

Abstract
The immune system defends the body from infectious and non-infectious threats. Distinct recognition strategies have evolved to generate antigen-specific immunity against pathogens or toxins versus antigen-independent tissue repair. Structural recognition, or the sensing of conserved motifs, guides the immune response to viruses, bacteria, fungi, and unicellular parasites. Functional recognition, which is sensing that is based on the activities of an input, guides antigen-independent tissue healing and antigen-specific Type 2 immunity to toxins, allergens, and helminth parasites. Damage-associated molecular patterns (DAMPs), released from damaged and dying cells, permit functional recognition by immune cells. However, the DAMP paradigm alone does not explain how functional recognition can lead to such disparate immune responses, namely wound healing and Type 2 immunity. Recent work established that sensory neurons release neuropeptides in response to a variety of toxins and allergens. These neuropeptides act on local innate immune cells, stimulating or inhibiting their activities. By integrating our knowledge on DAMP function with new information on the role of neuropeptides in innate immune activation in Type 2 immunity, we describe a decision tree model of functional recognition. In this model, neuropeptides complement or antagonize DAMPs to guide the development of antigen-specific Type 2 immunity through the activation of innate immune cells. We discuss why this decision tree system evolved and its implications to allergic diseases.
Keywords: allergy, innate immunology, neuroimmunology, neuropeptide
INTRODUCTION
The immune system evolved to combat pathogens, clear toxins and heal wounds. To do this, a network of innate immune cells such as macrophages, mast cells, innate lymphoid cells and dendritic cells rapidly detect these threats and initiate the proper inflammatory and tissue repair pathways to protect the host from harm. The type of threat encountered defines the immune response and innate immune cells activated. In general, Type 1 immune responses protect against intracellular bacteria or viral pathogens. Type 2 immune responses defend against toxins and multicellular helminth parasites, and are thought to be unintentionally triggered by allergens, which are non-infectious substances encountered in the environment. Type 3 immune responses guard against extracellular bacteria and fungi. Long lasting immune protection requires innate activation of the adaptive immune response, marked by the generation of antigen-specific T and B cells capable of specific pathogen and toxin clearance. Antigen-specific Type 1 and Type 3 immune responses are characterized by T helper type 1 (Th1) and Th17 cells, respectively, and B cells producing immunoglobulin (Ig)G subclasses associated with macrophage and complement activation. In contrast, antigen-specific Type 2 immune responses are characterized by Th2 cells and B cells producing IgE that can help to mediate mast cell and basophil degranulation. The combination of innate and adaptive immune mechanisms drives pathogen or toxin clearance and a return to homeostasis. But not all threats require adaptive immune activation. The wound response requires innate immune activation and tissue repair responses without the generation of adaptive, antigen-specific immunity that could target host antigens. The innate immune system must be able to identify and differentiate between these threats to host homeostasis.
Prokaryotic and viral pathogens are evolutionarily distant and distinct from the eukaryotic multicellular organisms that they infect. As such, they express specific, evolutionarily conserved molecular structures termed pathogen-associated molecular patterns (PAMPs) that bind to a limited number of host-expressed pattern recognition receptors (PRRs).1 Termed structural recognition, this sensing pathway leads to Type 1 immunity. Similarly, structural recognition is a key mechanism in the sensing of Type 3 immunogens such as complex eukaryotes including fungi. For example, the fungus Candida albicans is structurally recognized by the C-type lectin receptor Dectin-2 on dendritic cells and is required for Th17 differentiation.2 In contrast, damage to the host produces inflammation through the release of damage-associated molecular patterns (DAMPs) that bind to DAMP-specific receptors.1 DAMPs are host-derived molecules such as interleukin (IL)-1α and IL-33 that alert the innate immune system to tissue damage, initiating or enhancing the immune response. This mechanism is termed functional recognition because DAMPs signal the presence of a threat based on detecting its function or activity against the host. DAMPs are considered agnostic to the source of damage and can be released in response to sterile wounding, virulence factors released by bacteria or toxic venoms.1 DAMP signaling in the absence of PAMPs is thought to be a potent driver of both wound healing and Type 2 immune responses.3 This specific role for DAMPs in the functional recognition of allergens – as well as the few exceptions where pattern recognition receptors are important in Type 2 immune responses – has been reviewed previously.4 However, if DAMPs are released in response to multiple stimuli that require very different immune outcomes, what specifically guides adaptive Type 2 immunity?
The protein toxins, secreted helminth effector proteins and allergens that induce Type 2 immune responses are diverse in structure, evolutionary lineage and function. A commonality among these Type 2 immunogens is enzymatic activity, a trait that is essential for their immunogenicity as evidenced by the observation that heat or pharmacologic inactivation of their enzymatic activity abrogates their adjuvant activity.5,6 One method the host employs to functionally sense the enzymatic activity of Type 2 immunogens is the release of DAMPs. Type 2 immunogens cause the release of epithelial-derived IL-33, IL-25 and thymic stromal lymphopoietin (TSLP), which we classify here as DAMPs.5,7,8 Not only do both toxins and allergens rapidly elicit the release of IL-33 in affected tissues, but allergens can also further enhance the activity of released IL-33 through cleavage to its more active form.5,7,9 However, while important, IL-33 is not absolutely required for the activation of innate immune cells that leads to Th2 differentiation in response to toxins and allergens.5,10,11 Similarly, while TSLP can promote Th2 differentiation, in its absence Th2 differentiation to allergens was impaired, but not terminated.12 Likewise, IL-33 and the DAMP IL-25 contribute to the clearance of worms and Th2 differentiation upon infection with the helminth Nippostrongylus brasiliensis, but they are not required outright.13,14 These data suggest that epithelial DAMPs play an important role in augmenting and instructing, but not independently driving, antigen-specific Type 2 immunity. Thus, it is not clear that DAMPs alone can differentiate the requirement for antigen-independent tissue repair in the context of a sterile wound while also being solely responsible for antigen-specific Type 2 immunity in the context of toxins and allergens. Additional specialization in DAMPs may exist such that specific patterns of release promote local inflammation, while others promote the initiation of Type 2 immune responses. Nonetheless, it is also likely that there is an additional, non-redundant recognition strategy driving the response to Type 2 immunogens.
Allergic immune responses are considered to be maladaptive responses against non-infectious environmental substances. However, non-infectious does not necessarily mean innocuous. The ‘toxin hypothesis’ of allergy proposed that the Type 2 immune response commonly associated with allergen sensitization (i.e. the development of Th2 cells and IgE producing B cells) evolved to protect the host from repeated exposures to toxins.15 This theory of allergy was tested by the Galli and Medzhitov laboratories using venoms, defined as toxins produced by animals that are injected into other animals as a form of defense or to facilitate feeding. Venoms, such as those from stinging insects, are also common allergens. In support of the toxin hypothesis of allergy, Metz et al.16 determined that mast cells were required to defend against the sublethal and lethal effects of primary snake or honeybee envenomation. Subsequent studies found that a low-dose exposure to bee or snake venom protected from secondary challenge with a near-lethal dose of bee or snake venom in an IgE and FcεRI-dependent manner.5,17,18 Together, these data support that Type 2 immunity evolved to protect from primary and secondary exposures to toxins and may indicate that allergens trigger Type 2 immune responses because they are either toxins themselves or are associated with or linked to toxins.15 Understanding how toxins are sensed by the body provided key clues to an additional family of molecules important in the functional recognition strategy that toxins and allergens utilize to activate Type 2 immunity.
