

Abstract
Nonalcoholic steatohepatitis is a major public health concern and is characterized by the accumulation of triglyceride in hepatocytes and inflammation in the liver. Steatosis is caused by dysregulation of the influx and efflux of lipids, lipogenesis, and mitochondrial β-oxidation. Extracellular lysophosphatidic acid (LPA) regulates a broad range of cellular processes in development, tissue injury, and cancer. In the present study, we examined the roles of LPA in steatohepatitis induced by a methionine-choline-deficient (MCD) diet in mice. Hepatocytes express LPA receptor (Lpar) 1-3 mRNAs. Steatosis developed in mice fed the MCD diet was reduced by treatment with inhibitors for pan-LPAR or LPAR1. Hepatocyte-specific deletion of the Lpar1 gene also reduced the steatosis in the MCD model. Deletion of the Lpar1 gene in hepatocytes reduced expression of Cd36, a gene encoding a fatty acid transporter. Although LPA/LPAR1 signaling induces expression of Srebp1 mRNA in hepatocytes, LPA does not fully induce expression of SREBP1-target genes involved in lipogenesis. Human hepatocytes repopulated in chimeric mice are known to develop steatosis and treatment with an LPAR1 inhibitor reduces expression of CD36 mRNA and steatosis. Our data indicate that antagonism of LPAR1 reduces steatosis in mouse and human hepatocytes by down-regulation of Cd36.
Keywords: methionine-choline-deficient diet, LPA, LPAR1, SREBP1, steatohepatitis
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is a common cause of chronic liver disease in the Western world [1]. Nonalcoholic steatohepatitis (NASH) is the progressive form of NAFLD and is accompanied by steatosis, inflammation, cell ballooning of hepatocytes, and fibrosis [2, 3]. Some of the patients with NASH progress to cirrhosis, which is the end stage liver disease, and hepatocellular carcinoma. Currently, there is no FDA approved drugs for patients with NASH and cirrhosis. To reduce the number of patients with cirrhosis and their medical costs, the development of novel interventions for steatohepatitis is urgently needed. Toward this end, understanding cellular mechanisms underlying steatohepatitis is essential for identification of therapeutic targets for prevention of end-stage liver disease.
Steatosis is a protective mechanism of the liver from free fatty acids by storing them as neutral lipids in hepatocytes [4]. However, excessive storage of fatty acids in hepatocytes results in cell death by their lipotoxicity. Furthermore, damaged hepatocytes release damage-associated molecular patterns and induce inflammation in the liver [2]. Thus, reduction of excess hepatic steatosis is one of the therapeutic targets for NASH. Steatosis is caused by complex processes, including the dysregulation of lipid metabolism [2, 5]. CD36 is a scavenger receptor for uptake of fatty acids, oxidized low-density lipoprotein, and phospholipids. CD36 expressed on hepatocytes increases free fatty acid uptake and its expression is positively correlated with grades of steatosis in patients with NASH [6–8]. Hepatocyte-specific deletion of the Cd36 gene was shown to attenuate steatosis in mice induced by high fat diet [9], suggesting that CD36 promotes steatosis.
Sterol regulatory element binding transcription factor 1 (SREBP1) belongs to the family of bHLH-ZIP transcription factors and is a major regulator of de novo lipogenesis [10]. The SREBP1 gene has different transcripts, including Srebp1a and Srebp1c, and Srebp1c is involved in fatty acid synthesis [11]. Following proteolytic cleavage, mature SREBP1 up-regulates gene expression involved in fatty acid synthesis such as fatty acid synthase (Fasn) and diacylglycerol O-acyltransferase 1 (Dgat1) and promotes accumulation of triglycerides in hepatocytes. Steatosis is also caused by reduction of lipid β-oxidization in mitochondria that is facilitated by acyl-Coenzyme A oxidase 1 (ACOX1), carnitine palmitoyltransferase 1a (CPT1A), and peroxisome proliferator activated receptor (PPAR) α. Microsomal triglyceride transfer protein (Mttp) plays a role in the assembly and secretion of very low-density lipoproteins in the liver and its decreased secretion results in steatosis [12].
Lysophosphatidic acid (LPA) is a small phospholipid consisting of a glycerol backbone with fatty acid and phosphate groups. Extracellular LPA regulates cell proliferation, migration, and differentiation in development, tissue injury, and cancer [13, 14]. LPA is present at low concentrations in serum as an albumin (ALB)-bound form and elicits its biological effects via six different cell surface LPA receptors (LPAR) [15, 16]. LPAR1-3 are EDG family and LPAR4-6 are purinergic receptor family G-protein-coupled receptors. Upon LPA binding, LPARs induce the dissociation of GTP-bound Gα from Gβγ subunits and activate numerous down-stream signaling pathways. Extracellular LPA is synthesized by two major pathways: 1) from phosphatidic acid by secretory phospholipase A enzymes and 2) from lysophosphatidylcholine by ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2, so-called Autotaxin) [14, 16]. Enpp2 gene deficient mice are embryonic lethal with multiple abnormalities [17]. Given that Enpp2 gene heterozygous mice exhibit half of the normal plasma LPA level, ENPP2 is considered as a major enzyme for producing extracellular LPA. Hepatocyte-specific deletion of the Enpp2 gene or treatment with an ENPP2 inhibitor reduces liver fibrosis in mouse models [18, 19]. However, little is known about the role of LPA in steatohepatitis.
In the present study, we examined the role of extracellular LPA in steatohepatitis in mice fed a methionine-choline-deficient (MCD) diet and found that LPAR1 mediates steatosis. Hepatocyte-specific deletion of the Lpar1 gene resulted in reduction of MCD diet-induced steatosis via downregulation of Cd36 mRNA, a gene responsible for a hepatic fatty acid transporter. Antagonism of LPAR1 also reduced steatosis developed in human hepatocytes repopulated in immunodeficient mice. Our data indicate that LPA/LPAR1 signaling promotes steatosis in the liver.
2. Materials and method
2.1. Mice
Male C57BL/6 mice (5-8 weeks, Jackson Laboratory, Bar Harbor, ME) were used for cell isolation. To generate hepatocyte-specific deletion of the Lpar1 gene, we resuscitated Lpar1tm1a(EUCOMM)Wtsi mice at the UC Davis KOMP repository [20]. By crossing with Tg(Actb-Flpe) mouse expressing the FLP1 recombinase gene (Jackson Laboratory, #005703), we removed the LacZ and Neomycin selection genes and generated Lpar1flox mice in which the 3rd exon is designed to be removed by Cre/loxP recombination. To delete the Lpar1 gene in hepatocytes, we crossed Lpar1flox mice with AlbCre mice (Jackson Laboratory, #003574) and generated control Lpar1flox/flox and AlbCre; Lpar1flox/flox mice. Genotypes were determined by PCR from DNAs isolated from the tails using the following primers: Lpar1 (296 bp for wild type and 405 bp for a flox allele, 5’-TTT GAA ATG TTG GGC CTG AT and 5’-TCT CTG CTT GGT GGT CTG TG) and Cre (5’-ACC TGA AGA TGT TCG CGA TTA TC and 5’-ACC GTC AGT ACG TGA GAT ATC TT).
Mice were fed either a regular diet (710027, Dyets, Bethlehem, PA) or MCD diet (960439, MP Biomedicals, Irvine, CA) for 10 days [21, 22]. BrP-LPA (Echelon, Salt Lake City, UT) or AM095 (Apexbio, Houston, TX) were dissolved in dimethyl sulfoxide (DMSO, 10 mg/ml) [23, 24]. After dilution with PBS 10 times, we injected the BrP-LPA solution (7.5 mg/kg body weight), AM095 (10 mg/kg body weight), or control 10% DMSO intraperitoneally 3 times on day 3, 6, and 9 to mice.
We generated Alb promoter driven urokinase-type plasminogen activator cDNA (cDNA-uPA)/Severe combined immunodeficiency (Scid) mice in which the mouse liver was replaced by normal human hepatocytes as described previously [25]. The repopulation of human hepatocytes was estimated by measuring serum human ALB concentration in the mouse. The cDNA-uPA/Scid chimeric mice (male, 12 weeks old, n=3 for each group) were treated with AM095 (10 mg/kg body weight) every 3 days for a total of 3 times by intraperitoneal injection as described above. One day after the last injection, we collected the liver tissues for analyses. All animal experiments were performed in accordance with NIH guidelines or the Proper Conduct of Animal Experiments (Science Council of Japan) under the protocols approved by the IACUC at the University of Southern California or Phoenix Bio, respectively.
