

Abstract
The causative gene in cystic fibrosis (CF) was identified in 1989, 3 years before the publication of the first issue of Human Molecular Genetics. The cystic fibrosis transmembrane conductance regulator (CFTR) gene was among the first underlying a common inherited disorder to be cloned, and hence, its subsequent utilization toward a cure for CF provides a roadmap for other monogenic diseases. Over the past 30 years, the advances that built upon knowledge of the gene and the CFTR protein to develop effective therapeutics have been remarkable, and yet, the setbacks have also been challenging. Technological progress in other fields has often circumvented the barriers. This review focuses on key aspects of CF diagnostics and current approaches to develop new therapies for all CFTR mutations. It also highlights the major research advances that underpinned progress toward treatments and considers the remaining obstacles.
Introduction
Cystic fibrosis (CF) has long been regarded as a paradigm for the challenges of developing effective treatments or a cure for human monogenic disease. With an incidence close to 1 in 2500 live births in some populations, a frequently devastating phenotype when untreated, and until very recently, a life-limiting prognosis, CF has been the focus of a massive international research effort over the past 30 years.
Much has been written about the story of finding the causative gene in CF, the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which was published in 1989 (1–3). An earlier false start (4) delayed progress in finding the gene and set unrealistic expectations for the development of a cure for CF. Although gene therapy was a major early therapeutic focus (reviewed in 5), this approach encountered many insurmountable challenges at that time, in part, owing to a paucity of information on the relevant cellular targets and the difficulties in delivery to them. It took more than 20 years for the first effective pharmacological treatment for any CFTR mutation to reach the clinic, when the CFTR potentiator, ivacaftor (VX-770), was shown to improve lung function in CF patients with one G551D (the substitution of glycine by aspartic acid at amino acid 551) allele (6). Now, the majority of CF patients can benefit from therapeutic combinations of small molecule correctors of CFTR (reviewed in 7–11), where these are available.
Over the past 30 years, the successes of the effort to effectively treat CF have been driven by a broad multidisciplinary approach to the disease together with strong collaborations across diverse research teams and between academia and industry. It is not possible in a short review to cover all aspects of the work that paved the way for the development of effective CF therapies. Instead, I will focus upon more recent exciting advances in understanding the molecular genetic causes of CF and on novel approaches to developing treatments for the disease. I will also refer back to the foundational work upon which the current approaches are based.
Insights from Classical Genetics and Diagnostics
In the first year of Human Molecular Genetics (1992), several papers were published defining new mutations in the CFTR gene. The identification of the gene and the most common mutations opened a chapter of mutation-hunting in patients with CF and a number of other diagnoses, including pancreatitis (12–14) and congenital bilateral absence of the vas deferens (15). More than 2000 errors in CFTR are associated with CF www.genet.sickkids.on.ca, though the majority are not yet validated experimentally with respect to CF causation (https://cftr2.org).
In most countries with routine newborn testing for genetic diseases, this can be readily extended to include a diagnostic screen for CFTR mutations. Indeed, CFTR screening is now an integral part of the CF diagnosis (16), and early identification dramatically improves the patient outcomes. Most cases of CF occurring without a known family history of the disease are first detected by raised serum immunoreactive trypsinogen (IRT) tests on the Guthrie card blood sample taken at birth. Elevated IRT levels are seen in CF owing to early in utero damage to the pancreas and indicate a need for DNA-based diagnostic testing for mutations in the CFTR gene and subsequent sweat chloride tests (>60 mmol/l* is suggestive of CF). There are many robust approaches to perform mutation scanning across the CFTR locus, a service provided by commercial diagnostics laboratories or hospital-based genetics centers. In general, these tests screen for about 50 common errors within the coding sequence of CFTR, including splice sites, and known deep intronic pathogenic variants and may be tailored to the prevalence of mutations in specific populations (17). Identification of two mutant alleles is considered to be a positive diagnosis of CF, which is confirmed by a sweat test. A finding of only one mutant allele with an elevated IRT also warrants a sweat test, which, if positive*, confirms CF, but if normal, denotes a CF carrier. An intermediate sweat chloride with one CF mutant allele is considered an inconclusive diagnosis, called as CF-related metabolic syndrome (CRMS) (18) in the USA and as CF screen-positive, inconclusive diagnosis (CFSPID) (19,20) in Europe. The standard DNA tests detect the majority of mutations in CFTR, though there is still a need for advanced DNA analysis, which may include sequencing of the gene promoter, exons and intron/exon boundaries. In rare cases, whole locus/whole genome deep sequencing is required. The reliance on polymerase chain reaction (PCR) amplification for diagnostic testing has missed a significant number of mutations that involve large deletions or rearrangements in the CFTR locus, effectively producing a null allele (21,22). Specific deletions or duplications can be identified by microarray hybridization technology and may also be detected by examining read depth of deep-sequencing across the locus. The dramatic advances in functional genomics protocols based upon next-generation sequencing also facilitate screening for mutations that cause epigenetic changes at the CFTR locus. Although still too expensive for routine diagnostics, these approaches will likely extend the phenotypes associated with CFTR mutations.
CFTR, CF Modifier Genes and New Therapeutics
Seminal early work showed a chloride permeability defect in CF sweat gland ducts (23). Also, a novel small conductance anion channel was identified in human pancreatic duct cells, where it was predicted to drive bicarbonate secretion in parallel with a chloride bicarbonate exchanger (24,25). Later, the CFTR gene identified by positional cloning (3) was found to encode a small conductance anion channel (26–31). Together with biochemical evidence for the most common F508del mutation resulting in a misfolded protein (32,33), these data laid the foundation for in vitro cell culture systems to measure the properties of CFTR, the impact of mutations on the channel and the development of high throughput screens to develop novel therapeutics. Many research groups contributed to understanding the structure/function relationships of CFTR, a topic that is well reviewed elsewhere (34,35) and is still an active topic of investigation (36,37).
It became evident, relatively soon after the CFTR gene was cloned and the F508del mutation was identified, that patients with the same genotype could have very different disease outcomes. Many studies suggested a non-CFTR genetic component to this variability (reviewed in 38,39), which was confirmed by twin and sibling studies (40). These observations initiated an international effort to collect a sufficient number of CF patients for robust genome-wide association studies (GWAS) to find the potential genetic basis of this phenotypic diversity (41,42). GWAS identify regions of the genome that contain variants, which may influence loci associated with a phenotype, though it is often a long road to establish causality. The mechanisms underlying the impact of variants within the coding region of a gene may be relatively easily established. As for many other genetic diseases/traits, not all GWAS hits associated with CF lung disease severity have been readily associated with causative loci. Since many variants are intergenic, they are predicted to be regulatory, though the precise transcript, splice variant or cell type of importance may be obscure. Here, I will consider the high P-value SNPs associated with CF disease severity which are located on chromosomes 11, 5 and X alone.
Based upon the surrounding topological domain structure (43), it is probable that the strong association of CF lung disease severity with SNPs at chr11p13 is marking two epithelial ETS family transcription factor E47-like Ets transcription factor 5 (ELF5) (44–46) (Swahn et al., unpublished data) and Ets homologous factor (EHF) (47,48). However, the underlying causative mechanism has been hard to uncover. Though the transcriptional networks for both ELF5 and EHF have a pivotal role in maintaining lung epithelial integrity and its response to physical and environmental insults, it has not been possible to directly correlate high P-value SNPs 100 kb distal to these genes with their expression levels. Perhaps this lack of correlation is the result of very tight cell-type-specific expression of these two factors, particularly ELF5. Critical cell types may not be well represented in cultures of human airway epithelium or tissue samples where gene expression levels are assayed. An additional confounding variable is that ELF5 is a strong repressor of EHF and both genes apparently autoregulate (49), processes which could readily mask the functional association of a specific disease-associated SNP.
