

Abstract
The application of genomics to medicine has accelerated the discovery of mutations underlying disease and has enhanced our knowledge of the molecular underpinnings of diverse pathologies. As the amount of human genetic material queried via sequencing has grown exponentially in recent years, so too has the number of rare variants observed. Despite progress, our ability to distinguish which rare variants have clinical significance remains limited. Over the last decade, however, powerful experimental approaches have emerged to characterize variant effects orders of magnitude faster than before. Fueled by improved DNA synthesis and sequencing and, more recently, by CRISPR/Cas9 genome editing, multiplex functional assays provide a means of generating variant effect data in wide-ranging experimental systems. Here, I review recent applications of multiplex assays that link human variants to disease phenotypes and I describe emerging strategies that will enhance their clinical utility in coming years.
Introduction: The Challenge of Going from Variant to Function
Millions of human exomes and genomes have now been sequenced, yet we have only observed a small fraction of the rare variants in people alive today. Estimates of de novo mutation rates suggest that every single nucleotide variant (SNV) compatible with life occurs at least once per generation (1). In the genome aggregation database (gnomAD), comprising exomes and genomes from ~141 000 individuals, the majority of observed variants occur in exactly one individual and only 11.5% of possible synonymous SNVs occur at all (2). Improved sampling of genetically diverse populations will undoubtedly reveal new variants associated with phenotypes (3–5) but will also yield more rare variants whose phenotypic consequences are unknown. Similar to germline variants, large numbers of somatic mutations have been observed across cancer genomes (6,7). A small fraction occurs repeatedly, yet vastly more are unique variants with unknown effects on disease.
Through approaches ranging from direct-to-consumer genetic testing and liquid biopsies to whole-exome and whole-genome sequencing, more patients than ever before are receiving genetic test results (8). The value of identifying a causal germline variant has been well established for monogenic diseases (9–11). Furthermore, targeted therapies available to treat genetic diseases and cancers are giving clinicians the means to capitalize on the knowledge of variant effect more than ever before (12–16).
Yet, the translational potential of genomics remains limited largely by our inability to predict which variants observed in patients influence actionable phenotypes. For coding variants in genes commonly sequenced, this problem is manifest in hundreds of thousands of variants of uncertain significance (VUS) in databases such as ClinVar (17). These are often missense or splice variants that may alter a gene’s function in one of several ways (e.g. loss-of-function and gain-of-function), or have no discernible effect at all. In non-coding sequence, genome-wide association studies (GWAS) have linked thousands of loci to disease (18–20), yet pinpointing causal variants and discovering the precise mechanisms through which they act remain major bottlenecks to discovery (21). Missing heritability estimates suggest that rare variants of large effect are often missed by current ascertainment practices (22,23). This is further supported by less-than-predicted diagnostic yields when genetic testing is performed for many conditions (24,25).
The challenge of rare variant interpretation stems from an incomplete molecular accounting of how changes to deoxyribonucleic acid (DNA) sequence alter function on the molecular, cellular and organismal levels. Classical genetics approaches enable variant–phenotype associations without requiring knowledge of mechanism, but despite growing cohorts and genomic coverage, they still lack statistical power for most rare variants (26). Computational models that leverage, for instance, sequence conservation (27–29), epigenetic profiling (30,31) and/or biochemical properties of proteins (32,33) have improved, but they do not display the accuracy required for clinical variant classification without additional evidence.
Functional assays allow researchers to assess variant effects in isolation—e.g. determining how a missense mutation alters enzymatic activity, or how a promoter variant impacts gene expression. It is difficult to develop assays that guarantee high clinical impact, however, largely because variants may exert phenotypic effects through myriad molecular mechanisms. Furthermore, the incomplete penetrance, variable expressivity and pleiotropy observed across genetic disease highlight how complex making clinical predictions from molecular phenotypes can be. Despite these challenges, if a given element is linked to disease, functional data can be highly informative. Accordingly, American College of Medical Genetics and Genomics (ACMG) guidelines allow well-validated experimental data to serve as strong evidence of pathogenicity (34).
Until recently, efforts to classify variants experimentally have scaled poorly. The vast majority of variants observed in patients—even many known to be associated with human phenotypes—have never been tested in a laboratory setting. This may be starting to change, however, with the introduction of functional assays to measure variant effects at scale (35–37).
In this review, I describe how multiplex assays are enhancing our understanding of variant function in disease, with a focus on emerging strategies for increasing clinical impact. The challenge of variant interpretation often requires multiple assays that measure different molecular and cellular phenotypes to more fully unravel disease mechanisms. Relatedly, advances in genome editing are allowing variants to be assayed in their endogenous context more easily. Seamless integration of assay data with catalogs of human variation linked to phenotypic data will allow researchers to rapidly relate experimental findings to clinical significance. Therefore, as multiplex assays continue to shed light on the mechanisms underlying variant-phenotype associations, it follows that soon many more rare variants will become actionable, leading to tangible benefits for greater numbers of patients.
Multiplex assays: measuring variant effects with deep sequencing
Multiplex assays serve to reveal how each of many DNA sequences alter biological function. Sometimes broadly referred to as multiplex assays of variant effect (MAVEs) (37), these methods include deep mutational scanning (DMS), massively parallel reporter assays (MPRAs) and saturation genome editing (SGE), among others (35,38–40) (Fig. 1).
Figure 1 .
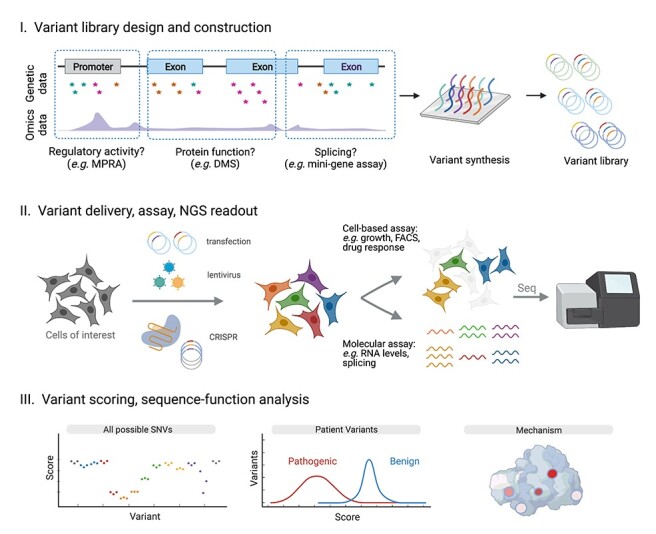
Principles of multiplex assays for interrogating human variant effects. Regions for mutagenesis are chosen from the genome with consideration of variants associated with disease and various omics data sets. In experimental design (Step I), variant alleles are cloned into an assay-specific construct, such as a reporter vector (MPRA), expression constructs (DMS), minigene cassettes (splice assays) or constructs to facilitate genome editing (SGE). Variants are then introduced to cells to create a diverse population in which each variant is present in many cells (Step II). Cell-based and molecular assays compatible with NGS are used to readout the effect of each variant in the pooled population. In analysis (Step III), sequencing counts are used to assign variants scores that can be compared to established pathogenic and benign variants. Integrated analysis with other sources of data (e.g. protein structure) can lead to mechanistic insights.