2.2. Liver cell isolation and culture
Hepatocytes, hepatic stellate cells, sinusoidal endothelial cells, and Kupffer cells were isolated from normal mouse livers at the Integrative Liver Cell Core of the Southern California Research Center for ALPD & Cirrhosis. Hepatocytes were isolated by collagenase perfusion followed by percoll centrifugation as previously described [26]. Nonparenchymal cells were isolated by perfusion of the liver using pronase (Roche, Indianapolis, IN) and collagenase (Sigma-Aldrich, St. Louis, MO) [27]. By discontinuous gradient ultracentrifugation, we obtained hepatic stellate cells and Kupffer cells as previously described [28, 29]. Sinusoidal endothelial cells were separated from the nonparenchymal cells by magnetic-activated cell sorting using anti-mouse CD146 microbeads (Miltenyi Biotech, Auburn, CA).
LPA (18:1; 1-oleoyl-2-hydroxy-sn-glycero-3-phosphate, Avanti, Alabaster, AL) was dissolved in PBS in the presence of fatty acid-free bovine serum ALB. Hepatocytes (3x106 cells) were cultured on 100 mm dishes in DMEM high glucose containing 10% FBS, 1 mM sodium pyruvate, and Antibiotic-Antimycotic (ThermoFisher Scientific, Waltham MA). After overnight culture, we replaced the culture medium without FBS for 6 hours, added LPA (10 μM), and cultured them for 2 days.
2.3. Quantitative reverse transcription followed by polymerase chain reaction (QPCR)
We isolated total RNAs from cells or liver tissues using RNAqueous Total RNA isolation kit (ThermoFisher Scientific). cDNA was synthesized using Maxima First Strand cDNA Synthesis kit as previously reported [28]. QPCR was performed with SYBR Green in a ViiA 7 Real-Time PCR System (ThermoFisher Scientific). The samples were run in triplicate. The relative mRNA levels per samples were calculated by subtracting the detection limit (40 Ct) from the cycle threshold value (Ct) of each gene in the same sample to obtain the ΔCt value. Taking the log2 of −ΔCt resulted in the relative expression value of each gene for each sample expressed in arbitrary units. Each value was normalized to Gapdh. Primer sequences used for mouse genes are: Enpp2 (5’-GTC AGA AAG GAA TGG GGT CA and 5’-GTC GGT GAG GAA GGA TGA AA), Lpar1 (5’-TCT TCT GGG CCA TTT TCA AC and 5’-TGC CTG AAG GTG GCG CTC AT), Lpar2 (5’-ACC ACA CTC AGC CTA GTC AAG AC and 5’-CTG AGT AAC GGG CAG ACT TG), Lpar3 (5’-ACA CCA GTG GCT CCA TCA G and 5’-GTT CAT GAC GGA GTT GAG CAG), Lpar4 (5’-AGG CAT GAG CAC ATT CTC TC and 5’-CAA CCT GGG TCT GAG ACT TG), Lpar5 (5’- CAA CAG GAC AGA TTC CAG AGG and 5’- GTA GGG AAC AAC AAG GTC AGA G), Lpar6 (5’-TGT TTC CAA CTG CTG CTT TG and 5’-GAG CAG TCC CAG TGG CTT AG), Acox1 (5’-TCG CAG ACC CTG AAG AAA TC and 5’-CCT GAT TCA GCA AGG TAG GG), Cd36 (5’-TGC ACC ACA TAT CTA CCA AA and 5’-TTG TAA CCC CAC AAG AGT TC), Cpt1a (5’-GCC CAT GTT GTA CAG CTT CC and 5’-AGT GGC CTC ACA GAC TCC AG), Dgat1 (5’-GAC GGC TAC TGG GAT CTG A and 5’-TCA CCA CAC ACC AAT TCA GG), Fasn (, 5’-GTC GTC TGC CTC CAG AGC and 5’-GTT GGC CCA GAA CTC CTG TA), Mttp (5’ - AAA GCA GAG CGG AGA CAG AG and 5 ’- TAT CGC TTT CTG GCT GAG GT), Ppara (5’- AGT TCG GGA ACA AGA CGT TG and 5’-CAG TGG GGA GAG AGG ACA GA), Pparg (5’-CTC CTG TTG ACC CAG AGC AT and 5’-AAT GCG AGT GGT CTT CCA TC), Srebp1a (5’-GGC TCT GGA ACA GAC ACT GG and 5’-TGG TTG TTG ATG AGC TGG AG), Srebp1c (5’-ATC GGC GCG GAA GCT GTC GGG GTA G and 5’-ACT GTC TTG GTT GTT GAT GAG CTG G), Srebp2 (5’-CAT CCA GCA GCC TTT GAT ATA CCA G and 5’-AGG ACC GGG ACC TGC TGC ACC TGT G), and tumor necrosis factor (Tnf, 5’-ACG GCA TGG ATC TCA AAG AC and 5’-AGA TAG CAA ATC GGC TGA CG). Primer sequences for Alb, Gapdh, neurotrophin receptor (Ntr), platelet and endothelial cell adhesion molecule 1 (Pecam1), and protein tyrosine phosphatase receptor type C (Ptprc) were described previously [30]. Primer sequences used for human genes are: CD36 (5’- CGG CTG CAG GTC AAC CTA TT and 5’- TTG GCC ACC CAG AAA CCA AT), and GAPDH (5’-CGT CCC GTA GAC AAA ATG GT and 5’-GAA TTT GCC GTG AGT GGA GT).
2.4. Oil red O staining
Liver tissues were fixed with 4% paraformaldehyde for 12 hours and then incubated with 15% and 30% sucrose for 4 h and 12 hours, respectively. Then, tissues were embedded in Tissue-Tek O.C.T. Compound (Sakura Finetech, Torrance, CA) and frozen in liquid nitrogen. Cryosections (7 μm) were stained with oil red O (Sigma-Aldrich) for 15 min. Images were randomly captured using a 10X objective with a 90i microscope and DS-Fi1 digital camera (Nikon, Melville, NY). From 10 images from each mouse, we quantified neutral lipids stained with oil red O by ImageJ software (NIH).
2.5. Biochemical analyses
Liver tissues were homogenized with a 20 volume of H2O containing 5% NP-40. Triglyceride levels in the liver tissues were measured using L-type triglyceride M kit (Fujifilm, Lexington, MA) and Varioskan LUX microplate reader (ThermoFisher Scientific). The plasma ALT values were measured using ALT reagent (Cliniqa, San Marcos, CA) and a PowerWave 200 spectrophotometer (BioTech, Winooski, VT).
2.6. Statistical analysis
Statistical significance was assessed by using a Student’s t-test between two samples or ANOVA followed by post-hoc Tukey HSD test among multiple samples. A P value of less than 0.05 was considered statistically significant.
3. Results
3.1. Expression patterns of six LPA receptors and Enpp2 mRNAs in different liver cell types in mice
Little is known about the expression profiles of the six Lpar mRNAs in liver cells and their roles in steatohepatitis. We isolated hepatocytes, hepatic stellate cells, sinusoidal endothelial cells, and Kupffer cells from normal adult mouse livers and measured these genes’ expression by QPCR. We confirmed selective expression of Alb in hepatocytes, Ntr in hepatic stellate cells, and Pecam1 in sinusoidal endothelial cells (Fig. 1A). Kupffer cells exclusively express Ptprc and also express Pecam1 mRNA. Hepatocytes express Lpar1 and Lpar2 mRNAs (Fig. 1B). Hepatocytes weakly express Lpar3 mRNA and are a major cell type expressing Enpp2 mRNA that encodes autotaxin responsible for generation of extracellular LPA. Hepatic stellate cells abundantly express Lpar1 and Lpar2 mRNAs (Fig. 1B). Sinusoidal endothelial cells express Lpar1-4 and Lpar6 mRNAs. Kupffer cells express Lpar1-6 mRNAs.
Fig. 1. Expression profiles of Lpar1-6 and Enpp2 genes in different liver cell types.