Anyone who has experienced toxin envenomation through a bee sting recalls the immediate sensations of pain and itch. This sensation occurs when unmyelinated C-type small diameter sensory neurons and Aδ fibers are activated by environmental disturbances such as the presence of a toxin. This leads to an action potential that courses up the axon of the pseudounipolar sensory neuron, which generally synapse with a second neuron located in the spinal cord. This signaling induces motor responses to protect the body from the harmful stimulus (i.e. scratching, withdrawal) and induces unpleasant sensations (i.e. itch, pain) to promote avoidance behaviors. These pathways are initiated nearly instantly, making sensory neurons uniquely poised to rapidly detect toxins. While envenomation by a bee sting is a classic example of sensory neurons acting as an immediate sensor of toxins, sensory neuronal activation is a common pathway activated by toxins derived from snakes, amphibians, fish, terrestrial and marine invertebrates, plants, fungi and bacteria.19–25 Similarly, allergens were found to directly elicit sensory neuron activation and to provoke itch and pain behaviors.26 These studies demonstrate that sensory neuronal detection may be a common mechanism of toxin and allergen sensing. But does sensory neuronal detection of allergens and toxins do more than promote avoidance behavior? Activated sensory neurons can communicate with proximally located cells through the release of neuropeptides from their free nerve endings. When acting upon endothelial cells this leads to vasodilation and extravasation, which causes the warm erythema associated with skin inflammation.27 Neuropeptides can also act on immune cells to promote or suppress immune function in a context dependent manner. The release of calcitonin-gene related peptide (CGRP) by sensory neurons in response to bacterial peptides and toxins suppresses neutrophil function.21,28 In contrast, fungal induced CGRP release promotes immune cell activation and anti-fungal responses.29,30 Likewise for Type 2 immunity, we and others recently found that protease allergen-activated sensory neurons release the neuropeptide substance P to drive inflammation.26,31 Together, these data support that sensory neurons are an immediate sensor of toxins and allergens and instruct immune responses through neuropeptides.
Given our growing understanding for the role of neuropeptides in the control of immune cells, in this review we describe the emerging literature that implicates neuropeptides as additional signaling molecules required to generate Type 2 immune responses to toxins and allergens. Neuropeptides share the qualities of quick release, promotion of local inflammation and a role in innate activation of adaptive immunity seen by DAMPs. Thus, from here we term these immune effecting peripheral neuropeptides as neuron-associated molecular patterns (NAMPs). This nomenclature is meant to draw a clear parallel with the concept of DAMP signaling in instructing immune responses. In contrast to the release of DAMPs from damaged or dying cells, NAMPs are actively secreted from pre-formed dense core vesicles and cell damage is not a prerequisite. Unlike neurotransmitters that undergo rapid reuptake after release, neuropeptides have a longer half-life, allowing for more persistent effects on both neurons and immune cells. Although neurotransmitters have recently been described as playing a role in immune cell function,32 in this review we will focus on the specific effects of sensory neuron-derived neuropeptides. We will examine the experimental evidence to support a new concept whereby the initiation of Type 2 immune responses to toxins or allergens requires an integrated exposure to DAMPs and NAMPs (Figure 1). With a focus on allergic responses, we will discuss how this additional signal in the functional recognition of toxins and allergens differs from the structural recognition that governs the sensing of Type 1 and Type 3 immunogens and what this implies about allergic diseases.
Figure 1.
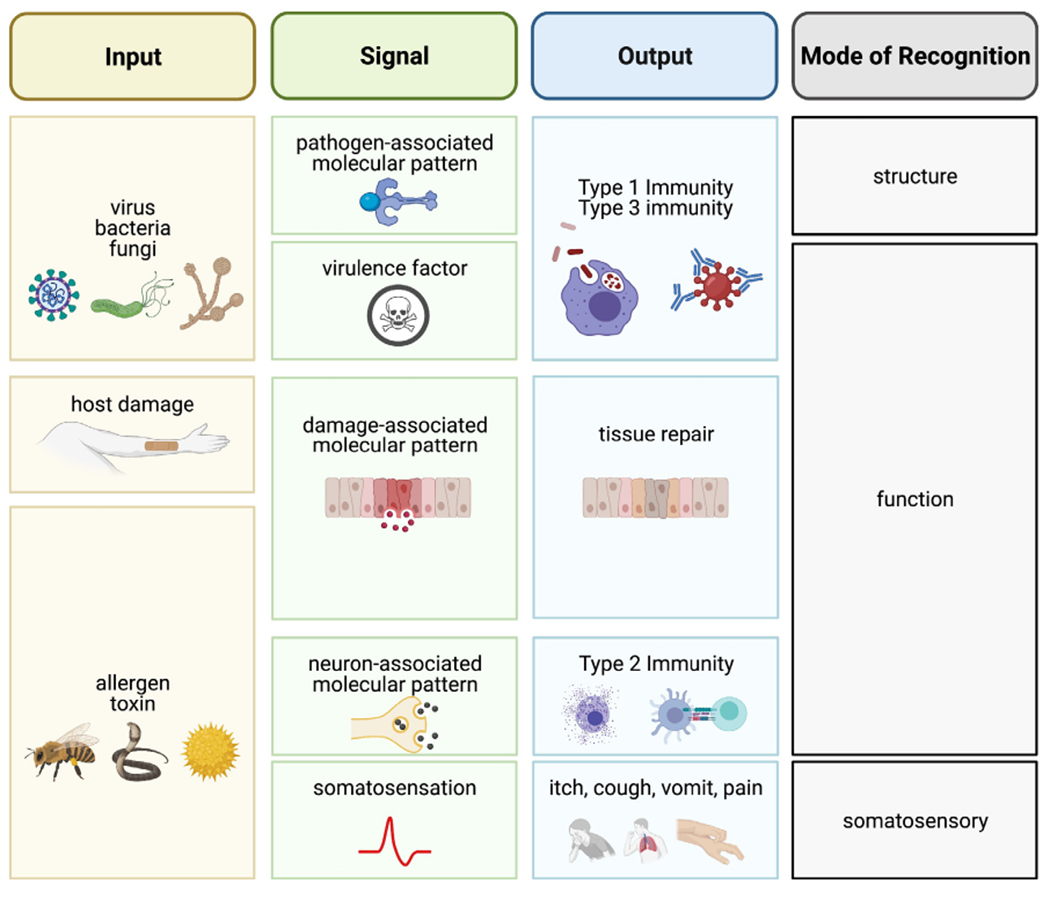
Recognition strategies that underlie antigen-specific immunity and antigen-independent wound healing. Inputs such as pathogens, host damage, and toxins or allergens are sensed by innate immune cells via distinct pathways that lead to divergent outputs. Pathogens are primarily sensed by their pathogen-associated molecular patterns with input of virulence factors. Thus, mainly structural recognition with some functional recognition generates Type 1 and Type 3 immunity. In contrast, sterile damage to the host produces damage-associated molecular patterns (DAMPs) that initiates tissue repair pathways under the umbrella of functional recognition. Meanwhile, toxins and allergens induce Type 2 immunity through the release of DAMPs and the potent activation of sensory neurons, leading to rapid somatosensory responses and the release of neuron-associated molecular patterns (NAMPs). Thus, while antigen-independent tissue repair and Type 2 immunity both rely on functional recognition in the absence of structural recognition, additional signals are required to escalate to an antigen-specific immune response.
