

Abstract
Huntington’s disease (HD) is a devastating neurogenetic disorder whose familial nature and progressive course were first described in the 19th century but for which no disease-modifying treatment is yet available. Through the active participation of HD families, this disorder has acted as a flagship for the application of human molecular genetic strategies to identify disease genes, understand pathogenesis and identify rational targets for development of therapies.
The Beginning of the Molecular Genetic Era
For Huntington’s disease (HD) (OMIM 143100) (1), the molecular genetic era began in 1983 (2). Previously, the focus in this late-onset neurodegenerative disease had been on description of its progressive clinical phenotype, which involves typically midlife onset of characteristic choreiform movements, accompanied or preceded by behavioral and cognitive disturbance, on the neuropathological description of neuronal loss that is most prominently visible in the striatum and on its autosomal dominant pattern of inheritance that is remarkable for showing ‘anticipation’ in which individuals with the disorder often display clinical onset earlier in life in successive generations (3). An extreme example of the latter involves onset of HD early in the juvenile years, typically inherited from a father, manifest as extreme rigidity with more widespread neuropathology (4).
By 1983, genetic linkage studies using the limited number of available expressed markers had failed to identify the chromosomal location of the HD gene (5). However, the capacity to monitor individual genomic sequence variants by their effect on DNA cleavage by sequence-specific bacterial restriction endonucleases had raised the possibility, if enough marker variants could be found, of scanning the entire genome, tracking the inheritance of individual chromosome regions and correlating them with the inheritance of HD. Genetic linkage analysis with DNA markers was rapidly successful (2,6), making the HD defect the first autosomal disease gene mapped using this strategy and prompting similar studies in many other disorders, along with lending support to suggestions for a concerted effort to generate and map polymorphic sequences across the human genome. The discovery of linked markers enabled for the first time predictive prenatal or presymptomatic genetic testing for HD, but this was only applicable to those with sufficient family members available and was inherently probabilistic, due to the potential for recombination (7). Given the psychiatric effects of the HD mutation in some individuals and the increased risk of suicide among mutation carriers, predictive testing was approached cautiously, with strong guidelines for accompanying genetic counseling (8).
The genetic markers assigned the HD gene to the terminal band of the chromosome 4 short arm, but knowledge of its neighborhood did not immediately identify the nature of the gene and its defect. It would be another decade before numerous technical improvements in DNA cloning, sequencing and physical mapping, many developed as part of the search, enabled the molecular cloning of the gene itself and discovery of the cause of HD (9). Again, this was accomplished through the use of DNA markers, in this instance to define a minimal candidate region through genetic association analysis that defined ancestral haplotypes to guide analysis of each of the genes being delineated (10). The HD defect was identified as an unstable expanded CAG trinucleotide repeat in a novel gene (9), whose symbol was subsequently changed from HD to HTT. The CAG repeat is located in the first exon of HTT, where it encodes a polyglutamine tract near the amino terminus of a large >350 kDa protein named huntingtin. The repeat shows length polymorphism in the general population that, when it exceeds ~35 CAGs, can cause HD (Fig. 1).
Figure 1 .
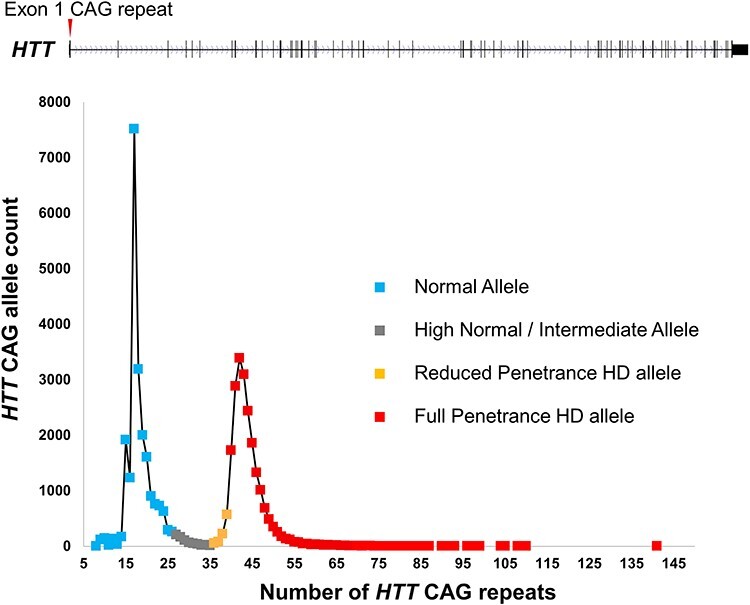
HTT CAG repeat distribution and relationship to HD. The 67-exon HTT gene and the location of its polymorphic CAG repeat (red arrow) in exon 1 are shown above a plot of the distribution of CAG allele lengths in >20 000 HD study participants (almost all HD heterozygotes) genotyped by the Molecular Neurogenetics Unit at the Massachusetts General Hospital. The distribution forms a continuum of allele lengths, with soft borders that distinguish allele lengths associated with phenotype. Normal alleles (approximately <27 CAGs), shown in blue, are not associated with a phenotype and are inherited in a stable manner. High normal alleles (shown in gray), sometimes called intermediate alleles (approximately >26 but < 36 CAGs), do not typically develop signs of HD but may show some degree of germline instability of the CAG repeat. Reduced penetrance alleles (roughly 36–39 CAGs and shown in orange) are associated with HD, but not all individuals with these alleles will develop clinical signs of the disorder. Full penetrance alleles (>39 CAGs, shown in red) are almost always associated with development of clinical signs and symptoms of HD. Of the expanded alleles (>35 CAGs) depicted, 4.4% are in the reduced penetrance range and 2% have >55 CAGs, in the range usually associated with very early onset of HD.
Genetic Characterization and Genotype–Phenotype Analysis
Two remarkable features of the HD mutation, apparent when it was first found and ultimately shown to be shared by a series of other neurological disorders caused by expanded CAG trinucleotide repeats, are its instability through meiotic transmission and the strong correlation of CAG repeat length with age at onset (9,11–13). In the families that contributed to the genetic linkage studies, analysis showed that, unlike its non-expanded counterpart, the expanded CAG repeat usually changes in size by one to a few units when passed from parent to child, with occasional larger increases usually, but not always, in the male germline (14–16). Indeed, size changes also occur occasionally for CAG alleles in the intermediate range, resulting in a ‘new mutation’ to HD (i.e. an affected individual with no family history of HD) when a CAG repeat now in the disease-causing range is received from a non-disease parent (17,18).
The length of the expanded CAG repeat, as estimated by a polymerase chain reaction amplification/sizing assay relative to a sequenced standard, is strongly correlated with age at onset of HD with longer repeats being associated with earlier onset (11–13). The combination of this genotype–phenotype correlation with the observance of larger meiotic CAG repeat increases preferentially in the male germline effectively explained the mystery of genetic anticipation in HD (15). The ability to define the mutant allele at the molecular level also confirmed the previous suggestion from family studies that, with respect to age at onset, HD is a complete dominant. Reported HD ‘homozygotes’ were found to have two expanded alleles and yet, as judged by the larger allele, to display an age at onset comparable to heterozygotes (19–22). Similarly, robust statistical analysis revealed no effect of the length of the non-expanded allele in heterozygotes (22). There has been debate concerning whether individuals with two expanded alleles display more rapid deterioration, but the most recent and largest study indicates that this is not the case (23,24).
