

Abstract
Background:
Epidemiological studies have shown Per- and polyfluoroalkyl substances (PFAS) to be associated with diseases of dysregulated lipid and sterol homeostasis such as steatosis and cardiometabolic disorders. However, the majority of mechanistic studies rely on single chemical exposures instead of identifying mechanisms related to the toxicity of PFAS mixtures.
Objectives:
The goal of the current study is to investigate mechanisms linking exposure to a PFAS mixture with alterations in lipid metabolism, including increased circulating cholesterol and bile acids.
Methods:
Male and female wild-type C57BL/6J mice were fed an atherogenic diet used in previous studies of pollutant-accelerated atherosclerosis and exposed to water containing a mixture of 5 PFAS representing legacy, replacement, and emerging subtypes (i.e., PFOA, PFOS, PFNA, PFHxS, and GenX), each at a concentration of 2 mg/L, for 12 weeks. Changes at the transcriptome and metabolome level were determined by RNA-seq and high-resolution mass spectrometry respectively.
Results:
We observed increased circulating cholesterol, sterol metabolites, and bile acids due to PFAS exposure, with some sexual dimorphic effects observed. PFAS exposure increased hepatic injury, demonstrated by increased liver weight, hepatic inflammation, and plasma alanine aminotransferase levels. Females displayed increased lobular and portal inflammation compared to the male PFAS-exposed mice. Hepatic transcriptomics analysis revealed PFAS exposure modulated multiple metabolic pathways, including those related to sterols, bile acids, and acyl carnitines, with multiple sex-specific differences observed. Finally, we show that hepatic and circulating levels of PFOA were increased in exposed females compared to males, but this sexual dimorphism was not the same for other PFAS examined.
Discussion:
Exposure of mice to a mixture of PFAS results in PFAS-mediated modulation of cholesterol levels, possibly through disruption of enterohepatic circulation.
Keywords: PFAS toxicity, Per- and polyfluoroalkyl substances, liver injury, cardiometabolic disease, hyperlipidemia, cholesterol, bile acids
Introduction
Per- and polyfluoroalkyl substances (PFAS) are commonly used synthetic chemicals that have been manufactured and used in numerous industries around the world since the 1940’s (Buck et al. 2011). Many PFAS are widely utilized for their surfactant properties, leading to their incorporation in to countless industrial and consumer products, such as cookware, food packaging, clothing, and carpets as well as in aqueous film forming foams used by firefighters (Clara et al. 2008; Nickerson et al. 2020; Trier et al. 2011). Over 4000 different versions of PFAS have been identified, with around 256 PFAS being commercially manufactured at globally relevant levels (Buck et al. 2021; OECD 2018). Long-chain “legacy” PFAS, have been used in products for decades, with two of the most well-studied being perfluorooctane sulfonic acid (PFOS) and perfluorooctanoic acid (PFOA). Due to the rising concern over the detrimental effects of PFOA and PFOS, these legacy pollutants were phased out 5-10 years ago. However, many legacy PFAS were replaced by alternatives, such as tridecafluorohexane-1-sulfonic (PFHxS), and even more recently by “emerging” PFAS, featuring shorter carbon chains or fluoroether replacements (e.g., hexafluoropropylene oxide dimer acid; GenX) (Brase et al. 2021; Dodds et al. 2020; Lindstrom et al. 2011; Wang et al. 2015).
Although extremely useful in the manufacturing of industrial and consumer products, the strength and stability of PFAS also results in their high resistance to degradation. Consequently, many legacy PFAS (e.g. PFOA and PFOS) have bioaccumulated in the environment, animals, and humans (Calafat et al. 2007; De Silva et al. 2021; Giesy and Kannan 2001). Consequently, mixtures of both legacy as well as alternative and emerging PFAS continue to be ubiquitously found throughout the environment, notably including food and water supplies all over the globe (Domingo and Nadal 2017, 2019). Furthermore, bioaccumulation of PFAS circulating in human blood has been widely reported, including detection in 98% of adult Americans ((CDC) 2005; Calafat et al. 2007). The biological half-lives for legacy PFAS in human serum have been determined to be around 4, 5, and 8.5 years for PFOA, PFOS, and perfluorohexane sulfonate (PFHxS), respectively (Olsen et al. 2007). Although less is known of the halflives of newer PFAS, evidence points to their excretion rates being considerably increased (Gannon et al. 2016).
Given the extensive bioaccumulation and persistence of PFAS, epidemiological studies investigating adverse health effects in humans due to PFAS exposure is a growing area of research, and numerous epidemiological studies have found PFAS exposure to be associated with chronic diseases such as dyslipidemia and cardiometabolic disorders (Blomberg et al. 2021; Darrow et al. 2016; Fragki et al. 2021; Jin et al. 2020; Liu et al. 2018; Winquist and Steenland 2014). Human studies have also established an association between elevated serum PFAS levels and liver injury, such as increased ALT levels, steatosis, and NAFLD severity (Darrow et al. 2016; Gallo et al. 2012; Gleason et al. 2015; Jain and Ducatman 2019a; Jin et al. 2020; Lin et al. 2010). Furthermore, human epidemiological studies focusing on exposure to several model PFAS have reproducibly found PFAS exposure to be associated with hypercholesteremia (Mora et al. 2018; Nelson et al. 2010; Sakr et al. 2007; Steenland et al. 2009). For example, in populations occupationally exposed to PFOA, with an average serum concentration of 1.13ug/ml PFOA, 1 ppm increase in serum PFOA was found to be associated with a 1.06 mg/dl increase in total cholesterol (Sakr et al. 2007). In communities exposed to PFOA via environmental contamination from PFAS manufacturing, people exhibited high mean serum concentrations of 80ng/ml PFOA (Steenland et al. 2009). These communities also demonstrated a positive association between PFAS exposure and total cholesterol, with a predicted increase in 11-12 mg/dl total cholesterol from lowest to highest decile of PFOA. When studying the general U.S. population, data from the 2003-2004 National Health and Nutrition Examination Survey (NHANES) determined a median serum concentration of 3.9 ng/ml PFOA and 21 ng/ml PFOS (Nelson et al. 2010). This study of “background” PFAS levels were found to show trends consistent with the previous human data, including a positive association between PFAS levels and total cholesterol. In fact, multiple studies show the dose-response for cholesterol is not linear, but rather log-linear or asymptotic, which would provide an explanation for rare and seemingly contradictory findings related to enrollment size and statistical power (Frisbee et al. 2010; Li et al. 2020; Steenland et al. 2009).
While there is large body of historical literature linking PFAS exposures to hypocholesterolemia in rodents (Butenhoff et al. 2012; Han et al. 2018; Loveless et al. 2006; Martin et al. 2007; Qazi et al. 2010; Seacat et al. 2002), more recent work has begun to uncover mechanisms linking single PFAS compounds to increased cholesterol levels and cardiovascular disease risk (Fragki et al. 2021; Rebholz et al. 2016; Schlezinger et al. 2020). These more recent studies often utilized models aimed at more closely mirroring human-relevant conditions. For example, a recent study using mice with liver-specific humanized PPARα demonstrated PFOA exposure to be positively associated with increased serum cholesterol levels in males (Schlezinger et al. 2020). Furthermore, there are interspecies differences in lipoprotein metabolism that also contribute to the discrepancies between mouse and human data. For example, cholesteryl ester transfer protein is completely absent in rodents and Apolipoprotein B is cleared much faster from rodent livers compared to humans, resulting in a higher ratio of HDL-C to LDL-C in rodents (Princen et al. 2016). The vast majority of the early rodent PFAS studies were conducted using a standard rodent chow diet containing low amounts of fat and negligible cholesterol. More recent studies have investigated the effects of PFAS in conjunction with diets containing high amounts of fat and/or cholesterol. One such study by Rebholz et al. found that mice fed a western diet and exposed to dietary PFOA resulted in elevated serum cholesterol levels in both male and female mice (Rebholz et al. 2016). Additional variables that may contribute to the apparent discrepancies between animal and human PFAS studies include single chemical exposures versus real world complex mixtures, chronic versus acute exposures, and interactions with other stressors such as diet.
Mechanistic toxicity studies that utilize mixtures of PFAS are understudied and may add additional evidence linking PFAS exposures to adverse health effects. In the present study, male and female C57BL/6J mice were fed an atherogenic diet and exposed to water containing a mixture of 5 environmentally relevant PFAS (PFOA, PFOS, perfluorononanoic acid (PFNA), PFHxS, and GenX) for 12 weeks. Exposure to our PFAS mixture resulted in elevated serum cholesterol levels as early as 5 weeks post exposure, which remained elevated at study conclusion, even with observed strong PPAR alpha activation. We then used unbiased approaches to examine transcriptome and metabolome level changes due to the PFAS mixture focusing on differences between sexes.