NAMP RELEASE IS A KEY MECHANISM OF TOXIN CLEARANCE AND ALLERGEN RECOGNITION
Mast cells
Mast cells play a central role in the clearance of toxins and are responsible for many of the trademark symptoms of allergies. Decades before the toxin hypothesis of allergy was proposed, Higginbotham found that subcutaneous injection of Russell’s Viper venom and honey bee venom induced mast cell release of heparin-containing granules, which could complex with venom components to reduce toxicity and permit phagocytosis and toxin removal.33,34 Although this suggested a role for mast cells in the detoxification of toxins, it was not until genetic models of mast cell deficiency were developed that it was experimentally proven. In 2006, the Galli laboratory illustrated that mast cells protected naïve mice from the pathologic effects of primary envenomation with snake and honeybee venom.16 To detoxify harmful substances, mast cells degranulate, releasing enzymes from pre-formed granules that proteolytically degrade toxins. Indeed, enzymatic components of mast cell granules such as carboxypeptidase A and chymase degrade sarafotoxin from snake venom and helodermin from the Gila monster, respectively.16,35,36 As discussed previously, upon secondary exposure to a venom, toxin-specific IgE and FcεRI on mast cells enhances the protection afforded by these cells.5,17,18 Although mast cell degranulation was induced by and protective against envenomation in all cases, how mast cells were activated in naïve mice that presumably did not contain venom-specific IgE, was unclear. The DAMPs IL-33 and TSLP do not induce mast cell degranulation on their own, but instead increase cytokine production.37 This suggests that an additional signal plays a more prominent role in mast cell degranulation.
Light and electron microscopy studies have shown that peripheral neurons envelop mast cells.35,38 These “intimate connections” led to the hypothesis that sensory neuronal-mast cell interactions were involved in a variety of inflammatory processes.35 Studies from the 1970s illustrated that mast cells treated in vitro with the NAMPs substance P and vasoactive intestinal peptide (VIP) released histamine, indicative of degranulation.36 In vivo, substance P appeared to induce mast cell degranulation in the skin when visualized by Giemsa stain.39 Using more sensitive measures of mast cell degranulation it was later demonstrated that substance P and VIP, but not CGRP, caused the release of β-hexosaminidase, an enzyme found in mast cell granules.40 Mechanistically, how do NAMPs elicit mast cell degranulation? The receptors for substance P include NK-1R, NK-2R, MRGPRA1 and MRGPRB2 in mice and MRGPRX2 in humans.41–43 Human mast cells express MRGPRX2, the human ortholog of mouse MRGPRB2, explaining how substance P evokes degranulation.44 These studies established that NAMPs can induce mast cell degranulation, but how this pathway functioned in the context of an immune response was not determined until 2019. Green et al.45 first determined that substance P acting on mast cells expressing MRGPRB2 mediated neurogenic inflammation and pain. In elegant studies, Serhan et al.31 extended NAMP-mast cell interactions to Type 2 immunity, finding that the common allergen house dust mite (HDM) activated sensory neurons and provoked their release of substance P. Sensory neuron-derived substance P acted on clusters of adjacent mast cells expressing MRGPRB2 to degranulate, ultimately responsible for the observed tissue swelling.31 This study illustrates that allergens elicit sensory neuron activation (NAMP release) to mobilize mast cells in the Type 2 immune response (Figure 2).
Figure 2.
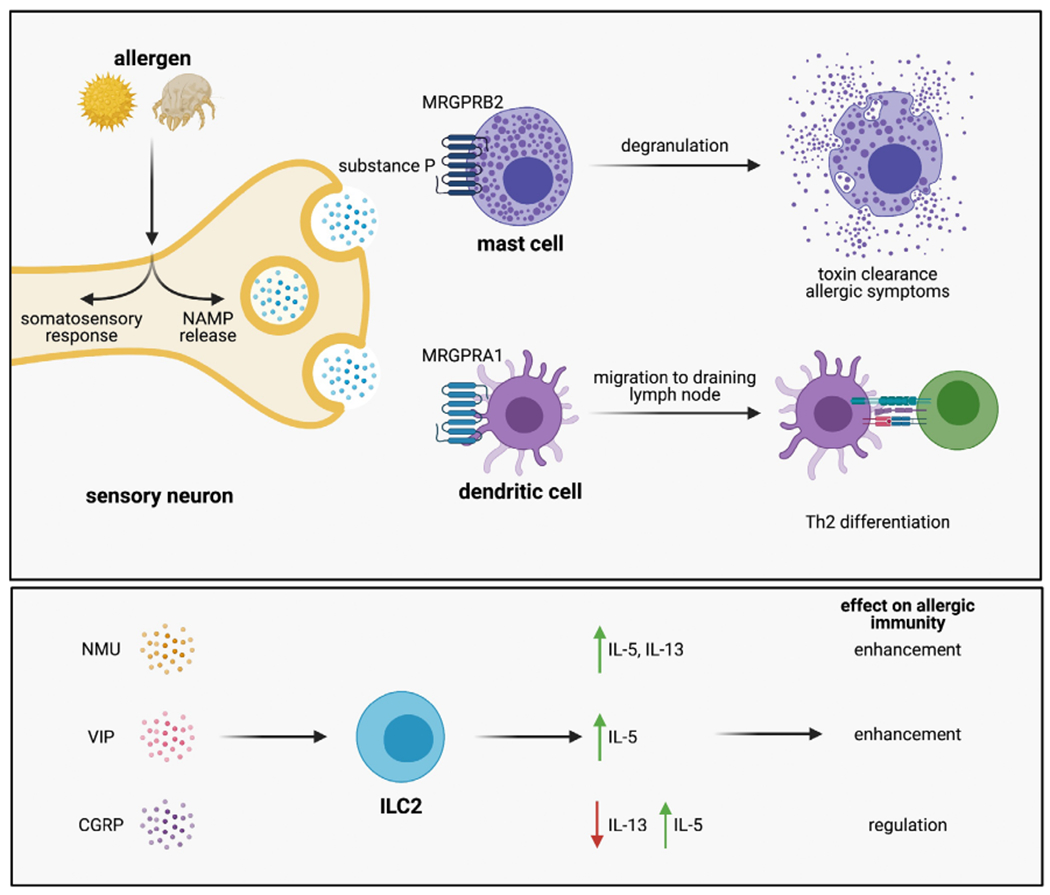
NAMPs control the generation of Type 2 immunity. Mast cells, group 2 innate lymphoid cells (ILC2s) and dendritic cells (DCs) are targets of NAMPs. Allergens cause sensory neurons to produce substance P, acting on local mast cells through MRGPRB2 to cause degranulation. Substance P also acts on proximally located CD301b+ dendritic cells through MRGPRA1, stimulating their migration to the draining lymph node where they initiate Th2 differentiation. Thus, NAMP release is required for rapid Type 2 effector responses and the initial sensitization to allergens. ILC2s respond to a variety of NAMPs including NMU, VIP and CGRP, which act on their own to stimulate ILC2 activation (NMU, VIP) or to regulate their activation by CGRP. While these pathways have only been studied in chronic allergic inflammation, they likely are involved in the initial clearance of toxins and sensing of allergens.