What makes multiplex assays highly scalable is that variants are engineered and tested in a pooled format, drastically reducing cost and minimizing sample processing. This is possible because next-generation sequencing (NGS) is used in a quantitative fashion to report on the functional effects of each variant in a pool—i.e. to ‘readout’ the assay. As standard NGS protocols can provide billions of sequencing reads (41), sequencing-based readouts have the statistical power to make hundreds of thousands of quantitative measurements of variant effect.
Since multiplex assays were first demonstrated over a decade ago, methods for engineering variant libraries and quantifying results with NGS have matured substantially (37,42). There are robust strategies to avoid experimental bottlenecks and analysis frameworks to faithfully extrapolate variant effect measurements from sequencing data (43–47). This has made it possible for small teams of scientists to quickly test thousands to hundreds of thousands of variants with minimal noise, though care must be taken to preserve data quality.
Because multiplex assays rely on NGS to measure variant effects, relatively simple assays have predominated to date. Common approaches to link variants to their effects rely on selection of phenotypes, such as cell growth (e.g. gene essentiality and drug resistance) (40,48–52), fluorescence (e.g. fluorescence-activated cell sorting (FACS)-based on target protein abundance or reporter expression) (53–56) or biochemical properties (57). Pools of cells are sampled corresponding to different timepoints, treatments or phenotypes, and sequencing of each pool enables comparison of variant frequencies across the experiment. For instance, if a variant becomes highly abundant after a drug treatment, we may infer it confers resistance. A common strategy for assays that look at regulatory element function or splicing is to assess transcript abundances through targeted RNA sequencing, often using molecular barcoding. A distinct advantage of multiplex assays is that experiments are internally controlled via inclusion of sequences with known effects, facilitating systematic comparisons of all variants in relation to those with established phenotypes.
Developing multiplex assays with high clinical relevance
The most immediate clinical impact of multiplex assays is being made via their application to coding sequences, largely because so many missense variants of unknown function have been encountered clinically. The groundwork for studying missense variants at scale was established in 2010. Fowler et al. (57) introduced deep mutational scanning to interrogate >600 000 variants in the human WW domain using phage display. Concurrently, Ernst et al. (58) used a similar strategy to test PDZ domain variants. Since then, over a million variants across dozens of different proteins have been engineered and assayed to examine effects on processes including protein stability, enzymatic activity, binding interactions, folding and structure, allostery and many more (51,55,59–65). The nuances of different assays have been reviewed elsewhere (42), and a thorough listing is available online (see https://www.mavedb.org) (66).
Recent deployments of functional assays to study protein variants have proven to be highly accurate at predicting pathogenicity when results are benchmarked to clinically established annotations. A yeast complementation assay was used to create a variant-effect map for CBS, the gene underlying classical homocystinuria (67). In addition to predicting pathogenic variants more accurately than computational models, the authors show that the degree of assay impairment correlates with the age of disease onset and severity in patients. Likewise, a study of >14 000 amyloid beta variants’ effects on aggregation enabled accurate identification of all 12 familial Alzheimer’s variants known to act dominantly despite the assay being performed in yeast (68). The first DMS of MSH2, a mismatch repair gene underlying Lynch syndrome in which >2000 VUS have been reported, achieved over 95% concordance with prior clinical interpretations of missense variants using 6-thioguanine selection (52). Our study of nearly 4000 BRCA1 variant effects using genome editing likewise showed >96% concordance with established variant annotations (69). These results underscore the value relatively simple assays can have for prospective variant classification when applied to genes with well-established phenotypes.
Searching for causal variants in non-coding sequences
Multiplex assays have been used to ask both which non-coding elements are functionally relevant and how specific variants alter function. In homage to the first saturation mutagenesis experiments studying the beta-globin promoter (70), MPRAs use reporter constructs to ask how DNA sequences function to initiate transcription. They are particularly useful for testing candidate regulatory elements nominated via association studies or biochemical annotation (71,72).
In 2009, one of the first multiplex assays to use NGS as a readout used in vitro transcription to test synthesized promoter fragments (73). Since then, several renditions of MPRAs have been performed using episomal- or integration-based cellular expression systems (74,75). Strategies for quantifying effects include expressed barcodes (38,39), reading out candidate elements from RNA directly (76) and using FACS to separate cell populations based on expression (77). MPRAs have also been carried out in primary cells, models of stem cell differentiation and in vivo (38,78–80).
Identifying causal variants in GWAS-implicated loci can provide new insights into underlying pathways that drive disease in humans (81). Once validated regulatory elements are linked to downstream gene targets, for instance, via clustered regularly interspaced short palindromic repeats (CRISPR) editing (82,83), further functional characterization may reveal drug targets (84). While there are relatively few regulatory regions known to harbor highly penetrant, pathogenic variants, MPRAs can identify which variants are critical in such regions. In one example, Doan et al. (85) use MPRAs to implicate homozygous variants in human-accelerated regions underlying autism cases. In an expansive effort, Kircher et al. (86) tested the functional effects of >30 000 point mutations across 20 non-coding regions implicated in disease, including the TERT and LDRL promoters and the SORT1-associated enhancer. This approach accurately identified causal variants across loci, thereby establishing the broad utility of MPRAs to aid classification of rare non-coding variants.
Multiplex assays are also showing great potential for identifying splice variants of strong effect. These assays have largely been performed using minigenes on plasmids transfected into human cell lines and have relied on transcribed barcodes, sequencing of variants from RNA or fluorescent reporter systems (56,87–90). In one powerful example, Rosenberg et al. (91) used splicing data from millions of degenerate sequences to train a highly accurate model for predicting splicing outcomes. A theme emerging from this work is that many splice-disruptive variants occur relatively far from canonical splice junctions, often extending deep into exons and introns. Notably, profiling >27 000 rare variants from human exomes revealed that nearly 4% disrupted splicing and that the vast majority of these occurred outside of canonical splice sites (56).
Beyond splicing, other multiplex assays to study RNA function include mutating 5′ untranslated regions (UTRs) (92) and synonymous codons (93) to study translation rates and mutating 3′ UTRs to assess messenger RNA (mRNA) stability (94). With more RNA sequencing and whole-genome sequencing being used clinically, these assays promise to illuminate additional mechanisms by which variants exert phenotypic effects in patients (95).
Emerging themes: integration of readouts from multiple functional assays achieves greater phenotypic depth
Assays with relatively simple readouts that are broadly generalizable across loci will prove valuable for scaling experiments to meet clinical demand. However, recent studies illustrate that more phenotypically detailed information can be gained by interrogating libraries with multiple functional readouts and in multiple cell types (Table 1). Cell-based approaches in which variant libraries are stably integrated allow cells to be expanded and assayed in multiple ways (40,50,96,97). One recent study tested the same MPRA library in five different cell lines to improve the identification of causal variants from GWAS data and to nominate cell-type-specific effects (94). Using cell-based assays, a deep mutational scan of the warfarin-target VKOR was used to readout both protein stability and enzymatic activity (61). This dual approach elucidated four transmembrane domains and key active site residues, while also providing clinical insights into variants that increase warfarin sensitivity.
Table 1.