Materials and Methods
Animal Experiments
C57BL/6J male and female mice at 7 weeks of age were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and were allowed to acclimate for 1 week upon arrival. Mice were randomly divided into 4 experimental groups: female+vehicle, female+PFAS, male+vehicle, and male+PFAS. After acclimation, animals were switched to the atherogenic diet for 1 week prior to administration of PFAS. The atherogenic diet (Clinton/Cybulsky low fat diet, 0.15% cholesterol) (Research Diets, New Brunswick, NJ, USA) is described in detail in Supplementary Table 5. and continued on the atherogenic diet throughout the study (Clinton/Cybulsky low fat diet, 0.15% cholesterol) (Research Diets, New Brunswick, NJ, USA). This lowfat atherogenic diet has been used in past studies of persistent organic pollutant toxicity (Petriello et al. 2018). The mice were fed ad libitum. The mice were exposed for 12 weeks via their drinking water to vehicle water alone or to a mixture of 5 PFAS: Perfluorooctane sulfonate (PFOS) (Sigma-Aldrich, St. Louis, MO, USA), perfluorooctanoic acid (PFOA) (Sigma-Aldrich, St. Louis, MO, USA), perfluorononanoic acid (PFNA) (Sigma-Aldrich, St. Louis, MO, USA), tridecafluorohexane-1-sulfonic (PFHxS) (J&K Scientific, Beijing, China) and ammonium perfluoro(2-methyl-3-oxahexanoate) (GenX) (Synquest Laboratories, Alachua, FL, USA). Each of the 5 PFAS was dissolved in water at a concentration of 2 mg/L and water bottles were changed weekly by Division of Laboratory Animal Resources staff. An average adult mouse drinks approximately 4 mL of water a day so this dosing strategy will result in approximately 8 micrograms of individual PFAS per day. For an average mouse of 25 grams, this would approximate to about 0.32 mg/kg per day of individual PFAS. This dosing regimen results in PFAS serum concentrations that mirror high occupational exposure to single PFAS, such as those found in workers at PFAS manufacturing plants, where studies have found average serum PFOA concentrations of 1.3-12.9ug/mL (Costa et al. 2009; Sakr et al. 2007). At study conclusion, mice were fasted overnight and humanely euthanized via carbon dioxide inhalation. The animal protocol was approved by the Wayne State University Institutional Animal Care and Use Committee.
Sample Collection
Weekly measurements were recorded of body weight per mouse and water and food intake per cage (i.e., per 5 mice). After week 6 for the females, and week 5 for the males, COVID-19 restrictions prevented the collection of weekly body weights, food and water intake, and blood collection until end of study. Plasma was collected longitudinally via the submandibular vein at the start of weeks 4, 6, and at study completion via cardiac puncture.
Body composition was measured via an EchoMRI Body Composition Analyzer (Echo Medical System, Houston, TX,) for 5 mice of each group at study conclusion. Tissues were harvested and stored at −80 °C prior to analysis or homogenized in TRI reagent (Thermo-Fisher Scientific). The activity of alanine aminotransferase was measured in the serum by using the Infinity ALT (GPT) Liquid Reagent (Thermo-Fisher Scientific, Waltham, MA). Hepatic triglycerides were determined as described previously. Briefly, lipids were extracted from the tissues using the Folch method as previously described (Folch et al. 1957). In brief, the dried lipids were reconstituted in chloroform and loaded onto a TLC plate. The lipids were separated in a hexane/diethyl ether/acetic acid (80/20/2, v/v/v) solution (Touchstone 1995). Spray the TLC plates lightly with primulin (0.005%) dissolved in acetone/water (80/20, v/v), then visualize bands under UV light. Sections of liver were fixed in 10% neutral-buffered formalin and embedded in paraffin. Sections of liver were analyzed via hematoxylin and eosin staining as well as terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and examined by a blinded hepatopathologist.
RNA-Sequencing
Total cellular RNA was extracted from frozen liver samples using the TRIzol reagent (Thermo Fisher Scientific). RNA purity and quantity were assessed via spectrophotometer (NanoDrop 2000, Thermo-Fisher Scientific). RNA Sequencing was performed by the Wayne State University Genome Sciences Core. The mRNA-seq library was prepared using the QuantSeq 3’ mRNA-Seq Library Prep Kit FWD (Lexogen). Libraries were assessed by the High Sensitivity D1000 (HS D1000) ScreenTape Assay (Agilent). Samples were sequenced on the NovaSeq system (illumina). Reads were aligned to the mouse genome (Build mm9) and tabulated for each gene region (Anders et al. 2015; Dobin et al. 2013). Differential gene expression analysis was used to compare transcriptome changes between treatment groups and are reported as the fold change between the specified conditions (sex, exposure, and interaction term; exposure x sex), p-value, the Benjamini-Hochberg (BH)-adjusted p-value, and the individual read counts by gene for each sample replicate. Significantly altered genes (|log fold change| ≥ 2; p-value ≤ 0.05) were used to identify affected pathways. Heatmaps were generated using the pheatmap package in R and refined in Adobe Illustrator. Both log2 read counts depicted in red and log2 fold change using a blue and yellow scale are depicted in the heatmaps.
Liver Metabolomic Profiling
Based on the enriched pathways discovered with RNAseq, we then utilized a non-targeted metabolomics approach performed by Metabolon Inc. (Durham, NC) and described previously (Miller et al. 2015). Here we report primarily on a subset of metabolites related to lipid and sterol homeostasis. Seven mice were chosen at random from each group, and corresponding liver and plasma samples from each mouse were shipped to Metabolon. Briefly, samples were prepared using the automated MicroLab STAR® system (Hamilton Company, Salt Lake City, UT) and proteins were precipitated with methanol. Ultrahigh performance liquid chromatography-tandem mass spectroscopy was carried out using a Waters ACQUITY ultra-performance liquid chromatography (UPLC) and a Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer interfaced with a heated electrospray ionization (HESI-II) source and Orbitrap mass analyzer operated at 35,000 mass resolution. Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities. Statistical analysis of fold changes by two-way ANOVA was used to identify metabolites that differed significantly (p<0.05) between experimental groups. Metabolon reported on 879 (liver) and 709 (plasma) compounds of known identity. Q-values for overall PFAS effect and PFAS-sex interaction are also reported. The q-value is calculated as an estimate of the false discovery rate to take into account the multiple comparisons that occur in metabolomic-based studies.
Validation of hepatic gene expression
Total cellular RNA was extracted from frozen liver samples using the TRIzol reagent (Thermo Fisher Scientific). RNA purity and quantity were assessed via spectrophotometer (NanoDrop 2000, Thermo-Fisher Scientific). cDNA was synthesized from the total RNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Taqman Fast Advanced Master Mix (Thermo Fisher Scientific) was used to perform real-time PCR. The following Taqman probes were used: Cyp7a1 (Mm00484152_m1), Cyp27a1 (Mm00470430_m1), Scl10a1 (Mm00441421_m1), Ephx1 (Mm00468752_m1), Abcc3 (Mm00551550_m1), and GAPDH (Mm99999915_g1) (Thermo Fisher Scientific).
Absolute quantitation of circulating PFAS
Plasma sample extraction was completed using an Agilent EMR-Captiva lipid extraction 96-well plate. The standard curve was extracted on the EMR-Captiva extraction plate using mouse plasma purchased from Sigma Aldrich. The following was done two rows at a time in order to keep the methanol:acetonitrile mixture cold. To begin, 500 μL of cold 15:85 methanol:acetonitrile was added to each well, followed by the PFAS surrogate standard solution to each well in the 96 well extraction plate. A sample size of 50 μL of plasma was added to its specified well, in which it was mixed by plunging up and down with the pipet for complete mixing. The plate was vortexed for 2 minutes at 1100 x g. Using a vacuum apparatus at 2-7 inHg for up to 5 min, the mixture was extracted into the deep well plate. When completely extracted, the plate was brought down to dryness using N2 gas. The plate was capped and stored the 96-well plate in the −80 freezer until ready for analysis. On day of analysis, samples were brought back up to a final volume of 300 μL using 50% methanol with 0.1% acetic acid spike with PFAS internal standard solution. Plate was capped and vortexed for 2 min at 1100 x g.
Samples were analyzed using a Thermo Scientific TSQ Altis with UltiMate 3000 UHPLC. Mobile phases used were A) 20 mM Ammonium acetate in LC-MS Grade Water and B) LC-MS Grade Acetonitrile. Two columns were utilized. A delay column (Thermo Scientific Acclaim RSLC C 18 2.1 x 50 mm, 2.2 μm) was placed after the high-pressure mixer in the system. The analytical column used is a Thermo Scientific Accucore C8 100 x 2.1 mm, 2.6 μm and was held at 40°C. The gradient started at 5% B from 0-1 min, increased to 30% B from 1-3 min, then to 65% B from 3-14 min. The column was rinsed at 98% B from 14-17.5 min and allowed to equilibrate at 5% B from 17.5-20 min. Total run time was 20 minutes. Injection volume was 5 μL. PFAS were detected in negative ion mode using electrospray ionization. Source settings were as follows: negative ion voltage at 1000 V, sheath gas 50 (arbitrary units), aux gas at 12 (arbitrary units), sweep gas at 0.5 (arbitrary units), ion transfer tube temperature at 275°C, and vaporizer temperature at 225°C. Limits of detection for the 5 PFAS were: HFPO-DA; 1 ng/mL, PFOA; 5 ng/mL, L-PFHxS; 0.47 ng/mL, PFNA; 5.0 ng/mL, L-PFOS; 0.48 ng/mL.
All solvents used for LC-MS analysis were LC-MS grade quality. LC-MS grade methanol and acetonitrile, and mouse plasma were purchased from Sigma Aldrich (Burlington, MA, USA). LC-MS grade water, acetic acid (1 mL glass vials), and ammonium acetate were purchased from Fisher Scientific (Waltham, MA, USA). Standards used for analytical determination of PFAS in mouse plasma were Native PFAS linear Primary dilution standard and Method 537 Internal standard mix from Wellington Laboratories, Inc. (Overland Park, KS, USA), and 13C6 PFHxS, 13C9 PFNA, 13C8 PFOS, and 13C8 PFOA were purchased from Cambridge Isotope Laboratories, Inc (Tewksbury, MA, USA).