Measurement of the HTT CAG repeat length also provided a direct test for prenatal or presymptomatic prediction or for differential diagnosis without the requirement for DNA from family members. Still, in the absence of an effective treatment, the uptake of genetic testing among ‘at-risk’ individuals has remained relatively low (25,26).
Model Systems
Prior to discovery of the HD mutation, studies of pathogenesis aimed to mimic the striatal neuropathology of HD through chemical toxicity (e.g. 3-nitropropionic acid, an inhibitor of the respiratory chain enzyme succinate dehydrogenase, or quinolinic acid, an N-methyl-D-aspartate receptor agonist). With the discovery of the HD defect, modeling studies shifted to an increasingly sophisticated array of gene-based models: exogenous transgene models, mutation-precise endogenous animal models and human patient cell models (27,28).
The first HD models galvanized the field and prompted a new direction that investigation of HD pathogenesis would take for the coming decades. The R6 series of HTT exon1 genomic DNA transgenic mice, with long 115–150 CAG repeats, had dramatic neurologic phenotypes and early death (29), but it was the observation of intranuclear inclusions in their brains (30) that propelled a fundamentally different view of HD pathogenesis into the mainstream. This came to be known as the ‘polyglutamine-aggregation hypothesis’. Based on the physical properties of polyglutamine, Perutz et al. (31) had theorized: ‘…extensions of their glutamine repeats may cause the affected proteins to agglomerate and precipitate in neurons; symptoms may set in when these precipitates have reached a critical size or have resulted in a critical number of neural blocks. This would explain better why symptoms appear earlier in life and become more severe the longer the extension of the glutamine repeats and why the main histological manifestation of HD consists in neural degeneration.’ Similar aggregates were also observed in human HD postmortem brain (32), adding them to the neuropathological hallmarks of HD, but not providing direct evidence of whether they were a cause or result of pathogenesis (i.e. ‘bullets or tombstones’).
This focus spawned a host of polyglutamine-fragment models, expressing polyglutamine alone or in the context of truncated HTT transgene products in cells from yeast to human, including neuronal cells in slices or culture, as well as in invertebrates, zebrafish, mice, rats, minipigs and non-human primates. In general, polyglutamine aggregation can but need not be toxic, altering gene-expression and a multitude of biological processes (33,34). Demonstration that conditional shutdown of an exon1 fragment transgene lessened its consequent phenotypes showed the polyglutamine-toxicity in the model to be dosage-dependent (35), supporting speculation that the HD mechanism would also be modifiable in this manner. Indeed, screens for polyglutamine-aggregation and polyglutamine-toxicity modulators identified numerous candidates (36,37), though many of these turned out to be assay-format specific. However, genetic experiments with endogenous CAG repeat knock-in and knock-out mouse models were revealing a more complex picture. Tests in these models of the criteria defined by human HD, complete dominance, increasing severity with CAG size and neuronal selectivity, were yielding results consistent with HD that are not easily explained by the polyglutamine-aggregation hypothesis.
Instead of the more rapid onset in homozygotes, predicted by a double dose of the mutation in the aggregation-hypothesis, the timing of pathogenic phenotypes in Htt CAG knock-in mice instead resembled that of heterozygotes (38,39). Nor, as might be expected in a dosage-dependent mechanism, did the presence or the absence of a normal allele alter timing of onset, which was comparable in Htt CAG heterozygotes and hemizygotes (38). These findings implied that an HTT gain of function not involving gene dosage is the initiator of HD pathogenesis. Indeed, these models revealed that genetic loss-of-function scenarios, either inactivation of the mutant allele itself or dominant negative loss of function of both alleles, are unlikely initiators. Htt CAG homozygotes did not exhibit the dramatic developmental abnormalities associated with null (40–42) and hypomorphic (43,44) Htt loss-of-function mutations. Moreover, these loss-of-function developmental blocks could be bypassed by either a single mutant or wild-type allele (43,44). Notably, it has been shown recently that in humans, mutations causing reduced huntingtin expression act in a recessive manner to cause a severe developmental disorder (Lopes-Maciel-Rodan syndrome) (45–47).
Though it received less attention at the time, the R6 HTT exon1 genomic fragment transgenic mouse series also fulfilled their original intent in providing models to study instability of the HD repeat mutation (48). This had not been prominent in transgenics with shorter repeats created for other repeat disorders. The exon1 CAG repeats changed in size gametically and somatically, with a pattern of expansion across brain regions and other tissues that implied both a length-requirement and the need for tissue-specific factors (48). The absence of instability for the non-expressing R6/0 line implied that transcription of the genomic DNA transgene may also be a necessary condition for somatic instability (48). Knock-in CAG repeat mice confirmed the length-dependency by showing progressively more somatic expansion with increasing pure CAG repeat length, starting from mild instability at 48 repeats (16) and, together with observations from other knock-in models, established that CAG and other trinucleotide repeats at distinct loci exhibit a similar pattern of instability across brain regions and other tissues (reviewed in 49).
The models confirmed and spurred investigation of instability in the human triplet repeat disorders (49). While gametic instability of inherited HD CAG repeats is frequent, changes in brain appear to be less frequent (15), although the ability to observe these events is compromised both by pathology at postmortem and the detection method, as shown by the extremely large repeat expansions observed in early-stage HD brain (50,51). Moreover, as forecast by the mouse models, the graded pattern of somatic repeat expansion across the (postmortem) brain and other tissues (greatest in neostriatum and liver, respectively) was found to be similar for the HTT-expanded CAG repeat associated with HD and the ATXN1-expanded CAG repeat associated with spinocerebellar ataxia 1 (SCA1), another of the ‘polyglutamine disorders’ (52).
The first genetic modifier of somatic triplet repeat expansion, DNA mismatch repair gene Msh2, was identified in crosses with the HTT exon1 genomic R6/1 mouse (53). This spurred genetic crosses with transgenics and endogenous knock-in models with pure repeats that led to the identification of other genes, including many (e.g. Pms1, Pms2, Mlh1, Mlh3 and Msh3), but not all (e.g. Msh6), mismatch repair proteins, involved in somatic instability (reviewed in 54). Endogenous models also linked repeat expansion to the timing of emergent phenotype (55,56), including demonstration with a naturally occurring Mlh1 variant that reduced expression levels decreased both the rate of expansion and the rate of striatal neuron pathology (57).