Strategies for combining data across multiplex assays to reveal mechanisms
| Strategy | Benefit | Examples |
|---|---|---|
| Combining readouts of protein function | ||
| Protein stability and enzymatic activity | Corroborating pathogenicity; nominating dominant negative variants | VKOR (61); PTEN (101); nudix hydrolase 15 (NUDT15) (97) |
| Specific protein function and cell survival | Corroborating pathogenicity; linking specific functions to cell-based phenotypes | BRCA1 (54) |
| Multiple drug treatments | Interrogating pathway dependencies; mapping resistance mutations | BCR-ABL (50); MCL1, BCL2L1 (117) |
| Analyzing splicing and protein function | ||
| RNA expression and cell survival | Improves clinical accuracy by identifying splice variants (including intronic) | BRCA1 (69); CARD11 (110) |
| Testing variants in multiple cell lines | ||
| Engineered genetic backgrounds | Discerns dominant versus recessive effects; assess epistasis | TP53 (99,100) |
| Different cell types | Reveals cell-type effects on gene regulation; explains mutational profiles in disease | DNA damage response pathway (118); several regulatory loci (86,94) |
| Cancer cell growth in vitro versus in vivo | Separating cell-intrinsic and cell-extrinsic variant effects | TP53 (98) |
Three mutational scans have been performed for TP53, the tumor suppressor gene most commonly mutated in human cancer. First, Kotler et al. (98) asked how mutations to the protein’s DNA-binding domain affect cell growth, both in culture and in tumor models. Whereas, hotspot mutations did not confer a growth advantage over null alleles in vitro, they did in vivo, a finding suggestive of potential gain-of-function effects. Meanwhile, Giacomelli et al. (99) leveraged CRISPR-screening data to devise assays in isogenic p53+ and p53-null lines using multiple drug treatments. The different combinations distinguished dominant negative variants from loss-of-function alleles. Finally, Boettcher et al. (100) used a leukemia line to show that hotspot TP53 mutations act as dominant negatives, a mechanism that fully explains the TP53 mutational landscape of acute myeloid leukemia (AML). Collectively, these papers illustrate how cellular context, genetic background and assay design can be crucial to elucidate disease mechanisms of variants.
Two groups have applied mutational scans to the tumor suppressor PTEN, studying variants’ effects on protein stability in human cells (55) and lipid phosphatase activity in yeast (59). Analyzing these data in conjunction with well-curated patient data revealed variants that increase autism spectrum disorder (ASD) risk and associate with early-onset cancer (101). Highlighting the value of integrating multiple assays, putative dominant negative variants were identified that retained stability but not enzymatic activity.
An alternative approach to layering multiple assays would be to use a single assay capable of capturing different functional classes of variants. Of note, one group has recently demonstrated using single-cell RNA-sequencing to interrogate patient variants observed in the oncogenes NRAS and MYC (102). Coupling expressed barcodes to each variant allowed cells to be genotyped and transcriptionally profiled, revealing distinct pathways activated by specific mutations. Though this implementation required sequencing >300 000 single cells to study 200 variants, single-cell readouts may prove advantageous for exploring variant effects with greater phenotypic depth going forward.
A common limitation to multiplex assays is the use of complementary DNA (cDNA) libraries that preclude discovery of splice-altering variants. Therefore, combining assays that assess splicing with those measuring effects on the protein level will be essential to achieve optimal clinical accuracy. Recently, >1000 variants in POU1F1 were assayed using a minigene reporter, and 113 were deemed splice-disruptive (90). Two of these co-segregated with disease in unsolved families with familial hypopituitarism. In our work on BRCA1, we were able to measure variant effects on both protein function and mRNA levels by using SGE. This implicated ~10% of loss-of-function missense variants as disrupting splicing (69). With whole-genome and RNA sequencing becoming more common clinically (95,103), there will be more opportunities to link splice-disruptive variants within introns to human phenotypes using multiplex assays.
Emerging themes: genome editing allows variants to be tested at endogenous loci with growing ease
As illustrated by variants impacting splicing, it is often advantageous to test variants at their endogenous loci. Apart from splicing, genomic context can be crucial for maintaining physiological protein expression levels and for assessing variants in regulatory elements. By way of example, a comparison of identical MPRAs performed on genome-integrated versus non-integrated constructs showed only a weak correlation in scores (104).
To our benefit, genome editing technologies have improved since the introduction of CRISPR/Cas9 to facilitate more efficient and precise editing in a wide variety of cell types (105) (Table 2). Several methods have been established to boost homology-directed repair (HDR) efficiencies, allowing more efficient integration of variants of interest (106). Haploid human lines (e.g. HAP1) can be edited to reveal variant effects that are recessive on the cellular level (69,107,108), though engineering polyploid cells to contain a single copy of a target locus may prove viable for more cell types (109). Likewise, a cloning-free SGE protocol was recently deployed in a diploid B cell lymphoma line to link dominant negative variants in CARD11 to rare immunodeficiencies (110).
Table 2.
Selected genome editing assays for testing human variant at scale
| Method | Paper | Description | Assay |
|---|---|---|---|
| SGE | (40) | HDR-mediated integration of variants at Cas9-targeted loci | Hexamer effects on splicing in HEK293 (n = 4048); DBR1 variant fitness in HAP1 (n = 365) |
| (69) | (as above) | BRCA1 variant effects on HAP1 fitness (n = 3893) and transcript levels (n = 2646) | |
| (110) | Cloning-free SGE with single-stranded DNA repair templates | CARD11 variant effects on TMD8 growth +/− ibrutinib and transcript levels (n = 2542) | |
| Base editor screens | (119) | gRNA libraries used with base editing to introduce specific variants | n = 745 gRNAs targeting all exons of BRCA1 for fitness effects in HAP1 |
| (117) | (as above) | n = 70 000+ gRNAs tested in various cell lines (HAP1, MELJUSO, A375 and HT29) and assays (drug sensitivity, resistance and fitness of 57 000+ ClinVar variants) | |
| (118) | (as above) | n = 50 000+ gRNAs to tile 86 DNA damage response genes, assaying essentiality and response to DNA damage drugs in MCF10A, MCF7 and HAP1 | |
| Saturation prime editing | (121) | Prime editing gRNAs designed to achieve saturation mutagenesis | Variant effects on lysosome trafficking (NPC1; n = 256) and growth (BRCA2; n = 465) in 293T |
Base editing and prime editing technologies have emerged as CRISPR-based alternatives for creating programmed variants and are continuing to improve through protein engineering (111–113). Base editors use Cas9 fused to cytosine or adenine deaminase domains to achieve targeted editing (111,114), whereas prime editing is accomplished via Cas9-directed reverse transcription to introduce programmed variants (112). One advantage of these systems is that highly specific variants are created without the need for double-stranded breaks and HDR, suggesting greater scalability may be possible (115,116).
Accordingly, the first large-scale base editor screens were recently published (117–119). Employing numerous growth-based assays in human cell lines, Hanna et al. assessed >52 000 ClinVar variants, discovering loss-of-function variants in disease genes and mapping protein residues where variants alter responses to targeted therapies. With similarly broad coverage, Cuella-Martin et al. used base editing to engineer missense variants across 86 DNA damage response genes, discovering functionally critical protein domains and providing evidence for VUS reclassification. Importantly, these screens required careful validation to confirm guide RNAs (gRNAs) scored as hits were creating the intended edits.