More clarity has been achieved by investigating modifiers of CF intestinal disease, particularly the bowel obstruction phenotype that may manifest at birth as meconium ileus. Notable are members of the solute carrier (SLC) family of genes, including SLC26A9 at chr1q32.1, SLC6A14 at chrXq23-34 and SLC9A3 at chr5p15.33 (50,51), which are strong candidates, though again causality is not fully established. SLC26A9 is an anion transporter and has a critical role in bicarbonate secretion from epithelial cells through chloride/bicarbonate exchange and chloride-independent release. Since one of the main functions of the pancreatic duct epithelium is the maintenance of very high luminal bicarbonate concentrations, a role for SLC26A9 as a modifier of CF digestive disease is pertinent. SLC6A14 encodes a basic and neutral amino acid transporter that is sodium- and chloride-dependent and has a well-studied role in the intestinal epithelium. SLC9A3 encodes a sodium–hydrogen exchanger that is important in the mouse intestinal epithelium. Of note, SLC9A3 is also involved in CF pathology in the airway epithelium (52), and another SLC family member, SLC26A6, was found to interact directly with CFTR in the mouse pancreas (53). Because of their mechanistic link with CFTR as an anion transporter, these SLCs are attractive candidates for novel therapeutics (54).
Approaches to find therapeutic modifiers of CF disease by targeting other ion channels, transporters or exchangers, to circumvent a defective CFTR protein, have also arisen independently of the GWAS. One notable example, which is currently in Phase 1 clinical trials, is a potentiator of the TMEM16A calcium-activated chloride channel (55–57) that is expressed in multiple cell types in the airway epithelium. Activation of TMEM16A in CF epithelia may enhance fluid secretion to a level that restores normal mucociliary clearance, though a direct effect on mucus secretion is not proven (58). Another therapeutic target is the epithelial sodium channel (ENaC) (59), which is pivotal in driving fluid transport in the lung epithelium and is influenced by CFTR (60,61). Loss of CFTR is associated with upregulation of ENaC, which in turn, reduces airway surface liquid and dehydrates mucus. Hence, ENaC inhibition is an attractive therapeutic approach that is currently in clinical trial (62).
Single Cell Atlases and CFTR Expression
Developing effective molecular therapeutics for CF may depend upon identifying the precise identity of the cell types in each tissue that expresses CFTR. Although there are few data examining the amount of CFTR mRNA and protein that is required to restore normal function to an epithelium, it is probable that expressing CFTR in the correct cell type may be advantageous. Data generated more than 20 years ago, showing tight cell-specific expression of CFTR mRNA by in situ hybridization (ISH) (63–65), have largely been confirmed by recent high-resolution single cell RNA sequencing (scRNA-seq) (66–68). ScRNA data generated from the pancreas (Fig. 1A and B) and small intestine (Fig. 1C and D) confirmed earlier mRNA ISH and immunocytochemistry results showing high levels of CFTR in pancreatic duct and intestinal crypt epithelial cells, respectively. In contrast, scRNA-seq data were less consistent with the observations of CFTR in ciliated cells in the lung epithelium (69). A rare high CFTR-expressing cell type that was observed but not characterized in earlier work was found to be the site of most abundant CFTR in the lung. This ‘pulmonary ionocyte’ has a unique gene expression signature driven by transcription factors, such as forkhead box I1 (FOXIi) and achaete-scute family BHLH transcription factor 3 (ASCL3). A rare high CFTR-expressing cell was also found much earlier by high-resolution immunostaining in the intestinal epithelium (70). Similarly, rare clear cells in the epididymis epithelium, which have a well-studied role in ion transport, also express high levels of CFTR and FOXI1 in addition to specific subunits of the H+-transporting vacuolar V-ATPase (Fig. 1E and F). Hence, clear cells and ‘pulmonary ionocytes’ may occupy a functionally similar niche in the epididymis and airway epithelium. CFTR is also robustly expressed in secretory cells in the efferent ducts of the male reproductive tract. The loss/obstruction of genital ducts in CF males is consistent with these findings.
Figure 1 .
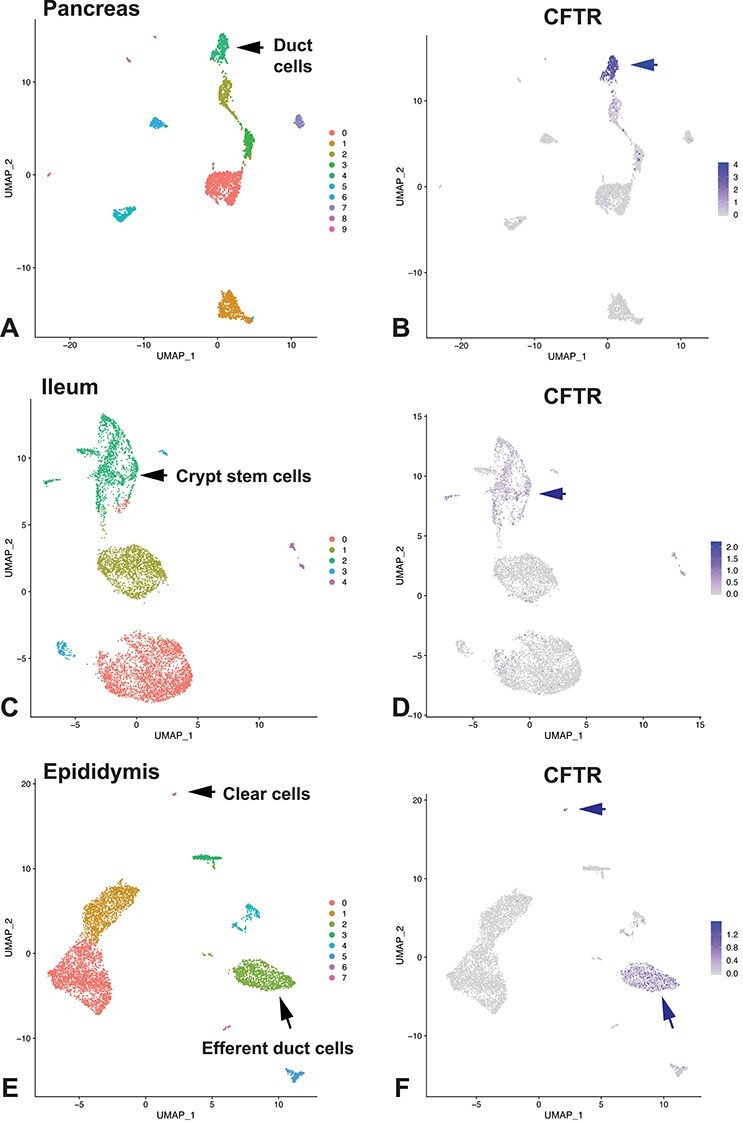
CFTR expression in individual cell types shown by scRNA-seq. All data generated by 10X Genomics Chromium platform. Dataset for pancreas GEO:GSE85241 (115); small intestine (ileum) GEO:GSE125970 (116); epididymis GEO:GSE148963 (117). (A), (C) and (E) show uniform manifold approximation and projection (UMAP) dimension reduction plots for cell clusters in pancreas, ileum and epididymis, respectively. CFTR expression in each tissue type shown by UMAP plots in corresponding images (B), (D) and (F). Markers used for cluster assignment: (A), (B), pancreatic duct: SLC family 4 member 4 (SLC4A4), which encodes a sodium bicarbonate co-transporter known to be involved in pancreatic duct bicarbonate secretion (118) and keratin 19 (KRT19); (C), (D), crypt stem cells: olfactome-din-4 (OLFM4); (F), (G), villin (VIL) in efferent duct, ATPase H+ transporting V1 subunit G3 (ATP6V1G3) in clear cells.