The finding in both in vivo models and humans that the rate of repeat expansion appears to be characteristic of a given neuronal (or other) cell population posed a conundrum. If expansion is linked to pathogenic vulnerability, then why is the neuronal cell type with the fastest rate of expansion (medium size spiny neurons in the neostriatum) only vulnerable in HD and not in every triplet repeat disorder? Other factors must explain this differential cell vulnerability. These may be cell type–specific factors critical to triggering a pathogenic process in the vulnerable cells, a phenomenon that has been explored in SCA1. A set of ATXN1 cDNA transgenic mouse models (58,59) reveals that in this system pathogenesis is not initiated by aggregation, by the mutant mRNA or by RAN-translation products but rather at the level of a Purkinje cell–specific ATXN1:Capicua transcription complex. Mutation of the ATXN1Q82 transgene that prevents ATXN1-binding to its Purkinje cell–specific transcription-complex partner Capicua also prevents Purkinje cell death, though mRNA and RAN-translation products remain unchanged (59). Moreover, gene expression signatures in the ATXN1Q82 mice, Atxn1 CAG knock-in mice and SCA1 patient cells, show that ATXN1Q82:Capicua complex induces pathogenic changes in Purkinje cells in a gain-of-function rather than a loss-of-function manner (59).
Human Molecular Genetics
Although animal models can help to refine hypotheses of pathogenesis, as they have in SCA1, they cannot be definitive in terms of the human condition, so the search for toxicity mechanisms in HD has also relied on further human genetic analysis. Capitalizing on the fact that, after controlling for inherited CAG repeat length, there is remaining heritable variance in age at onset (60,61), a series of candidate gene polymorphisms were tested as potential modifiers of HD pathogenesis. The candidate genes were chosen based upon a hypothesized mode of action thought to be relevant to HD (e.g. excitotoxicity, energy metabolism, growth factor and huntingtin-interactor) but they mainly yielded inconsistent results in small, underpowered and insufficiently controlled genetic association studies (62). Fortunately, the cloning of the HD gene in 1993 coincided with the beginning of a concerted effort on the part of the HD clinical research community (i.e. the Huntington Study Group in the U.S. and the European Huntington’s Disease Network and, more recently, the ENROLL-HD Platform) to embark upon large-scale cooperative natural history and clinical trial studies that would eventually overcome these obstacles (63–67). HD participant DNA samples from these and the historical HD studies (9,60) enabled the search for modifier loci through unbiased genome-wide association studies (GWAS) mainly in participants of European ancestry.
An initial combination of three GWAS comprising ~4000 subjects carried out by the GeM-HD Consortium identified genome-wide significant loci on chromosomes 8 and 15, with the latter displaying both onset-hastening and onset-delaying haplotypes (68). The modifier gene on chromosome 15 has been confirmed recently as FAN1, encoding a DNA endo- and exonuclease involved in the repair of DNA damage caused by cross-linking agents (69). These modifier alleles also influence onset of other CAG repeat disorders (70). In the mouse, targeted inactivation of Fan1 promotes somatic instability of trinucleotide repeats (71,72). The prime candidate at the onset-hastening chromosome 8 locus is RRM2B, which encodes the small subunit of a heterotetrameric ribonucleotide reductase that converts ribonucleoside diphosphates into deoxyribonucleoside diphosphates. Mutations in RRM2B are associated with autosomal recessive DNA depletion syndromes (OMIM 612075) and an autosomal dominant progressive external ophthalmoplegia (OMIM 613077).
Unlike GWAS designed to identify additive risk factors in common disease, the HD modifier GWAS sought to identify genetic interaction between the presence of an expanded repeat in HTT and naturally occurring polymorphisms in other genes. The lack of a discernable effect of the modifier alleles in the absence of an expanded CAG repeat was confirmed in non-HD participants of the PREDICT-HD study (73). In those with an expanded CAG repeat, the modifiers showed a significant influence on the earliest signs of HD pathogenesis that are evident at the group level ~15 years before clinical onset. However, the modifier alleles had distinct effects on measures of motor dysfunction, cognitive deficit and putamen volume, suggesting that they act by different mechanisms and/or in different cell types (73).
A third GWAS locus of suggestive significance on chromosome 3 was confirmed as genome-wide significant in targeted follow-up of the original study (74). Notably, this locus corresponds with the human orthologue of Mlh1, which had been revealed in the mouse studies to be a modifier of somatic CAG repeat expansion (57), adding to the findings from pathway analyses and analysis of FAN1 that DNA repair/maintenance mechanisms play an important role in HD (69,75,76). This notion was supported by a smaller GWAS of the TRACK-HD study participants, where intensive longitudinal phenotypic permitted development of an integrated measure of deterioration (77). This phenotype detected MSH3, another mismatch repair gene whose mouse ortholog is associated with CAG repeat instability (78), as a modifier of HD.
The most recent GeM-HD GWAS expanded the sample size to >9000 participants of European ancestry and resulted in the identification of three more DNA repair loci (PMS1, PMS2 and LIG1) as modifiers of onset, additional modifier haplotypes with opposing effects at MSH3, FAN1 and LIG1, and additional modifier loci on chromosomes 5 and 11 that might be indirectly associated with DNA maintenance/instability mechanisms or might influence HD through a different mechanism (75). Notably, the former harbors TCERG1, encoding a transcriptional regulator previously suggested from a candidate HD genetic study as a potential modifier based upon its interaction with huntingtin (79). The chromosome 11 locus is centered on CCDC82, which specifies a coiled-coil domain protein of unknown function. Although a smaller non-European ancestry GWAS of the participants in the U.S.-Venezuela Collaborative Research Project showed evidence of FAN1 as a modifier, the most significant locus was at SOSTDC1 (80). This gene encodes a bone morphogenetic protein antagonist suggesting that it may influence the toxicity mechanism, but its lack of significance in the European ancestry GWAS supports the possibility that it acts through a population-specific variant or environmental interaction.
HD Pathogenesis Involves Two Sequential Components
The broadly accepted expectation that polyglutamine toxicity drives HD onset has also been overturned by recent genetic studies. In the vast majority of HD individuals, the expanded CAG repeat is followed by CAACAG. Since both CAA and CAG codons specify glutamine, mutant huntingtin typically contains a polyglutamine tract that is greater by two than the pure CAG repeat. Rare HTT alleles that differ from this canonical structure and must be resolved by DNA sequencing had been reported in the ramp-up of molecular diagnosis (81,82). The two most frequent non-canonical variants, each representing <1% of HD chromosomes are, respectively, duplication of the CAACAG and loss of the interrupting CAA. These rare glutamine-encoding polymorphisms have revealed that the timing of HD onset is driven by a property of the pure CAG repeat, not by the polyglutamine in mutant huntingtin (Fig. 2) (75,83–85). Indeed, the CAACAG duplication alleles specify four more glutamines than CAA loss alleles, yet they are not associated with earlier onset.
Figure 2 .