Though currently limited by lower editing efficiencies, optimization of prime editing systems may further facilitate saturation mutagenesis of endogenous loci (120). A recent preprint describes ‘saturation prime editing’, using libraries of prime editing gRNAs to resolve pathogenicity for hundreds of variants in BRCA2 and NPC1 (121). In the future, coupling improved versions of these technologies with CRISPR tools to identify regulatory elements (122) will help reveal the functional impact of rare variants at regulatory loci. Overall, the rapid pace of improvement to CRISPR reagents suggests that, in coming years, engineering large numbers of variants in their endogenous context may become as easy as engineering them on plasmids.
Emerging themes: large genetic databases and multiplex assays synergistically improve variant classification
Multiplex assays are inherently orthologous to classical genetics approaches and computational predictors. Therefore, new experimental data sets can be benchmarked using established genotype–phenotype relationships. Majithia et al. (53) used a FACS-based assay to measure the effects of ~10 000 PPARG variants and found many of the lowest scoring alleles were exclusive to type 2 diabetes patients in a cohort of ~20 000 individuals. Applying SGE to BRCA1, we could immediately validate our results via comparison to hundreds of variant interpretations provided in ClinVar (69). Other groups have since used BRCA1 SGE data to reanalyze variants seen in hereditary cancer predisposition testing. In one cohort, clinical records were used to show that BRCA1 variants deemed loss-of-function by SGE confer a clinical risk indistinguishable from previously established pathogenic variants (123). This example illustrates how multiplex assays can be rapidly validated with pre-existing genetic data and subsequently used to reclassify variants observed in patients (Fig. 2).
Figure 2 .
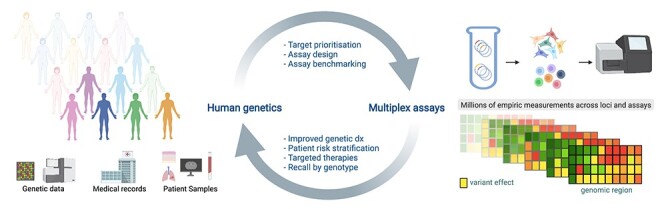
Integration of multiplex assays with genetic data from patients. Large numbers of VUS are observed in clinical testing and many loci associated with disease have yet to be functionally studied. These are priority targets to study using multiplex assays. Such assays can be rapidly validated via comparison to existing knowledge of variant effect and then integrated with large genetic data sets to improve diagnosis and help guide therapeutic strategies.
Large genetic databases lacking disease associations (e.g. gnomAD) also have utility for evaluating multiplex assays. Variants predicted to be deleterious by an assay should be seen less frequently in humans if they occur in genes under purifying selection, a trend now observed across multiple studies (52,69,101). Going forward, genetic data sets with deep phenotyping, such as the UK Biobank (124), will provide greater context for linking variant effects in vitro to human phenotypes. Recalling patients or accessing banked samples by genotype will enable rapid follow-up studies of variants deemed functionally relevant to disease.
Variant databases are increasingly incorporating results from functional assays (17,125) as well as predictions from machine learning methods (126–128). Models of variant effect built from multiplex assays have also been used to impute missing data and to predict deleterious variants genome-wide (44,129). As more experimental data are generated, such models will incrementally gain predictive power and may soon be able to accurately predict the effects of far more variants than can be assayed currently. To maximize the benefit of both multiplex assay data and improved computational models, we will require better tools for efficiently integrating multiple lines of evidence into clinical interpretation algorithms.
Conclusions and Future Challenges
In summary, multiplex assays have become a powerful means of generating variant effect data, and recent studies showcase how these technologies are starting to bring benefits to clinical variant interpretation.
Toward accurately reporting on the broad range of genetic effects throughout human development and disease, optimizing multiplex approaches in model systems, such as stem cells, organoids and in vivo models, will be a major challenge to overcome (130). Additionally, epistasis remains very difficult to test on the scale of individual variants owing to the immense number of potential interactions (131). Multiplex assays have the potential to improve variant interpretation for diverse populations that have been historically underrepresented in genetic studies (132). To make this work, however, we must ensure well-curated experimental data sets are widely available and take care to assess their utility across different populations. Global efforts to securely share genetic data will help facilitate this (133,134).
Despite these considerable challenges, the impact of these powerful technologies is already starting to be seen. By systematically testing large numbers of variants across numerous assays, we are building the basis for a more unbiased and quantitative understanding of genotype–phenotype relationships in humans. In coming years, multiplex assays will continue to reveal the genetic mechanisms underlying disease phenotypes, and in doing so, substantially improve the clinical utility of genetic data.
Supplementary Material
Acknowledgements
I thank Megan Buckley, Nicole Forrester and Joachim de-Jonghe for insightful discussions and feedback. Figures illustrations were made using BioRender software. Conflict of Interest statement. None declared.
Funding
Francis Crick Institute which receives its core funding from Cancer Research UK, the UK Medical Research Council and the Wellcome Trust (FC011142); Wellcome Trust (FC011142). Funding for open access charge: the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission.
References
- 1.Acuna-Hidalgo, R., Veltman, J.A. and Hoischen, A. (2016) New insights into the generation and role of de novo mutations in health and disease. Genome Biol., 17, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P. et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hindorff, L.A., Bonham, V.L., Brody, L.C., Ginoza, M.E.C., Hutter, C.M., Manolio, T.A. and Green, E.D. (2018) Prioritizing diversity in human genomics research. Nat. Rev. Genet., 19, 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergström, A., McCarthy, S.A., Hui, R., Almarri, M.A., Ayub, Q., Danecek, P., Chen, Y., Felkel, S., Hallast, P., Kamm, J. et al. (2020) Insights into human genetic variation and population history from 929 diverse genomes. Science, 367, eaay5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taliun, D., Harris, D.N., Kessler, M.D., Carlson, J., Szpiech, Z.A., Torres, R., Taliun, S.A.G., Corvelo, A., Gogarten, S.M., Kang, H.M. et al. (2021) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature, 590, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerami, E., Gao, J., Dogrusoz, U., Gross, B.E., Sumer, S.O., Aksoy, B.A., Jacobsen, A., Byrne, C.J., Heuer, M.L., Larsson, E. et al. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov., 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium (2020) Pan-cancer analysis of whole genomes. Nature, 578, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shendure, J., Findlay, G.M. and Snyder, M.W. (2019) Genomic medicine-progress, pitfalls, and promise. Cell, 177, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ACMG Board of Directors (2015) Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet. Med., 17, 505–507. [DOI] [PubMed] [Google Scholar]