Data Analysis
Statistical analyses of RNA sequencing gene expression data was performed using R Statistical Software. Statistical analyses of animal measurement data and gene expression data analyzed in Figure 1 were performed using the SigmaPlot version 14 (Systat Software Inc, San Jose, CA, USA). Linear regression analysis was performed using JMP Statistical Software (SAS Institute). Multiple group data were compared using Two Way ANOVA, followed by the Holm-Sidak method post-hoc test for multiple comparisons. Graphed values are expressed as mean ± SEM. A probability value of p ≤ 0.05 was considered statistically significant.
Figure 1. PFAS exposure induces sex-dependent liver injury.
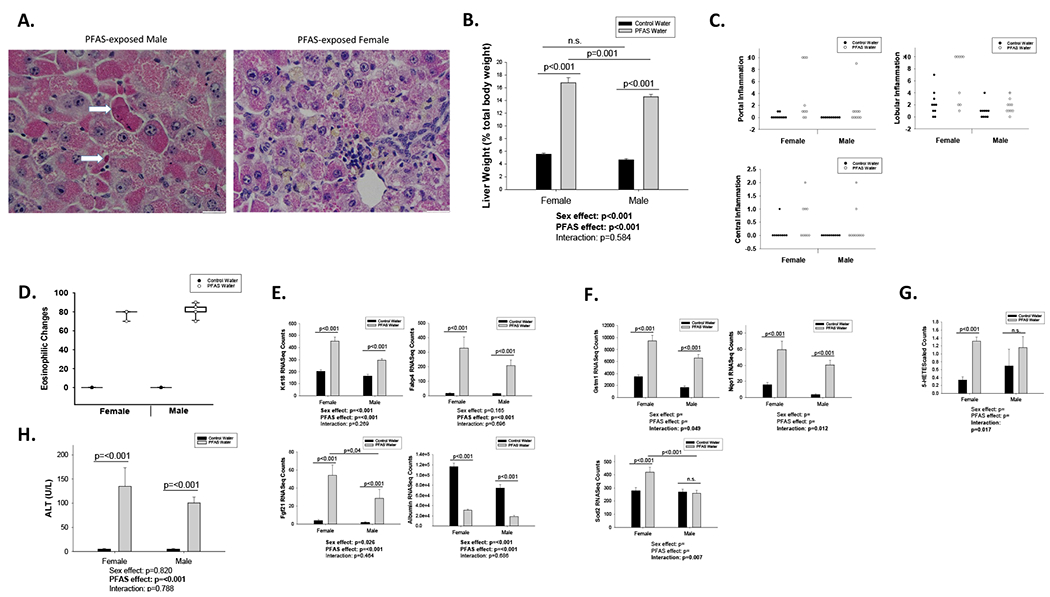
Male and female C57BL/6J mice were exposed to vehicle water or the PFAS mixture for 12 weeks: female+vehicle (n=10), female+PFAS (n=9), male+vehicle (n=10), and male+PFAS (n=10). (A) Male and female liver sections were stained for H&E. Male PFAS mouse: The hepatocytes are swollen with eosinophilic cytoplasm, indicating hepatocyte injury. Scattered acidophil bodies (hepatocyte necrosis) are also present (arrows). Female PFAS mouse: the portal triad demonstrates mild chronic inflammation with lymphocytes. The hepatocytes are swollen with eosinophilic cytoplasm, consistent with injury. In this mouse, hepatocellular and canalicular cholestasis (yellow pigment) is also present. (B) Liver weight given as a percentage of total body weight. (C) Liver sections were analyzed and quantified for number of inflammatory foci or eosinophilic cells per field of view. (D) Percentage of hepatocytes demonstrating eosinophilic cytoplasm (E) Effects of PFAS exposure on hepatic gene expression of liver injury- and toxicant-associated fatty liver disease-related markers. (F) Effects of PFAS exposure on hepatic gene expression of oxidative stress-related markers. (G) Effects of PFAS on 5-Hete metabolite levels. Bars represent mean ± S.E.M. of ten mice (RNA sequencing) or seven mice (Metabolomics Scaled Counts) in each group. Statistical significance for all (p<0.05) was determined by two-way ANOVA analysis and post-hoc comparisons by Holm-sidak method.
Results
3.1. PFAS exposure induces sex-dependent anthropometric changes, oxidative stress, and liver injury
To begin to characterize PFAS mixture toxicity, we measured impacts on body weight and composition as well as glucose and cholesterol. Male mice exposed to PFAS gained ~30% more body weight than male controls during weeks 1-3 of exposure (p=0.039), but this weight gain was not observed in females (p=0.596) (overall exposure effect; p=0.067, interaction; p=0.264) (Supplementary Figure 1D). This was not due to increased food consumption in the mice during these weeks (Supplementary Figure 1C). However, this observation was not retained throughout the duration of the study. Comparing weights at euthanasia to baseline, female control mice body weights increased ~26% and PFAS-exposed females decreased ~18% (p<0.001), while male control mice increased ~37% and PFAS-exposed males decreased ~30% from baseline (p<0.001) (interaction; p<0.001) (Supplementary Figure 1A&D). Additionally, water intake was significantly increased due to PFAS exposure only in female mice during all weeks with available consumption data (i.e., Weeks 1-5; p<0.01), while there was no significant changes in the males (Supplementary Figure 1B). Water intake values were not recorded after Week 4 for males due to COVID-19 restrictions. At euthanasia, body composition was determined by echoMRI. In agreement with the body weight loss, percent body fat was also significantly decreased in the mice exposed to the PFAS mixture for 12 weeks compared to vehicle. The percent body fat of the females decreased by ~45% (p=0.036) after 12 weeks PFAS compared to female controls, while the males decreased by ~72% (p<0.001) compared to male controls (overall exposure effect; p<0.001, interaction; p=0.123) (Supplementary Figure 1E). At the same time, percent lean body mass was not significantly affected in the PFAS-exposed male or female mice compared to their respective control mice, although the overall effect from PFAS exposure regardless of sex was significantly increased (overall exposure effect; p=0.035, interaction; p=0.644) (Supplementary Figure 1F). Fasted glucose levels at study conclusion were significantly decreased by 29% in PFAS-exposed females compared to corresponding vehicle (p<0.001) and decreased by 49% in PFAS-exposed males compared to their corresponding vehicle controls (p<0.001) (interaction; p=0.007) (Supplementary Figure 1G). Total cholesterol levels measured in unfasted PFAS-exposed females at week 4 of the study (3 weeks of PFAS exposure) demonstrated no significant difference compared to vehicle, with cholesterol levels measuring 130 and 126 mg/dl in vehicle and PFAS-exposed female mice, respectively (p=0.68) (Supplementary Figure 1H). At this same timepoint total cholesterol levels in males were also not significantly affected by PFAS exposure, with male total cholesterol levels of 69 mg/dl in male vehicle mice and 78 mg/dl in male PFAS-exposed mice (p=0.29) (Supplementary Figure 1I). However, total cholesterol levels measured in unfasted females after 5 weeks of PFAS exposure demonstrated significantly higher levels in the PFAS-exposed female mice, at 97 mg/dl, compared with vehicle treated female mice, at 54 mg/dl (p<0.001) (Supplementary Figure 1H). Corresponding male samples for this time point were not available.
To characterize the hepatic insult elicited from a mixture of five PFAS (PFOA, PFOS, PFNA, PFHxS, and GenX) in male and female mice after 12 weeks of exposure, liver injury and inflammatory responses were analyzed. Hematoxylin and eosin (H&E) stained liver sections of both male and female mice exposed to either the vehicle water or the PFAS mixture were examined by a hepatopathologist (Figure 1A). Liver weight (given as a percentage of body weight) was significantly increased after 12 weeks of PFAS exposure in both females and males, with females increasing by ~3-fold (p<0.001) and males increasing by ~3.1-fold (p<0.001) over corresponding controls of the same sex (overall exposure effect; p<0.001, interaction; p=0.584) (Figure 1B). Exposure of mice to the PFAS mixture resulted in hepatocyte hypertrophy and injury, as indicated by the enlargement of the hepatocytes, increased eosinophilic cytoplasm, focal cholestasis, and rare acidophil bodies (hepatocyte necrosis) compared to vehicle mice irrespective of sex (Fig.1A&D). In the female PFAS-exposed mice, the portal triad also demonstrates mild chronic inflammation with lymphocytes, as well as hepatocellular and canalicular cholestasis (Fig. 1A). Furthermore, PFAS exposed females appeared to display increased lobular and portal inflammation compared to the male PFAS-exposed mice (Figure 1C). TUNEL staining of liver sections revealed no increase in apoptosis due to PFAS exposure in either sex (results not shown). Finally, circulating plasma levels of alanine aminotransferase (ALT) significantly increased from 5.2U/L to 134.8U/L in PFAS-exposed females compared to vehicle (p<0.001) and 5.2U/L to 100.6U/L in PFAS-exposed males compared to vehicle (p<0.001) (exposure effect; p<0.001, interaction; p=0.788) (Figure 1H).