In addition to its abundance in pulmonary ionocytes, CFTR is expressed at lower levels in certain secretory epithelial cells in the lung epithelium (71). When human bronchial epithelial (HBE) cells are isolated from the lung and are grown at air/liquid interface (a classical model for CFTR functional assays), subpopulations of secretory cells are seen by scRNA-seq to express CFTR. To what extent these CFTR-expressing cells have adapted to the in vitro culture conditions and how closely they resemble the differentiated properties of secretory epithelial cell populations in the intact lung is currently the focus of intense investigation. Together, with recent data showing that CF proximal lungs show altered cellularity (72), the results will be of great importance in designing effective cell-targeted therapeutics.
CRISPR/Cas9 and Other Gene Editing Therapeutics to Correct CFTR
Earlier attempts at CF gene therapy, aimed at delivering a normal copy of CFTR to the airway epithelium, were therapeutically ineffective. Inefficient liposome-mediated delivery, unacceptable risks associated with viral vectors and gene promoter silencing all contributed to this failure. These approaches will not be considered further here, as they were the topic of a 2019 review in the journal.
The advent of CRISPR/Cas9 and other gene editing technologies has opened a new chapter that may result in more effective approaches to cure rather than just treat CF. Two major opportunities to correct a mutant CFTR gene are immediately apparent. The first uses direct gene editing to repair errors involving one or a few bases in the coding region of CFTR. The second involves replacing the coding region of a defective CFTR gene with a perfect copy. Efficient repair of the F508del mutation was first achieved by CRISPR/Cas9 homology-directed repair in the intestinal stem cells, which were subsequently expanded into colon organoids (73); then by zinc finger nuclease (ZFN)-mediated replacement of F508del in airway basal cells (74); and most recently, by base editing in intestinal stem cell (75). The main limitation with base editing protocols is the need to generate new therapeutics for the majority of the >2000 predicted mutations in CFTR. Attempts to insert a correct cDNA into a 5′ exon or intron of CFTR are challenging for two main reasons: first, the lentivirus vectors optimized for effective delivery cannot accommodate the full CFTR cDNA; and second, the introduced cDNA may alter the higher order chromatin structure around the CFTR locus that is required for its normal regulation (76,77). Proof-of-principle studies validated this approach, when a partial cDNA (exons 9–27) was inserted into intron 8 of the CFTR locus by ZFNs, shown to splice normally to exon 8, and to restore a normal CFTR protein to F508del airway basal cells (74). Moreover, this insertion did not disrupt the 3D architecture of the endogenous CFTR locus. Another recent approach to circumvent the size limitations of therapeutic delivery of CFTR used two adeno-associated viral vectors sequentially, each containing half of the CFTR cDNA, to insert a correct full-length cDNA into exon 1 of the locus. This partially restored CFTR function in cultured HBE cells carrying a number of different mutations in CFTR (78). Whether this approach will result in stable long-term correction of CFTR mutations in vivo is not yet clear. Of note, gene replacement methods that use the endogenous CFTR promoter to drive cDNA expression may be most effective in bronchial epithelial cells, where CFTR enhancers are located upstream of the gene promoter (at −35 and −44 kb) (Fig. 2). Epithelial cells in the intestine utilize different intronic enhancers to drive the CFTR expression, including ones in the 1st and 11th introns (legacy nomenclature). In these cells, enhancer/promoter looping could be disrupted by intronic or exonic insertion and the expression of the CFTR cDNA. In the light of controversies over potential off-target actions of the CRISPR/Cas9 editing machinery (79,80), there may be some delay in considering exonic or intronic gene replacement as an in vivo therapeutic for CF. Particularly so for a disease that is life-limiting but not lethal and which now has effective pharmacological treatments for most mutations.
Figure 2 .

CFTR regulatory elements and therapeutic approaches. Cell-type-specific chromatin structure at the CFTR locus. UCSC genome browser graphic showing the genomic location of CFTR and adjacent genes (chr 7) (top) and a subset of key cis-regulatory elements for the CFTR locus (below, numbered according to location). Open chromatin mapped by DNase-seq and 4C-seq interaction profiles with a CFTR promoter viewpoint are shown for Caco2 (colon carcinoma), Calu-3 (lung adenocarcinoma) and skin fibroblasts (no CFTR expression). 4C-seq data have two parts. The upper part indicates the main trend of the contact profile using a 5 kb window size. Relative interactions are normalized to the strongest point (which is set to 1) within each image. The lower part is a domainogram with color-coded intensity values to show relative interactions with window sizes varying from 2 to 50 kb. Arrows denote key interactions: −80.1 and +48.9 kb mark the topological domain boundaries; −20.9 and +6.8 kb are key CTCF-binding insulator elements; intron 11 (legacy) harbors a strong intestinal enhancer; −44 and −35 kb contain strong enhancers in airway cell types (reviewed in 119).
Other Approaches to Restore Functional CFTR
Targeting splice-site mutations with antisense oligonucleotides (ASOs)
As for other monogenic diseases, a proportion of mutations are found within the splicing machinery for the CFTR mRNA. The variants may reduce efficacy of splice donor or acceptor sequences, alter splice site enhancers in the locus or generate novel intronic splice sites, which recruit non-coding sequences into the mRNA. Dramatic clinical success in the use of ASO therapeutics for spinal muscular atrophy (SMA) (reviewed in 81,82) has encouraged the ASO approach to correct splicing mutations in CFTR. There are several common splicing mutations in CFTR, which together contribute about 15% of known disease-associated mutations in the genes. These include c.1680-886A>G (1811+1634 G>A) and c.3718-2477C>T (3849+10 kb C>T), a deep intronic mutation (in intron 22) that creates a novel 5′ splice site causing inclusion of a cryptic exon containing a termination codon. An ASO approach has recently proven to be effective at repressing the mutation and blocking aberrant splicing in HBE cells (83). In addition, earlier work suggested that the c.3718-2477 C>T and c.2647+5G>A mutations in CFTR were also amenable to an ASO therapeutic approach (84,85). Further recent enhancements in the chemistry and bioavailability of ASOs developed for other diseases (reviewed in 86) will speed the progress of these therapies, which may be long-lasting. A remaining challenge is delivering the ASOs to the cells that express CFTR in the human airway epithelium and other relevant sites in vivo. However, there is optimism that these hurdles will be overcome.
Targeting stop mutations
Among other common errors in the CFTR gene are several stop mutations, including G542X (c.1624G>T), R553X (c.1657C>T) and W1282X (c.3846G>A). Since these mutant alleles do not produce a protein, the current pharmacological approaches using correctors and potentiator drugs are not effective therapies. As for stop mutations in other disease-associated loci, a huge effort has gone into to developing compounds that not only promote read-through of the stop signal, by enhancing recruitment of alternative amino acids, but also therapeutics that inhibit nonsense-mediated mRNA decay, which degrades transcripts containing premature termination codons. Though aminoglycoside antibiotics are effective at promoting read-through of stop mutations (87,88), in their current formulations, they are too toxic to be utilized as a viable therapeutic. A non-antibiotic aminoglycoside analog, ELX-02, appears to be less toxic, is effective at facilitating read-through of the G542X mutation and restores CFTR-dependent forskolin-induced swelling in CF patient-derived intestinal organoids (89). Currently, a number of other approaches to circumvent stop mutations are in pre-clinical testing (https://www.cff.org/trials/pipeline), including delivery of suppressor tRNAs (90), direct delivery of mRNA to the airway epithelium (reviewed in 91) or more conventional gene replacement therapies utilizing adeno-associated virus (reviewed in 92).