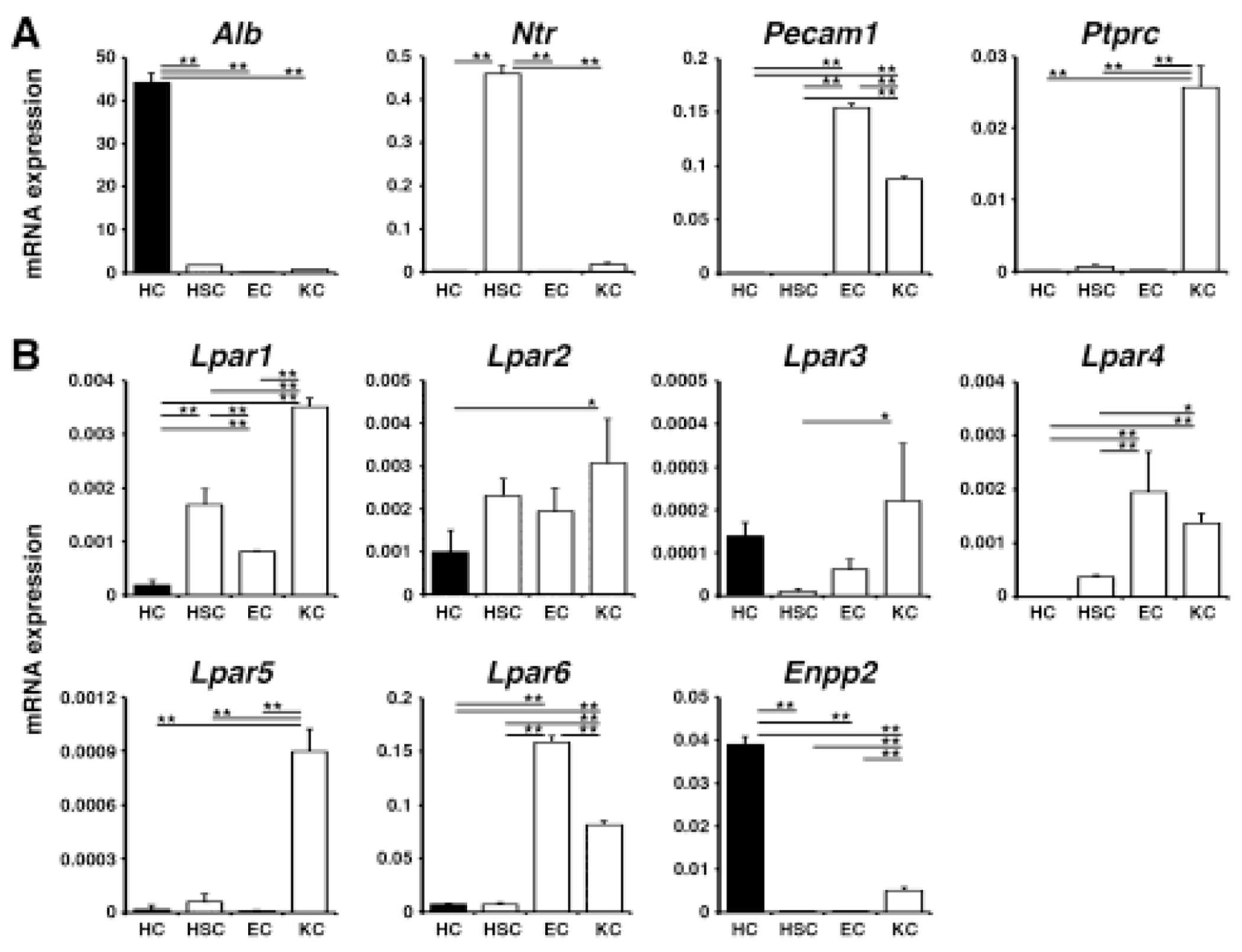
QPCR of hepatocytes (HC), hepatic stellate cells (HSC), sinusoidal endothelial cells (EC), and Kupffer cells (KC) isolated from normal mouse livers. (A) Expression of marker genes for each cell type. (B) Expression of Lpar1-6 and Enpp2 mRNAs in different liver cell types. mRNA expression values were normalized against Gapdh. Each value is the mean ± SD of triplicate measurements. *, p < 0.05; **, p <0.01.
3.2. Blocking LPAR1 reduces MCD-induced steatosis
To examine the role of LPA in the initiation of steatohepatitis, we fed mice with the MCD diet for 10 days [21, 31]. By oil red O staining, we confirmed induction of steatosis in the liver by the MCD diet (Fig. 2A,B). BrP-LPA is an antagonist for LPAR1-4 and ENPP2 [24]. After intraperitoneal injection of BrP-LPA on day 3, 6, and 9 to mice fed the MCD diet, we observed reduction of steatosis in the liver as revealed by oil red O staining (Fig. 2A,B). Hepatocytes mainly express Lpar1 and Lpar2 mRNAs (Fig. 1B). LPAR1 is broadly expressed in different organs and its knockout causes lethality in many neonatal mice due to abnormal suckling behavior while the survivors show severe growth retardation [32]. In contrast, Lpar2 knockout mice are viable and show no abnormal phenotype [33]. Although no abnormalities were described about the liver morphogenesis in Lpar1 and Lpar2-null mice, LPAR1 has been shown to involved in hepatitis C virus replication in hepatocytes and development of hepatocellular carcinoma [34, 35]. Since nothing was known about the role of LPAR1 in hepatocytes in steatohepatitis, we examined its role in the MCD-fed mouse liver using AM095, an inhibitor for LPAR1. Similar to BrP-LPA, treatment with AM095 reduced steatosis (Fig. 2A,B). We confirmed that BrP-LPA or AM095 treatment reduces triglyceride levels in the liver (Fig. 2C). On the other hand, these antagonists did not reduce alanine aminotransferase levels induced by the MCD diet (Fig. 2D). These data suggest that LPA induces steatosis caused by the MCD diet mainly via LPAR1 in the liver.
Fig. 2. Antagonism of LPAR1 signaling ameliorates MCD-induced steatosis in mice.
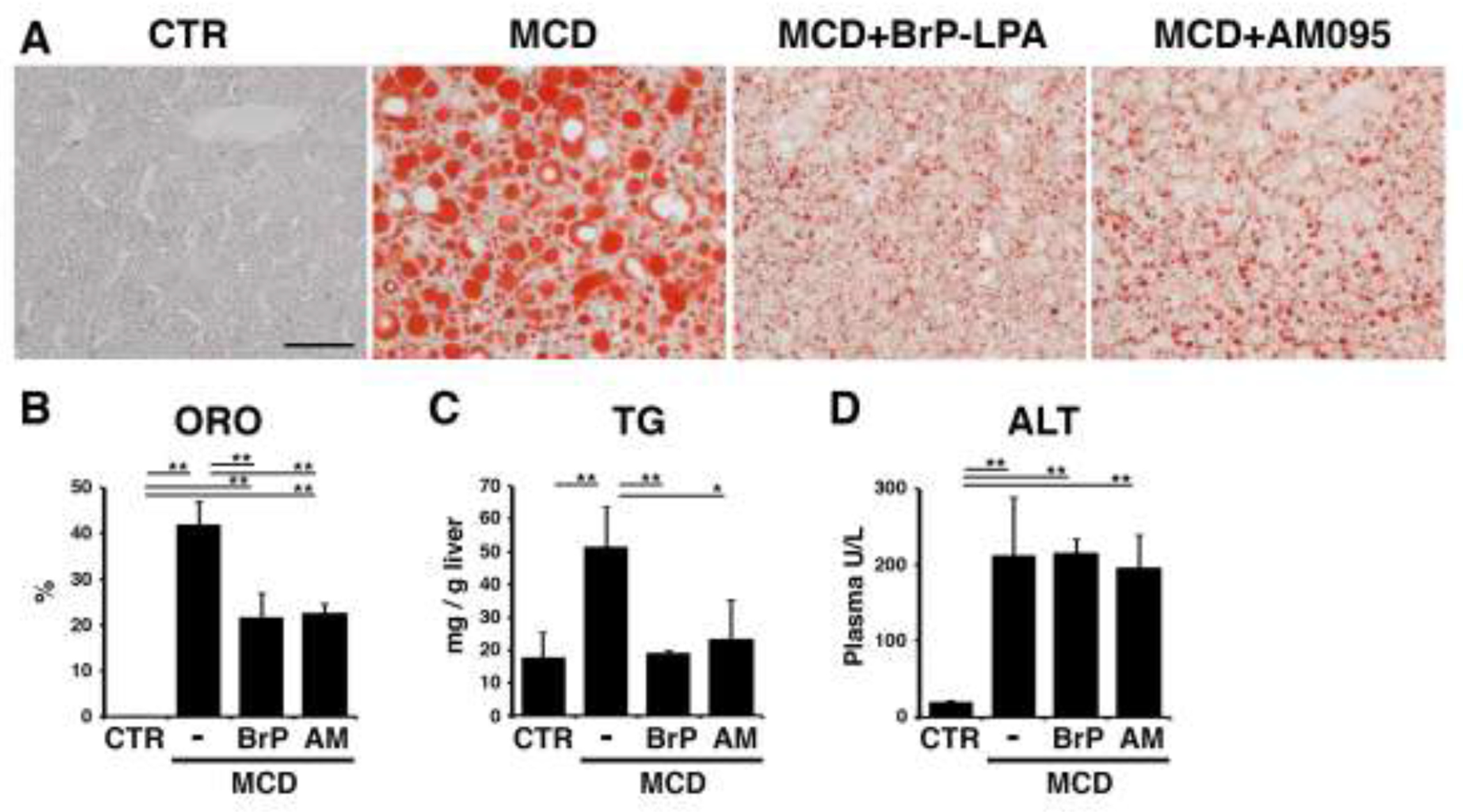
Mice fed the MCD diet for 10 days were treated with BrP-LPA (a broad LPAR antagonist), AM095 (an LPAR1 antagonist), or control DMSO on day 3, 6, and 9 (male, n=3 for each group). (A) Oil red O (ORO) staining of the livers. MCD-induced steatosis is ameliorated by BrP-LPA or AM095 treatment. (B) Quantification of ORO staining in each group. (C) Measurement of triglyceride (TG) levels in the livers. (D) Plasma aminotransferase (ALT) levels. *, p < 0.05; **, p < 0.01.