We then utilized results from RNA-seq analyses to examine transcriptional changes due to PFAS exposure focusing on a subset of genes related to toxicant-associated fatty liver disease (TAFLD) (Baselli et al. 2020; Cave et al. 2016; Coilly et al. 2019; He et al. 2021). Hepatic mRNA levels of keratin 18 (Krt18) were upregulated 2.2-fold in PFAS-exposed females compared to vehicle (p<0.001) and 1.8-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; p<0.001, interaction; p=0.269) (Figure 1E). Hepatic mRNA levels of fatty acid binding protein 4 (Fabp4) were upregulated 19.8-fold in PFAS-exposed females compared to vehicle (p<0.001) and 15-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; p<0.001, interaction; p=0.696) (Figure 1E). Hepatic mRNA levels of fibroblast growth factor 21 (Fgf21) were upregulated 13.5-fold in PFAS-exposed females compared to vehicle (p<0.001) and 15.1-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; p<0.001, interaction; p=0.464) (Figure 1E). Hepatic mRNA levels of albumin (Alb) were downregulated 3.78-fold in PFAS-exposed females compared to vehicle (p<0.001) and 4-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; p<0.001, interaction; p=0.686) (Figure 1E). In addition, results from our RNA-seq analyses revealed transcriptional changes due to PFAS exposure in a subset of genes related to oxidative stress, many of which displayed a significant interaction between sex and PFAS exposure (Figure 1F). For example, hepatic mRNA levels of glutathione s-transferase mu 1 (Gstm1) were upregulated by 2.7-fold in PFAS-exposed females compared to vehicle (p<0.001) and 3.9-fold in PFAS-exposed males compared to vehicle (p<0.001) (interaction; p=0.049) (Figure 1F). Hepatic mRNA levels of NAD(P)H quinone dehydrogenase 1 (Nqo1) were upregulated by 3.8-fold in females compared to vehicle (p<0.001) and 11.3-fold in PFAS-exposed males compared to vehicle (p<0.001) (interaction; p=0.012) (Figure 1F). Hepatic mRNA levels of superoxide dismutase 2 (Sod2) were upregulated by 1.5-fold in PFAS exposed females compared to corresponding vehicle (p<0.001), while there was no significant change in males due to PFAS exposure (interaction; p=0.007) (Figure 1F). Finally, to investigate if the upregulated oxidative stress-induced genes correspond to increased biomarkers of oxidative stress, we examined the hepatic and circulating metabolome and determined that hepatic levels of 5-hydroxyeicosatetraenoic acid (5-HETE) were increased by ~3.9-fold (p<0.001) in PFAS exposed females, but no significant differences in 5-HETE were observed between male groups (interaction; p=0.017) (Figure 1G).
3.2. PFAS exposure modulates hepatic and circulating bile acid and sterol levels
To begin to investigate the observation that PFAS may impact on cholesterol homeostasis and flux, we completed metabolomic analysis on hepatic tissue and plasma for both sexes. Clear differences were observed between PFAS-exposed and vehicle mice in both the liver and plasma, indicating substantial alteration in the metabolic profile in the mice in both liver and plasma after PFAS exposure (Table 1). Except for taurohyocholate, all measured bile acids (primary, conjugated, and secondary) which had significant fold changes were decreased in the livers of PFAS-exposed mice, while all the primary bile acids showed a significant increase in the plasma. The full list of sterols, primary bile acids, and secondary bile acid metabolites measured in liver tissue and plasma are included in Supplementary Tables 1 and 2, respectively. Table 1 shows relative fold change increases or decreases in several common sterols, primary bile acids, and secondary bile acids, both in the liver tissue and in the circulating plasma. At euthanasia, fasted plasma cholesterol levels in PFAS-treated mice were increased 1.62-fold in females (p=0.0017) and 1.33-fold in males (p=0.048) compared to their controls (overall exposure effect; q<0.01, interaction; n.s.) (Table 1). Hepatic cholesterol levels, however, were not significantly altered females (p=0.78) or males (p=0.51) (overall exposure effect; q=0.14, interaction; q=0.43). Hepatic triglyceride levels were also measured using thin-layer chromatography and both sexes exhibited decreased total triglyceride levels (Supplementary Figure 6). Notably, hepatic levels of the sterol metabolite 4-cholesten-3-one was increased by 5.23-fold in PFAS-exposed females compared to vehicle (p<0.001) and 15.41-fold in PFAS-exposed males compared to vehicle (p<0.001) (interaction; q=0.01) (Table 1). Correspondingly, plasma levels of 4-cholesten-3-one increased by 6.06-fold in PFAS-exposed females compared to vehicle (p<0.001) and 16.51-fold in PFAS-exposed males compared to vehicle (p<0.001) (interaction; q<0.01) (Table 1). Hepatic levels of the sterol metabolite cholesterol sulfate were reduced 0.26-fold in PFAS-exposed females compared to vehicle (p<0.001) and 0.28-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; q<0.01, interaction; q=0.48). Plasma levels of the sterol metabolite cholesterol sulfate were not significantly altered in females (p=0.13) or males (p=0.62) (overall exposure effect; q<0.05, interaction; q=0.70). Hepatic levels of the sterol metabolite 7-alpha-hydroxy-3-oxo-4-cholestenoate (7-Hoca) were reduced 0.51-fold (p=0.003) in PFAS-exposed females compared to vehicle, while the effect on males was not significant (p=0.27) (overall exposure effect; q<0.01, interaction; p=0.20). However, plasma levels of 7-Hoca were increased 2.22-fold (p=0.002) in PFAS-exposed males compared to vehicle, while the effect on females was not significant (p=0.72) (overall exposure effect; q<0.01, interaction; p=0.18) (Table 1).
Table 1. PFAS exposure modulates hepatic and circulating sterol and bile acid levels.
Hepatic protein and plasma samples from n=7 mice from each treatment group were analyzed by a non-targeted metabolomics approach using LC-MS by Metabolon Inc. Relative fold change between various treatment groups is given for hepatic sterols and bile acid metabolites. Statistical significance for the interaction and main effects of PFAS exposure and gender was determined by two-way ANOVA analysis. Red indicates a significant relative fold change increase (p<0.05). Blue indicates a significant relative fold change reduction (p<0.05). q-values for overall PFAS treatment and PFAS-sex interaction are displayed in the right columns, with significant (q<0.05) q-values indicated in bold.
| Biochemical Name | Fold of Change PFAS Female vs. PFAS Male | Fold of Change PFAS Female vs. Vehicle Female | Fold of Change PFAS Male vs. Vehicle Male | Overall PFAS Treatment q-value | Sex-PFAS Treatment Interaction q-value |
|---|---|---|---|---|---|
| Hepatic Sterols and Bile Acids | |||||
| Sterols | |||||
| cholesterol | 1.01 | 1.02 | 0.95 | q=0.14 | q=0.43 |
| cholesterol sulfate | 1.29 | 0.26 | 0.28 | q<0.01 | q=0.48 |
| 7-alpha-hydroxy-3-oxo-4-cholestenoate (7-Hoca) | 1.10 | 0.51 | 0.81 | q<0.01 | q=0.20 |
| 4-cholesten-3-one | 1.02 | 5.23 | 15.41 | q<0.01 | q=0.01 |
| Primary Bile Acid Metabolism | |||||
| cholate | 0.82 | 0.37 | 0.49 | q=0.01 | q=0.49 |
| chenodeoxycholate | 0.97 | 0.34 | 0.21 | q<0.01 | q=0.37 |
| beta-muricholate | 1.09 | 0.23 | 0.17 | q<0.01 | q=0.33 |
| alpha-muricholate | 1.60 | 0.12 | 0.12 | q<0.01 | q=0.30 |
| tauro-beta-muricholate | 1.24 | 0.52 | 0.95 | q<0.05 | q=0.33 |
| Secondary Bile Acid Metabolism | |||||
| deoxycholate | 2.30 | 0.17 | 0.16 | q<0.01 | q=0.58 |
| taurodeoxycholate | 0.93 | 0.04 | 0.14 | q<0.01 | q=0.01 |
| ursodeoxycholate | 1.13 | 0.11 | 0.08 | q<0.01 | q=0.32 |
| tauroursodeoxycholate | 1.25 | 0.23 | 0.43 | q<0.01 | q=0.28 |
| taurohyocholate | 0.91 | 1.07 | 2.82 | q<0.01 | q=0.15 |
| taurochenodeoxycholic acid (7 or 27)-sulfate | 1.00 | 0.21 | 0.03 | q<0.01 | q<0.01 |
| Plasma Sterols and Bile Acids | |||||
| Sterols | |||||
| cholesterol | 0.75 | 1.62 | 1.33 | q<0.01 | q=0.60 |
| cholesterol sulfate | 0.80 | 0.86 | 0.96 | q<0.05 | q=0.70 |
| 7-alpha-hydroxy-3-oxo-4-cholestenoate (7-Hoca) | 0.79 | 1.11 | 2.22 | q<0.01 | q=0.18 |
| 4-cholesten-3-one | 0.72 | 6.06 | 16.51 | q<0.01 | q<0.01 |
| Primary Bile Acid Metabolism | |||||
| cholate | 0.93 | 2.86 | 12.43 | q<0.01 | q=0.21 |
| chenodeoxycholate | 1.39 | 2.44 | 2.75 | q<0.01 | q=0.89 |
| beta-muricholate | 1.01 | 2.32 | 6.92 | q<0.01 | q=0.24 |
| Secondary Bile Acid Metabolism | |||||
| deoxycholate | 0.84 | 0.37 | 2.66 | q=0.12 | q<0.01 |
| taurodeoxycholate | 0.95 | 0.14 | 1.27 | q<0.01 | q<0.01 |
| ursodeoxycholate | 0.96 | 0.70 | 3.02 | q<0.05 | q=0.34 |
| tauroursodeoxycholate | 0.83 | 3.36 | 11.48 | q<0.01 | q=0.10 |
| hyocholate | 0.83 | 4.85 | 20.98 | q<0.01 | q=0.14 |
As the main route of cholesterol metabolism and turnover is through conversion to bile acids (di Gregorio et al. 2021), we next report impacts of PFAS exposure on bile acid profiles. Hepatic metabolomics revealed that the majority of primary bile acids were significantly reduced by PFAS exposure in both males and females, indicating possible changes in bile acid synthesis, excretion, export, or reuptake (Table 1). Hepatic levels of chenodeoxycholate were reduced by 0.34-fold in PFAS-exposed females compared to vehicle (p=0.009) and reduced by 0.21-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; q<0.01, interaction; q=0.37). Hepatic levels of β-muricholate were reduced by 0.23-fold in PFAS-exposed females compared to vehicle (p<0.001) and reduced by 0.17-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; p<0.01, interaction; p=0.33). Hepatic levels of α-muricholate were reduced by 0.12-fold in PFAS-exposed females compared to vehicle (p<0.001) and reduced by 0.12-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; q<0.01, interaction; q=0.30). However, hepatic levels of the primary bile acid cholate was not significantly affected in males (p=0.18) or females (p=0.054) (overall exposure effect; q<0.01, interaction; q=0.49).
