTHL1 tumor syndrome. In GeneReviews® [Internet]. University of Washington, Seattle. [PubMed] [Google Scholar]
- 206.Nielsen, M., Infante, E. and Brand, R. (2012) Polyposis. In Adam, M.P., Ardinger, H.H., Pagon, R.A. et al. (eds), GeneReviews. University of Washington, Seattle, Seattle (WA). [Google Scholar]
- 207.Yehia, L. and Eng, C. (2001) Hamartoma tumor syndrome. In Adam, M.P., Ardinger, H.H., Pagon, R.A. et al. (eds), GeneReviews. University of Washington, Seattle, Seattle (WA). [Google Scholar]
- 208.Jasperson, K.W., Tuohy, T.M., Neklason, D.W. and Burt, R.W. (2010) Hereditary and familial colon cancer. Gastroenterology, 138, 2044–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.American Cancer Society . American Cancer Society. https://www.cancer.org (accessed Jun 30, 2021).
- 210.Shiovitz, S., Copeland, W.K., Passarelli, M.N., Burnett-Hartman, A.N., Grady, W.M., Potter, J.D., Gallinger, S., Buchanan, D.D., Rosty, C., Win, A.K. et al. (2014) Characterisation of familial colorectal cancer type X, Lynch syndrome, and non-familial colorectal cancer. Br. J. Cancer, 111, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Steinke, V., Holzapfel, S., Loeffler, M., Holinski-Feder, E., Morak, M., Schackert, H.K., Görgens, H., Pox, C., Royer-Pokora, B., von Knebel-Doeberitz, M. et al. (2014) Evaluating the performance of clinical criteria for predicting mismatch repair gene mutations in Lynch syndrome: a comprehensive analysis of 3,671 families. Int. J. Cancer, 135, 69–77. [DOI] [PubMed] [Google Scholar]
- 212.Antelo, M., Golubicki, M., Roca, E., Mendez, G., Carballido, M., Iseas, S., Cuatrecasas, M., Moreira, L., Sanchez, A., Carballal, S. et al. (2019) Lynch-like syndrome is as frequent as Lynch syndrome in early-onset nonfamilial nonpolyposis colorectal cancer. Int. J. Cancer, 145, 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Chika, N., Eguchi, H., Kumamoto, K., Suzuki, O., Ishibashi, K., Tachikawa, T., Akagi, K., Tamaru, J.-I., Okazaki, Y. and Ishida, H. (2017) Prevalence of Lynch syndrome and Lynch-like syndrome among patients with colorectal cancer in a Japanese hospital-based population. Jpn. J. Clin. Oncol., 47, 108–117. [DOI] [PubMed] [Google Scholar]
- 214.Jung, B., Staudacher, J.J. and Beauchamp, D. (2017) Transforming growth factor β superfamily signaling in development of colorectal cancer. Gastroenterology, 152, 36–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Salvatore, L., Calegari, M.A., Loupakis, F., Fassan, M., Di Stefano, B., Bensi, M., Bria, E. and Tortora, G. (2019) PTEN in colorectal cancer: shedding light on its role as predictor and target. Cancer, 11, 1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Liu, D., Keijzers, G. and Rasmussen, L.J. (2017) DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res., 773, 174–187. [DOI] [PubMed] [Google Scholar]
- 217.Zhan, T., Rindtorff, N. and Boutros, M. (2017) Wnt signaling in cancer. Oncogene, 36, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Beard, W.A., Horton, J.K., Prasad, R. and Wilson, S.H. (2019) Eukaryotic base excision repair: new approaches shine light on mechanism. Annu. Rev. Biochem., 88, 137–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Pavlov, Y.I., Zhuk, A.S. and Stepchenkova, E.I. (2020) DNA polymerases at the eukaryotic replication fork thirty years after: connection to cancer. Cancer, 12, 3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Shackelford, D.B. and Shaw, R.J. (2009) The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer, 9, 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Nishisho, I., Nakamura, Y., Miyoshi, Y., Miki, Y., Ando, H., Horii, A., Koyama, K., Utsunomiya, J., Baba, S. and Hedge, P. (1991) Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science, 253, 665–669. [DOI] [PubMed] [Google Scholar]
- 222.Joslyn, G., Carlson, M., Thliveris, A., Albertsen, H., Gelbert, L., Samowitz, W., Groden, J., Stevens, J., Spirio, L. and Robertson, M. (1991) Identification of deletion mutations and three new genes at the familial polyposis locus. Cell, 66, 601–613. [DOI] [PubMed] [Google Scholar]