It is tempting to speculate that toxins, like allergens, directly activate neurons, leading to NAMP release and to mast cell activation. If so, then it is reasonable to conclude that this neuro-immune interaction that evolved to degrade toxins is responsible for many of the cardinal symptoms of allergic disease. But how are these interactions regulated? Could NAMPs other than substance P play synergistic or antagonistic roles? Interestingly, Caughey et al.46 found that the proteases of degranulated mast cells could degrade substance P and VIP, implicating a possible negative feedback loop to halt excess inflammation following allergen recognition. Vice versa, Meixiong et al.47 illustrated that tryptase release from degranulated mast cells activated local neurons leading to itch. Whether this also promotes substance P release is unknown, but these examples show the potential for robust feedback loops altering inflammatory responses to allergens.47
Group 2 innate lymphoid cells
Group 2 innate lymphoid cells (ILC2s) are a rare population of helper-like lymphocytes that are enriched in peripheral tissues. There is evidence that ILC2s play a role in toxin and allergic immune responses, especially before chronic inflammation is established.48 While lymphocytic in origin, these cells are unlike T and B cells in that they do not express antigen-specific receptors and instead respond to environmental cues through germline-encoded receptors. ILC2s respond to IL-33, TSLP and IL-25 by producing the characteristic Th2 cytokines IL-5 and IL-13, thereby acting as an early source of these Type 2 cytokines.7,49 This may allow ILC2s to both promote early allergic inflammation as well as to direct the development of adaptive immune responses to allergens. As an example, ILC2-derived IL-13 promotes DC migration to the draining lymph node and is thus necessary for optimal Th2 differentiation in response to allergens.10 In addition, activation of ILC2s by IL-33 is important for Th2 differentiation in response to bee venom.5 While DAMPs are sufficient to induce robust ILC2 activation, recently NAMPs have emerged as additional regulators of ILC2 function. Although much of the work on NAMPs affecting ILC2 function was done in the setting of chronic allergic inflammation, given the essential role for ILC2s in generating Type 2 immune responses it is possible that early release of NAMPs could dictate ILC2-mediated pathways in toxin clearance and allergen sensitization.
In 2017, single cell and bulk RNA sequencing performed by three independent laboratories identified the Nmur1 gene highly enriched and uniquely expressed by ILC2s.50–52 Nmur1 encodes the receptor for the neuropeptide neuromedin U (NMU), which suggested that this NAMP might direct ILC2s during the early phase of the Type 2 immune response. Indeed, ILC2s responded directly to NMU by increasing the production of IL-5 and IL-13, while NMU further enhanced DAMP-induced IL-5 and IL-13.50–52 Wallrapp et al.52 found that in Nmu deficient mice, ILC2s failed to upregulate IL-5 and IL-13 to the same degree as in wild-type mice sensitized and challenged with HDM. However, the impact of this deficiency in ILC2 activation on overall inflammation was partial, suggesting that a second signal complements NMU control of ILC2 function in the setting of chronic allergic inflammation. In addition to NMU, VIP has also been described to directly induce IL-5 production in ILC2s,53,54 but at present it is unknown how VIP-ILC2 signaling might shape toxin clearance or allergic sensitization. While the aforementioned studies suggested a pro-inflammatory role for NMU and VIP in ILC2 activation, a second series of studies in 2019 found an immunomodulatory role for CGRP in ILC2 function. Indeed, CGRP was found to reduce IL-13 and to increase IL-5 production by lung or gut ILC2s.55–57 In vivo, exogenous CGRP or lack of Ramp1 (part of the CGRP receptor) reduced IL-33-driven ILC2 activation and downstream inflammation in the lung, suggesting that CGRP could play an immunomodulatory role during the early Type 2 immune response.55,56 In an ovalbumin model of food allergy, exogenous CGRP given during allergen challenge was sufficient to dampen ILC2 and mast cell expansion, key features of disease.57 Together with data showing a role for CGRP in inhibiting local innate inflammatory responses to bacteria and fungi, this may indicate a global immunoregulatory function for CGRP. While these rigorous studies clearly identify NAMPs as key regulators of ILC2 function, it remains unknown whether toxins or allergens directly induce the release of NAMPs like NMU or VIP from sensory neurons, nor how that signaling would impact ILC2-mediated Type 2 immune responses.
Dendritic cells
Dendritic cells (DCs) are professional antigen presenting cells that bridge the gap between innate and adaptive immunity. Immature DCs constantly sample their environment, but once they are activated by innate immune stimuli, DCs undergo maturation. Mature DCs downregulate phagocytosis and upregulate their surface expression of antigen and CCR7, a chemokine receptor that guides their migration to the draining lymph node.58,59 Thus, maturation fixes the presented antigens in the draining lymph node to those encountered at the time of DC activation. In the structural recognition paradigm, PAMPs bind to PRRs expressed by DCs, inducing stereotypical DC maturation. In response to Type 2 immunogens including allergens, DCs are required for the generation of antigen-specific Th2 cells59,60 and T follicular helper cells (Tfh).61,62 In vivo, allergens readily induce DC migration to the draining lymph node, but DCs treated with allergen in vitro fail to upregulate CCR7 and show no evidence of activation.5,26,59 This observation illustrates that DCs do not directly detect Type 2 immunogens by structural recognition, suggesting that functional recognition underlies their sensing of toxins and allergens. DAMPs, the key signaling molecules in functional recognition, play only a partial role in the development of antigen-specific immunity to allergens. In the absence of the DAMPs IL-33 or TSLP, DC migration and Th2 differentiation is reduced, but not abolished, suggesting that an additional signal is required for antigen-specific immune responses to toxins and allergens.10–12
Recent work indicates that NAMPs function in parallel with DAMPs to direct DCs to generate antigen-specific Type 2 immunity. Using whole mount immunostaining, Veres et al.63 showed that airway CD11c-eYFP+ cells, most likely DCs, were closely associated with protein gene product (PGP) 9.5+ neurons expressing CGRP and substance P. This study suggested that DCs may be a target of substance P and CGRP during allergen sensitization due to their close proximity to these NAMP-expressing neurons. Indeed, lung DCs were found to express the CGRP receptor genes (Calcrl associated with Ramp1 or Ramp3).64 While CGRP inhibited LPS-induced bone marrow-derived DC (BMDC) maturation in vitro, CGRP treated BMDCs also prevented the features of allergic airway inflammation when transferred to recipient mice.64 Kashem et al.29 found that the fungus Candida albicans directly induced the release of CGRP from sensory neurons. This NAMP signaling induced IL-23 release from dermal CD301b+ DCs that directed dermal γδ T cells to produce IL-17A, which was required for innate resistance to the fugus.29 These data suggested, as has been shown with respect to innate inflammation and ILC2 function, that except for in the setting of Candida infection, CGRP generally acts to suppress immune function.
In contrast to the immunosuppressive effects of CGRP, Substance P may have immunostimulatory effects. Our group found that sensory neurons treated with the model allergen papain potently secreted substance P.26 Sensory neuron-derived substance P acted upon local CD301b+ DCs, the subset in the skin required for Th2 differentiation,59,60 stimulating their migration to the draining lymph node by signaling through MRGPRA1.26 This substance P-MRGPRA1 signaling pathway was required for allergen-induced Th2 differentiation, suggesting that activation of sensory neurons is an essential requirement in the generation of Type 2 immunity to toxins and allergens26 (Figure 2). However, substance P is not sufficient; injection of substance P alone led to CD301b+ DC migration, but not to Th2 differentiation. This indicates that another factor, possibly a DAMP, is necessary in combination with Substance P to induce allergen-specific adaptive immunity.