3.3. Reduction of Cd36 mRNA by an LPAR1 antagonist in MCD-induced steatohepatitis
To determine how LPA signaling mediates steatosis, we measured the expression of genes involved in the pathogenesis of steatosis. CD36 is a fatty acid transporter [36] and its mRNA expression was decreased by BrP-LPA or AM095 treatment in the MCD-fed mice (Fig. 3). SREBP1 is a major regulator of de novo lipogenesis [10]. Both Srebp1a and Srebp1c isoforms were down-regulated by BrP-LPA or AM095 treatment in the MCD-fed mice (Fig. 3). Srebp2 mRNA was partially suppressed by BrP-LPA. However, genes positively regulated by SREBP1, such as Dgat1 and Fasn, were not down-regulated by these antagonists (Fig. 3). Hepatic expression of Pparg and Mttp were not changed by these antagonists. Genes involved in mitochondrial β-oxidation such as Acox1, Cpt1a, and Ppara were not changed by BrP-LPA or AM095. Hepatic expression of Tnf was not changed by these antagonists. The MCD diet significantly increased the expression of Lpar1 and Lpar4 in the liver (Fig. 3). Although BrP-LPA or AM095 treatment tended to decrease these mRNAs, the differences were not statistically significant. These results suggest that blocking of LPA/LPAR1 signaling reduces steatosis mainly via reduction of Cd36 expression in the liver.
Fig. 3. Antagonism of LPAR1 signaling down-regulates Cd36 mRNA expression in the MCD-fed mouse liver.
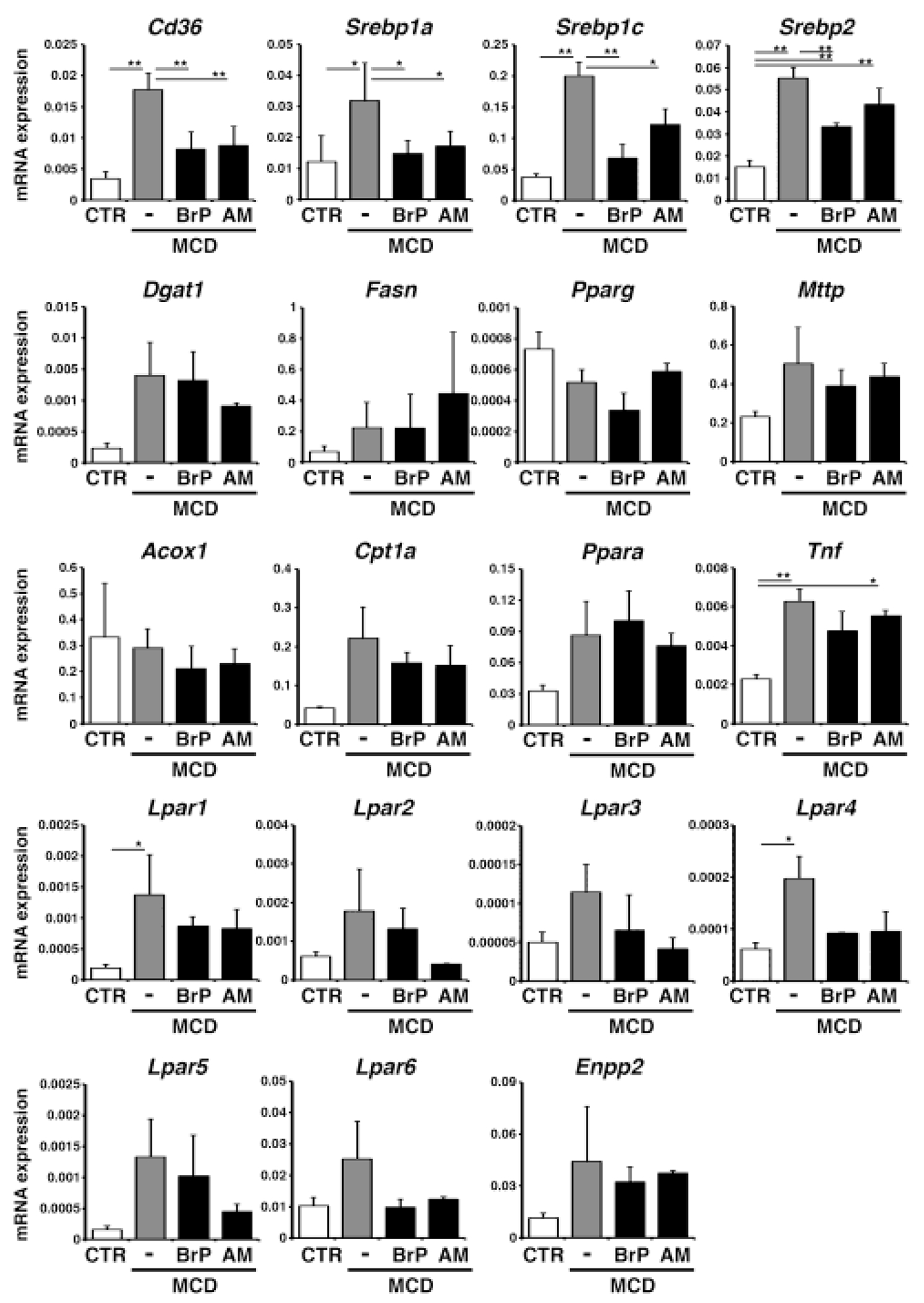
Mice fed the MCD diet for 10 days were treated with BrP-LPA (BrP: broad LPAR antagonist), AM095 (AM: LPAR1 antagonist), or control DMSO on day 5, 7, and 9 (male, n=3 for each group). mRNA expression of the liver tissues was analyzed by QPCR. mRNA expression values were normalized against Gapdh. Each value is the mean ± SD of triplicate measurements. *, p < 0.05; **, p<0.01.
3.4. Deletion of Lpar1 gene down-regulates Cd36 mRNA in hepatocytes
Lpar1 is broadly expressed in different liver cell types (Fig. 1B) and the AM095 treatment might reduce steatosis in the MCD-fed mice by changing the phenotypes of nonparenchymal cells. To determine the role of Lpar1 in hepatocytes, we deleted Lpar1 gene only in hepatocytes using AlbCre and Lpar1flox mice. Both control Lpar1flox/flox and AlbCre; Lpar1flox/flox (Lpar1ΔHep) were viable and showed no abnormal phenotype. We isolated hepatocytes from the control and Lpar1ΔHep mouse livers and confirmed around 90% reduction of Lpar1 mRNA expression in hepatocytes from Lpar1ΔHep compared to those from the control (Fig. 4A). The expression of Lpar2-6 and Enpp2 was not changed by the deletion of Lpar1 in hepatocytes. Lpar1-null weakly reduced Cd36 mRNA expression (Fig. 4A). Although Lpar1-null hepatocytes reduced expression of Srebp1c, its target gene, Fasn, was not down-regulated in hepatocytes.
Fig. 4. Down-regulation of Cd36 mRNA expression in Lpar1-null hepatocytes.