- 10.Godard, B., ten Kate, L., Evers-Kiebooms, G. and Aymé, S. (2003) Population genetic screening programmes: principles, techniques, practices, and policies. Eur. J. Hum. Genet., 11, S49–S87. [DOI] [PubMed] [Google Scholar]
- 11.Yurgelun, M.B. and Hampel, H. (2018) Recent advances in lynch syndrome: diagnosis, treatment, and cancer prevention. Am. Soc. Clin. Oncol. Educ. Book, 38, 101–109. [DOI] [PubMed] [Google Scholar]
- 12.Baudino, T.A. (2015) Targeted cancer therapy: the next generation of cancer treatment. Curr. Drug Discov. Technol., 12, 3–20. [DOI] [PubMed] [Google Scholar]
- 13.Demarest, S.T. and Brooks-Kayal, A. (2018) From molecules to medicines: the dawn of targeted therapies for genetic epilepsies. Nat. Rev. Neurol., 14, 735–745. [DOI] [PubMed] [Google Scholar]
- 14.Quon, B.S. and Rowe, S.M. (2016) New and emerging targeted therapies for cystic fibrosis. BMJ, 352, i859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashworth, A. and Lord, C.J. (2018) Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol., 15, 564–576. [DOI] [PubMed] [Google Scholar]
- 16.June, C.H., O’Connor, R.S., Kawalekar, O.U., Ghassemi, S. and Milone, M.C. (2018) CAR T cell immunotherapy for human cancer. Science, 359, 1361–1365. [DOI] [PubMed] [Google Scholar]
- 17.Landrum, M.J., Lee, J.M., Benson, M., Brown, G., Chao, C., Chitipiralla, S., Gu, B., Hart, J., Hoffman, D., Hoover, J. et al. (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res., 44, D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maurano, M.T., Humbert, R., Rynes, E., Thurman, R.E., Haugen, E., Wang, H., Reynolds, A.P., Sandstrom, R., Qu, H., Brody, J. et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science, 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Degner, J.F., Pai, A.A., Pique-Regi, R., Veyrieras, J.-B., Gaffney, D.J., Pickrell, J.K., De Leon, S., Michelini, K., Lewellen, N., Crawford, G.E. et al. (2012) DNase I sensitivity QTLs are a major determinant of human expression variation. Nature, 482, 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang, F. and Lupski, J.R. (2015) Non-coding genetic variants in human disease. Hum. Mol. Genet., 24, R102–R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cano-Gamez, E. and Trynka, G. (2020) From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet., 11, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schubert, S.A., Morreau, H., de Miranda, N.F.C.C. and van Wezel, T. (2020) The missing heritability of familial colorectal cancer. Mutagenesis, 35, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuk, O., Schaffner, S.F., Samocha, K., Do, R., Hechter, E., Kathiresan, S., Daly, M.J., Neale, B.M., Sunyaev, S.R. and Lander, E.S. (2014) Searching for missing heritability: designing rare variant association studies. Proc. Natl. Acad. Sci. U. S. A., 111, E455–E464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stefanski, A., Calle-López, Y., Leu, C., Pérez-Palma, E., Pestana-Knight, E. and Lal, D. (2021) Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: a systematic review and meta-analysis. Epilepsia, 62, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lionel, A.C., Costain, G., Monfared, N., Walker, S., Reuter, M.S., Hosseini, S.M., Thiruvahindrapuram, B., Merico, D., Jobling, R., Nalpathamkalam, T. et al. (2018) Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet. Med., 20, 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barton, A.R., Sherman, M.A., Mukamel, R.E. and Loh, P.-R. (2021) Whole-exome imputation within UK Biobank powers rare coding variant association and fine-mapping analyses. Nat. Genet., 53, 1260–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng, P.C. and Henikoff, S. (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res., 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper, G.M., Stone, E.A., Asimenos, G., NISC Comparative Sequencing Program, Green, E.D., Batzoglou, S. and Sidow, A. (2005) Distribution and intensity of constraint in mammalian genomic sequence. Genome Res., 15, 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollard, K.S., Hubisz, M.J., Rosenbloom, K.R. and Siepel, A. (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res., 20, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, D., Gorkin, D.U., Baker, M., Strober, B.J., Asoni, A.L., McCallion, A.S. and Beer, M.A. (2015) A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet., 47, 955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beer, M.A. (2017) Predicting enhancer activity and variant impact using gkm-SVM. Hum. Mutat., 38, 1251–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glusman, G., Rose, P.W., Prlić, A., Dougherty, J., Duarte, J.M., Hoffman, A.S., Barton, G.J., Bendixen, E., Bergquist, T., Bock, C. et al. (2017) Mapping genetic variations to three-dimensional protein structures to enhance variant interpretation: a proposed framework. Genome Med., 9, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adzhubei, I., Jordan, D.M. and Sunyaev, S.R. (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet., 76, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W.W., Hegde, M., Lyon, E., Spector, E. et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fowler, D.M. and Fields, S. (2014) Deep mutational scanning: a new style of protein science. Nat. Methods, 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoue, F. and Ahituv, N. (2015) Decoding enhancers using massively parallel reporter assays. Genomics, 106, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gasperini, M., Starita, L. and Shendure, J. (2016) The power of multiplexed functional analysis of genetic variants. Nat. Protoc., 11, 1782–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patwardhan, R.P., Hiatt, J.B., Witten, D.M., Kim, M.J., Smith, R.P., May, D., Lee, C., Andrie, J.M., Lee, S.-I., Cooper, G.M. et al. (2012) Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol., 30, 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melnikov, A., Murugan, A., Zhang, X., Tesileanu, T., Wang, L., Rogov, P., Feizi, S., Gnirke, A., Callan, C.G., Jr., Kinney, J.B. et al. (2012) Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol., 30, 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Findlay, G.M., Boyle, E.A., Hause, R.J., Klein, J.C. and Shendure, J. (2014) Saturation editing of genomic regions by multiplex homology-directed repair. Nature, 513, 120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Modi, A., Vai, S., Caramelli, D. and Lari, M. (2021) The Illumina Sequencing Protocol and the NovaSeq 6000 System. In Mengoni, A., Bacci, G. and Fondi, M. (eds), (eds)Bacterial Pangenomics: Methods and Protocols. Springer US, New York, NY, pp. 15–42. [DOI] [PubMed] [Google Scholar]