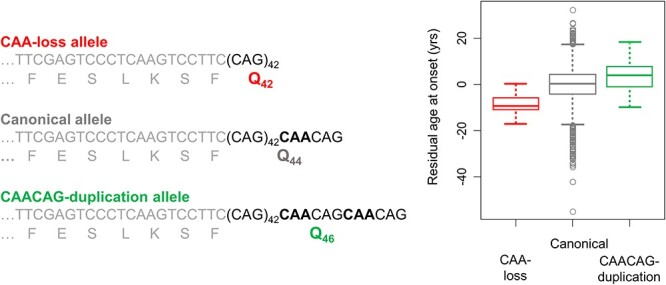
The timing of HD onset is driven by a property of CAG repeat length, not polyglutamine. Canonical HTT CAG expansion alleles associated with HD have repeats with a penultimate CAA interruption. Since both CAG and CAA are codons that specify glutamine, this results in a huntingtin protein with two more glutamines in its polyglutamine repeat than there are consecutive CAGs in the DNA. This figure depicts the effect of two minor variant alleles, one in which the CAA is lost (red) and one in which a second CAA results in effective duplication of the terminal CAACAG segment (green). To the left, CAA-loss, canonical and CAACAG-duplication alleles, all with pure CAG tracts of 43 units, are compared, showing that each specifies a different number of glutamines. On the right, a box plot of residual age at onset (i.e. the difference between observed age at onset and that expected based upon the uninterrupted CAG repeat length) for the set of participants in the GeM-HD GWAS studies shows that, despite having the longest polyglutamine length, those with CAACAG-duplication (N = 69) alleles do not show earlier onset. In contrast, the CAA-loss allele carriers (N = 21) show earlier onset than canonical (N = 7724; eight most frequent haplotypes) and CAACAG-duplication carriers despite having the shortest polyglutamine tracts.
The lack of cumulative dose-dependent damage predicted by HD homozygotes, the existence of a length-dependent property of the uninterrupted CAG repeat that drives onset and the preponderance of DNA maintenance genes among HD onset modifiers, all point to HD pathogenesis consisting of two sequential components (Fig. 3): (1) somatic expansion of the inherited expanded CAG allele occurs in target cells at a rate influenced by the DNA maintenance modifiers, and (2) when some threshold repeat length is achieved, toxicity is triggered and the target cell suffers damage, dysfunction and death (75,83). The mechanism underlying this second toxicity component remains to be determined, perhaps through the identification of additional genetic modifiers that bring the process into focus. It might involve a de novo action at the threshold CAG length or an ongoing action that exceeds cellular coping mechanisms. It might involve full-length mutant huntingtin since, from biochemical studies, the polyglutamine tract size alters the structure, phosphorylation pattern and activities of the protein (an alpha-helical solenoid-like scaffold), which impacts on a wide variety of functions including chromatin regulation, vesicle trafficking, mitochondrial function and translation (27,34,86–88). Alternatively, toxicity might result from incomplete HTT exon1-2 splicing such that polyglutamine–exon1 fragment is produced (89,90). Mechanistic gain of function at level of the DNA or mRNA rather than protein is also possible (91–93) but each remains to be more fully explored. A similar two-component model likely applies to other diseases involving expanded CAG and potentially other repeat diseases, with the first step involving somatic expansion of the repeat and the second involving a different mechanism of toxicity peculiar to the locus and its target cells.
Figure 3 .
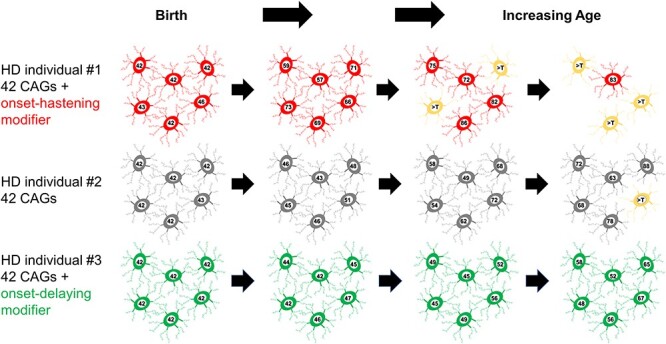
Genetic modification of the rate driver of HD pathogenesis. This figure provides an illustration of the phases of the two-component model of HD pathogenesis that has emerged from human molecular genetic studies. The timing of HD onset is driven by the uninterrupted CAG repeat length and is influenced by genetic modifiers that are associated with somatic expansion of trinucleotide repeats. Depicted are sets of medium-spiny striatal neurons at birth and with increasing age in individuals who inherited 42 uninterrupted HTT CAG repeats but with different modifier alleles. With progressive age, the CAG length shows variable degrees of expansion, with the greatest and least occurring in the individual with a strong onset-hastening modifier (red) and with a strong onset-delaying modifier (green), respectively. When the repeat in a given neuron reaches a threshold repeat length (noted as >T, a length that is currently not known but is assumed for this presentation to be >90 CAGs), toxic damage occurs through a mechanism that is not yet certain, resulting in neuronal dysfunction (yellow) and eventual death (disappearance).
Conclusion
Human genetic analysis has already demonstrated that the rate driver for HD onset (i.e. somatic CAG repeat expansion) is modifiable and has pointed to therapeutic targets that are currently being prosecuted. There is as yet no corresponding convergence of DNA modifiers on the toxicity mechanism, although RRM2B, TCERG1, CCDC82 and/or SOSTDC1 could well operate at this level. The GWAS studies of HD are currently being expanded in size to achieve greater power, to include more participants of non-European ancestry and to address additional disease phenotypes, with the hope that modifiers of the toxicity mechanism and the cellular response to it will emerge and that the processes responsible will constitute novel in-human validated therapeutic targets. Treatments are badly needed to delay or prevent onset of HD and/or deterioration in manifest individuals. The best hope for a disease-modifying intervention to date has been lowering the expression of mutant huntingtin by any of a variety of methods (94,95). Although this approach has generated great excitement, in a major disappointment to the HD community, the largest and most advanced Phase 3 clinical trial testing antisense oligonucleotide (ASO) suppression of both mutant and normal huntingtin (96) was halted recently based upon the overall risk/benefit assessment of an independent data monitoring committee, although no new safety signals emerged. It remains unclear whether this failure casts doubt on the validity of full-length huntingtin as a target in individuals who already exhibit manifest disease. It is conceivable that suppression of mutant huntingtin must be accomplished earlier, to a greater extent and/or in different cell types or in an allele-specific manner, that suppression must target exon 1 rather than full-length protein, or that toxicity is not precipitated at the protein level. Continued identification of genetic modifiers of HD and the stage of disease at which they act by investigating the human disease patients themselves may not only help to answer this question but will also provide alternative routes to understanding the mechanisms underlying human HD and to developing effective disease-modifying treatments.
Acknowledgements
We thank the many Huntington’s disease individuals and families for their participation in research and the HD clinicians and clinical researchers who have enabled that participation to advance understanding of HD.
Conflict of Interest statement. J.F.G. is a Scientific Advisory Board member and has a financial interest in Triplet Therapeutics, Inc. His NIH-funded project is using genetic and genomic approaches to uncover other genes that significantly influence when diagnosable symptoms emerge and how rapidly they worsen in Huntington’s disease. The company is developing new therapeutic approaches to address triplet repeat disorders such Huntington’s disease, myotonic dystrophy and spinocerebellar ataxias. His interests were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict of interest policies. He has also been a consultant for Wave Life Sciences USA. J-M.L. is on the Scientific Advisory Board of GenEdit, Inc. M.E.M. declares no conflicting interests.