The last frontier: developmental aspects of CF and the need for model systems
The thick, sticky mucus that is a characteristic feature of CF underlies the pathology in many organ systems. The specific mucin glycoproteins involved may vary between tissues (93,94). However, the underlying deficit directly relates to loss of CFTR, which causes altered bicarbonate secretion (95), abnormal hydration (96–98) and an altered luminal milieu (99,100) on epithelial surfaces. Importantly, these changes alter the unpacking of mucus granules and the biophysics and clearance of the mucus layer (101–104) and may be a very early event in CF lung disease (105).
For the past 30 years, the main focus of developing new therapies for CF has been toward correcting lung disease, the major cause of morbidity. Though many of the therapeutics will have systemic effects, they will not be able to undo the pathology caused by loss of CFTR in utero. Particularly important is the damage to the pancreas, which is evident by the second trimester of gestation and results in pancreatic duct obstruction and destruction of acinar tissue. The cystic spaces which replace normal acinar tissue gave rise to the original name for the disease ‘cystic fibrosis of the pancreas’, first defined by Dorothy Anderson in 1938 (106–108). Similarly, some children with CF have early onset liver disease, which initiates through gall bladder obstruction during development, and males with CF usually lack intact genital ducts (vas deferens and epididymis) at birth. Since developmental deficits cannot be examined in humans, CF animal models have an additional valuable contribution to make. All the models have provided some insights into CF pathology (109), though the nuances or major divergence in anatomy and physiology between species make some models more useful than others to answer specific biological questions (110). Though the G551D CF ferret showed partial phenotypic correction of CF pathologies following sustained in utero administration of the CFTR potentiator VX-770 (ivacaftor) (111), at the time of writing, there are no published data on F508del or pan-mutation in utero correction. From a translational point of view, developmental aspects of CF are best studied in an animal model that mirrors human CF pathology in utero, which has a similar gestation and developmental profile as human and where pregnancies usually involve only one or two fetuses. These criteria are well met in the sheep models of CF (112) (Viotti Perisse et al. 2021, in revision); moreover, developmental physiology of the sheep lung is very well established (reviewed in 113). Like other large animal models of CF (114), the sheep has a profound intestinal phenotype at birth, which is much more severe than the presentation of meconium ileus in some babies with CF. However, the causes of this intestinal obstruction likely coincide with the equivalent human phenotype, specifically, early pancreatic duct obstruction and resulting pancreatic autolysis (Fig. 3), combined with characteristic liver and intestinal phenotypes. It is not yet clear why the large animal intestinal phenotype is so severe in both omnivores and herbivores, though it provides a clear endpoint to evaluate the efficacy of in utero therapeutics.
Figure 3 .
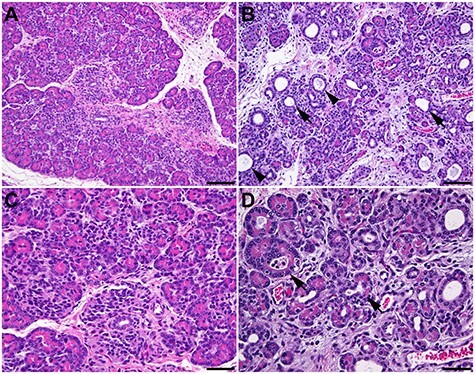
Pancreatic pathology of a newborn CFTR −/− lamb. (A) and (C) Histology of the normal pancreas of a control lamb. (B) and (D) CFTR −/− lamb. Pancreatic acinar atrophy with stromal collapse and dilatation of acini are evident (arrow). Mucus is present in some duct lumens (arrowhead). Hematoxylin and eosin staining. (A) and (B) 200×. Bar = 100 μm. (C) and (D) 400×. Bar = 50 μm.
In summary, 30 years of research into the molecular basis of CF disease has been remarkably productive. Although it has taken much longer than optimistic predictions, for most individuals with CF (and access to the new CFTR modulator drugs), key aspects of the disease are now effectively treated. In the spirit of ‘no-one left behind’, we must redouble our efforts to treat or correct the currently intractable mutations. Furthermore, an integrated, translational approach to utilize available animal models to better understand and prevent early lesions in CF, which may have life-limiting sequelae, is definitely warranted. Stem cell and organoid protocols may also make a substantial contribution in this area. These combined approaches will dovetail with advances in state of the art gene replacement/correction therapies, which will are likely to yield significant clinical benefits.
Acknowledgements
I thank Shiyi Yin for bioinformatics analysis of the data shown in Figure 1; Drs Arnaud Van Wettere and Irina Polejaeva for Figure 3; Drs Johanna Rommens, Gunnar Hansson and Rebecca Darrah for helpful discussion. Many scientists have contributed to the research progress over the past 30 years which has enabled the development of effective therapeutics for CF. Unfortunately, in a short review article, it is not possible to reference all relevant papers; so, with apologies, I acknowledge all contributors whose work I may not have specifically cited.
Conflict of Interest statement. None declared.
Funding
Our work is supported by the National Institutes of Health (R01HL094585 and R01HD068901) and the Cystic Fibrosis Foundation (Harris 15/17XX0, Harris18P0, Poleja18G0 and Davis 19XX0).