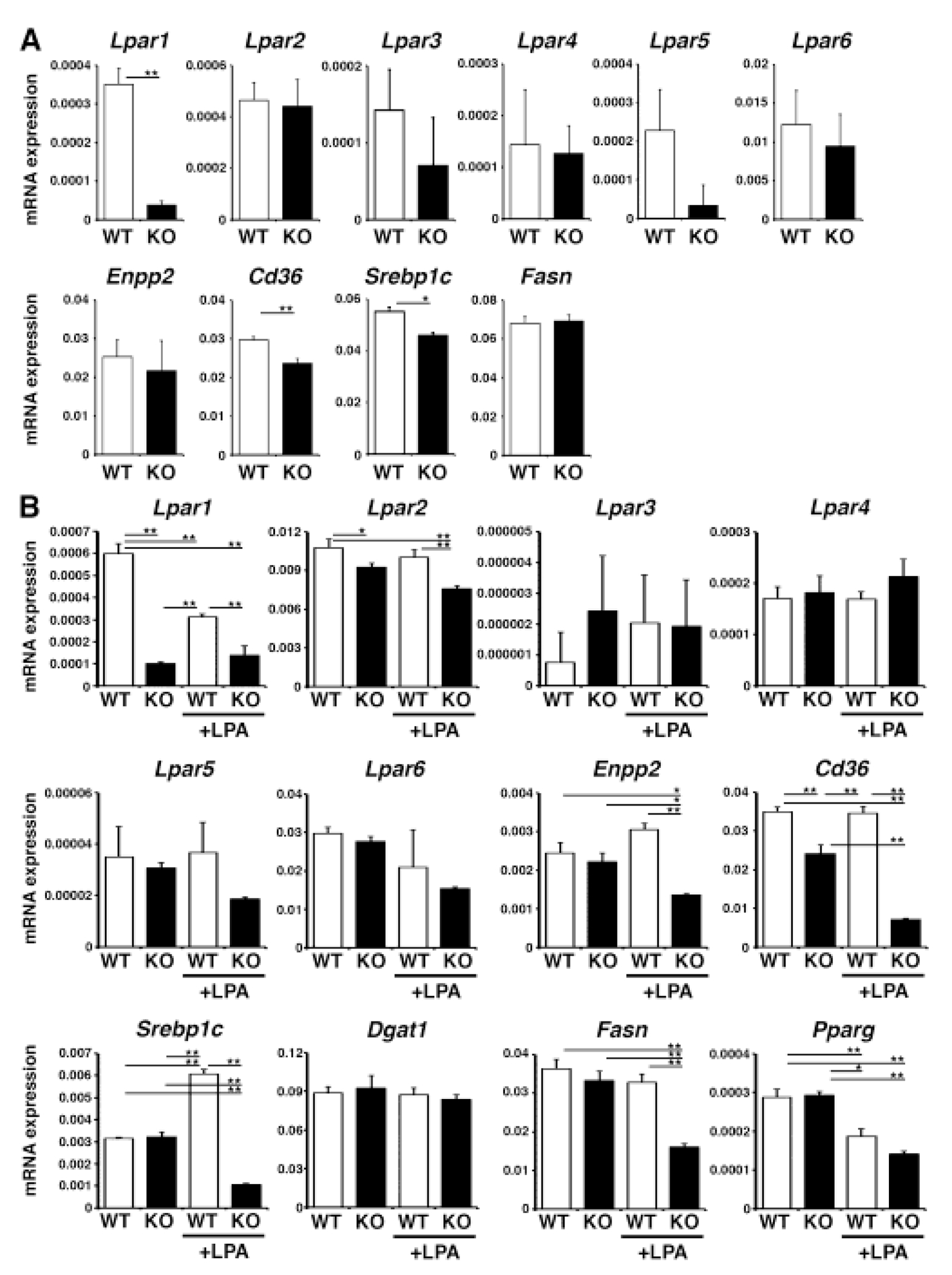
The Lpar1 gene was deleted in hepatocytes in AlbCre; Lpar1fkox/flox (Lpar1ΔHep) mice. (A) QPCR of hepatocytes isolated from the wild type Lpar1flox/flox (WT) and Lpar1ΔHep (KO) mice. Note the efficient deletion of Lpar1 mRNA in KO. (B) QPCR of cultured hepatocytes treated with LPAfor 2 days. *, p < 0.05; **, p < 0.01.
To examine the role of LPA in hepatocytes, we isolated them from the wild type and Lpar1ΔHep mouse livers and plated them on dishes in DMEM containing 10% FBS overnight. Because FBS contains LPA, we pretreated hepatocytes in DMEM without FBS for 6 hours and then treated them with 10 μM of LPA for 2 days. QPCR showed that LPA decreases the expression of Lpar1 mRNA in wild type hepatocytes (Fig. 4B), implying a negative feedback regulation. Compared to Lpar1, expression of Lpar2 was not sensitive to LPA and its gene was constantly expressed in hepatocytes. Expression of Lpar3-6 mRNAs was not changed significantly by deletion of Lpar1 gene and addition of LPA in hepatocytes. Addition of LPA to Lpar1-null hepatocytes resulted in reduction of Enpp2 gene, implying that Lpar1 gene is necessary to maintain Enpp2 expression upon stimulation with LPA.
LPA treatment strongly reduced the expression of Cd36 mRNA in Lpar1-null hepatocytes. LPA treatment increased the expression of Srebp1c mRNA in the control hepatocytes and its induction was strongly suppressed in Lpar1-null hepatocytes. Among the Srebp1c-target genes, Fasn, but not Dgat1, was down-regulated in Lpar1-null hepatocytes in the presence of LPA. Expression of Pparg mRNA was weakly suppressed by LPA in both wild type and Lpar1-null hepatocytes. Our data indicate that LPA/LPAR1 signaling maintains expression of Cd36 mRNA in hepatocytes.
3.5. Deletion of Lpar1 gene in hepatocytes reduces MCD-induced steatosis
Next, we fed control Lpar1flox/flox and Lpar1ΔHep mice with the MCD diet for 10 days. Compared to the control mice, Lpar1ΔHep mice showed less oil red O staining in the liver by feeding with the MCD diet (Fig 5A,B). We confirmed the reduction of triglycerides in the Lpar1ΔHep liver compared to the control in mice fed the MCD diet (Fig. 5C). QPCR showed that Cd36 mRNA expression was reduced in Lpar1ΔHep liver compared to the control liver fed with the MCD diet (Fig. 5D). The expression of Srebp1c was increased in the Lpar1ΔHep mice fed the control diet, but its expression was downregulated in both the control and Lpar1ΔHep mice fed the MCD diet. Dgat1 and Fasn mRNAs were not changed by the deletion of Lpar1 or the MCD diet.
Fig. 5. Hepatocyte-specific deletion of Lpar1 gene reduces steatosis in mice fed the MCD diet.