Contributor Information
James F Gusella, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Medical and Population Genetics Program, The Broad Institute of M.I.T. and Harvard, Cambridge, MA 02142, USA; Department of Genetics, Blavatnik Institute, Harvard Medical School, Boston, MA 02115, USA.
Jong-Min Lee, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Medical and Population Genetics Program, The Broad Institute of M.I.T. and Harvard, Cambridge, MA 02142, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA.
Marcy E MacDonald, Molecular Neurogenetics Unit, Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA 02114, USA; Medical and Population Genetics Program, The Broad Institute of M.I.T. and Harvard, Cambridge, MA 02142, USA; Department of Neurology, Harvard Medical School, Boston, MA 02115, USA.
Funding
National Institutes of Health (NS091161, NS082079, NS016367 and NS105709); an Anonymous Donor and the CHDI Foundation.
References
- 1.Huntington, G. (1872) On chorea. Med. Surg. Reporter of Philadelphia, 26, 317–321. [Google Scholar]
- 2.Gusella, J.F., Wexler, N.S., Conneally, P.M., Naylor, S.L., Anderson, M.A., Tanzi, R.E., Watkins, P.C., Ottina, K., Wallace, M.R., Sakaguchi, A.Y. et al. (1983) A polymorphic DNA marker genetically linked to Huntington's disease. Nature, 306, 234–238. [DOI] [PubMed] [Google Scholar]
- 3.Conneally, P.M. (1984) Huntington disease: genetics and epidemiology. Am. J. Hum. Genet., 36, 506–526. [PMC free article] [PubMed] [Google Scholar]
- 4.Myers, R.H., Madden, J.J., Teague, J.L. and Falek, A. (1982) Factors related to onset age of Huntington disease. Am. J. Hum. Genet., 34, 481–488. [PMC free article] [PubMed] [Google Scholar]
- 5.Pericak-Vance, M.A., Conneally, P.M., Merritt, A.D., Roos, R., Norton, J.A., Jr. and Vance, J.M. (1978) Genetic linkage studies in Huntington disease. Cytogenet. Cell Genet., 22, 640–645. [DOI] [PubMed] [Google Scholar]
- 6.Gusella, J.F., Tanzi, R.E., Anderson, M.A., Hobbs, W., Gibbons, K., Raschtchian, R., Gilliam, T.C., Wallace, M.R., Wexler, N.S. and Conneally, P.M. (1984) DNA markers for nervous system diseases. Science, 225, 1320–1326. [DOI] [PubMed] [Google Scholar]
- 7.Meissen, G.J., Myers, R.H., Mastromauro, C.A., Koroshetz, W.J., Klinger, K.W., Farrer, L.A., Watkins, P.A., Gusella, J.F., Bird, E.D. and Martin, J.B. (1988) Predictive testing for Huntington's disease with use of a linked DNA marker. N. Engl. J. Med., 318, 535–542. [DOI] [PubMed] [Google Scholar]
- 8.Went, L. (1990) Ethical issues policy statement on Huntington's disease molecular genetics predictive test. International Huntington Association. World Federation of Neurology. J. Med. Genet., 27, 34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- 10.MacDonald, M.E., Novelletto, A., Lin, C., Tagle, D., Barnes, G., Bates, G., Taylor, S., Allitto, B., Altherr, M., Myers, R. et al. (1992) The Huntington's disease candidate region exhibits many different haplotypes. Nat. Genet., 1, 99–103. [DOI] [PubMed] [Google Scholar]
- 11.Duyao, M., Ambrose, C., Myers, R., Novelletto, A., Persichetti, F., Frontali, M., Folstein, S., Ross, C., Franz, M., Abbott, M. et al. (1993) Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat. Genet., 4, 387–392. [DOI] [PubMed] [Google Scholar]
- 12.Snell, R.G., MacMillan, J.C., Cheadle, J.P., Fenton, I., Lazarou, L.P., Davies, P., MacDonald, M.E., Gusella, J.F., Harper, P.S. and Shaw, D.J. (1993) Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington's disease. Nat. Genet., 4, 393–397. [DOI] [PubMed] [Google Scholar]
- 13.Andrew, S.E., Goldberg, Y.P., Kremer, B., Telenius, H., Theilmann, J., Adam, S., Starr, E., Squitieri, F., Lin, B., Kalchman, M.A. et al. (1993) The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat. Genet., 4, 398–403. [DOI] [PubMed] [Google Scholar]
- 14.Laccone, F. and Christian, W. (2000) A recurrent expansion of a maternal allele with 36 CAG repeats causes Huntington disease in two sisters. Am. J. Hum. Genet., 66, 1145–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacDonald, M.E., Barnes, G., Srinidhi, J., Duyao, M.P., Ambrose, C.M., Myers, R.H., Gray, J., Conneally, P.M., Young, A., Penney, J. et al. (1993) Gametic but not somatic instability of CAG repeat length in Huntington's disease. J. Med. Genet., 30, 982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wheeler, V.C., Auerbach, W., White, J.K., Srinidhi, J., Auerbach, A., Ryan, A., Duyao, M.P., Vrbanac, V., Weaver, M., Gusella, J.F. et al. (1999) Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum. Mol. Genet., 8, 115–122. [DOI] [PubMed] [Google Scholar]
- 17.Goldberg, Y.P., Kremer, B., Andrew, S.E., Theilmann, J., Graham, R.K., Squitieri, F., Telenius, H., Adam, S., Sajoo, A., Starr, E. et al. (1993) Molecular analysis of new mutations for Huntington's disease: intermediate alleles and sex of origin effects. Nat. Genet., 5, 174–179. [DOI] [PubMed] [Google Scholar]
- 18.Myers, R.H., MacDonald, M.E., Koroshetz, W.J., Duyao, M.P., Ambrose, C.M., Taylor, S.A., Barnes, G., Srinidhi, J., Lin, C.S., Whaley, W.L. et al. (1993) De novo expansion of a (CAG)n repeat in sporadic Huntington's disease. Nat. Genet., 5, 168–173. [DOI] [PubMed] [Google Scholar]
- 19.Wexler, N.S., Young, A.B., Tanzi, R.E., Travers, H., Starosta-Rubinstein, S., Penney, J.B., Snodgrass, S.R., Shoulson, I., Gomez, F., Ramos Arroyo, M.A. et al. (1987) Homozygotes for Huntington's disease. Nature, 326, 194–197. [DOI] [PubMed] [Google Scholar]
- 20.Myers, R.H., Leavitt, J., Farrer, L.A., Jagadeesh, J., McFarlane, H., Mastromauro, C.A., Mark, R.J. and Gusella, J.F. (1989) Homozygote for Huntington disease. Am. J. Hum. Genet., 45, 615–618. [PMC free article] [PubMed] [Google Scholar]
- 21.Durr, A., Hahn-Barma, V., Brice, A., Pecheux, C., Dode, C. and Feingold, J. (1999) Homozygosity in Huntington's disease. J. Med. Genet., 36, 172–173. [PMC free article] [PubMed] [Google Scholar]
- 22.Lee, J.M., Ramos, E.M., Lee, J.H., Gillis, T., Mysore, J.S., Hayden, M.R., Warby, S.C., Morrison, P., Nance, M., Ross, C.A. et al. (2012) CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology, 78, 690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Squitieri, F., Gellera, C., Cannella, M., Mariotti, C., Cislaghi, G., Rubinsztein, D.C., Almqvist, E.W., Turner, D., Bachoud-Levi, A.C., Simpson, S.A. et al. (2003) Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain, 126, 946–955. [DOI] [PubMed] [Google Scholar]