Contrary to the liver, metabolomic analysis of mouse plasma revealed that circulating levels of the majority of primary bile acids were significantly increased by PFAS exposure in both males and females compared to vehicle. Plasma levels of cholate were elevated 2.86-fold in PFAS-exposed female compared to vehicle (p=0.025), while levels in PFAS-exposed males were elevated 12.43-fold compared to vehicle (p<0.001) (overall exposure effect; q<0.01, interaction; q=0.21). Plasma levels of β-muricholate were increased by 2.32-fold in PFAS-exposed females compared to vehicle (p=0.021) and 6.92-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; q<0.01, interaction; q=0.24). Although the measured levels of secondary bile acids reported in Table 1 represent hepatic tissue levels as opposed to those of the small intestine, some insight can be gained by analysis of the measured levels in the liver and plasma. Hepatic levels of deoxycholate, ursodeoxycholate, and tauroursodeoxycholate were all significantly decreased, ranging from 0.08-fold to 0.43-fold reduced (p<0.01), in PFAS-exposed males or females compared to their respective vehicles (overall exposure effect; q<0.01 for all). Hepatic levels of taurodeoxycholate decreased by 0.04-fold in PFAS-exposed females compared to vehicle (p<0.001) and decreased by 0.14-fold in PFAS-exposed males compared to vehicle (p<0.001) (interaction; q=0.01). Hepatic levels of taurochenodeoxycholic acid (7 or 27)-sulfate decreased by 0.21-fold in PFAS-exposed females compared to vehicle (p=0.37) and decreased by 0.03-fold in PFAS-exposed males compared to vehicle (p=0.12) (interaction; q<0.01). Interestingly, hepatic levels of the secondary bile acid taurohyocholate, produced by taurine conjugation of hyocholic acid, showed a 2.82-fold increase in PFAS-exposed males compared to vehicle (p=0.006) and no significant change in PFAS-exposed females compared to vehicle (p=0.63) (overall exposure effect; q<0.01, interaction; q=0.15). As opposed to the liver, plasma levels of tauroursodeoxycholate were increased 3.36-fold in PFAS-exposed females compared to vehicle (p=0.002) and 11.48-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; q<0.01, interaction; q=0.10). Plasma levels of hyocholate were increased 4.85-fold in PFAS-exposed females compared to vehicle (p=0.003) and 20.98-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; q<0.01, interaction; q=0.14). Notably, levels of taurodeoxycholate were significantly reduced 0.14-fold only in PFAS-exposed females compared to vehicle (p<0.001), while levels in males were not significantly affected (p=0.91) (interaction; q<0.01). Deoxycholate was significantly reduced by 0.37-fold in PFAS-exposed females compared to vehicle (p=0.003), while levels in PFAS-exposed males were increased by 2.66-fold compared to vehicle (p=0.002) (interaction; q<0.01).
3.4. Acyl carnitine levels are dysregulated due to PFAS in sex specific ways
Given the increased markers of oxidative stress we observed in the livers of PFAS-exposed mice, we next examined whether PFAS exposure increased mitochondrial fatty acid oxidation, which has been proposed as a pathway leading to reactive oxygen species production (Zorov et al. 2014). Our metabolomics results revealed relative changes in the levels of ketone bodies and several acyl carnitines in the liver and plasma, as shown in Supplementary Table 3. At euthanasia, relative hepatic levels of acetylcarnitine were not significantly altered in PFAS-exposed females compared to vehicle (p=0.053), but were increased 8.21-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.05). Hepatic levels of hexanoylcarnitine were increased by 1.58-fold in PFAS-exposed females compared to vehicle (p<0.05) and 11.54-fold in PFAS-exposed males compared to vehicle (p<0.001) (interaction; q=0.041). Hepatic levels of octanoylcarnitine were increased by 7.33-fold in PFAS-exposed males compared to vehicle (p<0.001), while females showed no differences (p=0.21) (interaction; q=0.006). Hepatic levels of decanoylcarnitine were increased by 1.38-fold in PFAS-exposed females compared to vehicle (p=0.035) and 7.49-fold in PFAS-exposed males compared to corresponding vehicle (p<0.001) (interaction; q=0.001). Hepatic levels of laurylcarnitine were increased by 1.37-fold in PFAS-exposed females compared to vehicle (p=0.011) and 8.97-fold in PFAS-exposed males compared to vehicle (p<0.001) (interaction; q=0.032). Hepatic levels of myristoylcarnitine were increased by 2.04-fold in PFAS-exposed males compared to vehicle (p=0.004), while females showed no significant differences (p=0.96) (overall exposure effect; q<0.01, interaction; q=0.085). Hepatic levels of stearoylcarnitine were increased by 1.7-fold in PFAS-exposed females compared to vehicle (p=0.001) and 5.43-fold in PFAS-exposed males compared to vehicle (p<0.001) (interaction; q=0.006). Hepatic levels of arachidoylcarnitine were decreased by 0.19-fold in PFAS-exposed females compared to vehicle (p<0.001), while males were not significantly affected (p=0.36) (interaction; q<0.001). Hepatic levels of behenoylcarnitine were decreased by 0.35-fold in PFAS-exposed males compared to vehicle (p<0.001), while females were not significantly affected (p=0.17) (overall exposure effect; q<0.01, interaction; q=0.065). Hepatic levels of lignoceroylcarnitine were decreased by 0.58-fold in PFAS-exposed females compared to vehicle (p<0.001) and 0.50-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; q<0.01, interaction; q=0.35).
Conversely, the relative circulating levels of acylcarnitines were largely decreased in the plasma of PFAS-exposed mice (Supplementary Table 3). Plasma levels of hexanoylcarnitine were decreased by 0.48-fold in PFAS-exposed females compared to vehicle (p=0.023), while males showed no significant differences (p=0.27) (overall exposure effect; q<0.01, interaction; q=0.64). Plasma levels of octanoylcarnitine were decreased by 0.39-fold in PFAS-exposed females compared to vehicle (p=0.002) and 0.51-fold in PFAS-exposed males compared to vehicle (p=0.013) (overall exposure effect; q<0.01, interaction; q=0.78). Plasma levels of laurylcarnitine were decreased by 0.45-fold in PFAS-exposed females compared to vehicle (p=0.007) and 0.57-fold in PFAS-exposed males compared to vehicle (p=0.022) (overall exposure effect; q<0.01, interaction; q=0.87). Plasma levels of myristoylcarnitine were decreased by 0.37-fold in PFAS-exposed females compared to vehicle (p<0.001) and 0.62-fold in PFAS-exposed males compared to vehicle (p=0.040) (overall exposure effect; q<0.01, interaction; q=0.52). Plasma levels of palmitoylcarnitine were decreased by 0.50-fold in PFAS-exposed females compared to vehicle (p<0.001) and 0.67-fold in PFAS-exposed males compared to vehicle (p=0.016) (overall exposure effect; q<0.01, interaction; q=0.66). Plasma levels of stearoylcarnitine were decreased by 0.37-fold in PFAS-exposed females compared to vehicle (p<0.001) and 0.43-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; q<0.01, interaction; q=0.81). Plasma levels of arachidoylcarnitine were decreased by 0.69-fold in PFAS-exposed males compared to vehicle (p=0.005), while females showed no significant differences (p=0.83) (overall exposure effect; q<0.01, interaction; q=0.24).
Hepatic levels of the ketone body 3-hydroxybutyrate (BHBA), which is produced via hepatic fatty acid oxidation and circulate to other organs during gluconeogenesis, was increased in the liver by 3.35-fold in PFAS-exposed females compared to vehicle (p<0.001) and increased by 3.50-fold in PFAS-exposed males compared to vehicle (p<0.001) (overall exposure effect; q<0.01, interaction; q=0.58). In the plasma, relative levels of BHBA were also increased 3.94-fold in PFAS-exposed females compared to vehicle (p=0.008) and increased by 4.62-fold in PFAS-exposed males compared to vehicle (p=0.005) (overall exposure effect; q<0.01, interaction; q=0.91).
3.3. PFAS effects on expression of genes related to metabolism of bile acids and fatty acids
To identify possible transcriptional mechanisms that may explain the observed metabolomics results we performed hepatic RNA sequencing. Gene ontology enrichment analyses were performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) software, and heat maps were created for genes enriched for multiple pathways related to lipid and sterol homeostasis. Figure 2A depicts the heatmap generated using genes identified as part of the sterol or bile acid biosynthetic, metabolic, or catabolic pathways. We examined the following 6 genes related to sterol metabolic processes (GO:0016125~sterol metabolic process): Nsdhl, Apoa4, Fgf1, Ebp, Cebpa, and Vldlr. mRNA levels of Nsdhl, Fgf1, Ebp, and Cebpa all markedly decreased by 0.18-0.42 fold change in the PFAS-exposed mice compared to vehicle (overall exposure effect; p<0.01). The mRNA of Apoa4 markedly increased by 3.83-fold change in the PFAS-exposed mice compared to vehicle (overall exposure effect; p<0.01). mRNA levels of Vldlr demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels increased by 4.57-fold in PFAS-exposed females compared to vehicle (p<0.05) and increased by 57.2-fold in PFAS-exposed males compared to corresponding vehicle treated controls (p<0.05) (interaction; p<0.01) (Figure 2A and Table 2).