References
- 1.Kerem, B., Rommens, J.M., Buchanan, J.A., Markiewicz, D., Cox, T.K., Chakravarti, A., Buchwald, M. and Tsui, L.C. (1989) Identification of the cystic fibrosis gene: genetic analysis. Science, 245, 1073–1080. [DOI] [PubMed] [Google Scholar]
- 2.Rommens, J.M., Iannuzzi, M.C., Kerem, B., Drumm, M.L., Melmer, G., Dean, M., Rozmahel, R., Cole, J.L., Kennedy, D., Hidaka, N. et al. (1989) Identification of the cystic fibrosis gene: chromosome walking and jumping. Science, 245, 1059–1065. [DOI] [PubMed] [Google Scholar]
- 3.Riordan, J.R., Rommens, J.M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak, Z., Zielenski, J., Lok, S., Plavsic, N., Chou, J.L. et al. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science, 245, 1066–1073. [DOI] [PubMed] [Google Scholar]
- 4.Estivill, X., Farrall, M., Scambler, P.J., Bell, G.M., Hawley, K.M., Lench, N.J., Bates, G.P., Kruyer, H.C., Frederick, P.A., Stanier, P. et al. (1987) A candidate for the cystic fibrosis locus isolated by selection for methylation-free islands. Nature, 326, 840–845. [DOI] [PubMed] [Google Scholar]
- 5.Hart, S.L. and Harrison, P.T. (2017) Genetic therapies for cystic fibrosis lung disease. Curr. Opin. Pharmacol., 34, 119–124. [DOI] [PubMed] [Google Scholar]
- 6.Ramsey, B.W., Davies, J., McElvaney, N.G., Tullis, E., Bell, S.C., Drevinek, P., Griese, M., McKone, E.F., Wainwright, C.E., Konstan, M.W. et al. (2011) A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med., 365, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mijnders, M., Kleizen, B. and Braakman, I. (2017) Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis. Curr. Opin. Pharmacol., 34, 83–90. [DOI] [PubMed] [Google Scholar]
- 8.Boyle, M.P. and De Boeck, K. (2013) A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir. Med., 1, 158–163. [DOI] [PubMed] [Google Scholar]
- 9.Wainwright, C.E., Elborn, J.S., Ramsey, B.W., Marigowda, G., Huang, X., Cipolli, M., Colombo, C., Davies, J.C., De Boeck, K., Flume, P.A. et al. (2015) Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med., 373, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Veit, G., Roldan, A., Hancock, M.A., Da Fonte, D.F., Xu, H., Hussein, M., Frenkiel, S., Matouk, E., Velkov, T. and Lukacs, G.L. (2020) Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight, 5(18), e139983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bear, C.E. (2020) A therapy for most with cystic fibrosis. Cell, 180, 211. [DOI] [PubMed] [Google Scholar]
- 12.Ooi, C.Y. and Durie, P.R. (2012) Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in pancreatitis. J. Cyst. Fibros., 11, 355–362. [DOI] [PubMed] [Google Scholar]
- 13.Schneider, A., Larusch, J., Sun, X., Aloe, A., Lamb, J., Hawes, R., Cotton, P., Brand, R.E., Anderson, M.A., Money, M.E. et al. (2011) Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology, 140, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomez Lira, M., Patuzzo, C., Castellani, C., Bovo, P., Cavallini, G., Mastella, G. and Pignatti, P.F. (2001) CFTR and cationic trypsinogen mutations in idiopathic pancreatitis and neonatal hypertrypsinemia. Pancreatology, 1, 538–542. [DOI] [PubMed] [Google Scholar]
- 15.Dumur, V., Gervais, R., Rigot, J.-M., Delomel-Vinner, E., Decaestecker, B., Lafitte, J.-J. and Roussel, P. (1996) Congenital bilateral absence of the vas deferens (CBAVD) and cystic fibrosis transmembrane regulator (CFTR): correlation between genotype and phenotype. Hum. Genet., 97, 7–10. [DOI] [PubMed] [Google Scholar]
- 16.Farrell, P.M., White, T.B., Ren, C.L., Hempstead, S.E., Accurso, F., Derichs, N., Howenstine, M., McColley, S.A., Rock, M., Rosenfeld, M. et al. (2017) Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J. Pediatr., 181S, S4–S15 e11. [DOI] [PubMed] [Google Scholar]
- 17.Stafler, P., Mei-Zahav, M., Wilschanski, M., Mussaffi, H., Efrati, O., Lavie, M., Shoseyov, D., Cohen-Cymberknoh, M., Gur, M., Bentur, L. et al. (2016) The impact of a national population carrier screening program on cystic fibrosis birth rate and age at diagnosis: implications for newborn screening. J. Cyst. Fibros., 15, 460–466. [DOI] [PubMed] [Google Scholar]
- 18.Terlizzi, V., Claut, L., Tosco, A., Colombo, C., Raia, V., Fabrizzi, B., Lucarelli, M., Angeloni, A., Cimino, G., Castaldo, A. et al. (2021) A survey of the prevalence, management and outcome of infants with an inconclusive diagnosis following newborn bloodspot screening for cystic fibrosis (CRMS/CFSPID) in six Italian centres. J. Cyst. Fibros., 21, S1569-1993(21)00097-7. [DOI] [PubMed] [Google Scholar]
- 19.Munck, A., Bourmaud, A., Bellon, G., Picq, P., Farrell, P.M. and Group, D.S. (2020) Phenotype of children with inconclusive cystic fibrosis diagnosis after newborn screening. Pediatr. Pulmonol., 55, 918–928. [DOI] [PubMed] [Google Scholar]
- 20.Ooi, C.Y., Sutherland, R., Castellani, C., Keenan, K., Boland, M., Reisman, J., Bjornson, C., Chilvers, M.A., van Wylick, R., Kent, S. et al. (2019) Immunoreactive trypsinogen levels in newborn screened infants with an inconclusive diagnosis of cystic fibrosis. BMC Pediatr., 19, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferec, C., Casals, T., Chuzhanova, N., Macek, M., Jr., Bienvenu, T., Holubova, A., King, C., McDevitt, T., Castellani, C., Farrell, P.M. et al. (2006) Gross genomic rearrangements involving deletions in the CFTR gene: characterization of six new events from a large cohort of hitherto unidentified cystic fibrosis chromosomes and meta-analysis of the underlying mechanisms. Eur. J. Hum. Genet., 14, 567–576. [DOI] [PubMed] [Google Scholar]
- 22.Hantash, F.M., Redman, J.B., Starn, K., Anderson, B., Buller, A., McGinniss, M.J., Quan, F., Peng, M., Sun, W. and Strom, C.M. (2006) Novel and recurrent rearrangements in the CFTR gene: clinical and laboratory implications for cystic fibrosis screening. Hum. Genet., 119, 126–136. [DOI] [PubMed] [Google Scholar]
- 23.Quinton, P.M. (1983) Chloride impermeability in cystic fibrosis. Nature, 301, 421–422. [DOI] [PubMed] [Google Scholar]
- 24.Gray, M.A., Harris, A., Coleman, L., Greenwell, J.R. and Argent, B.E. (1989) Two types of chloride channel on duct cells cultured from human fetal pancreas. Am. J. Phys., 257, C240–C251. [DOI] [PubMed] [Google Scholar]
- 25.Gray, M.A., Pollard, C.E., Harris, A., Coleman, L., Greenwell, J.R. and Argent, B.E. (1990) Anion selectivity and block of the small-conductance chloride channel on pancreatic duct cells. Am. J. Phys., 259, C752–C761. [DOI] [PubMed] [Google Scholar]
- 26.Drumm, M.L., Pope, H.A., Cliff, W.H., Rommens, J.M., Marvin, S.A., Tsui, L.C., Collins, F.S., Frizzell, R.A. and Wilson, J.M. (1990) Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell, 62, 1227–1233. [DOI] [PubMed] [Google Scholar]
- 27.Gregory, R.J., Cheng, S.H., Rich, D.P., Marshall, J., Paul, S., Hehir, K., Ostedgaard, L., Klinger, K.W., Welsh, M.J. and Smith, A.E. (1990) Expression and characterization of the cystic fibrosis transmembrane conductance regulator. Nature, 347, 382–386. [DOI] [PubMed] [Google Scholar]
- 28.Rich, D.P., Anderson, M.P., Gregory, R.J., Cheng, S.H., Paul, S., Jefferson, D.M., McCann, J.D., Klinger, K.W., Smith, A.E. and Welsh, M.J. (1990) Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature, 347, 358–363. [DOI] [PubMed] [Google Scholar]
- 29.Bear, C.E., Duguay, F., Naismith, A.L., Kartner, N., Hanrahan, J.W. and Riordan, J.R. (1991) Cl-channel activity in Xenopus oocytes expressing the cystic fibrosis gene. J. Biol. Chem., 266, 19142–19145. [PubMed] [Google Scholar]