- 223.Nieminen, T.T., Abdel-Rahman, W.M., Ristimäki, A., Lappalainen, M., Lahermo, P., Mecklin, J.-P., Järvinen, H.J. and Peltomäki, P. (2011) BMPR1A mutations in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology, 141, e23–e26. [DOI] [PubMed] [Google Scholar]
- 224.Bellido, F., Sowada, N., Mur, P., Lázaro, C., Pons, T., Valdés-Mas, R., Pineda, M., Aiza, G., Iglesias, S., Soto, J.L. et al. (2018) Association between germline mutations in BRF1, a subunit of the RNA polymerase III transcription complex, and hereditary colorectal cancer. Gastroenterology, 154, 181–194.e20. [DOI] [PubMed] [Google Scholar]
- 225.Borck, G., Hög, F., Dentici, M.L., Tan, P.L., Sowada, N., Medeira, A., Gueneau, L., Thiele, H., Kousi, M., Lepri, F. et al. (2015) BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies. Genome Res., 25, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chan, T.L., Yuen, S.T., Kong, C.K., Chan, Y.W., Chan, A.S.Y., Ng, W.F., Tsui, W.Y., Lo, M.W.S., Tam, W.Y., Li, V.S.W. et al. (2006) Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat. Genet., 38, 1178–1183. [DOI] [PubMed] [Google Scholar]
- 227.Ligtenberg, M.J.L., Kuiper, R.P., Chan, T.L., Goossens, M., Hebeda, K.M., Voorendt, M., Lee, T.Y.H., Bodmer, D., Hoenselaar, E., Hendriks-Cornelissen, S.J.B. et al. (2009) Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet., 41, 112–117. [DOI] [PubMed] [Google Scholar]
- 228.Sivagnanam, M., Mueller, J.L., Lee, H., Chen, Z., Nelson, S.F., Turner, D., Zlotkin, S.H., Pencharz, P.B., Ngan, B.-Y., Libiger, O. et al. (2008) Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology, 135, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Troelstra, C., van Gool, A., de Wit, J., Vermeulen, W., Bootsma, D. and Hoeijmakers, J.H. (1992) ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell, 71, 939–953. [DOI] [PubMed] [Google Scholar]
- 230.Mallery, D.L., Tanganelli, B., Colella, S., Steingrimsdottir, H., van Gool, A.J., Troelstra, C., Stefanini, M. and Lehmann, A.R. (1998) Molecular analysis of mutations in the CSB (ERCC6) gene in patients with Cockayne syndrome. Am. J. Hum. Genet., 62, 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Bonjoch, L., Franch-Expósito, S., Garre, P., Belhadj, S., Muñoz, J., Arnau-Collell, C., Díaz-Gay, M., Gratacós-Mulleras, A., Raimondi, G., Esteban-Jurado, C. et al. (2020) Germline mutations in FAF1 are associated with hereditary colorectal cancer. Gastroenterology, 159, 227–240.e7. [DOI] [PubMed] [Google Scholar]
- 232.Seguí, N., Mina, L.B., Lázaro, C., Sanz-Pamplona, R., Pons, T., Navarro, M., Bellido, F., López-Doriga, A., Valdés-Mas, R., Pineda, M. et al. (2015) Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology, 149, 563–566. [DOI] [PubMed] [Google Scholar]
- 233.Zhou, W., Otto, E.A., Cluckey, A., Airik, R., Hurd, T.W., Chaki, M., Diaz, K., Lach, F.P., Bennett, G.R., Gee, H.Y. et al. (2012) FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet., 44, 910–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Guda, K., Moinova, H., He, J., Jamison, O., Ravi, L., Natale, L., Lutterbaugh, J., Lawrence, E., Lewis, S., Willson, J.K.V. et al. (2009) Inactivating germ-line and somatic mutations in polypeptide N-acetylgalactosaminyltransferase 12 in human colon cancers. Proc. Natl. Acad. Sci. U. S. A., 106, 12921–12925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Jaeger, E., Leedham, S., Lewis, A., Segditsas, S., Becker, M., Cuadrado, P.R., Davis, H., Kaur, K., Heinimann, K., Howarth, K. et al. (2012) Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat. Genet., 44, 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sanders, M.A., Chew, E., Flensburg, C., Zeilemaker, A., Miller, S.E., Al Hinai, A.S., Bajel, A., Luiken, B., Rijken, M., Mclennan, T. et al. (2018) MBD4 guards against methylation damage and germ line deficiency predisposes to clonal hematopoiesis and early-onset AML. Blood, 132, 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Palles, C., Chew, E., Grolleman, J.E., Galavotti, S., Flensburg, C., Jansen, E.A.M., Curley, H., Chegwidden, L., Barnes, E.A., Bajel, A. et al. (2021) Germline loss-of-function variants in the base-excision repair gene MBD4 cause a Mendelian recessive syndrome of adenomatous colorectal polyposis and acute myeloid leukaemia. BioRxiv preprint, April 28, 2021. 10.1101/2021.04.27.441137 preprint: not peer reviewed. [DOI] [Google Scholar]