Data generated in our laboratory revealed a specific effect of substance P on CD301b+ DCs based on their expression of MRGPRA1, but other studies have shown that BMDCs generated with granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 expressed the neurokinin 1 receptor (NK-1R), one of the four substance P receptors in addition to NK-2R, MRGPRA1, and MRGPRB2.65 These BMDCs treated with NK-1R agonists were found to enhance immune responses associated with pathogen clearance, but at this time there is no known role for DC-expressed NK-1R in Type 2 immune responses.66 While MRGPRA1 expression was not evaluated in these studies, the four substance P receptors may be expressed by different DC subsets, be tissue dependent, or emerge in specific cytokine environments during maturation. Additional studies of these mechanisms are needed as different substance P receptor expression patterns may dictate how DCs function during an immune response. In addition to a clear role for substance P in directing DCs to generate Type 2 immunity, human monocyte-derived DCs were found to dose-dependently migrate to gradients of neuropeptide Y (NPY), and mice deficient in Npy had less robust DC migration to the draining lymph node and decreased features of allergic inflammation.67,68 Pituitary adenylate cyclase-activating polypeptide (PACAP) was similarly found to promote DC maturation and migration in a model of contact hypersensitivity to a hapten, suggesting that there may be many additional NAMPs that control the functions of DCs.69 Together, these data indicate that NAMPs can direct the movement of DCs, but additional studies are warranted as this may not be their only function during Type 2 immune responses.
A DECISION TREE OF FUNCTIONAL RECOGNITION FOR THE GENERATION OF TYPE 2 IMMUNITY
Structural recognition of PAMPs is unique to the Type 1 and Type 3 immune responses. In contrast, functional recognition guides antigen-independent tissue repair and antigen-specific Type 2 immunity, while also augmenting Type 2 immune responses.1 Previously, it was thought that the major signaling molecules of the functional recognition pathway were DAMPs. But it was never clear how, in the absence of an additional recognition pathway initiating Type 2 immunity, DAMPs alone could specify between the induction of antigen-independent wound healing or antigen-specific Type 2 immunity. We hypothesize that the combinatorial processing of competing DAMP and NAMP signals instructs the immune response. Thus, we propose a decision tree model of functional recognition integrating DAMP and NAMP signals that is required for the innate immune system to discriminate between inputs that require antigen-independent tissue repair versus antigen-specific Type 2 immunity.
In the decision tree of functional recognition, the activities of innate immune cells are directed by the integration of signals from DAMPs and NAMPs, which can be regulatory or activating. In the absence of DAMPs or NAMPs, tissue homeostasis is maintained. If NAMPs are released without concomitant DAMP release, a regulatory NAMP will result in local immunosuppression, while an activating NAMP will result in a local, short-lived Type 2 effector response (i.e. mast cell degranulation). If an input results in the release of a DAMP but no NAMP, then tissue reparative pathways are initiated that do not result in adaptive Type 2 immunity. In each of these cases, single exposure to DAMP or NAMP will not be sufficient to induce an adaptive immune response. Similarly, the combination of a DAMP with a regulatory NAMP will not be sufficient to induce adaptive Type 2 immunity, but it will shape the Type 2 effector response (i.e. altered ILC2 activation). However, when an input results in the presence of both a DAMP and an activating NAMP, this combination will license the development of antigen specificity (i.e. DC-induced Th2 differentiation) along with local effector responses. This decision tree paradigm integrating DAMPs and NAMPs in the functional recognition paradigm is summarized in Figure 3.
Figure 3.
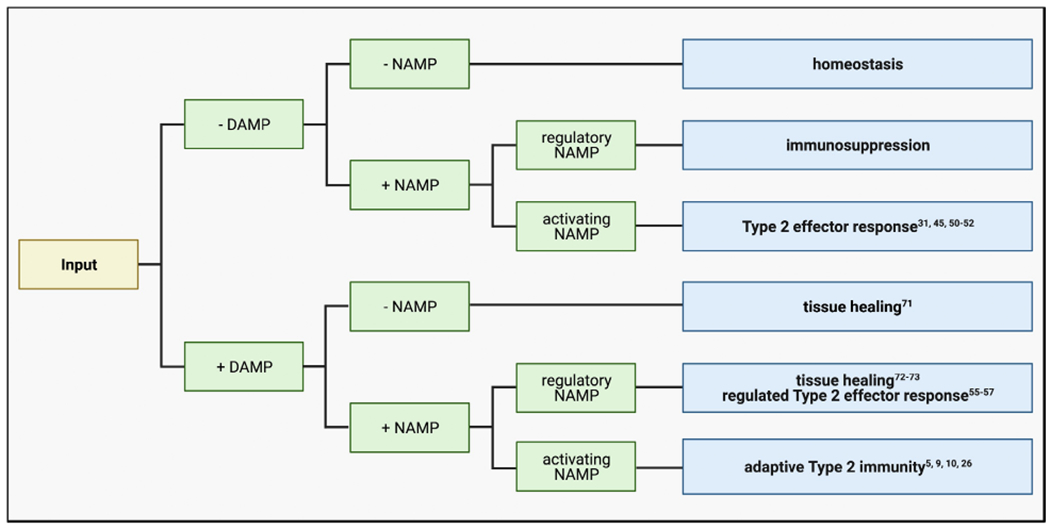
A decision tree model of functional recognition governs Type 2 immunity. Because functional recognition underlies both antigen-independent wound healing and antigen-specific Type 2 immunity, innate immune cells must discriminate when to mount each response. The decision tree model of functional recognition proposes that alone, DAMPs initiate tissue repair pathways. Only when in combination with a NAMP can DAMPs instruct innate immune cells to generate antigen-specific Type 2 immunity. Thus, antigen-specific Type 2 immune responses are guarded by an extra layer of signaling that is required to generate antigen specificity. This decision tree model explains how functional recognition can govern such disparate immune responses.
We developed the decision tree model of functional recognition as a testable hypothesis to explain how innate immune cells can differentiate between signals that require antigen-specific immune responses and antigen-independent reparative responses. In this context it is exciting to consider how such a model could explain Treg cell development as well as the generation of specialized Tfh cells. Although it is beyond the scope of this current review to consider the generation of these specialized Th cells, we expect that DAMP and NAMP signaling pathways may be utilized to activate diverse Th cell subsets. Indeed, a recent study found that Type 2 immunogens uniquely generated Tfh cells that produce IL-13 and are required for the generation of high affinity IgE.70 It would be exciting to determine whether these specialized Tfh cells require the same DAMP and NAMP signals that instruct Th2 differentiation.