Figure 2. PFAS exposure modulates genes related to metabolism of bile acids and fatty acids.
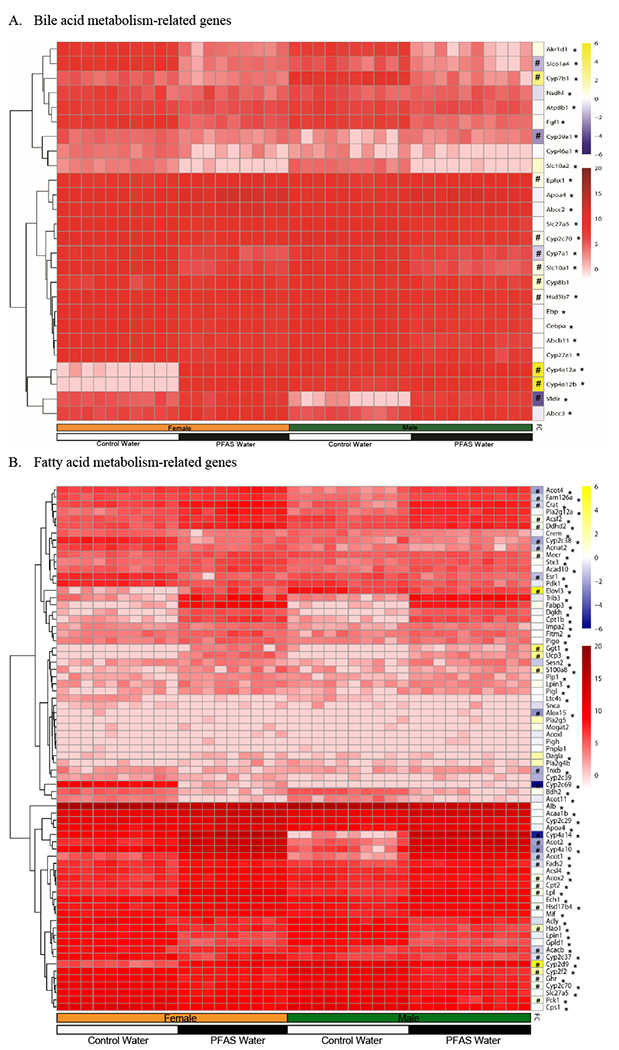
Hepatic mRNA was isolated and analyzed via RNA sequencing. Mice in each treatment group: female+vehicle (n=10), female+PFAS (n=9), male+vehicle (n=10), and male+PFAS (n=10). (A) Heatmap of PFAS-induced effects and sex-related effects on hepatic gene expression pattern for bile acid metabolism-related genes. (B) Heatmap of PFAS and sex-related effects on hepatic gene expression pattern for fatty acid metabolism-related genes. Red = indicates scaled raw counts. Yellow/Blue = indicates fold-change (FC) of the interaction between PFAS exposure and sex. #Statistically significant interaction (p<0.05) between PFAS exposure and sex was determined by Two-Way ANOVA. *Statistically significant effect from PFAS exposure.
Table 2. PFAS exposure modulates genes related to metabolism of bile acids and fatty acids.
Liver samples from each treatment group were analyzed by RNA sequencing. Mice in each treatment group: female+vehicle (n=10), female+PFAS (n=9), male+vehicle (n=10), and male+PFAS (n=10). List of hepatic bile acid metabolism- and fatty acid metabolism-associated genes and their fold change due to overall PFAS exposure. Significant p-values and significant Benjamini-Hochberg (BH)-adjusted p-values are denoted in bold (p<0.05) for overall PFAS exposure and PFAS-sex interaction.
| Gene name | Overall PFAS Exposure Fold Change | Overall PFAS Exposure p-value | Overall PFAS Exposure BH-adjusted p-value | PFAS-Sex Interaction p-value | PFAS-Sex Interaction BH-adjusted p-value |
|---|---|---|---|---|---|
| GO:0016125~sterol metabolic process | |||||
| Nsdhl | 0.41 | p<0.001 | p<0.001 | p=0.003 | p=0.42 |
| Apoa4 | 3.83 | p<0.001 | p<0.001 | p=0.26 | p=0.72 |
| Fgf1 | 0.18 | p<0.001 | p<0.001 | p=0.40 | p=0.91 |
| Ebp | 0.38 | p<0.001 | p<0.001 | p=0.27 | p=0.74 |
| Cebpa | 0.42 | p<0.001 | p<0.001 | p=0.14 | p=0.52 |
| Vldlr | 4.57 | p<0.001 | p<0.001 | p<0.001 | p<0.001 |
| GO:0008206~bile acid metabolic process | |||||
| Cyp46a1 | 0.22 | p<0.001 | p=0.002 | p=1.0 | p=1.0 |
| Cyp7b1 | 0.33 | p<0.001 | p<0.001 | p<0.001 | p<0.001 |
| Slc27a5 | 0.36 | p<0.001 | p<0.001 | p=0.53 | p=1.0 |
| Cyp7a1 | 0.14 | p<0.001 | p<0.001 | p=0.01 | p=0.09 |
| Cyp27a1 | 0.56 | p<0.001 | p<0.001 | p=0.25 | p=0.70 |
| Akr1d1 | 0.06 | p<0.001 | p<0.001 | p=0.14 | p=0.52 |
| Cyp39a1 | 0.40 | p<0.001 | p=0.009 | p<0.001 | p<0.001 |
| Hsd3b7 | 0.79 | p=0.005 | p=0.06 | p=0.01 | p=0.10 |
| Cyp4a12a | 1762.4 | p<0.001 | p<0.001 | p<0.001 | p<0.001 |
| Cyp4a12b | 2454.0 | p<0.001 | p<0.001 | p<0.001 | p<0.001 |
| Cyp8b1 | 1.24 | p=0.15 | p=0.54 | p<0.001 | p=0.005 |
| GO:0015721~bile acid and bile salt transport | |||||
| Atp8b1 | 0.47 | p<0.001 | p<0.001 | p=0.57 | p=1.0 |
| Slco1a4 | 0.05 | p<0.001 | p<0.001 | p<0.001 | p=0.005 |
| Slc10a1 | 0.15 | p<0.001 | p<0.001 | p=0.002 | p=0.03 |
| Slc10a2 | 0.05 | p<0.001 | p<0.001 | p=0.34 | p=0.84 |
| Ephx1 | 6.14 | p<0.001 | p<0.001 | p<0.001 | p<0.001 |
| Abcc2 | 2.18 | p<0.001 | p<0.001 | p=0.12 | p=0.48 |
| Abcc3 | 3.85 | p<0.001 | p<0.001 | p=0.07 | p=0.32 |
| Cyp2c70 | 0.27 | p<0.001 | p<0.001 | p=0.002 | p=0.02 |
| Abcb11 | 0.35 | p<0.001 | p<0.001 | p=0.59 | p=1.0 |
| GO:0019395~fatty acid oxidation | |||||
| Bdh2 | 0.13 | p<0.001 | p<0.001 | p=0.33 | p=0.81 |
| Acacb | 0.46 | p<0.001 | p<0.001 | p=0.001 | p=0.02 |
| Ech1 | 3.66 | p<0.001 | p<0.001 | p=0.23 | p=0.67 |
| Acoxl | 2.32 | p=0.18 | p=0.59 | p=0.30 | p=0.78 |
| Hsd17b4 | 3.57 | p<0.001 | p<0.001 | p=0.01 | p=0.11 |
| Sesn2 | 2.08 | p=0.03 | p=0.18 | p=0.07 | p=0.33 |
| Acox2 | 3.08 | p<0.001 | p<0.001 | p<0.001 | p<0.001 |
| Cpt1b | 26.5 | p<0.001 | p<0.001 | p=0.81 | p=1.0 |
| Acad10 | 2.30 | p<0.001 | p<0.001 | p=0.93 | p=1.0 |
| Acaa1b | 5.80 | p<0.001 | p<0.001 | p=0.96 | p=1.0 |
| GO:0046486~glycerolipid metabolic process | |||||
| Dgkh | 29.2 | p<0.001 | p<0.001 | p=0.79 | p=1.0 |
| Fitm2 | 2.92 | p<0.001 | p<0.001 | p=0.49 | p=1.0 |
| Mogat2 | 2.23 | p=0.20 | p=0.64 | p=0.83 | p=1.0 |
| Lpin3 | 2.44 | p=0.003 | p=0.04 | p=0.47 | p=0.98 |
| Acsl4 | 3.85 | p<0.001 | p<0.001 | p=0.22 | p=0.65 |
| Pnpla1 | 0.42 | p=0.11 | p=0.45 | p=1.0 | p=1.0 |
| Lpl | 18.2 | p<0.001 | p<0.001 | p<0.001 | p<0.001 |
| Tnxb | 0.38 | p=0.01 | p=0.09 | p=0.008 | p=0.07 |
| Gpld1 | 0.12 | p<0.001 | p<0.001 | p=0.79 | p=1.0 |
| Slc27a5 | 0.36 | p<0.001 | p<0.001 | p=0.53 | p=1.0 |
| Cps1 | 0.22 | p<0.001 | p<0.001 | p=0.93 | p=1.0 |
| Pck1 | 0.36 | p<0.001 | p<0.001 | p=0.01 | p=0.10 |
| Lpin1 | 0.20 | p<0.001 | p<0.001 | p=0.25 | p=0.71 |
| Ddhd2 | 4.94 | p<0.001 | p<0.001 | p=0.01 | p=0.09 |
| Dagla | 3.45 | p=0.03 | p=0.21 | p=0.07 | p=0.34 |
| Esr1 | 0.17 | p<0.001 | p<0.001 | p<0.001 | p<0.001 |
We next examined the following 11 genes related to bile acid biosynthetic processes (GO:0008206~bile acid metabolic process): Cyp46a1, Cyp7b1, Slc27a5, Cyp7a1, Cyp27a1, Akr1d1, Cyp39a1, Hsd3b7, Cyp4a12a, Cyp4a12b, and Cyp8b1 (Figure 2 and Table 2). The mRNA of Cyp46a1, Slc27a5, Cyp7a1, Cyp27a1, and Akr1d1 all markedly decreased by 0.06-0.56 fold change in the PFAS-exposed mice compared to vehicle overall (overall exposure effect; p<0.01). mRNA levels of Cyp7b1 demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels decreased by 0.32-fold in PFAS-exposed females compared to vehicle (p<0.05) and decreased by 0.03-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.01). mRNA levels of Cyp39a1 demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels decreased by 0.38-fold in PFAS-exposed females compared to vehicle (p<0.05), while males were not significantly affected (interaction; p<0.01). Furthermore, mRNA levels of Cyp4a12a and Cyp4a12b, both involved in the synthesis of hyocholic acid, also demonstrated a significant interaction between PFAS exposure and sex. The hepatic mRNA levels of Cyp4a12a increased by 2158.5-fold in PFAS-exposed females compared to vehicle (p<0.05) and increased by 1.77-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.01). The hepatic mRNA levels of Cyp4a12b increased by 5897.8-fold in PFAS-exposed females compared to vehicle (p<0.05) and increased by 1.94-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.01). mRNA levels of Cyp8b1 demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels decreased by 0.47-fold in PFAS-exposed males compared to vehicle (p<0.05), while females showed no significant differences (interaction; p<0.01).