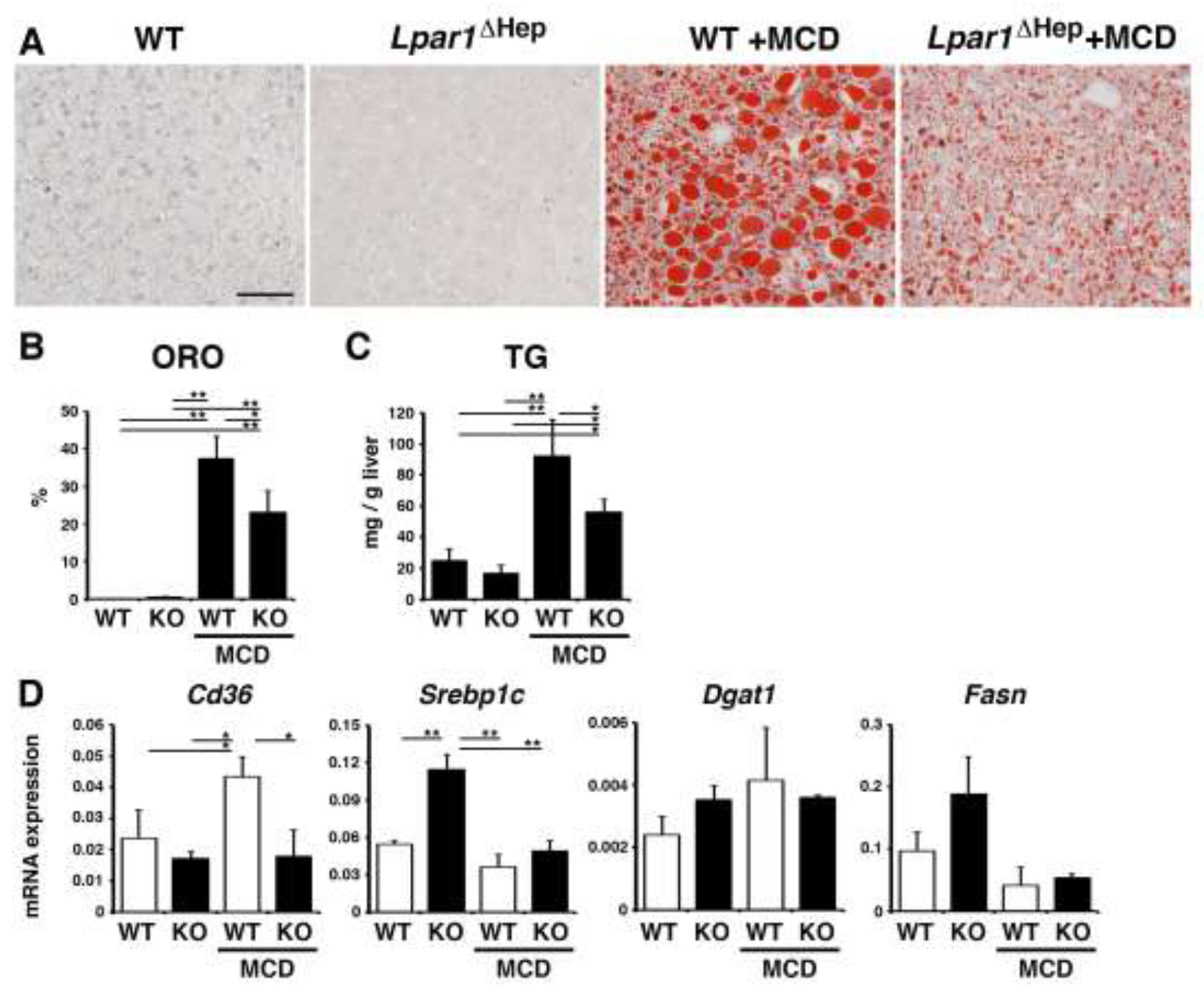
Control Lpar1flox/flox (WT) and Lpar1ΔHep (KO) mice were fed with the MCD diet for 10 days (male, n=3 for each group). (A) Oil red O staining of the liver tissues from the WT and Lpar1ΔHep mice fed with the control or MCD diet. After the MCD diet, the Lpar1ΔHep liver shows less accumulation of lipids compared to the WT liver. (B) Quantification of oil red O (ORO) staining in each group. (C) Measurement of triglyceride levels (TG) in the livers. (D) mRNA expression of the liver tissues was analyzed by QPCR. mRNA expression values were normalized against Gapdh. Each value is the mean ± SD of triplicate measurements. *, p < 0.05; **, p < 0.01.
3.6. Reduction of steatosis in chimeric mice bearing human hepatocytes by blocking LPAR1
Human hepatocytes transplanted into cDNA-uPA/Scid mice are known to repopulate in the injured mouse liver [25]. Chimeric mice replaced by human hepatocytes develop steatosis due to the inability of mouse growth hormone to activate its human receptor [37]. Using this chimeric mouse, we tested whether LPAR1 is involved in steatosis developed in human hepatocytes. Chimeric mice bearing human hepatocytes showed high concentrations of human ALB (10.4-11.9 mg/ml, n=6) in the mouse sera that validated successful replacement of the mouse liver by human hepatocytes by over 80% [25]. We treated the chimeric mice with AM095 (10 mg/g body weight) every 3 days for a total of 3 times. Oil red O staining showed the presence of steatosis in the control chimeric mice treated with the control DMSO (Fig. 6A). AM095-treated chimeric mice showed reduction of steatosis (Fig. 6A,B). The hepatic triglyceride level was reduced by the AM095 treatment in the chimeric mice (Fig. 6C). QPCR showed that the expression of CD36 mRNA is reduced in the chimeric mouse liver by the AM095 treatment (Fig. 6D). These data suggest that antagonism of LPAR1 ameliorates steatosis in human hepatocytes.
Fig. 6. Reduction of steatosis in chimeric mice bearing human hepatocytes by antagonism of LPAR1.
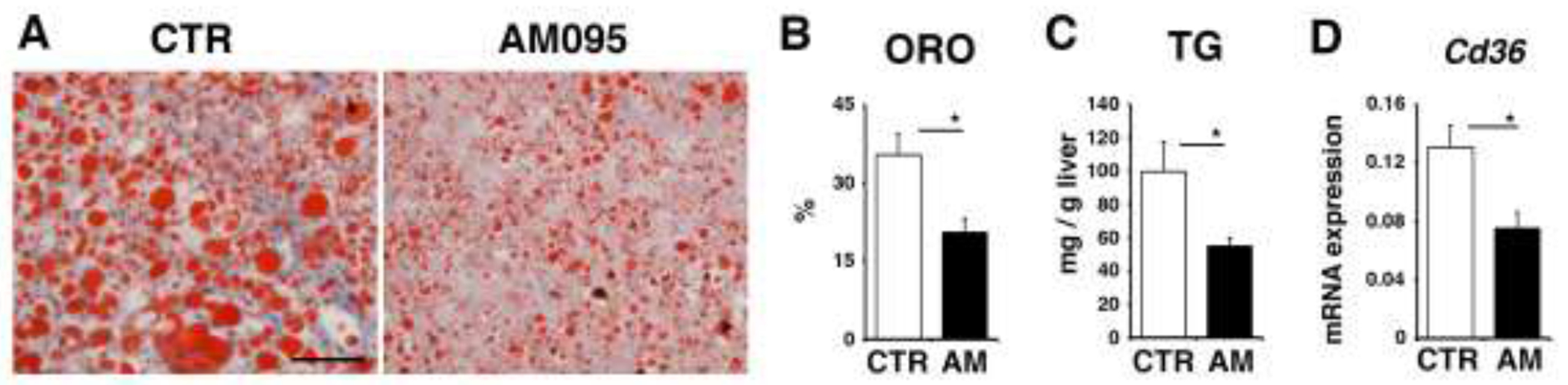
. cDNA-uPA/Scid mice with humanized livers were treated with control DMSO or AM095 every 3 days for a total of 3 times (male, n=3 for each group) and the livers were analyzed. (A) Oil red O staining of the liver tissues from the chimeric mice with AM095 or control DMSO. (B) Quantification of oil red O (ORO) staining. (C) Measurement of triglyceride levels (TG) in the livers. (D) CD36 mRNA expression of the liver tissues analyzed by QPCR. mRNA expression values were normalized against Gapdh. Each value is the mean ± SD of triplicate measurements. *, p < 0.05.
4. Discussion
The global prevalence of NASH is increasing and development of new drugs is needed to reduce the number of patients with NASH. In the present study, we found that antagonism of LPAR1 reduces steatosis in mice caused by the MCD diet. After treatment with BrP-LPA or AM095, we observed reduced steatosis and expression of Cd36 mRNA in the mouse liver fed the MCD diet. CD36 acts as a free fatty acid translocase in hepatocytes and participates in steatosis [7]. In mouse NASH models, deletion of Cd36 gene is known to reduce steatosis [9, 36]. Taken together, our data suggest that downregulation of Cd36 gene is one of the mechanisms of the reduction of steatosis caused by the MCD diet.
Deletion of the Lpar1 gene in hepatocytes results in down-regulation of Cd36 mRNA expression in hepatocytes. Although treatment with LPA did not induce its expression in cultured hepatocytes, expression in Lpar1-null hepatocytes was strongly reduced, suggesting that LPA/LPAR1 signaling is necessary to maintain the expression level of Cd36 mRNA in hepatocytes. Expression of Cd36 gene is positively regulated by PPARγ, liver X receptor, and pregnant X receptor [36]. In our study, deletion of Lpar1 gene did not change expression of Pparg mRNA in hepatocytes. In addition, treatment with LPA reduced the Pparg mRNA expression in both wild type and Lpar1-null hepatocytes and we did not observe a correlation between Cd36 and Pparg mRNA expression by deletion of the Lpar1 gene or treatment with LPA. Upon binding to LPA, LPAR1 activates Ras and PI3K pathways through Gαi/o, a PLC pathway through Gαq/11, and a Rho pathway through Gα12/13 [15, 16]. LPAR1 also suppresses Gαs-mediated activation of PKA via Gαi/o. It remains to be clarified how LPAR1-mediated signaling pathways regulate expression of Cd36 gene in hepatocytes. In addition to the role of LPAR1 in maintaining the expression of Cd36 mRNA in hepatocyte, other LPARs, such as LPAR2, might negatively regulate its expression. Since primary hepatocytes rapidly lose their function in culture, further studies using genetic mouse models will be necessary to determine how LPAR1 and other LPARs regulate Cd36 gene expression in hepatocytes and participate in steatosis.