- 30.Bear, C.E., Li, C.H., Kartner, N., Bridges, R.J., Jensen, T.J., Ramjeesingh, M. and Riordan, J.R. (1992) Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell, 68, 809–818. [DOI] [PubMed] [Google Scholar]
- 31.Hasegawa, H., Skach, W., Baker, O., Calayag, M.C., Lingappa, V. and Verkman, A.S. (1992) A multifunctional aqueous channel formed by CFTR. Science, 258, 1477–1479. [DOI] [PubMed] [Google Scholar]
- 32.Cheng, S.H., Gregory, R.J., Marshall, J., Paul, S., Souza, D.W., White, G.A., O'Riordan, C.R. and Smith, A.E. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell, 63, 827–834. [DOI] [PubMed] [Google Scholar]
- 33.Lukacs, G.L., Mohamed, A., Kartner, N., Chang, X.B., Riordan, J.R. and Grinstein, S. (1994) Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J., 13, 6076–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukacs, G.L. and Verkman, A.S. (2012) CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol. Med., 18, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skach, W.R. (2006) CFTR: new members join the fold. Cell, 127, 673–675. [DOI] [PubMed] [Google Scholar]
- 36.Zhang, Z., Liu, F. and Chen, J. (2017) Conformational changes of CFTR upon phosphorylation and ATP binding. Cell, 170, 483–491.e488. [DOI] [PubMed] [Google Scholar]
- 37.Zhang, Z., Liu, F. and Chen, J. (2018) Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. U. S. A., 115, 12757–12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrari, M. and Cremonesi, L. (1996) Genotype-phenotype correlation in cystic fibrosis patients. Ann Biol Clin (Paris), 54, 235–241. [PubMed] [Google Scholar]
- 39.Salvatore, F., Scudiero, O. and Castaldo, G. (2002) Genotype-phenotype correlation in cystic fibrosis: the role of modifier genes. Am. J. Med. Genet., 111, 88–95. [DOI] [PubMed] [Google Scholar]
- 40.Vanscoy, L.L., Blackman, S.M., Collaco, J.M., Bowers, A., Lai, T., Naughton, K., Algire, M., McWilliams, R., Beck, S., Hoover-Fong, J. et al. (2007) Heritability of lung disease severity in cystic fibrosis. Am. J. Respir. Crit. Care Med., 175, 1036–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wright, F.A., Strug, L.J., Doshi, V.K., Commander, C.W., Blackman, S.M., Sun, L., Berthiaume, Y., Cutler, D., Cojocaru, A., Collaco, J.M. et al. (2011) Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat. Genet., 43, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corvol, H., Blackman, S.M., Boelle, P.Y., Gallins, P.J., Pace, R.G., Stonebraker, J.R., Accurso, F.J., Clement, A., Collaco, J.M., Dang, H. et al. (2015) Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun., 6, 8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stolzenburg, L.R., Yang, R., Kerschner, J.L., Fossum, S., Xu, M., Hoffmann, A., Lamar, K.M., Ghosh, S., Wachtel, S., Leir, S.H. et al. (2017) Regulatory dynamics of 11p13 suggest a role for EHF in modifying CF lung disease severity. Nucleic Acids Res., 45, 8773–8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalyuga, M., Gallego-Ortega, D., Lee, H.J., Roden, D.L., Cowley, M.J., Caldon, C.E., Stone, A., Allerdice, S.L., Valdes-Mora, F., Launchbury, R. et al. (2012) ELF5 suppresses estrogen sensitivity and underpins the acquisition of antiestrogen resistance in luminal breast cancer. PLoS Biol., 10, e1001461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piggin, C.L., Roden, D.L., Gallego-Ortega, D., Lee, H.J., Oakes, S.R. and Ormandy, C.J. (2016) ELF5 isoform expression is tissue-specific and significantly altered in cancer. Breast Cancer Res., 18, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piggin, C.L., Roden, D.L., Law, A.M.K., Molloy, M.P., Krisp, C., Swarbrick, A., Naylor, M.J., Kalyuga, M., Kaplan, W., Oakes, S.R. et al. (2020) ELF5 modulates the estrogen receptor cistrome in breast cancer. PLoS Genet., 16, e1008531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fossum, S.L., Mutolo, M.J., Tugores, A., Ghosh, S., Randell, S.H., Jones, L.C., Leir, S.H. and Harris, A. (2017) Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J. Biol. Chem., 292, 10938–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fossum, S.L., Mutolo, M.J., Yang, R., Dang, H., O'Neal, W.K., Knowles, M.R., Leir, S.H. and Harris, A. (2014) Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res., 42, 13588–13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swahn, H., Sabith Ebron, J., Lamar, K.M., Yin, S., Kerschner, J.L., NandyMazumdar, M., Coppola, C., Mendenhall, E.M., Leir, S.H. and Harris, A. (2019) Coordinate regulation of ELF5 and EHF at the chr11p13 CF modifier region. J. Cell. Mol. Med., 23, 7726–7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gong, J., Wang, F., Xiao, B., Panjwani, N., Lin, F., Keenan, K., Avolio, J., Esmaeili, M., Zhang, L., He, G. et al. (2019) Genetic association and transcriptome integration identify contributing genes and tissues at cystic fibrosis modifier loci. PLoS Genet., 15, e1008007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strug, L.J., Gonska, T., He, G., Keenan, K., Ip, W., Boelle, P.Y., Lin, F., Panjwani, N., Gong, J., Li, W. et al. (2016) Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics. Hum. Mol. Genet., 25, 4590–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dorfman, R., Taylor, C., Lin, F., Sun, L., Sandford, A., Pare, P., Berthiaume, Y., Corey, M., Durie, P. and Zielenski, J. (2011) Modulatory effect of the SLC9A3 gene on susceptibility to infections and pulmonary function in children with cystic fibrosis. Pediatr. Pulmonol., 46, 385–392. [DOI] [PubMed] [Google Scholar]
- 53.Wang, Y., Soyombo, A.A., Shcheynikov, N., Zeng, W., Dorwart, M., Marino, C.R., Thomas, P.J. and Muallem, S. (2006) Slc26a6 regulates CFTR activity in vivo to determine pancreatic duct HCO3− secretion: relevance to cystic fibrosis. EMBO J., 25, 5049–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strug, L.J., Stephenson, A.L., Panjwani, N. and Harris, A. (2018) Recent advances in developing therapeutics for cystic fibrosis. Hum. Mol. Genet., 27, R173–R186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caputo, A., Caci, E., Ferrera, L., Pedemonte, N., Barsanti, C., Sondo, E., Pfeffer, U., Ravazzolo, R., Zegarra-Moran, O. and Galietta, L.J. (2008) TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science, 322, 590–594. [DOI] [PubMed] [Google Scholar]
- 56.Benedetto, R., Ousingsawat, J., Wanitchakool, P., Zhang, Y., Holtzman, M.J., Amaral, M., Rock, J.R., Schreiber, R. and Kunzelmann, K. (2017) Epithelial chloride transport by CFTR requires TMEM16A. Sci. Rep., 7, 12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Danahay, H.L., Lilley, S., Fox, R., Charlton, H., Sabater, J., Button, B., McCarthy, C., Collingwood, S.P. and Gosling, M. (2020) TMEM16A potentiation: a novel therapeutic approach for the treatment of cystic fibrosis. Am. J. Respir. Crit. Care Med., 201, 946–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Danahay, H., Fox, R., Lilley, S., Charlton, H., Adley, K., Christie, L., Ansari, E., Ehre, C., Flen, A., Tuvim, M.J. et al. (2020) Potentiating TMEM16A does not stimulate airway mucus secretion or bronchial and pulmonary arterial smooth muscle contraction. FASEB Bioadv, 2, 464–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Canessa, C.M., Schild, L., Buell, G., Thorens, B., Gautschi, I., Horisberger, J.D. and Rossier, B.C. (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature, 367, 463–467. [DOI] [PubMed] [Google Scholar]