We subsequently examined the following 9 genes related to bile acid transport (GO:0015721~bile acid and bile salt transport): Atp8b1, Slco1a4, Slc10a1, Slc10a2, Ephx1, Abcc2, Abcc3, Cyp2c70, and Abcb11 (Figure 2A and Figure 3). mRNA levels of Abcc2 increased by 2.18-fold change and Abcc3 increased by 3.85-fold change in the PFAS-exposed mice compared to vehicle overall (overall exposure effect; p<0.01). Hepatic mRNA levels of Atp8b1, Slc10a2, and Abcb11 all decreased by 0.05-0.47-fold change in PFAS-exposed mice compared to vehicle overall (overall exposure effect; p<0.01). mRNA levels of Slco1a4 demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels decreased by 0.05-fold in PFAS-exposed females compared to vehicle (p<0.05) and decreased by 0.12-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.01). Hepatic mRNA levels levels of Slc10a1 demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels decreased by 0.15-fold in PFAS-exposed females compared to vehicle (p<0.05) and 0.08-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.05). Hepatic mRNA levels of Cyp2c70 demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels decreased by 0.26-fold in PFAS-exposed females compared to vehicle (p<0.05) and 0.16-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.05). mRNA levels of Ephx1 demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels increased by 6.04-fold in PFAS-exposed females compared to vehicle (p<0.05) and 3.22-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.01).
Figure 3. Effects of PFAS exposure and sex on the enterohepatic circulation.
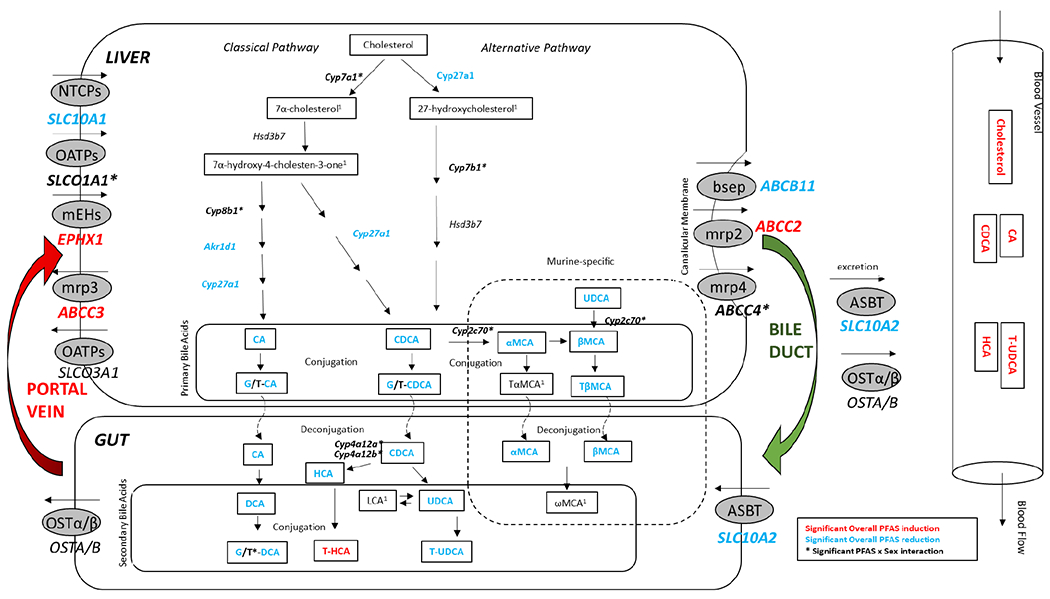
This schematic depicts pathways related to cholesterol and bile acid metabolism and transport. Perturbations in these pathways due to PFAS exposure, or the interaction of PFAS exposure and gender, are indicated by color. Red: Significant PFAS induction. Blue: Significant PFAS reduction. Bolded Black*: Significant PFAS x Sex interaction. Black: No significant effect. 1Not measured. Changes in gene expression patterns analyzed by RNA sequencing are shown in italics. Significance is determined at p-values adjusted for multiple comparisons (q<0.05). Changes in metabolic signature are shown outlined in black. Genes and metabolites shown in the Gut portion of the schematic are inferred from hepatic results.
Another heatmap was generated using genes identified as part of the fatty acid biosynthetic, metabolic, or transport pathways (Figure 2B). Of the many genes included, 10 genes related to fatty acid beta-oxidation processes were analyzed (GO:0019395~fatty acid oxidation), as follows: Bdh2, Acacb, Ech1, Acoxl, Hsd17b4, Sesn2, Acox2, Cpt1b, Acad10, and Acaa1b (Figure 2B). Hepatic mRNA levels of Ech1, Acoxl, Sesn2, Cpt1b, Acad10, Hsd17b4, and Acaa1b all increased by 2.08-26.5-fold change in PFAS-exposed mice compared to vehicle overall (overall exposure effect; p<0.01). The mRNA of Bdh2 decreased by 0.13-fold change in PFAS-exposed mice compared to vehicle overall (overall exposure effect; p<0.01). mRNA levels of Acacb demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels decreased by 0.44-fold in PFAS-exposed females compared to vehicle (p<0.05), while males showed no significant differences (interaction; p<0.05). mRNA levels of Acox2 demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels increased by 2.99-fold in PFAS-exposed females compared to vehicle (p<0.05) and 1.52-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.01).
Another 17 genes related to acylglycerol metabolic processes (GO:0046486~glycerolipid metabolic process) were analyzed, as follows: Dgkh, Fitm2, Mogat2, Lpin3, Acsl4, Pnpla1, Lpl, Tnxb, Gpld1, Slc27a5, Apoa4, Cps1, Pck1, Lpin1, Ddhd2, Dagla, and Esr1 (Figure 2B). Hepatic mRNA of Dgkh, Fitm2, Lpin3, Acsl4, Apoa4, Mogat2, and Ddhd2 were all increased by 2.23-29.2 fold change in PFAS-exposed mice compared to vehicle overall (overall exposure effect; p<0.01). mRNA levels of Gpld1, Slc27a5, Cps1, Pck1, and Lpin1 were all decreased by 0.12-0.36-fold change in PFAS-exposed mice compared to vehicle overall (overall exposure effect; p<0.01). Hepatic mRNA levels of Lpl demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels increased by 17.3-fold in PFAS-exposed females compared to vehicle (p<0.05) and 8.2-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.01). mRNA levels of Esr1 demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels decreased by 0.17-fold in PFAS-exposed females compared to vehicle (p<0.05) and 0.42-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.01) (Table 2 and Figure 2).
Real-time PCR was used for the validation of expression levels of several key genes: Cyp7a1, Cyp27a1, Slc10a1, Ephx1, and Abcc3 (Supplementary Figure 5).