EXPERIMENTAL EVIDENCE OF THE DECISION TREE OF FUNCTIONAL RECOGNITION TO ELICIT TYPE 2 IMMUNE RESPONSES OR TISSUE HEALING
Emerging literature demonstrates the decision tree of functional recognition in dictating Type 2 immune responses (Table 1). The activating NAMP substance P induces mast cell degranulation through the receptor MRGPRB2, an example of an activating NAMP causing a Type 2 effector response.31,45 Molofsky et al. previously reviewed the ability of IL-33 to induce tissue repair,71 an example of a DAMP promoting antigen-independent healing in the absence of NAMPs. Upon damaging overexposure to UV radiation, sensory neurons produce TAFA4 that induces macrophage secretion of IL-10, a pathway required to stimulate tissue healing.72 Given that overexposure to UV radiation also induces the release of the DAMP high mobility group box 1 (HMGB1),73 this is an example of a DAMP and regulatory NAMP together promoting tissue healing. When compared with exposure to IL-33 alone, dual exposure to IL-33 and the regulatory NAMP CGRP reduced IL-13 and increased IL-5 production by ILC2s.55–57 This is an example of a regulatory NAMP tuning DAMP-induced innate immune activation, as the decision tree of functional recognition illustrates. The ultimate purpose of our model is to define the unique signals required to generate antigen-specific Type 2 immunity. We recently showed that the activating NAMP substance P is required for Th2 differentiation in response to IL-33 inducing protease allergens.26 At the same time as predicted by the decision tree, neither the DAMP IL-33 nor the activating NAMP substance P are alone sufficient to induce Th2 differentiation.5,10,11,26 Based on these compelling data, in Figure 3, we hypothesize that the dual presence of a DAMP and activating NAMP is the signal that is required to license antigen-specific Type 2 immunity.
Table 1.
Experimental evidence of the decision tree of functional recognition
| NAMP | DAMP | Output | Reference |
|---|---|---|---|
| Substance P | - | Mast cell degranulation through MRGPRB2 | 31,45 |
| NMU | - | ILC2 activation through NMUR1 | 50–52 |
| - | IL-33 | Induction of tissue repair | 71 |
| TAFA4 | HMGB1 | Tissue healing to UV overexposure through macrophage IL-10 | 72,73 |
| CGRP | IL-33 | Regulation of ILC2 cytokine production | 55–57 |
| Substance P | IL-33 | Th2 differentiation dependent on both, but neither are sufficient alone | 5,10,11,26 |
Based on this decision tree model of functional recognition, it is reasonable to conclude that the type of NAMP, when in combination with a DAMP, determines whether antigen-independent tissue healing or antigen-specific Type 2 immunity is generated. Indeed, experimental evidence suggests that in addition to TAFA4 release upon UV overexposure, the regulatory NAMP CGRP is similarly induced and while this promotes wound healing,74 it prevents the development of DC-dependent contact hypersensitivity.75 These results suggest that regulatory NAMPs such as CGRP or TAFA4 limit the development of fully competent, antigen-specific Type 2 immune responses when signaling in combination with a DAMP. In contrast, the decision tree model proposes that an activating NAMP such as substance P or NMU is a unique signal that escalates DAMP-induced tissue healing to antigen-specific Type 2 immunity. Thus, the proposed framework predicts that the type of NAMP released dictates how functional recognition can lead to divergent immune outcomes. In the current model, it is not known how the kinetics of DAMP and NAMP release impacts the broad outputs we define. In addition, our knowledge of the NAMPs that signal in the functional recognition decision tree is limited to only a few well described pathways. Thus, unbiased approaches are needed to fully delineate the DAMPs and NAMPs released by different toxins and allergens. Studies such as these will allow for further iteration of our proposed conceptual framework of functional recognition.
IMPLICATIONS OF THE DECISION TREE OF FUNCTIONAL RECOGNITION IN TYPE 2 IMMUNITY
Why did toxin and allergen sensing evolve to require multiple signals (i.e. DAMPs and NAMPs) from multiple sources in order to elicit Type 2 immunity? In the structural recognition paradigm, pathogens are sensed by evolutionarily conserved structures that are vital to pathogen survival and unlikely to mutate through evolutionary pressures. Expression of these PAMPs can be shared amongst similar species, meaning that a relatively small number of PRRs expressed on immune cells can recognize structural motifs from a vast number of pathogens. While the PAMP activates the local innate immune response to promote rapid pathogen clearance, it also acts as the adjuvant, promoting DC maturation. Because the adjuvant is normally linked to the antigen, with only one signal the activated DC is permitted to stimulate the development of antigen-specific immunity to the pathogen. In contrast, toxins and allergens are incredibly diverse and do not share common structural features that would permit structural recognition. Instead, their toxic nature induces DAMP production and activates neuropeptide release from sensory neurons. The DC detects these DAMPs and NAMPs, not the toxin itself leading to the risk of off target immune activation. This separation may be beneficial, as it may promote the generation of immune responses to proteins that associate with toxic small molecules. Indeed, there is experimental evidence that separating the detection of antigen and adjuvant can enhance Type 2 immune protection against toxins. Starkl et al.18 found that sensitization to an irrelevant antigen was sufficient to protect from a secondary, lethal exposure to Russell’s Viper venom if the irrelevant antigen was supplied upon primary envenomation. In this way, Type 2 immune responses evolved to recognize antigens that are commonly associated with toxins, but not necessarily toxins themselves.
Separating the detection of adjuvant and antigen to generate Type 2 immunity, however, is problematic in the context of modern allergic diseases. On the one hand, the decision tree for Type 2 immunity is cautious, requiring at least two signals to escalate from DAMP-induced tissue repair to generation of antigen-specific immunity. This provides a level of protection to Type 2 immunity, meaning that only signals strong enough to elicit both DAMPs and NAMPs could generate antigen specificity. On the other hand, this method of recognition can present real danger to the host. In structural recognition, the antigen and adjuvant signals are integrated together in the same cell (DC), meaning that it is highly unlikely for antigen-specific immunity to be generated against a bystander antigen in the near vicinity.76 However, since toxins and allergens elicit their adjuvant activity through DAMP and NAMP release, the detection of the adjuvant and antigen are separated. This increases the likelihood that a bystander antigen is recognized by a DC in the context of DAMP and NAMP signaling, generating antigen-specific immunity against the incorrect antigen. Could this explain polysensitization, the concept that allergic individuals are often sensitized to more than one family of allergens? Or could this explain ‘allergen creep’, or the propensity that over time, sensitized individuals are more likely to acquire antigen-specific immunity to additional allergens compared with non-sensitized individuals?
Toxins, and by extension, allergens, require a much more robust and rapid innate immune response than a pathogen since poisons represent an immediate and potentially fatal threat to the host. Thus, a rapid and sometimes extreme response (anaphylaxis) to toxins is acceptable to the host since the absence of a rapid and robust detoxification method could be fatal. NAMP-induced robust innate immune cell activation for toxins is thus ideal, but in the context of allergic diseases robust Type 2 immune responses to environmental antigens results in persistent, harmful inflammation that in certain cases is fatal.
While the emerging literature described here demonstrates that NAMPs play a complementary role with DAMPs in initiating Type 2 immune responses, it is possible that additional, as yet unknown, signals are required to fully elicit antigen-specific Type 2 immunity. It is likely that upon the exposure to a toxin or allergen, innate immune cells integrate signals from a variety of DAMPs and NAMPs in their microenvironment before antigen-independent pathways escalate to antigen-specific Type 2 immunity. For example, sensory neurons release CGRP in response to pore forming toxins and this inhibits inflammatory cell infiltration and function in the context of bacterial infection with PAMP signaling.21,22,28 In contrast, allergens cause sensory neurons to potently release substance P that promotes Type 2 immune responses by eliciting mast cell degranulation or CD301b+ DC migration to the draining lymph node.26,31 Interestingly, the model allergen papain induced substance P release while completely abolishing homeostatic CGRP release from sensory neurons.26 This suggests that innate immune cells may respond not to a single DAMP or NAMP, but rather sense the patterns of release of a variety of DAMPs and NAMPs before developing Type 2 immune responses.