SREBP1 acts as a transcription factor for induction of de novo lipogenesis by induction of Dgat1 and Fasn genes. We observed that the MCD diet induces expression of Srebp1a and Srebp1c mRNAs in the liver and these genes were suppressed by co-treatment with BrP-LPA or AM095. In addition, we observed induction of a mature form of SREBP1 protein in the liver by the MCD diet and its induction was suppressed by BrP-LPA or AM095 (data not shown). From these results, we assumed that LPAR1/SREBP1 is responsible for induction of steatosis in the liver. However, we did not observe down-regulation of Dgat1 and Fasn mRNAs by BrP-LPA or AM095 in the MCD-fed mouse liver. Although hepatocyte-specific deletion of Lpar1 caused reduction of the expression of Srebp1c in the liver, no significant changes in Dgat1 and Fasn were observed by the deletion of Lpar1. In culture, treatment of LPA induced Srebp1c mRNA expression in hepatocytes and its expression was strongly suppressed in Lpar1-null hepatocytes. Although LPA similarly down-regulated Fasn mRNA in Lpar1-null hepatocytes, Dgat1 mRNA was not suppressed. These data suggest that LPA/LPAR1/SREBP1 pathway is not a major regulator for inducing steatosis by de novo lipogenesis.
Chimeric cDNA-uPA/Scid mice replaced by human hepatocytes have been used for testing drugs and hepatitis virus infection [25]. Transplanted human hepatocytes replace the injured mouse liver caused by the overexpression of uPA in the mouse hepatocytes and exhibit functions similar to humans. However, repopulated human hepatocytes develop steatosis in the mouse liver due to the inability of human growth hormone receptor to be stimulated by endogenous mouse growth hormone [37]. In fact, treatment with human growth hormone attenuates steatosis in human hepatocytes in the chimeric mice. In the present study, we found that antagonism of LPAR1 reduces steatosis in human hepatocytes in the chimeric mice. Similar to our mouse data, we observed reduction of CD36 mRNA in the chimeric mice by treatment with AM095. A recent study reported that treatment of growth hormone attenuates steatosis by downregulation of Cd36 mRNA expression in low-density lipoprotein receptor-deficient mice fed with a high fat diet [38]. Thus, LPAR1 will be a therapeutic target for suppression of steatosis by suppression of Cd36 in hepatocytes.
AM095 is a potent inhibitor for LPAR1. We found that treatment with AM095 reduces steatosis caused by the MCD diet in mice. In the liver, LPAR1 is broadly expressed in hepatocytes, hepatic stellate cells, sinusoidal endothelial cells, and Kupffer cells and the antagonism by AM095 may suppress steatosis in hepatocytes via nonparenchymal cells. However, hepatocyte-specific deletion of Lpar1 also showed the reduction of steatosis by the MCD diet, indicating that LPAR1 expressed in hepatocytes is responsible for steatosis caused by the MCD diet. Hepatic stellate cells, sinusoidal endothelial cells, and Kupffer cells express Lpar1 and other Lpar mRNAs, but their roles remain elusive in liver injury. Both LPA and ENPP2 have short half-lives in mice and are rapidly cleared from the circulation within minutes by sinusoidal endothelial cells in the liver [39, 40]. Plasma ENPP2 levels are increased in patients with NASH and fibrosis [19, 41, 42]. Hepatocyte-specific deletion of Enpp2 gene was shown to reduce liver fibrosis in mice treated with CCl4 [19]. In addition, a selective inhibitor for ENPP2 showed moderate suppression of steatosis in mouse NASH models [18]. A recent study suggests that LPAR1 mediates the activation of HSCs that localize near the central vein in injured mouse liver [43]. Further studies are necessary to understand the roles of different types of LPARs in different liver cell types in the progression of NASH.
Highlights.
Hepatocytes express Lpar1-3 mRNAs.
Antagonism of LPAR1 reduces liver steatosis in the mouse fed the MCD diet.
Deletion of Lpar1 gene in hepatocytes reduces steatosis in the mouse liver.
LPA/LPAR1 signaling maintains expression of Cd36 mRNA in hepatocytes.
Antagonism of LPAR1 reduces steatosis in human hepatocytes repopulated in mouse livers.
Acknowledgement
We thank the Integrative Liver Cell Core and Animal Core of the Southern California Research Center for ALPD & Cirrhosis for isolation of liver cells and mouse models and Drs. Jerold Chun and Richard Rivera at the Scripps Research Institute for their advice on Lpar1 knockout mice.
Grant support
This work was supported by National Institutes of Health Grants (R24AA012885, R21AA027222) and Alcohol and Health in Suntory Global Innovation Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S, Nonalcoholic steatohepatitis: a review, JAMA, 323 (2020) 1175–1183. [DOI] [PubMed] [Google Scholar]
- [2].Schuster S, Cabrera D, Arrese M, Feldstein AE, Triggering and resolution of inflammation in NASH, Nat Rev Gastroenterol Hepatol, 15 (2018) 349–364. [DOI] [PubMed] [Google Scholar]
- [3].Anstee QM, Day CP, The genetics of NAFLD, Nat Rev Gastroenterol Hepatol, 10 (2013) 645–655. [DOI] [PubMed] [Google Scholar]
- [4].Malhi H, Gores GJ, Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease, Semin Liver Dis, 28 (2008) 360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chakravarthy MV, Neuschwander-Tetri BA, The metabolic basis of nonalcoholic steatohepatitis, Endocrinol Diabetes Metab, 3 (2020) e00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Koonen DP, Jacobs RL, Febbraio M, Young ME, Soltys CL, Ong H, Vance DE, Dyck JR, Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity, Diabetes, 56 (2007) 2863–2871. [DOI] [PubMed] [Google Scholar]
- [7].Rada P, González-Rodríguez Á, García-Monzón C, Valverde Á M, Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver?, Cell Death Dis, 11 (2020) 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Miquilena-Colina ME, Lima-Cabello E, Sánchez-Campos S, García-Mediavilla MV, Fernández-Bermejo M, Lozano-Rodríguez T, Vargas-Castrillón J, Buqué X, Ochoa B, Aspichueta P, González-Gallego J, García-Monzón C, Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in nonalcoholic steatohepatitis and chronic hepatitis C, Gut, 60 (2011) 1394–1402. [DOI] [PubMed] [Google Scholar]
- [9].Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M, Weiss EJ, Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice, Endocrinology, 157 (2016) 570–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Horton JD, Goldstein JL, Brown MS, SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver, J Clin Invest, 109 (2002) 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ferre P, Foufelle F, Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c, Diabetes Obes Metab, 12Suppl 2 (2010) 83–92. [DOI] [PubMed] [Google Scholar]
- [12].Mirandola S, Realdon S, Iqbal J, Gerotto M, Dal Pero F, Bortoletto G, Marcolongo M, Vario A, Datz C, Hussain MM, Alberti A, Liver microsomal triglyceride transfer protein is involved in hepatitis C liver steatosis, Gastroenterology, 130 (2006) 1661–1669. [DOI] [PubMed] [Google Scholar]
- [13].Moolenaar WH, Perrakis A, Insights into autotaxin: how to produce and present a lipid mediator, Nat Rev Mol Cell Biol, 12 (2011) 674–679. [DOI] [PubMed] [Google Scholar]
- [14].Knowlden S, Georas SN, The autotaxin-LPA axis emerges as a novel regulator of lymphocyte homing and inflammation, J Immunol, 192 (2014) 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yanagida K, Ishii S, Non-Edg family LPA receptors: the cutting edge of LPA research, J Biochem, 150 (2011) 223–232. [DOI] [PubMed] [Google Scholar]