- 60.Ismailov, I.I., Awayda, M.S., Jovov, B., Berdiev, B.K., Fuller, C.M., Dedman, J.R., Kaetzel, M.A. and Benos, D.J. (1996) Regulation of epithelial sodium channels by the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem., 271, 4725–4732. [DOI] [PubMed] [Google Scholar]
- 61.Stutts, M.J., Rossier, B.C. and Boucher, R.C. (1997) Cystic fibrosis transmembrane conductance regulator inverts protein kinase A-mediated regulation of epithelial sodium channel single channel kinetics. J. Biol. Chem., 272, 14037–14040. [DOI] [PubMed] [Google Scholar]
- 62.Goss, C.H., Jain, R., Seibold, W., Picard, A.C., Hsu, M.C., Gupta, A. and Fajac, I. (2020) An innovative phase II trial to establish proof of efficacy and optimal dose of a new inhaled epithelial sodium channel inhibitor BI 1265162 in adults and adolescents with cystic fibrosis: BALANCE-CF(TM) 1. ERJ Open Res, 6, 00395–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trezise, A.E., Chambers, J.A., Wardle, C.J., Gould, S. and Harris, A. (1993) Expression of the cystic fibrosis gene in human foetal tissues. Hum. Mol. Genet., 2, 213–218. [DOI] [PubMed] [Google Scholar]
- 64.Hyde, K., Reid, C.J., Tebbutt, S.J., Weide, L., Hollingsworth, M.A. and Harris, A. (1997) The cystic fibrosis transmembrane conductance regulator as a marker of human pancreatic duct development. Gastroenterology, 113, 914–919. [DOI] [PubMed] [Google Scholar]
- 65.Engelhardt, J.F., Yankaskas, J.R., Ernst, S.A., Yang, Y., Marino, C.R., Boucher, R.C., Cohn, J.A. and Wilson, J.M. (1992) Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat. Genet., 2, 240–248. [DOI] [PubMed] [Google Scholar]
- 66.Montoro, D.T., Haber, A.L., Biton, M., Vinarsky, V., Lin, B., Birket, S.E., Yuan, F., Chen, S., Leung, H.M., Villoria, J. et al. (2018) A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature, 560, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Plasschaert, L.W., Zilionis, R., Choo-Wing, R., Savova, V., Knehr, J., Roma, G., Klein, A.M. and Jaffe, A.B. (2018) A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature, 560, 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deprez, M., Zaragosi, L.E., Truchi, M., Becavin, C., Ruiz Garcia, S., Arguel, M.J., Plaisant, M., Magnone, V., Lebrigand, K., Abelanet, S. et al. (2020) A single-cell atlas of the human healthy airways. Am. J. Respir. Crit. Care Med., 202, 1636–1645. [DOI] [PubMed] [Google Scholar]
- 69.Kreda, S.M., Mall, M., Mengos, A., Rochelle, L., Yankaskas, J., Riordan, J.R. and Boucher, R.C. (2005) Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell, 16, 2154–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ameen, N.A., Ardito, T., Kashgarian, M. and Marino, C.R. (1995) A unique subset of rat and human intestinal villus cells express the cystic fibrosis transmembrane conductance regulator. Gastroenterology, 108, 1016–1023. [DOI] [PubMed] [Google Scholar]
- 71.Okuda, K., Dang, H., Kobayashi, Y., Carraro, G., Nakano, S., Chen, G., Kato, T., Asakura, T., Gilmore, R.C., Morton, L.C. et al. (2020) Secretory cells dominate airway CFTR expression and function in human airway superficial epithelia. Am. J. Respir. Crit. Care Med., 203, 275–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carraro, G., Langerman, J., Sabri, S., Lorenzana, Z., Purkayastha, A., Zhang, G., Konda, B., Aros, C.J., Calvert, B.A., Szymaniak, A. et al. (2021) Transcriptional analysis of cystic fibrosis airways at single-cell resolution reveals altered epithelial cell states and composition. Nat. Med., 7, 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schwank, G., Koo, B.K., Sasselli, V., Dekkers, J.F., Heo, I., Demircan, T., Sasaki, N., Boymans, S., Cuppen, E., van der Ent, C.K. et al. (2013) Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell, 13, 653–658. [DOI] [PubMed] [Google Scholar]
- 74.Suzuki, S., Crane, A.M., Anirudhan, V., Barilla, C., Matthias, N., Randell, S.H., Rab, A., Sorscher, E.J., Kerschner, J.L., Yin, S. et al. (2020) Highly efficient gene editing of cystic fibrosis patient-derived airway basal cells results in functional CFTR correction. Mol. Ther., 28, 1684–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Geurts, M.H., de Poel, E., Amatngalim, G.D., Oka, R., Meijers, F.M., Kruisselbrink, E., van Mourik, P., Berkers, G., de Winter-de Groot, K.M., Michel, S. et al. (2020) CRISPR-based adenine editors correct nonsense mutations in a cystic fibrosis organoid biobank. Cell Stem Cell, 26, 503–510.e507. [DOI] [PubMed] [Google Scholar]
- 76.NandyMazumdar, M., Yin, S., Paranjapye, A., Kerschner, J.L., Swahn, H., Ge, A., Leir, S.H. and Harris, A. (2020) Looping of upstream cis-regulatory elements is required for CFTR expression in human airway epithelial cells. Nucleic Acids Res., 48, 3513–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang, R., Kerschner, J.L., Gosalia, N., Neems, D., Gorsic, L.K., Safi, A., Crawford, G.E., Kosak, S.T., Leir, S.H. and Harris, A. (2016) Differential contribution of cis-regulatory elements to higher order chromatin structure and expression of the CFTR locus. Nucleic Acids Res., 44, 3082–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vaidyanathan, S., Baik, R., Chen, L., Bravo, D.T., Suarez, C.J., Abazari, S.M., Salahudeen, A.A., Dudek, A.M., Teran, C.A., Davis, T.H. et al. (2021) Targeted replacement of full-length CFTR in human airway stem cells by CRISPR-Cas9 for pan-mutation correction in the endogenous locus. Mol. Ther., On line ahead of print., 21, S1525-0016(21)00152-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kosicki, M., Tomberg, K. and Bradley, A. (2018) Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol., 36, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leibowitz, M.L., Papathanasiou, S., Doerfler, P.A., Blaine, L.J., Sun, L., Yao, Y., Zhang, C.Z., Weiss, M.J. and Pellman, D. (2021) Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet., 53, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rigo, F., Hua, Y., Krainer, A.R. and Bennett, C.F. (2012) Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol., 199, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bennett, C.F., Krainer, A.R. and Cleveland, D.W. (2019) Antisense oligonucleotide therapies for neurodegenerative diseases. Annu. Rev. Neurosci., 42, 385–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Michaels, W.E., Bridges, R.J. and Hastings, M.L. (2020) Antisense oligonucleotide-mediated correction of CFTR splicing improves chloride secretion in cystic fibrosis patient-derived bronchial epithelial cells. Nucleic Acids Res., 48, 7454–7467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Friedman, K.J., Kole, J., Cohn, J.A., Knowles, M.R., Silverman, L.M. and Kole, R. (1999) Correction of aberrant splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by antisense oligonucleotides. J. Biol. Chem., 274, 36193–36199. [DOI] [PubMed] [Google Scholar]
- 85.Igreja, S., Clarke, L.A., Botelho, H.M., Marques, L. and Amaral, M.D. (2016) Correction of a cystic fibrosis splicing mutation by antisense oligonucleotides. Hum. Mutat., 37, 209–215. [DOI] [PubMed] [Google Scholar]
- 86.Silva, A.C., Lobo, D.D., Martins, I.M., Lopes, S.M., Henriques, C., Duarte, S.P., Dodart, J.C., Nobre, R.J. and Pereira de Almeida, L. (2020) Antisense oligonucleotide therapeutics in neurodegenerative diseases: the case of polyglutamine disorders. Brain, 143, 407–429. [DOI] [PubMed] [Google Scholar]