CONCLUSIONS
The reason that the immune system generates vigorous responses to allergens has long plagued immunologists, especially because there was an incomplete understanding of how allergens were recognized by the innate immune system. By viewing allergen recognition pathways in the context of the toxin hypothesis of allergy, it was hypothesized that signals from additional cellular sources were required to complement DAMPs in inducing Type 2 immune responses. This additional family of signaling molecules was found to be neuropeptides, referred to in this review as NAMPs. Here we propose a decision tree model of functional recognition that guides Type 2 immunity, wherein innate immune cells distinguish when to escalate antigen-independent tissue repair pathways to antigen-specific Type 2 immune responses by integrating DAMP and NAMP signals. In this decision tree model of functional recognition, the generation of an antigen-specific Type 2 immune response requires signals from at least one DAMP and at least one activating NAMP. The decision tree paradigm reduces the risk of generating antigen-specific immunity against host antigens upon sterile wounding and DAMP release. However, it could explain polysensitization and allergen creep, since innate immune cells do not detect the antigen and adjuvant together. By better understanding the mechanisms that underlie the decision tree for Type 2 immunity, novel therapeutics can be developed to target both DAMP and NAMP immune interactions. Prospective therapeutics targeting DAMPs and NAMPs may better modulate allergic diseases than the current standard of care.
ACKNOWLEDGMENTS
We thank members of the Sokol laboratory for their feedback and suggestions. This work was supported by NIH K08AI121421 (to CLS), T32HL116275 (to CHF), DFG PE2864/3-1 (to CP), Massachusetts General Hospital Transformative Scholar Award (to CLS), AAAAI Foundation and Dr Donald YM Leung/JACI Editors Faculty Development Award (to CLS), and Food Allergy Science Initiative (to CLS). Figures were generated using BioRender.
CONFLICT OF INTEREST
CLS is a paid consultant for Bayer and Merck. CLS receives sponsored research support from GSK.
REFERENCES
- 1.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol 2015; 16: 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saijo S, Ikeda S, Yamabe K, et al. Dectin-2 recognition of α-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 2010; 32: 681–691. [DOI] [PubMed] [Google Scholar]
- 3.Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol 2013; 13:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palm NW, Rosenstein RK, Medzhitov R. Allergic host defences. Nature 2012; 484: 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palm NW, Rosenstein RK, Yu S, Schenten DD, Florsheim E, Medzhitov R. Bee venom phospholipase A2 induces a primary type 2 response that is dependent on the receptor ST2 and confers protective immunity. Immunity 2013; 39: 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sokol CL, Barton GM, Farr AG, Medzhitov R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol 2008; 9: 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halim Timotheus YF, Krauß Ramona H, Sun Ann C, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 2012; 36: 451–463. [DOI] [PubMed] [Google Scholar]
- 8.Kouzaki H, Tojima I, Kita H, Shimizu T. Transcription of interleukin-25 and extracellular release of the protein is regulated by allergen proteases in airway epithelial cells. Am J Respir Cell Mol Biol 2013; 49: 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cayrol C, Duval A, Schmitt P, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol 2018; 19: 375–385. [DOI] [PubMed] [Google Scholar]
- 10.Halim TYF, Steer CA, Mathä L, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014; 40: 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Besnard A-G, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B. IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur J Immunol 2011; 41: 1675–1686. [DOI] [PubMed] [Google Scholar]
- 12.Tang H, Cao W, Kasturi SP, et al. The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nat Immunol 2010; 11: 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010; 464: 1367–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN. T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med 2000; 191: 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Profet M The function of allergy: immunological defense against toxins. Q Rev Biol 1991; 66: 23–62. [DOI] [PubMed] [Google Scholar]
- 16.Metz M, Piliponsky AM, Chen C-C, et al. Mast cells can enhance resistance to snake and honeybee venoms. Science 2006; 313: 526. [DOI] [PubMed] [Google Scholar]
- 17.Marichal T, Starkl P, Reber LL, et al. A beneficial role for immunoglobulin E in host defense against honeybee venom. Immunity 2013; 39: 963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Starkl P, Marichal T, Gaudenzio N, et al. IgE antibodies, FcεRIα, and IgE-mediated local anaphylaxis can limit snake venom toxicity. J Allergy Clin Immunol 2016; 137: 246–257. e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohlen CJ, Chesler AT, Sharif-Naeini R, et al. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature 2011; 479: 410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siemens J, Zhou S, Piskorowski R, et al. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 2006; 444: 208–212. [DOI] [PubMed] [Google Scholar]
- 21.Chiu IM, Heesters BA, Ghasemlou N, et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013; 501: 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blake KJ, Baral P, Voisin T, et al. Staphylococcus aureus produces pain through pore-forming toxins and neuronal TRPV1 that is silenced by QX-314. Nat Commun 2018; 9: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vriens J, Nilius B, Vennekens R. Herbal compounds and toxins modulating TRP channels. Curr Neuropharmacol 2008; 6: 79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du Y-R, Xiao Y, Lu Z-M, et al. Melittin activates TRPV1 receptors in primary nociceptive sensory neurons via the phospholipase A2 cascade pathways. Biochem Biophys Res Commun 2011; 408: 32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chahl LA, Kirk EJ. Toxins which produce pain. Pain 1975; 1: 3–49. [DOI] [PubMed] [Google Scholar]
- 26.Perner C, Flayer CH, Zhu X, et al. Substance P release by sensory neurons triggers dendritic cell migration and initiates the type-2 immune response to allergens. Immunity 2020; 53: 1063–1077. e1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bull HA, Hothersall J, Chowdhury N, Cohen J, Dowd PM. Neuropeptides induce release of nitric oxide from human dermal microvascular endothelial cells. J Invest Dermatol 1996; 106: 655–660. [DOI] [PubMed] [Google Scholar]
- 28.Pinho-Ribeiro FA, Baddal B, Haarsma R, et al. Blocking neuronal signaling to immune cells treats Streptococcal invasive infection. Cell 2018; 173: 1083–1097. e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, Kaplan DH. Nociceptive sensory fibers drive interleukin-23 production from cd301b+ dermal dendritic cells and drive protective cutaneous immunity. Immunity 2015; 43: 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maruyama K, Takayama Y, Kondo T, et al. Nociceptors boost the resolution of fungal osteoinflammation via the TRP channel-CGRP-Jdp2 axis. Cell Rep 2017; 19: 2730–2742. [DOI] [PubMed] [Google Scholar]
- 31.Serhan N, Basso L, Sibilano R, et al. House dust mites activate nociceptor-mast cell clusters to drive type 2 skin inflammation. Nat Immunol 2019; 20: 1435–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moriyama S, Brestoff JR, Flamar AL, et al. β2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 2018; 359: 1056–1061. [DOI] [PubMed] [Google Scholar]
- 33.Higginbotham RD. Mast cells and local resistance to Russell’s viper venom. J Immunol 1965; 95: 867–875. [PubMed] [Google Scholar]
- 34.Higginbotham RD, Karnella S. The significance of the mast cell response to bee venom. J Immunol 1971; 106: 233. [PubMed] [Google Scholar]