- 24.Cubo, E., Martinez-Horta, S.I., Santalo, F.S., Descalls, A.M., Calvo, S., Gil-Polo, C., Munoz, I., Llano, K., Mariscal, N., Diaz, D. et al. (2019) Clinical manifestations of homozygote allele carriers in Huntington disease. Neurology, 92, e2101–e2108. [DOI] [PubMed] [Google Scholar]
- 25.Quaid, K.A. (2017) Genetic testing for Huntington disease. Handb. Clin. Neurol., 144, 113–126. [DOI] [PubMed] [Google Scholar]
- 26.Anderson, K.E., Eberly, S., Marder, K.S., Oakes, D., Kayson, E., Young, A., Shoulson, I. and Investigators, P. (2019) The choice not to undergo genetic testing for Huntington disease: Results from the PHAROS study. Clin. Genet., 96, 28–34. [DOI] [PubMed] [Google Scholar]
- 27.Kolobkova, Y.A., Vigont, V.A., Shalygin, A.V. and Kaznacheyeva, E.V. (2017) Huntington's disease: calcium dyshomeostasis and pathology models. Acta Naturae, 9, 34–46. [PMC free article] [PubMed] [Google Scholar]
- 28.Zhunina, O.A., Yabbarov, N.G., Orekhov, A.N. and Deykin, A.V. (2019) Modern approaches for modelling dystonia and Huntington's disease in vitro and in vivo. Int. J. Exp. Pathol., 100, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies, S.W. et al. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell, 87, 493–506. [DOI] [PubMed] [Google Scholar]
- 30.Davies, S.W., Turmaine, M., Cozens, B.A., DiFiglia, M., Sharp, A.H., Ross, C.A., Scherzinger, E., Wanker, E.E., Mangiarini, L. and Bates, G.P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell, 90, 537–548. [DOI] [PubMed] [Google Scholar]
- 31.Perutz, M.F., Johnson, T., Suzuki, M. and Finch, J.T. (1994) Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. USA, 91, 5355–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DiFiglia, M., Sapp, E., Chase, K.O., Davies, S.W., Bates, G.P., Vonsattel, J.P. and Aronin, N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science, 277, 1990–1993. [DOI] [PubMed] [Google Scholar]
- 33.Ross, C.A. and Poirier, M.A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med., 10, S10–S17. [DOI] [PubMed] [Google Scholar]
- 34.Dickey, A.S. and La Spada, A.R. (2018) Therapy development in Huntington disease: From current strategies to emerging opportunities. Am. J. Med. Genet. A, 176, 842–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto, A., Lucas, J.J. and Hen, R. (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell, 101, 57–66. [DOI] [PubMed] [Google Scholar]
- 36.Trepte, P., Strempel, N. and Wanker, E.E. (2014) Spontaneous self-assembly of pathogenic huntingtin exon 1 protein into amyloid structures. Essays Biochem., 56, 167–180. [DOI] [PubMed] [Google Scholar]
- 37.Takeuchi, T. and Nagai, Y. (2017) Protein misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Sci., 7, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wheeler, V.C., Gutekunst, C.A., Vrbanac, V., Lebel, L.A., Schilling, G., Hersch, S., Friedlander, R.M., Gusella, J.F., Vonsattel, J.P., Borchelt, D.R. et al. (2002) Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum. Mol. Genet., 11, 633–640. [DOI] [PubMed] [Google Scholar]
- 39.Kumar, A., Zhang, J., Tallaksen-Greene, S., Crowley, M.R., Crossman, D.K., Morton, A.J., Van Groen, T., Kadish, I., Albin, R.L., Lesort, M. et al. (2016) Allelic series of Huntington's disease knock-in mice reveals expression discorrelates. Hum. Mol. Genet., 25, 1619–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duyao, M.P., Auerbach, A.B., Ryan, A., Persichetti, F., Barnes, G.T., McNeil, S.M., Ge, P., Vonsattel, J.P., Gusella, J.F., Joyner, A.L. et al. (1995) Inactivation of the mouse Huntington's disease gene homolog Hdh. Science, 269, 407–410. [DOI] [PubMed] [Google Scholar]
- 41.Zeitlin, S., Liu, J.P., Chapman, D.L., Papaioannou, V.E. and Efstratiadis, A. (1995) Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat. Genet., 11, 155–163. [DOI] [PubMed] [Google Scholar]
- 42.Nasir, J., Floresco, S.B., O'Kusky, J.R., Diewert, V.M., Richman, J.M., Zeisler, J., Borowski, A., Marth, J.D., Phillips, A.G. and Hayden, M.R. (1995) Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell, 81, 811–823. [DOI] [PubMed] [Google Scholar]
- 43.Murthy, V., Tebaldi, T., Yoshida, T., Erdin, S., Calzonetti, T., Vijayvargia, R., Tripathi, T., Kerschbamer, E., Seong, I.S., Quattrone, A. et al. (2019) Hypomorphic mutation of the mouse Huntington's disease gene orthologue. PLoS Genet., 15, e1007765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White, J.K., Auerbach, W., Duyao, M.P., Vonsattel, J.P., Gusella, J.F., Joyner, A.L. and MacDonald, M.E. (1997) Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat. Genet., 17, 404–410. [DOI] [PubMed] [Google Scholar]
- 45.Jung, R., Lee, Y., Barker, D., Correia, K., Shin, B., Loupe, J., Collins, R.L., Lucente, D., Ruliera, J., Gillis, T. et al. (2021) Mutations causing Lopes-Maciel-Rodan Syndrome are huntingtin hypomorphs. Hum. Mol. Genet., 30, 135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodan, L.H., Cohen, J., Fatemi, A., Gillis, T., Lucente, D., Gusella, J. and Picker, J.D. (2016) A novel neurodevelopmental disorder associated with compound heterozygous variants in the huntingtin gene. Eur. J. Hum. Genet., 24, 1826–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lopes, F., Barbosa, M., Ameur, A., Soares, G., de Sa, J., Dias, A.I., Oliveira, G., Cabral, P., Temudo, T., Calado, E. et al. (2016) Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet., 53, 190–199. [DOI] [PubMed] [Google Scholar]
- 48.Bates, G.P., Mangiarini, L., Mahal, A. and Davies, S.W. (1997) Transgenic models of Huntington's disease. Hum. Mol. Genet., 6, 1633–1637. [DOI] [PubMed] [Google Scholar]
- 49.Monckton, D.G. (2021) The contribution of somatic expansion of the CAG repeat to symptomatic development in Huntington's disease: a historical perspective. J. Huntington's Dis., 10, 7–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kennedy, L., Evans, E., Chen, C.M., Craven, L., Detloff, P.J., Ennis, M. and Shelbourne, P.F. (2003) Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet., 12, 3359–3367. [DOI] [PubMed] [Google Scholar]