- 238.Wu, Y., Berends, M.J., Sijmons, R.H., Mensink, R.G., Verlind, E., Kooi, K.A., van der Sluis, T., Kempinga, C. and van dDer Zee, A. G., Hollema, H., et al. (2001) A role for MLH3 in hereditary nonpolyposis colorectal cancer. Nat. Genet., 29, 137–138. [DOI] [PubMed] [Google Scholar]
- 239.Chubb, D., Broderick, P., Dobbins, S.E., Frampton, M., Kinnersley, B., Penegar, S., Price, A., Ma, Y.P., Sherborne, A.L., Palles, C. et al. (2016) Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun., 7, 11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Stewart, G.S., Maser, R.S., Stankovic, T., Bressan, D.A., Kaplan, M.I., Jaspers, N.G., Raams, A., Byrd, P.J., Petrini, J.H. and Taylor, A.M. (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell, 99, 577–587. [DOI] [PubMed] [Google Scholar]
- 241.Nicolaides, N.C., Papadopoulos, N., Liu, B., Wei, Y.F., Carter, K.C., Ruben, S.M., Rosen, C.A., Haseltine, W.A., Fleischmann, R.D. and Fraser, C.M. (1994) Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature, 371, 75–80. [DOI] [PubMed] [Google Scholar]
- 242.Spier, I., Holzapfel, S., Altmüller, J., Zhao, B., Horpaopan, S., Vogt, S., Chen, S., Morak, M., Raeder, S., Kayser, K. et al. (2015) Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int. J. Cancer, 137, 320–331. [DOI] [PubMed] [Google Scholar]
- 243.Robles-Espinoza, C.D., Harland, M., Ramsay, A.J., Aoude, L.G., Quesada, V., Ding, Z., Pooley, K.A., Pritchard, A.L., Tiffen, J.C., Petljak, M. et al. (2014) POT1 loss-of-function variants predispose to familial melanoma. Nat. Genet., 46, 478–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Shi, J., Yang, X.R., Ballew, B., Rotunno, M., Calista, D., Fargnoli, M.C., Ghiorzo, P., Bressac-de Paillerets, B., Nagore, E., Avril, M.F. et al. (2014) Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat. Genet., 46, 482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Bainbridge, M.N., Armstrong, G.N., Gramatges, M.M., Bertuch, A.A., Jhangiani, S.N., Doddapaneni, H., Lewis, L., Tombrello, J., Tsavachidis, S., Liu, Y. et al. (2015) Germline mutations in shelterin complex genes are associated with familial glioma. J. Natl. Cancer Inst., 107, 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Calvete, O., Martinez, P., Garcia-Pavia, P., Benitez-Buelga, C., Paumard-Hernández, B., Fernandez, V., Dominguez, F., Salas, C., Romero-Laorden, N., Garcia-Donas, J. et al. (2015) A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li-Fraumeni-like families. Nat. Commun., 6, 8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Li, J., Yen, C., Liaw, D., Podsypanina, K., Bose, S., Wang, S.I., Puc, J., Miliaresis, C., Rodgers, L., McCombie, R. et al. (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science, 275, 1943–1947. [DOI] [PubMed] [Google Scholar]
- 248.Gala, M.K., Mizukami, Y., Le, L.P., Moriichi, K., Austin, T., Yamamoto, M., Lauwers, G.Y., Bardeesy, N. and Chung, D.C. (2014) Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology, 146, 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Schulz, E., Klampfl, P., Holzapfel, S., Janecke, A.R., Ulz, P., Renner, W., Kashofer, K., Nojima, S., Leitner, A., Zebisch, A. et al. (2014) Germline variants in the SEMA4A gene predispose to familial colorectal cancer type X. Nat. Commun., 5, 5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Abid, A., Ismail, M., Mehdi, S.Q. and Khaliq, S. (2006) Identification of novel mutations in the SEMA4A gene associated with retinal degenerative diseases. J. Med. Genet., 43, 378–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Yu, C.E., Oshima, J., Fu, Y.H., Wijsman, E.M., Hisama, F., Alisch, R., Matthews, S., Nakura, J., Miki, T., Ouais, S. et al. (1996) Positional cloning of the Werner’s syndrome gene. Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]