- [16].Yung YC, Stoddard NC, Chun J, LPA receptor signaling: pharmacology, physiology, and pathophysiology, J Lipid Res, 55 (2014) 1192–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van Meeteren LA, Ruurs P, Stortelers C, Bouwman P, van Rooijen MA, Pradere JP, Pettit TR, Wakelam MJ, Saulnier-Blache JS, Mummery CL, Moolenaar WH, Jonkers J, Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development, Mol Cell Biol, 26 (2006) 5015–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bain G, Shannon KE, Huang F, Darlington J, Goulet L, Prodanovich P, Ma GL, Santini AM, Stein AJ, Lonergan D, King CD, Calderon I, Lai A, Hutchinson JH, Evans JF, Selective inhibition of Autotaxin is efficacious in mouse models of liver fibrosis, J Pharmacol Exp Ther, 360 (2017) 1–13. [DOI] [PubMed] [Google Scholar]
- [19].Kaffe E, Katsifa A, Xylourgidis N, Ninou I, Zannikou M, Harokopos V, Foka P, Dimitriadis A, Evangelou K, Moulas AN, Georgopoulou U, Gorgoulis VG, Dalekos GN, Aidinis V, Hepatocyte autotaxin expression promotes liver fibrosis and cancer, Hepatology, 65 (2017) 1369–1383. [DOI] [PubMed] [Google Scholar]
- [20].Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A, A conditional knockout resource for the genome-wide study of mouse gene function, Nature, 474 (2011) 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rinella ME, Green RM, The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance, J Hepatol, 40 (2004) 47–51. [DOI] [PubMed] [Google Scholar]
- [22].Ibrahim SH, Hirsova P, Malhi H, Gores GJ, Animal models of nonalcoholic steatohepatitis: eat, delete, and inflame, Dig Dis Sci, 61 (2016) 1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Swaney JS, Chapman C, Correa LD, Stebbins KJ, Broadhead AR, Bain G, Santini AM, Darlington J, King CD, Baccei CS, Lee C, Parr TA, Roppe JR, Seiders TJ, Ziff J, Prasit P, Hutchinson JH, Evans JF, Lorrain DS, Pharmacokinetic and pharmacodynamic characterization of an oral lysophosphatidic acid type 1 receptor-selective antagonist, J Pharmacol Exp Ther, 336 (2011)693–700. [DOI] [PubMed] [Google Scholar]
- [24].Jiang G, Xu Y, Fujiwara Y, Tsukahara T, Tsukahara R, Gajewiak J, Tigyi G, Prestwich GD, Alpha-substituted phosphonate analogues of lysophosphatidic acid (LPA) selectively inhibit production and action of LPA, ChemMedChem, 2 (2007) 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tateno C, Kawase Y, Tobita Y, Hamamura S, Ohshita H, Yokomichi H, Sanada H, Kakuni M, Shiota A, Kojima Y, Ishida Y, Shitara H, Wada NA, Tateishi H, Sudoh M, Nagatsuka S, Jishage K, Kohara M, Generation of novel chimeric mice with humanized livers by using hemizygous cDNA-uPA/SCID mice, PLoS One, 10 (2015) e0142145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wu R, Murali R, Kabe Y, French SW, Chiang YM, Liu S, Sher L, Wang CC, Louie S, Tsukamoto H, Baicalein targets GTPase-mediated autophagy to eliminate liver tumor-initiating stem cell-like cells resistant to mTORCl inhibition, Hepatology, 68 (2018) 1726–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lua I, Li Y, Zagory JA, Wang KS, French SW, Sevigny J, Asahina K, Characterization of hepatic stellate cells, portal fibroblasts, and mesothelial cells in normal and fibrotic livers, J Hepatol, 64 (2016) 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li Y, Wang J, Asahina K, Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury, Proc Natl Acad Sci U S A, 110 (2013) 2324–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yang MD, Chiang YM, Higashiyama R, Asahina K, Mann DA, Mann J, Wang CC, Tsukamoto H, Rosmarinic acid and baicalin epigenetically derepress peroxisomal proliferator-activated receptor gamma in hepatic stellate cells for their antifibrotic effect, Hepatology, 55 (2012) 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Asahina K, Tsai SY, Li P, Ishii M, Maxson RE Jr., Sucov HM, Tsukamoto H, Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development, Hepatology, 49 (2009) 998–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tosello-Trampont AC. Landes SG. Nguyen V, Novobrantseva TI. Hahn YS. Kupffer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-a production. J Biol Chem. 287 (2012) 40161–40172, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Contos JJ, Fukushima N, Weiner JA, Kaushal D, Chun J, Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior, Proc Natl Acad Sci U S A, 97 (2000)13384–13389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Contos JJ, Ishii I, Fukushima N, Kingsbury MA, Ye X, Kawamura S, Brown JH, Chun J, Characterization of lpa2 (Edg4) and lpal/lpa2 (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa2, Mol Cell Biol, 22 (2002) 6921–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nakagawa S, Wei L, Song WM, Higashi T, Ghoshak S, Kim RS, Bian CB, Yamada S, Sun X, Venkatesh A, Goossens N, Bain G, Lauwers GY, Koh AP, El-Abtah M, Ahmad NB, Hoshida H, Erstad DJ, Gunasekaran G, Lee Y, Yu ML, Chuang WL, Dai CY, Kobavashi M, Kumada H, Beppu T, Baba H, Mahaian M, Nair VD, Lanuti M, Villanueva A, Sangiovanni A, Iavarone M, Colombo M, Llovet JM, Subramanian A, Tager AM, Friedman SL, Baumert TF, Schwarz ME, Chung RT, Tanabe KK, Zhang B, Fuchs BC, Hoshida Y. Molecular liver cancer prevention in cirrhosis by organ transcriptome analysis and lysophosphatidic acid pathway inhibition. Cancer Cell. 30 (2016) 879–890, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Farquhar MJ, Humphreys IS, Rudge SA, Wilson GK, Bhattacharya B, Ciaccia M, Hu K, Zhang Q, Mailly L, Reynolds GM, Ashcroft M, Balfe P, Baumert TF, Roessler S, Wakelam MJO, McKeating JA, Autotaxin-lvsophosphatidic acid receptor signalling regulates hepatitis C virus replication, J Hepatol, 66 (2017) 919–929. [DOI] [PubMed] [Google Scholar]
- [36].Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, Lee JH, Khadem S, Ren S, Li S, Silverstein RL, Xie W, Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARg in promoting steatosis, Gastroenterology, 134 (2008) 556–567 [DOI] [PubMed] [Google Scholar]
- [37].Tateno C, Kataoka M, Utoh R, Tachibana A, Itamoto T, Asahara T, Miya F, Tsunoda T, Yoshizato K, Growth hormone-dependent pathogenesis of human hepatic steatosis in a novel mouse model bearing a human hepatocyte-repopulated liver, Endocrinology, 152 (2011) 1479–1491. [DOI] [PubMed] [Google Scholar]
- [38].Jang HS, Kim K, Lee MR, Kim SH, Choi JH, Park MJ, Treatment of growth hormone attenuates hepatic steatosis in hyperlipidemic mice via downregulation of hepatic CD36 expression, Anim Cells Syst, 24 (2020) 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jansen S, Andries M, Vekemans K, Vanbilloen H, Verbruggen A, Bollen M, Rapid clearance of the circulating metastatic factor autotaxin by the scavenger receptors of liver sinusoidal endothelial cells, Cancer Lett, 284 (2009) 216–221. [DOI] [PubMed] [Google Scholar]
- [40].Albers HM, Dong A, van Meeteren LA, Egan DA, Sunkara M, van Tilburg EW, Schuurman K, van Tellingen O, Morris AJ, Smyth SS, Moolenaar WH, Ovaa H, Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPAin the circulation, Proc Natl Acad Sci U S A, 107 (2010) 7257–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nakagawa H, Ikeda H, Nakamura K, Ohkawa R, Masuzaki R, Tateishi R, Yoshida H, Watanabe N, Tejima K, Kume Y, Iwai T, Suzuki A, Tomiya T, Inoue Y, Nishikawa T, Ohtomo N, Tanoue Y, Omata M, Igarashi K, Aoki J, Koike K, Yatomi Y, Autotaxin as a novel serum marker of liver fibrosis, Clin Chim Acta, 412 (2011) 1201–1206. [DOI] [PubMed] [Google Scholar]
- [42].Pleli T, Martin D, Kronenberger B, Brunner F, Koberle V, Grammatikos G, Farnik H, Martinez Y, Finkelmeier F, Labocha S, Ferreiros N, Zeuzem S, Piiper A, Waidmann O, Serum autotaxin is a parameter for the severity of liver cirrhosis and overall survival in patients with liver cirrhosis--a prospective cohort study, PLoS One, 9 (2014) e103532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, P Matchett K, Portman JR, Wallenborg K, Picelli S, Zagorska A, Pendem SV, Hudson TE, Wu MM, Budas GR, Breckenridge DG, Harrison EM, Mole DJ, Wigmore SJ, Ramachandran P, Ponting CP, Teichmann SA, Marioni JC, Henderson NC, Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis, Cell Rep, 29 (2019) 1832–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]