- 42.Weile, J. and Roth, F.P. (2018) Multiplexed assays of variant effects contribute to a growing genotype-phenotype atlas. Hum. Genet., 137, 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hiatt, J.B., Patwardhan, R.P., Turner, E.H., Lee, C. and Shendure, J. (2010) Parallel, tag-directed assembly of locally derived short sequence reads. Nat. Methods, 7, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weile, J., Sun, S., Cote, A.G., Knapp, J., Verby, M., Mellor, J.C., Wu, Y., Pons, C., Wong, C., van Lieshout, N. et al. (2017) A framework for exhaustively mapping functional missense variants. Mol. Syst. Biol., 13, 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bloom, J.D. (2015) Software for the analysis and visualization of deep mutational scanning data. BMC Bioinformatics, 16, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rubin, A.F., Gelman, H., Lucas, N., Bajjalieh, S.M., Papenfuss, A.T., Speed, T.P. and Fowler, D.M. (2017) A statistical framework for analyzing deep mutational scanning data. Genome Biol., 18, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faure, A.J., Schmiedel, J.M., Baeza-Centurion, P. and Lehner, B. (2020) DiMSum: an error model and pipeline for analyzing deep mutational scanning data and diagnosing common experimental pathologies. Genome Biol., 21, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hietpas, R.T., Jensen, J.D. and Bolon, D.N.A. (2011) Experimental illumination of a fitness landscape. Proc. Natl. Acad. Sci. U. S. A., 108, 7896–7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitzman, J.O., Starita, L.M., Lo, R.S., Fields, S. and Shendure, J. (2015) Massively parallel single-amino-acid mutagenesis. Nat. Methods, 12, 203–206 4 p following 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma, L., Boucher, J.I., Paulsen, J., Matuszewski, S., Eide, C.A., Ou, J., Eickelberg, G., Press, R.D., Zhu, L.J., Druker, B.J. et al. (2017) CRISPR-Cas9-mediated saturated mutagenesis screen predicts clinical drug resistance with improved accuracy. Proc. Natl. Acad. Sci. U. S. A., 114, 11751–11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahler, E., Register, A.C., Chakraborty, S., Fang, L., Dieter, E.M., Sitko, K.A., Vidadala, R.S.R., Trevillian, B.M., Golkowski, M., Gelman, H. et al. (2019) A combined approach reveals a regulatory mechanism coupling Src’s kinase activity, localization, and phosphotransferase-independent functions. Mol. Cell, 74, 393–408.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jia, X., Burugula, B.B., Chen, V., Lemons, R.M., Jayakody, S., Maksutova, M. and Kitzman, J.O. (2021) Massively parallel functional testing of MSH2 missense variants conferring Lynch syndrome risk. Am. J. Hum. Genet., 108, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Majithia, A.R., Tsuda, B., Agostini, M., Gnanapradeepan, K., Rice, R., Peloso, G., Patel, K.A., Zhang, X., Broekema, M.F., Patterson, N. et al. (2016) Prospective functional classification of all possible missense variants in PPARG. Nat. Genet., 48, 1570–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Starita, L.M., Islam, M.M., Banerjee, T., Adamovich, A.I., Gullingsrud, J., Fields, S., Shendure, J. and Parvin, J.D. (2018) A multiplex homology-directed DNA repair assay reveals the impact of more than 1,000 BRCA1 missense substitution variants on protein function. Am. J. Hum. Genet., 103, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matreyek, K.A., Starita, L.M., Stephany, J.J., Martin, B., Chiasson, M.A., Gray, V.E., Kircher, M., Khechaduri, A., Dines, J.N., Hause, R.J. et al. (2018) Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet., 50, 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheung, R., Insigne, K.D., Yao, D., Burghard, C.P., Wang, J., Hsiao, Y.-H.E., Jones, E.M., Goodman, D.B., Xiao, X. and Kosuri, S. (2019) A multiplexed assay for exon recognition reveals that an unappreciated fraction of rare genetic variants cause large-effect splicing disruptions. Mol. Cell, 73, 183–194.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fowler, D.M., Araya, C.L., Fleishman, S.J., Kellogg, E.H., Stephany, J.J., Baker, D. and Fields, S. (2010) High-resolution mapping of protein sequence-function relationships. Nat. Methods, 7, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ernst, A., Gfeller, D., Kan, Z., Seshagiri, S., Kim, P.M., Bader, G.D. and Sidhu, S.S. (2010) Coevolution of PDZ domain-ligand interactions analyzed by high-throughput phage display and deep sequencing. Mol. BioSyst., 6, 1782–1790. [DOI] [PubMed] [Google Scholar]
- 59.Mighell, T.L., Evans-Dutson, S. and O’Roak, B.J. (2018) A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am. J. Hum. Genet., 102, 943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harris, D.T., Wang, N., Riley, T.P., Anderson, S.D., Singh, N.K., Procko, E., Baker, B.M. and Kranz, D.M. (2016) Deep mutational scans as a guide to engineering high affinity T cell receptor interactions with peptide-bound major histocompatibility complex. J. Biol. Chem., 291, 24566–24578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chiasson, M.A., Rollins, N.J., Stephany, J.J., Sitko, K.A., Matreyek, K.A., Verby, M., Sun, S., Roth, F.P., DeSloover, D., Marks, D.S. et al. (2020) Multiplexed measurement of variant abundance and activity reveals VKOR topology, active site and human variant impact. elife, 9, e58026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koenig, P., Lee, C.V., Walters, B.T., Janakiraman, V., Stinson, J., Patapoff, T.W. and Fuh, G. (2017) Mutational landscape of antibody variable domains reveals a switch modulating the interdomain conformational dynamics and antigen binding. Proc. Natl. Acad. Sci. U. S. A., 114, E486–E495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Starr, T.N., Greaney, A.J., Hilton, S.K., Ellis, D., Crawford, K.H.D., Dingens, A.S., Navarro, M.J., Bowen, J.E., Tortorici, M.A., Walls, A.C. et al. (2020) Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell, 182, 1295–1310.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmiedel, J.M. and Lehner, B. (2019) Determining protein structures using deep mutagenesis. Nat. Genet., 51, 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCormick, J.W., Russo, M.A.X., Thompson, S., Blevins, A. and Reynolds, K.A. (2021) Structurally distributed surface sites tune allosteric regulation. elife, 10, e68346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Esposito, D., Weile, J., Shendure, J., Starita, L.M., Papenfuss, A.T., Roth, F.P., Fowler, D.M. and Rubin, A.F. (2019) MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol., 20, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun, S., Weile, J., Verby, M., Wu, Y., Wang, Y., Cote, A.G., Fotiadou, I., Kitaygorodsky, J., Vidal, M., Rine, J. et al. (2020) A proactive genotype-to-patient-phenotype map for cystathionine beta-synthase. Genome Med., 12, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seuma, M., Faure, A.J., Badia, M., Lehner, B. and Bolognesi, B. (2021) The genetic landscape for amyloid beta fibril nucleation accurately discriminates familial Alzheimer’s disease mutations. elife, 10, e63364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Findlay, G.M., Daza, R.M., Martin, B., Zhang, M.D., Leith, A.P., Gasperini, M., Janizek, J.D., Huang, X., Starita, L.M. and Shendure, J. (2018) Accurate classification of BRCA1 variants with saturation genome editing. Nature, 562, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Myers, R.M., Tilly, K. and Maniatis, T. (1986) Fine structure genetic analysis of a β-globin promoter. Science, 232, 613–618. [DOI] [PubMed] [Google Scholar]
- 71.GTEx Consortium (2020) The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science, 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.ENCODE Project Consortium, Moore, J.E., Purcaro, M.J., Pratt, H.E., Epstein, C.B., Shoresh, N., Adrian, J., Kawli, T., Davis, C.A., Dobin, A. et al. (2020) Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature, 583, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patwardhan, R.P., Lee, C., Litvin, O., Young, D.L., Pe’er, D. and Shendure, J. (2009) High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol., 27, 1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trauernicht, M., Martinez-Ara, M. and van Steensel, B. (2020) Deciphering gene regulation using massively parallel reporter assays. Trends Biochem. Sci., 45, 90–91. [DOI] [PubMed] [Google Scholar]