- 87.Clancy, J.P., Bebok, Z., Ruiz, F., King, C., Jones, J., Walker, L., Greer, H., Hong, J., Wing, L., Macaluso, M. et al. (2001) Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med., 163, 1683–1692. [DOI] [PubMed] [Google Scholar]
- 88.Wilschanski, M., Yahav, Y., Yaacov, Y., Blau, H., Bentur, L., Rivlin, J., Aviram, M., Bdolah-Abram, T., Bebok, Z., Shushi, L. et al. (2003) Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N. Engl. J. Med., 349, 1433–1441. [DOI] [PubMed] [Google Scholar]
- 89.Crawford, D.K., Mullenders, J., Pott, J., Boj, S.F., Landskroner-Eiger, S. and Goddeeris, M.M. (2021) Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J. Cyst. Fibros., 20, 436–442. [DOI] [PubMed] [Google Scholar]
- 90.Lueck, J.D., Yoon, J.S., Perales-Puchalt, A., Mackey, A.L., Infield, D.T., Behlke, M.A., Pope, M.R., Weiner, D.B., Skach, W.R., McCray, P.B., Jr. et al. (2019) Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun., 10, 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kowalski, P.S., Rudra, A., Miao, L. and Anderson, D.G. (2019) Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther., 27, 710–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang, D., Tai, P.W.L. and Gao, G. (2019) Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov., 18, 358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rose, M.C. and Voynow, J.A. (2006) Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol. Rev., 86, 245–278. [DOI] [PubMed] [Google Scholar]
- 94.Reid, C.J., Hyde, K., Ho, S.B. and Harris, A. (1997) Cystic fibrosis of the pancreas: involvement of MUC6 mucin in obstruction of pancreatic ducts. Mol. Med., 3, 403–411. [PMC free article] [PubMed] [Google Scholar]
- 95.Quinton, P.M. (2008) Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet, 372, 415–417. [DOI] [PubMed] [Google Scholar]
- 96.Boucher, R.C. (2007) Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med., 261, 5–16. [DOI] [PubMed] [Google Scholar]
- 97.Matsui, H., Wagner, V.E., Hill, D.B., Schwab, U.E., Rogers, T.D., Button, B., Taylor, R.M., 2nd, Superfine, R., Rubinstein, M., Iglewski, B.H. et al. (2006) A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc. Natl. Acad. Sci. U. S. A., 103, 18131–18136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith, J.J., Karp, P.H. and Welsh, M.J. (1994) Defective fluid transport by cystic fibrosis airway epithelia. J. Clin. Invest., 93, 1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith, J.J., Travis, S.M., Greenberg, E.P. and Welsh, M.J. (1996) Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell, 85, 229–236. [DOI] [PubMed] [Google Scholar]
- 100.Zabner, J., Smith, J.J., Karp, P.H., Widdicombe, J.H. and Welsh, M.J. (1998) Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Mol. Cell, 2, 397–403. [DOI] [PubMed] [Google Scholar]
- 101.Gustafsson, J.K., Ermund, A., Ambort, D., Johansson, M.E., Nilsson, H.E., Thorell, K., Hebert, H., Sjovall, H. and Hansson, G.C. (2012) Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J. Exp. Med., 209, 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schutte, A., Ermund, A., Becker-Pauly, C., Johansson, M.E., Rodriguez-Pineiro, A.M., Backhed, F., Muller, S., Lottaz, D., Bond, J.S. and Hansson, G.C. (2014) Microbial-induced meprin beta cleavage in MUC2 mucin and a functional CFTR channel are required to release anchored small intestinal mucus. Proc. Natl. Acad. Sci. U. S. A., 111, 12396–12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Henderson, A.G., Ehre, C., Button, B., Abdullah, L.H., Cai, L.H., Leigh, M.W., DeMaria, G.C., Matsui, H., Donaldson, S.H., Davis, C.W. et al. (2014) Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J. Clin. Invest., 124, 3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xie, Y., Lu, L., Tang, X.X., Moninger, T.O., Huang, T.J., Stoltz, D.A. and Welsh, M.J. (2020) Acidic submucosal gland pH and elevated protein concentration produce abnormal cystic fibrosis mucus. Dev. Cell, 54, 488–500.e485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Esther, C.R., Jr., Muhlebach, M.S., Ehre, C., Hill, D.B., Wolfgang, M.C., Kesimer, M., Ramsey, K.A., Markovetz, M.R., Garbarine, I.C., Forest, M.G. et al. (2019) Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci. Transl. Med., 11(486), eaav3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Andersen, D.H. (1938) Cystic fibrosis of the pancreas and its relation to celiac disease. Am. J. Dis. Child., 56, 344–399. [Google Scholar]
- 107.Andersen, D.H. (1949) Therapy and prognosis of fibrocystic disease of the pancreas. Pediatrics, 3, 406–417. [PubMed] [Google Scholar]
- 108.Andersen, D.H. (1958) Cystic fibrosis of the pancreas. J. Chronic Dis., 7, 58–90. [DOI] [PubMed] [Google Scholar]
- 109.Semaniakou, A., Croll, R.P. and Chappe, V. (2018) Animal models in the pathophysiology of cystic fibrosis. Front. Pharmacol., 9, 1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McCarron, A., Donnelley, M. and Parsons, D. (2018) Airway disease phenotypes in animal models of cystic fibrosis. Respir. Res., 19, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sun, X., Yi, Y., Yan, Z., Rosen, B.H., Liang, B., Winter, M.C., Evans, T.I.A., Rotti, P.G., Yang, Y., Gray, J.S. et al. (2019) In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci. Transl. Med., 11(485), eaau7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fan, Z., Perisse, I.V., Cotton, C.U., Regouski, M., Meng, Q., Domb, C., Van Wettere, A.J., Wang, Z., Harris, A., White, K.L. et al. (2018) A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. JCI Insight, 3(19), e123529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Harris, A. (1997) Towards an ovine model of cystic fibrosis. Hum. Mol. Genet., 6, 2191–2194. [DOI] [PubMed] [Google Scholar]
- 114.Rogers, C.S., Stoltz, D.A., Meyerholz, D.K., Ostedgaard, L.S., Rokhlina, T., Taft, P.J., Rogan, M.P., Pezzulo, A.A., Karp, P.H., Itani, O.A. et al. (2008) Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science, 321, 1837–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Muraro, M.J., Dharmadhikari, G., Grun, D., Groen, N., Dielen, T., Jansen, E., van Gurp, L., Engelse, M.A., Carlotti, F., de Koning, E.J. et al. (2016) A single-cell transcriptome atlas of the human pancreas. Cell Syst, 3, 385–394.e383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang, Y., Song, W., Wang, J., Wang, T., Xiong, X., Qi, Z., Fu, W., Yang, X. and Chen, Y.G. (2020) Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. J. Exp. Med., 217, e20191130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leir, S.H., Yin, S., Kerschner, J.L., Cosme, W. and Harris, A. (2020) An atlas of human proximal epididymis reveals cell-specific functions and distinct roles for CFTR. Life Sci Alliance, 3, e202000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Abuladze, N., Lee, I., Newman, D., Hwang, J., Boorer, K., Pushkin, A. and Kurtz, I. (1998) Molecular cloning, chromosomal localization, tissue distribution, and functional expression of the human pancreatic sodium bicarbonate cotransporter. J. Biol. Chem., 273, 17689–17695. [DOI] [PubMed] [Google Scholar]
- 119.Swahn, H. and Harris, A. (2019) Cell-selective regulation of CFTR gene expression: relevance to gene editing therapeutics. Genes (Basel), 10, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]