- 51.Shelbourne, P.F., Keller-McGandy, C., Bi, W.L., Yoon, S.R., Dubeau, L., Veitch, N.J., Vonsattel, J.P., Wexler, N.S., Group, U.S.-V.C.R., Arnheim, N. et al. (2007) Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum. Mol. Genet., 16, 1133–1142. [DOI] [PubMed] [Google Scholar]
- 52.Mouro Pinto, R., Arning, L., Giordano, J.V., Razghandi, P., Andrew, M.A., Gillis, T., Correia, K., Mysore, J.S., Grote Urtubey, D.M., Parwez, C.R. et al. (2020) Patterns of CAG repeat instability in the central nervous system and periphery in Huntington's disease and in spinocerebellar ataxia type 1. Hum. Mol. Genet., 29, 2551–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manley, K., Shirley, T.L., Flaherty, L. and Messer, A. (1999) Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Genet., 23, 471–473. [DOI] [PubMed] [Google Scholar]
- 54.Wheeler, V.C. and Dion, V. (2021) Modifiers of CAG/CTG Repeat Instability: Insights from Mammalian Models. J. Huntington's Dis., 10, 123–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kovalenko, M., Dragileva, E., St Claire, J., Gillis, T., Guide, J.R., New, J., Dong, H., Kucherlapati, R., Kucherlapati, M.H., Ehrlich, M.E. et al. (2012) Msh2 acts in medium-spiny striatal neurons as an enhancer of CAG instability and mutant huntingtin phenotypes in Huntington's disease knock-in mice. PLoS One, 7, e44273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Budworth, H., Harris, F.R., Williams, P., Lee, D.Y., Holt, A., Pahnke, J., Szczesny, B., Acevedo-Torres, K., Ayala-Pena, S. and McMurray, C.T. (2015) Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington's disease. PLoS Genet., 11, e1005267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pinto, R.M., Dragileva, E., Kirby, A., Lloret, A., Lopez, E., St Claire, J., Panigrahi, G.B., Hou, C., Holloway, K., Gillis, T. et al. (2013) Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington's disease mice: genome-wide and candidate approaches. PLoS Genet., 9, e1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burright, E.N., Clark, H.B., Servadio, A., Matilla, T., Feddersen, R.M., Yunis, W.S., Duvick, L.A., Zoghbi, H.Y. and Orr, H.T. (1995) SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell, 82, 937–948. [DOI] [PubMed] [Google Scholar]
- 59.Rousseaux, M.W.C., Tschumperlin, T., Lu, H.C., Lackey, E.P., Bondar, V.V., Wan, Y.W., Tan, Q., Adamski, C.J., Friedrich, J., Twaroski, K. et al. (2018) ATXN1-CIC complex is the primary driver of cerebellar pathology in spinocerebellar ataxia type 1 through a gain-of-function mechanism. Neuron, 97, 1235–1243 e1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li, J.L., Hayden, M.R., Almqvist, E.W., Brinkman, R.R., Durr, A., Dode, C., Morrison, P.J., Suchowersky, O., Ross, C.A., Margolis, R.L. et al. (2003) A genome scan for modifiers of age at onset in Huntington disease: The HD MAPS study. Am. J. Hum. Genet., 73, 682–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wexler, N.S., Lorimer, J., Porter, J., Gomez, F., Moskowitz, C., Shackell, E., Marder, K., Penchaszadeh, G., Roberts, S.A., Gayan, J. et al. (2004) Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc. Natl. Acad. Sci. USA, 101, 3498–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gusella, J.F., MacDonald, M.E. and Lee, J.M. (2014) Genetic modifiers of Huntington's disease. Mov. Disord., 29, 1359–1365. [DOI] [PubMed] [Google Scholar]
- 63.Huntington Study Group Pharos Investigators (2006) At risk for Huntington disease: the PHAROS (Prospective Huntington At Risk Observational Study) cohort enrolled. Arch. Neurol., 63, 991–996. [DOI] [PubMed] [Google Scholar]
- 64.Paulsen, J.S., Hayden, M., Stout, J.C., Langbehn, D.R., Aylward, E., Ross, C.A., Guttman, M., Nance, M., Kieburtz, K., Oakes, D. et al. (2006) Preparing for preventive clinical trials: the predict-HD study. Arch. Neurol., 63, 883–890. [DOI] [PubMed] [Google Scholar]
- 65.Huntington Study Group Cohort Investigators and Dorsey, E (2012) Characterization of a large group of individuals with huntington disease and their relatives enrolled in the COHORT study. PLoS One, 7, e29522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Orth, M., Network, E.H.'s.D., Handley, O.J., Schwenke, C., Dunnett, S., Wild, E.J., Tabrizi, S.J. and Landwehrmeyer, G.B. (2011) Observing Huntington's disease: the European Huntington's Disease Network's REGISTRY. J. Neurol. Neurosurg. Psychiatry, 82, 1409–1412. [DOI] [PubMed] [Google Scholar]
- 67.Landwehrmeyer, G.B., Fitzer-Attas, C.J., Giuliano, J.D., Goncalves, N., Anderson, K.E., Cardoso, F., Ferreira, J.J., Mestre, T.A., Stout, J.C. and Sampaio, C. (2017) Data analytics from enroll-HD, a global clinical research platform for Huntington's disease. Mov. Disord. Clin. Pract., 4, 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Genetic Modifiers of Huntington's Disease Consortium (2015) Identification of genetic factors that modify clinical onset of Huntington's disease. Cell, 162, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim, K.H., Hong, E.P., Shin, J.W., Chao, M.J., Loupe, J., Gillis, T., Mysore, J.S., Holmans, P., Jones, L., Orth, M. et al. (2020) Genetic and functional analyses point to FAN1 as the source of multiple Huntington disease modifier effects. Am. J. Hum. Genet., 107, 96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bettencourt, C., Hensman-Moss, D., Flower, M., Wiethoff, S., Brice, A., Goizet, C., Stevanin, G., Koutsis, G., Karadima, G., Panas, M. et al. (2016) DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol., 79, 983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Loupe, J.M., Pinto, R.M., Kim, K.H., Gillis, T., Mysore, J.S., Andrew, M.A., Kovalenko, M., Murtha, R., Seong, I., Gusella, J.F. et al. (2020) Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington's disease knock-in mice is blocked by Mlh1 knock-out. Hum. Mol. Genet., 29, 3044–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao, X.N. and Usdin, K. (2018) FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair (Amst.), 69, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Long, J.D., Lee, J.M., Aylward, E.H., Gillis, T., Mysore, J.S., Abu Elneel, K., Chao, M.J., Paulsen, J.S., MacDonald, M.E. and Gusella, J.F. (2018) Genetic modification of Huntington disease acts early in the prediagnosis