- 75.Gasperini, M., Tome, J.M. and Shendure, J. (2020) Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet., 21, 292–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arnold, C.D., Gerlach, D., Stelzer, C., Boryń, Ł.M., Rath, M. and Stark, A. (2013) Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science, 339, 1074–1077. [DOI] [PubMed] [Google Scholar]
- 77.Townshend, B., Kennedy, A.B., Xiang, J.S. and Smolke, C.D. (2015) High-throughput cellular RNA device engineering. Nat. Methods, 12, 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bourges, C., Groff, A.F., Burren, O.S., Gerhardinger, C., Mattioli, K., Hutchinson, A., Hu, T., Anand, T., Epping, M.W., Wallace, C. et al. (2020) Resolving mechanisms of immune-mediated disease in primary CD4 T cells. EMBO Mol. Med., 12, e12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Inoue, F., Kreimer, A., Ashuach, T., Ahituv, N. and Yosef, N. (2019) Identification and massively parallel characterization of regulatory elements driving neural induction. Cell Stem Cell, 25, 713–727.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farley, E.K., Olson, K.M., Zhang, W., Brandt, A.J., Rokhsar, D.S. and Levine, M.S. (2015) Suboptimization of developmental enhancers. Science, 350, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tewhey, R., Kotliar, D., Park, D.S., Liu, B., Winnicki, S., Reilly, S.K., Andersen, K.G., Mikkelsen, T.S., Lander, E.S., Schaffner, S.F. et al. (2016) Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell, 165, 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klein, J.C., Keith, A., Rice, S.J., Shepherd, C., Agarwal, V., Loughlin, J. and Shendure, J. (2019) Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun., 10, 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gasperini, M., Hill, A. J., McFaline-Figueroa, J. L., Martin, B., Kim, S., Zhang, M. D., Jackson, D., Leith, A., Schreiber, J., Noble, W. S., et al. (2019) A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell, 176, 377–390.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi, J., Zhang, T., Vu, A., Ablain, J., Makowski, M.M., Colli, L.M., Xu, M., Hennessey, R.C., Yin, J., Rothschild, H. et al. (2020) Massively parallel reporter assays of melanoma risk variants identify MX2 as a gene promoting melanoma. Nat. Commun., 11, 2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Doan, R.N., Bae, B.-I., Cubelos, B., Chang, C., Hossain, A.A., Al-Saad, S., Mukaddes, N.M., Oner, O., Al-Saffar, M., Balkhy, S. et al. (2016) Mutations in human accelerated regions disrupt cognition and social behavior. Cell, 167, 341–354.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kircher, M., Xiong, C., Martin, B., Schubach, M., Inoue, F., Bell, R.J.A., Costello, J.F., Shendure, J. and Ahituv, N. (2019) Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun., 10, 3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ke, S., Shang, S., Kalachikov, S.M., Morozova, I., Yu, L., Russo, J.J., Ju, J. and Chasin, L.A. (2011) Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res., 21, 1360–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Julien, P., Miñana, B., Baeza-Centurion, P., Valcárcel, J. and Lehner, B. (2016) The complete local genotype–phenotype landscape for the alternative splicing of a human exon. Nat. Commun., 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ke, S., Anquetil, V., Zamalloa, J.R., Maity, A., Yang, A., Arias, M.A., Kalachikov, S., Russo, J.J., Ju, J. and Chasin, L.A. (2018) Saturation mutagenesis reveals manifold determinants of exon definition. Genome Res., 28, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gergics, P., Smith, C., Bando, H., Jorge, A.A.L., Rockstroh-Lippold, D., Vishnopolska, S., Castinetti, F., Maksutova, M., Carvalho, L.R.S., Hoppmann, J. et al. (2021) High-throughput splicing assays identify missense and silent splice-disruptive POU1F1 variants underlying pituitary hormone deficiency. Am. J. Hum. Genet., 108, 1526–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rosenberg, A.B., Patwardhan, R.P., Shendure, J. and Seelig, G. (2015) Learning the sequence determinants of alternative splicing from millions of random sequences. Cell, 163, 698–711. [DOI] [PubMed] [Google Scholar]
- 92.Sample, P.J., Wang, B., Reid, D.W., Presnyak, V., McFadyen, I.J., Morris, D.R. and Seelig, G. (2019) Human 5’ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol., 37, 803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmitz, A. and Zhang, F. (2021) Massively parallel gene expression variation measurement of a synonymous codon library. BMC Genomics, 22, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Griesemer, D., Xue, J.R., Reilly, S.K., Ulirsch, J.C., Kukreja, K., Davis, J., Kanai, M., Yang, D.K., Montgomery, S.B., Novina, C.D. et al. (2021) Genome-wide functional screen of 3’UTR variants uncovers causal variants for human disease and evolution. bioRxiv, 424697. doi: 10.1101/2021.01.13.424697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gonorazky, H.D., Naumenko, S., Ramani, A.K., Nelakuditi, V., Mashouri, P., Wang, P., Kao, D., Ohri, K., Viththiyapaskaran, S., Tarnopolsky, M.A. et al. (2019) Expanding the boundaries of RNA sequencing as a diagnostic tool for rare Mendelian disease. Am. J. Hum. Genet., 104, 466–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Matreyek, K.A., Stephany, J.J., Chiasson, M.A., Hasle, N. and Fowler, D.M. (2019) An improved platform for functional assessment of large protein libraries in mammalian cells. Nucleic Acids Res., 48, e1–e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Suiter, C.C., Moriyama, T., Matreyek, K.A., Yang, W., Scaletti, E.R., Nishii, R., Yang, W., Hoshitsuki, K., Singh, M., Trehan, A. et al. (2020) Massively parallel variant characterization identifies NUDT15 alleles associated with thiopurine toxicity. Proc. Natl. Acad. Sci. U. S. A., 117, 5394–5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kotler, E., Shani, O., Goldfeld, G., Lotan-Pompan, M., Tarcic, O., Gershoni, A., Hopf, T.A., Marks, D.S., Oren, M. and Segal, E. (2018) A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol. Cell, 71, 178–190.e8. [DOI] [PubMed] [Google Scholar]
- 99.Giacomelli, A.O., Yang, X., Lintner, R.E., McFarland, J.M., Duby, M., Kim, J., Howard, T.P., Takeda, D.Y., Ly, S.H., Kim, E. et al. (2018) Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet., 50, 1381–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Boettcher, S., Miller, P.G., Sharma, R., McConkey, M., Leventhal, M., Krivtsov, A.V., Giacomelli, A.O., Wong, W., Kim, J., Chao, S. et al. (2019) A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science, 365, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mighell, T.L., Thacker, S., Fombonne, E., Eng, C. and O’Roak, B.J. (2020) An integrated deep-mutational-scanning approach provides clinical insights on PTEN genotype-phenotype relationships. Am. J. Hum. Genet., 106, 818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ursu, O., Neal, J.T., Shea, E., Thakore, P.I., Jerby-Arnon, L., Nguyen, L., Dionne, D., Diaz, C., Bauman, J., Mosaad, M.M. et al. (2020) Massively parallel phenotyping of variant impact in cancer with Perturb-seq reveals a shift in the spectrum of cell states induced by somatic mutations. bioRxiv, 383307. doi: 10.1101/2020.11.16.383307. [DOI] [Google Scholar]