- 35.Heine H, Förster FJ. Histophysiology of mast cells in skin and other organs. Arch Dermatol Res 1975; 253: 225–228. [DOI] [PubMed] [Google Scholar]
- 36.Johnson AR, Erdös EG. Release of histamine from mast cells by vasoactive peptides. Proc Soc Exp Biol Med 1973; 142: 1252–1256. [DOI] [PubMed] [Google Scholar]
- 37.Rönnberg E, Ghaib A, Ceriol C, et al. Divergent effects of acute and prolonged interleukin 33 exposure on mast cell IgE-mediated functions. Front Immunol 2019; 10: 1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Egan CL, Viglione-Schneck MJ, Walsh LJ, et al. Characterization of unmyelinated axons uniting epidermal and dermal immune cells in primate and murine skin. J Cutan Pathol 1998; 25: 20–29. [DOI] [PubMed] [Google Scholar]
- 39.Matsuda H, Kawakita K, Kiso Y, Nakano T, Kitamura Y. Substance P induces granulocyte infiltration through degranulation of mast cells. J Immunol 1989; 142: 927–931. [PubMed] [Google Scholar]
- 40.Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology 2008; 123: 398–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNeil BD, Pundir P, Meeker S, et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015; 519: 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Azimi E, Reddy VB, Shade K-TC, et al. Dual action of neurokinin-1 antagonists on Mas-related GPCRs. JCI Insight 2016; 1: e89362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Azimi E, Reddy VB, Pereira PJS, Talbot S, Woolf CJ, Lerner EA. Substance P activates Mas-related G protein-coupled receptors to induce itch. J Allergy Clin Immunol 2017; 140: 447–453. e443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tatemoto K, Nozaki Y, Tsuda R, et al. Immunoglobulin E-independent activation of mast cell is mediated by Mrg receptors. Biochem Biophys Res Commun 2006; 349: 1322–1328. [DOI] [PubMed] [Google Scholar]
- 45.Green DP, Limjunyawong N, Gour N, Pundir P, Dong X. A mast-cell-specific receptor mediates neurogenic inflammation and pain. Neuron 2019; 101: 412–420. e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caughey GH, Leidig F, Viro NF, Nadel JA. Substance P and vasoactive intestinal peptide degradation by mast cell tryptase and chymase. J Pharmacol Exp Ther 1988; 244: 133–137. [PubMed] [Google Scholar]
- 47.Meixiong J, Anderson M, Limjunyawong N, et al. Activation of mast-cell-expressed Mas-related G-protein-coupled receptors drives non-histaminergic itch. Immunity 2019; 50: 1163–1171. e1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Artis D, Spits H. The biology of innate lymphoid cells. Nature 2015; 517: 293–301. [DOI] [PubMed] [Google Scholar]
- 49.Barlow JL, Bellosi A, Hardman CS, et al. Innate IL-13–producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol 2012; 129: 191–198. e194. [DOI] [PubMed] [Google Scholar]
- 50.Cardoso V, Chesné J, Ribeiro H, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 2017; 549: 277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klose CSN, Mahlakõiv T, Moeller JB, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 2017; 549: 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wallrapp A, Riesenfeld SJ, Burkett PR, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017; 549: 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nussbaum JC, Van Dyken SJ, von Moltke J, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 2013; 502: 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Talbot S, Abdulnour R-EE, Burkett PR, et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron 2015; 87: 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nagashima H, Mahlakõiv T, Shih HY, et al. Neuropeptide CGRP limits group 2 innate lymphoid cell responses and constrains type 2 inflammation. Immunity 2019; 51: 682–695. e686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wallrapp A, Burkett PR, Riesenfeld SJ, et al. Calcitonin gene-related peptide negatively regulates alarmin-driven type 2 innate lymphoid cell responses. Immunity 2019; 51: 709–723. e706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu H, Ding J, Porter CBM, et al. Transcriptional atlas of intestinal immune cells reveals that neuropeptide α-CGRP modulates group 2 innate lymphoid cell responses. Immunity 2019; 51: 696–708. e699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohl L, Mohaupt M, Czeloth N, et al. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 2004; 21: 279–288. [DOI] [PubMed] [Google Scholar]
- 59.Sokol CL, Camire RB, Jones MC, Luster AD. The chemokine receptor CCR8 promotes the migration of dendritic cells into the lymph node parenchyma to initiate the allergic immune response. Immunity 2018; 49: 449–463. e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kumamoto Y, Linehan M, Weinstein Jason S, Laidlaw Brian J, Craft Joseph E, Iwasaki A. CD301b+ dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity 2013; 39: 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi Youn S, Kageyama R, Eto D, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 2011; 34: 932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goenka R, Barnett LG, Silver JS, et al. Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J Immunol 2011; 187: 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Veres TZ, Rochlitzer S, Shevchenko M, et al. Spatial interactions between dendritic cells and sensory nerves in allergic airway inflammation. Am J Respir Cell Mol Biol 2007; 37: 553–561. [DOI] [PubMed] [Google Scholar]
- 64.Rochlitzer S, Veres TZ, Kühne K, et al. The neuropeptide calcitonin gene-related peptide affects allergic airway inflammation by modulating dendritic cell function. Clin Exp Allergy 2011; 41: 1609–1621. [DOI] [PubMed] [Google Scholar]
- 65.Janelsins BM, Mathers AR, Tkacheva OA, et al. Proinflammatory tachykinins that signal through the neurokinin 1 receptor promote survival of dendritic cells and potent cellular immunity. Blood 2009; 113: 3017–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Janelsins BM, Sumpter TL, Tkacheva OA, et al. Neurokinin-1 receptor agonists bias therapeutic dendritic cells to induce type 1 immunity by licensing host dendritic cells to produce IL-12. Blood 2013; 121: 2923–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buttari B, Profumo E, Domenici G, et al. Neuropeptide Y induces potent migration of human immature dendritic cells and promotes a Th2 polarization. FASEB J 2014; 28: 3038–3049. [DOI] [PubMed] [Google Scholar]
- 68.Oda N, Miyahara N, Taniguchi A, et al. Requirement for neuropeptide Y in the development of type 2 responses and allergen-induced airway hyperresponsiveness and inflammation. Am J Physiol Lung Cell Mol Physiol 2019; 316: L407–L417. [DOI] [PubMed] [Google Scholar]
- 69.Yamamoto Y, Otsuka A, Ishida Y, et al. Pituitary adenylate cyclase-activating polypeptide promotes cutaneous dendritic cell functions in contact hypersensitivity. J Allergy Clin Immunol 2021 10.1016/j.jaci.2021.02.005. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 70.Gowthaman U, Chen JS, Zhang B, et al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science 2019; 365: eaaw6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity 2015; 42: 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoeffel G, Debroas G, Roger A, et al. Sensory neuron-derived TAFA4 promotes macrophage tissue repair functions. Nature 2021; 594: 94–99. [DOI] [PubMed] [Google Scholar]
- 73.Johnson KE, Wulff BC, Oberyszyn TM, Wilgus TA. Ultraviolet light exposure stimulates HMGB1 release by keratinocytes. Arch Dermatol Res 2013; 305: 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sleijffers A, Herreilers M, van Loveren H, Garssen J. Ultraviolet B radiation induces upregulation of calcitonin gene-related peptide levels in human Finn chamber skin samples. J Photochem Photobiol B Biol 2003; 69: 149–152. [DOI] [PubMed] [Google Scholar]
- 75.Niizeki H, Alard P, Streilein JW. Calcitonin gene-related peptide is necessary for ultraviolet B-impaired induction of contact hypersensitivity. J Immunol 1997; 159: 5183–5186. [PubMed] [Google Scholar]
- 76.Chaturvedi A, Pierce SK. How location governs toll-like receptor signaling. Traffic 2009; 10: 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]