phase. Am. J. Hum. Genet., 103, 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee, J.M., Chao, M.J., Harold, D., Abu Elneel, K., Gillis, T., Holmans, P., Jones, L., Orth, M., Myers, R.H., Kwak, S. et al. (2017) A modifier of Huntington's disease onset at the MLH1 locus. Hum. Mol. Genet., 26, 3859–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Genetic Modifiers of Huntington's Disease Consortium (2019) CAG repeat not polyglutamine length determines timing of Huntington's disease onset. Cell, 178, 887–900 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goold, R., Flower, M., Moss, D.H., Medway, C., Wood-Kaczmar, A., Andre, R., Farshim, P., Bates, G.P., Holmans, P., Jones, L. et al. (2019) FAN1 modifies Huntington's disease progression by stabilizing the expanded HTT CAG repeat. Hum. Mol. Genet., 28, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moss, D.J.H., Pardinas, A.F., Langbehn, D., Lo, K., Leavitt, B.R., Roos, R., Durr, A., Mead, S., Track-HD investigators, Registry investigators, R. et al. (2017) Identification of genetic variants associated with Huntington's disease progression: a genome-wide association study. Lancet Neurol., 16, 701–711. [DOI] [PubMed] [Google Scholar]
- 78.Dragileva, E., Hendricks, A., Teed, A., Gillis, T., Lopez, E.T., Friedberg, E.C., Kucherlapati, R., Edelmann, W., Lunetta, K.L., MacDonald, M.E. et al. (2009) Intergenerational and striatal CAG repeat instability in Huntington's disease knock-in mice involve different DNA repair genes. Neurobiol. Dis., 33, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holbert, S., Denghien, I., Kiechle, T., Rosenblatt, A., Wellington, C., Hayden, M.R., Margolis, R.L., Ross, C.A., Dausset, J., Ferrante, R.J. et al. (2001) The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis. Proc. Natl. Acad. Sci. USA, 98, 1811–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chao, M.J., Kim, K.H., Shin, J.W., Lucente, D., Wheeler, V.C., Li, H., Roach, J.C., Hood, L., Wexler, N.S., Jardim, L.B. et al. (2018) Population-specific genetic modification of Huntington's disease in Venezuela. PLoS Genet., 14, e1007274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gellera, C., Meoni, C., Castellotti, B., Zappacosta, B., Girotti, F., Taroni, F. and DiDonato, S. (1996) Errors in Huntington disease diagnostic test caused by trinucleotide deletion in the IT15 gene. Am. J. Hum. Genet., 59, 475–477. [PMC free article] [PubMed] [Google Scholar]
- 82.Pecheux, C., Mouret, J.F., Durr, A., Agid, Y., Feingold, J., Brice, A., Dode, C. and Kaplan, J.C. (1995) Sequence analysis of the CCG polymorphic region adjacent to the CAG triplet repeat of the HD gene in normal and HD chromosomes. J. Med. Genet., 32, 399–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hong, E.P., MacDonald, M.E., Wheeler, V.C., Jones, L., Holmans, P., Orth, M., Monckton, D.G., Long, J.D., Kwak, S., Gusella, J.F. et al. (2021) Huntington's disease pathogenesis: two sequential components. J. Huntington's Dis., 10, 35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ciosi, M., Maxwell, A., Cumming, S.A., Hensman Moss, D.J., Alshammari, A.M., Flower, M.D., Durr, A., Leavitt, B.R., Roos, R.A.C., team, T.-H.D. et al. (2019) A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine, 48, 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wright, G.E.B., Collins, J.A., Kay, C., McDonald, C., Dolzhenko, E., Xia, Q., Becanovic, K., Drogemoller, B.I., Semaka, A., Nguyen, C.M. et al. (2019) Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am. J. Hum. Genet., 104, 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bates, G.P., Dorsey, R., Gusella, J.F., Hayden, M.R., Kay, C., Leavitt, B.R., Nance, M., Ross, C.A., Scahill, R.I., Wetzel, R. et al. (2015) Huntington disease. Nat. Rev. Dis. Primers., 1, 15005. [DOI] [PubMed] [Google Scholar]
- 87.Bassi, S., Tripathi, T., Monziani, A., Di Leva, F. and Biagioli, M. (2017) Epigenetics of Huntington's disease. Adv. Exp. Med. Biol., 978, 277–299. [DOI] [PubMed] [Google Scholar]
- 88.Jung, T., Shin, B., Tamo, G., Kim, H., Vijayvargia, R., Leitner, A., Marcaida, M.J., Astorga-Wells, J., Jung, R., Aebersold, R. et al. (2020) The polyglutamine expansion at the N-terminal of Huntingtin protein modulates the dynamic configuration and phosphorylation of the C-terminal HEAT domain. Structure, 28, 1035–1050 e1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sathasivam, K., Neueder, A., Gipson, T.A., Landles, C., Benjamin, A.C., Bondulich, M.K., Smith, D.L., Faull, R.L., Roos, R.A., Howland, D. et al. (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA, 110, 2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gipson, T.A., Neueder, A., Wexler, N.S., Bates, G.P. and Housman, D. (2013) Aberrantly spliced HTT, a new player in Huntington's disease pathogenesis. RNA Biol., 10, 1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schwartz, J.L., Jones, K.L. and Yeo, G.W. (2021) Repeat RNA expansion disorders of the nervous system: post-transcriptional mechanisms and therapeutic strategies. Crit. Rev. Biochem. Mol. Biol., 56, 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang, X., Goodrich, K.J., Conlon, E.G., Gao, J., Erbse, A.H., Manley, J.L. and Cech, T.R. (2019) C9orf72 and triplet repeat disorder RNAs: G-quadruplex formation, binding to PRC2 and implications for disease mechanisms. RNA, 25, 935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bruneau, B.G. and Nora, E.P. (2018) Chromatin domains go on repeat in disease. Cell, 175, 38–40. [DOI] [PubMed] [Google Scholar]
- 94.Marxreiter, F., Stemick, J. and Kohl, Z. (2020) Huntingtin lowering strategies. Int. J. Mol. Sci., 21, 2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shin, J.W., Kim, K.H., Chao, M.J., Atwal, R.S., Gillis, T., MacDonald, M.E., Gusella, J.F. and Lee, J.M. (2016) Permanent inactivation of Huntington's disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet., 25, 4566–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tabrizi, S.J., Leavitt, B.R., Landwehrmeyer, G.B., Wild, E.J., Saft, C., Barker, R.A., Blair, N.F., Craufurd, D., Priller, J., Rickards, H. et al. (2019) Targeting Huntingtin expression in patients with Huntington's disease. N. Engl. J. Med., 380, 2307–2316. [DOI] [PubMed] [Google Scholar]