- 103.Montalban, G., Bonache, S., Moles-Fernández, A., Gisbert-Beamud, A., Tenés, A., Bach, V., Carrasco, E., López-Fernández, A., Stjepanovic, N., Balmaña, J. et al. (2019) Screening of BRCA1/2 deep intronic regions by targeted gene sequencing identifies the first germline BRCA1 variant causing pseudoexon activation in a patient with breast/ovarian cancer. J. Med. Genet., 56, 63–74. [DOI] [PubMed] [Google Scholar]
- 104.Inoue, F., Kircher, M., Martin, B., Cooper, G.M., Witten, D.M., McManus, M.T., Ahituv, N. and Shendure, J. (2017) A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res., 27, 38–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pickar-Oliver, A. and Gersbach, C.A. (2019) The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol., 20, 490–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu, M., Rehman, S., Tang, X., Gu, K., Fan, Q., Chen, D. and Ma, W. (2018) Methodologies for improving HDR efficiency. Front. Genet., 9, 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carette, J.E., Raaben, M., Wong, A.C., Herbert, A.S., Obernosterer, G., Mulherkar, N., Kuehne, A.I., Kranzusch, P.J., Griffin, A.M., Ruthel, G. et al. (2011) Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature, 477, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Erwood, S., Brewer, R.A., Bily, T.M.I., Maino, E., Zhou, L., Cohn, R.D. and Ivakine, E.A. (2019) Modeling Niemann-Pick disease type C in a human haploid cell line allows for patient variant characterization and clinical interpretation. Genome Res., 29, 2010–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Essletzbichler, P., Konopka, T., Santoro, F., Chen, D., Gapp, B.V., Kralovics, R., Brummelkamp, T.R., Nijman, S.M.B. and Bürckstümmer, T. (2014) Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res., 24, 2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meitlis, I., Allenspach, E.J., Bauman, B.M., Phan, I.Q., Dabbah, G., Schmitt, E.G., Camp, N.D., Torgerson, T.R., Nickerson, D.A., Bamshad, M.J. et al. (2020) Multiplexed functional assessment of genetic variants in CARD11. Am. J. Hum. Genet., 107, 1029–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Komor, A.C., Kim, Y.B., Packer, M.S., Zuris, J.A. and Liu, D.R. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature, 533, 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Anzalone, A.V., Randolph, P.B., Davis, J.R., Sousa, A.A., Koblan, L.W., Levy, J.M., Chen, P.J., Wilson, C., Newby, G.A., Raguram, A. et al. (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 576, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Anzalone, A.V., Koblan, L.W. and Liu, D.R. (2020) Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol., 38, 824–844. [DOI] [PubMed] [Google Scholar]
- 114.Gaudelli, N.M., Komor, A.C., Rees, H.A., Packer, M.S., Badran, A.H., Bryson, D.I. and Liu, D.R. (2017) Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature, 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yu, J.S.L. and Yusa, K. (2019) Genome-wide CRISPR-Cas9 screening in mammalian cells. Methods, 164–165, 29–35. [DOI] [PubMed] [Google Scholar]
- 116.Ma, Y., Zhang, J., Yin, W., Zhang, Z., Song, Y. and Chang, X. (2016) Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods, 13, 1029–1035. [DOI] [PubMed] [Google Scholar]
- 117.Hanna, R.E., Hegde, M., Fagre, C.R., DeWeirdt, P.C., Sangree, A.K., Szegletes, Z., Griffith, A., Feeley, M.N., Sanson, K.R., Baidi, Y. et al. (2021) Massively parallel assessment of human variants with base editor screens. Cell, 184, 1064–1080.e20. [DOI] [PubMed] [Google Scholar]
- 118.Cuella-Martin, R., Hayward, S.B., Fan, X., Chen, X., Huang, J.-W., Taglialatela, A., Leuzzi, G., Zhao, J., Rabadan, R., Lu, C. et al. (2021) Functional interrogation of DNA damage response variants with base editing screens. Cell, 184, 1081–1097.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kweon, J., Jang, A.-H., Shin, H.R., See, J.-E., Lee, W., Lee, J.W., Chang, S., Kim, K. and Kim, Y. (2020) A CRISPR-based base-editing screen for the functional assessment of BRCA1 variants. Oncogene, 39, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim, H.K., Yu, G., Park, J., Min, S., Lee, S., Yoon, S. and Kim, H.H. (2020) Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol., 39, 198–206. [DOI] [PubMed] [Google Scholar]
- 121.Erwood, S., Bily, T.M.I., Lequyer, J., Yan, J., Gulati, N., Brewer, R.A., Zhou, L., Pelletier, L., Ivakine, E.A. and Cohn, R.D. (2021) Saturation variant interpretation using CRISPR prime editing. bioRxiv, (2021), 443710. doi: 10.1101/2021.05.11.443710. [DOI] [PubMed] [Google Scholar]
- 122.Kampmann, M. (2018) CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem. Biol., 13, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wan, Q., Hu, L., Ouyang, T., Li, J., Wang, T., Fan, Z., Fan, T., Lin, B., Xu, Y. and Xie, Y. (2021) Clinical phenotypes combined with saturation genome editing identifying the pathogenicity of BRCA1 variants of uncertain significance in breast cancer. Familial Cancer, 20, 85–95. [DOI] [PubMed] [Google Scholar]
- 124.Bycroft, C., Freeman, C., Petkova, D., Band, G., Elliott, L.T., Sharp, K., Motyer, A., Vukcevic, D., Delaneau, O., O’Connell, J. et al. (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature, 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cline, M.S., Liao, R.G., Parsons, M.T., Paten, B., Alquaddoomi, F., Antoniou, A., Baxter, S., Brody, L., Cook-Deegan, R., Coffin, A. et al. (2018) BRCA challenge: BRCA exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet., 14, e1007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rentzsch, P., Witten, D., Cooper, G.M., Shendure, J. and Kircher, M. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res., 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sundaram, L., Gao, H., Padigepati, S.R., McRae, J.F., Li, Y., Kosmicki, J.A., Fritzilas, N., Hakenberg, J., Dutta, A., Shon, J. et al. (2018) Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet., 50, 1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J.F., Darbandi, S.F., Knowles, D., Li, Y.I., Kosmicki, J.A., Arbelaez, J., Cui, W., Schwartz, G.B. et al. (2019) Predicting splicing from primary sequence with deep learning. Cell, 176, 535–548.e24. [DOI] [PubMed] [Google Scholar]
- 129.Gray, V.E., Hause, R.J., Luebeck, J., Shendure, J. and Fowler, D.M. (2018) Quantitative missense variant effect prediction using large-scale mutagenesis data. Cell Syst, 6, 116–124.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yu, K., Lin, C.-C.J., Hatcher, A., Lozzi, B., Kong, K., Huang-Hobbs, E., Cheng, Y.-T., Beechar, V.B., Zhu, W., Zhang, Y. et al. (2020) PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nature, 578, 166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Han, K., Jeng, E.E., Hess, G.T., Morgens, D.W., Li, A. and Bassik, M.C. (2017) Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol., 35, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ndugga-Kabuye, M.K. and Issaka, R.B. (2019) Inequities in multi-gene hereditary cancer testing: lower diagnostic yield and higher VUS rate in individuals who identify as Hispanic, African or Asian and Pacific Islander as compared to European. Familial Cancer, 18, 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Philippakis, A.A., Azzariti, D.R., Beltran, S., Brookes, A.J., Brownstein, C.A., Brudno, M., Brunner, H.G., Buske, O.J., Carey, K., Doll, C. et al. (2015) The matchmaker exchange: a platform for rare disease gene discovery. Hum. Mutat., 36, 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Molnár-Gábor, F. and Korbel, J.O. (2020) Genomic data sharing in Europe is stumbling-could a code of conduct prevent its fall? EMBO Mol. Med., 12, e11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.