3.5. Increases in circulating and hepatic relative PFAS levels after PFAS exposure and activation of PPARα target genes
Because of the increase in circulating cholesterol levels seen in both our male and female mice exposed to our PFAS mixture, and the effect sex had on the relative cholesterol levels, we next analyzed the relative hepatic and plasma PFAS levels. The metabolomic platform leveraged for our analyses included three of the five PFAS within the mixture. PFAS were observed in all mice, and interestingly, female controls displayed significantly lower PFAS compared to male controls (~0.3-fold) (p<0.05) (Table 3A). As expected, 12 weeks of PFAS exposure led to increased hepatic and circulating PFAS levels in mice, but there were distinct compound-specific differences. Hepatic levels of PFOS increased by 386.3-fold in PFAS-exposed females compared to vehicle (p<0.05) and 127.2-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.05) (Table 3A). Hepatic levels of PFOA increased by 4693.4-fold in PFAS-exposed females compared to vehicle (p<0.05) and 571.9-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.05). Hepatic levels of PFHxS increased by 2035.7-fold in PFAS-exposed females compared to vehicle (p<0.05) and 387.1-fold in PFAS-exposed males compared to vehicle (p<0.05) (overall exposure effect; p<0.05, interaction; 0.05<p<0.10). No significant correlations were observed between hepatic PFAS levels and circulating cholesterol levels (data not shown). As opposed to hepatic levels, circulating plasma levels of PFOS increased by 545.2-fold in PFAS-exposed females compared to vehicle (p<0.05) and 708.9-fold in PFAS-exposed males compared to vehicle (p<0.05) (overall exposure effect; p<0.05, interaction; 0.05<p<0.10) (Table 3A). Plasma levels of PFOA increased by 10883.6-fold in PFAS-exposed females compared to vehicle (p<0.05) and 6936.8-fold in PFAS-exposed males compared to vehicle (p<0.05) (overall exposure effect; p<0.05). Plasma levels of PFHxS increased by 6491-fold in PFAS-exposed females compared to vehicle (p<0.05) and 5414.2-fold in PFAS-exposed males compared to vehicle (p<0.05) (overall exposure effect; p<0.05, interaction; n.s.). Significant positive correlations were observed in the female PFAS-exposed mice between circulating cholesterol levels and plasma levels of PFOA and PFHxS (Supplementary Figure 3). Only plasma levels of PFOA displayed a positive significant correlation with circulating cholesterol levels in the male PFAS-exposed mice. Finally, in remaining available plasma samples we measured absolute concentrations of all 5 PFAS by electrospray ionization mass spectrometry (Table 3B). Although the exposure mixture was made up of equal concentrations of the 5 PFAS, circulating levels differed with PFNA>PFHxS>PFOS>PFOA>GenX. HFPO-DA (GenX) levels were a magnitude lower than the more persistent PFAS with observed concentrations that averaged ~100 ppb. As observed with the untargeted metabolomics, PFOA was significantly higher in exposed females and PFOS was significantly higher in exposed males.
Table 3. Gender specific changes in PFAS levels.
A. Liver and circulating plasma samples from n=7 mice from each treatment group was analyzed by a non-targeted metabolomics approach by Metabolon Inc. Three of the five PFAS included in the PFAS mixture were analyzed using LC-MS. Relative fold change between various treatment groups is given for hepatic and plasma levels of PFOS, PFOA, and PFHxS. Statistical significance for the interaction and main effects of PFAS exposure and gender was determined by two-way ANOVA analysis. B. Absolute plasma concentrations (ug/mL) of 5 PFAS included in the exposure mixture. Plasma was extracted from male and female mice (n=3 per group) exposed to PFOS, PFOA, PFNA, PFHxS, and GenX and levels were quantitated by electrospray ionization mass spectrometry. Red indicates a significant relative fold change increase (p<0.05); Blue indicates a significant relative fold change reduction (p<0.05).
| A. | |||||
|---|---|---|---|---|---|
| Relative PFAS Levels | |||||
| Biochemical Name | Fold Change Vehicle Female vs. Vehicle Male | Fold Change PFAS Female vs. PFAS Male | Fold Change PFAS vs. Vehicle | Overall PFAS Treatment q-value | Sex-PFAS Treatment Interaction q-value |
| Relative Hepatic PFAS Levels | |||||
| Perfluorooctanesulfonate (PFOS) | 0.30 | 0.92 | 188.03 | q<0.01 | q<0.01 |
| Perfluorooctanoate (PFOA) | 0.30 | 2.43 | 1513.94 | q<0.01 | q<0.01 |
| Perfluorohexanesulfonic acid (PFHxS) | 0.25 | 1.30 | 705.46 | q<0.01 | q=0.12 |
| Relative Plasma PFAS Levels | |||||
| Perfluorooctanesulfonate (PFOS) | 1.00 | 0.77 | 627.07 | q<0.01 | q=0.28 |
| Perfluorooctanoate (PFOA) | 1.25 | 1.96 | 10270.75 | q<0.01 | q=0.40 |
| Perfluorohexanesulfonic acid (PFHxS) | 0.80 | 0.96 | 6629.5 | q<0.01 | q=0.88 |
| B. | |||||
| Absolute Plasma PFAS Levels | |||||
| Biochemical Name | PFAS Female Average Concentration (ug/mL) | PFAS male Average Concentration (ug/mL) | Overall Average Concentration (ug/mL) | Fold Change PFAS Female vs. PFAS Male | p-value |
| PFOA | 23.4 +/− 9.0 | 7.3 +/− 1.8 | 15.4 +/− 10.5 | 3.2 | p=0.04 |
| L-PFOS | 13.8 +/− 2.3 | 22.5 +/− 4.1 | 18.1 +/− 5.6 | 0.6 | p=0.03 |
| L-PFHxS | 22.0 +/−8.3 | 25.6 +/− 5.8 | 23.8 +/−6.7 | 0.9 | p=0.57 |
| PFNA | 33.7 +/− 7.6 | 40.9 +/− 10.6 | 37.3 +/− 9.1 | 0.8 | p=0.40 |
| HFPO-DA (GenX) | 0.19 +/− 0.11 | 0.10 +/− 0.04 | 0.14 +/− 0.09 | 1.8 | p=0.27 |
As PFAS are known agonists of PPARα, our RNA sequencing analysis revealed significant alterations in the regulation of numerous PPARα target genes (Table 4). The mRNA of Fgf21 and Hmgcs2 were increased by 13.62- and 2.71-fold change, respectively, in PFAS-exposed mice compared to vehicle overall (overall exposure effect; p<0.05). The mRNA levels of Cd36 demonstrated a significant interaction between PFAS exposure and sex, with hepatic levels increased by 6.58-fold in PFAS-exposed females compared to vehicle (p<0.05) and 9.18-fold in PFAS-exposed males compared to vehicle (p<0.05) (interaction; p<0.02). In addition to being a PPARα target gene, Hmgcs2 is also the first enzyme in the cholesterol biosynthesis pathway from acetoacetyl-CoA. Although Hmgcs2 is upregulated by PFAS exposure, the subsequent cholesterol biosynthesis pathway enzymes, including Hmgcr, Mvk, Pmvk, and Mvd, were shown to be downregulated (Supplementary Table 4).
Table 4. PFAS exposure modulates hepatic PPARα target gene expression levels.
Liver samples from each treatment group were analyzed by RNA sequencing. Mice in each treatment group: female+vehicle (n=10), female+PFAS (n=9), male+vehicle (n=10), and male+PFAS (n=10). List of hepatic Pparα target genes and their fold change due to overall PFAS exposure. Significant p-values and significant Benjamini-Hochberg p-values are denoted in bold (p<0.05) for overall PFAS exposure and PFAS-sex interaction.
| Hepatic PPARα Target Gene | Overall PFAS Exposure Fold Change | Overall PFAS Exposure p-value | Overall PFAS Exposure Benjamini-Hochberg adjusted p-value | PFAS-Sex Interaction p-value | PFAS-Sex Interaction Benjamini-Hochberg-adjusted p-value |
|---|---|---|---|---|---|
| Pparα | 0.78 | p=0.002 | p=0.03 | p=0.64 | p=1.00 |
| Lipid/Hormone Transport | |||||
| Cd36 | 6.71 | p<0.001 | p=0.02 | p=0.001 | p=0.02 |
| Slc27a1 | 17.07 | p<0.001 | p<0.001 | p=0.41 | p=0.92 |
| Acyl-CoA Formation/Hydrolysis/Binding | |||||
| Acsl1 | 2.79 | p<0.001 | p<0.001 | p=0.50 | p=1.00 |
| Fabp1 | 2.18 | p<0.001 | p<0.001 | p=0.01 | p=0.09 |
| Mitochondrial-Oxidation/Oxidative Phosphorylation | |||||
| Acaa2 | 1.54 | p<0.001 | p<0.001 | p=0.09 | p=0.39 |
| Acadm | 3.78 | p<0.001 | p<0.001 | p=0.53 | p=1.00 |
| Cpt1a | 1.67 | p<0.001 | p<0.001 | p=0.48 | p=1.00 |
| Cpt2 | 3.11 | p<0.001 | p<0.001 | p=0.004 | p=0.04 |
| Slc25a20 | 3.96 | p<0.001 | p<0.001 | p=0.28 | p=0.74 |
| Ketogenesis/Ketolysis | |||||
| Fgf21 | 13.62 | p<0.001 | p<0.001 | p=0.65 | p=1.00 |
| Hmgcs2 | 2.71 | p<0.001 | p<0.001 | p=0.24 | p=0.70 |
| Peroxisomal-Oxidation | |||||
| Acox1 | 5.63 | p<0.001 | p<0.001 | p=0.02 | p=0.17 |
| Abcd3 | 2.49 | p<0.001 | p<0.001 | p=0.70 | p=1.00 |
| Acaa1a | 4.22 | p<0.001 | p<0.001 | p=0.36 | p=0.85 |
| Hsd17b4 | 3.57 | p<0.001 | p<0.001 | p=0.01 | p=0.11 |
| Lipogenesis | |||||
| Agpat2 | 0.84 | p=0.026 | p=0.18 | p<0.001 | p=0.001 |
| Fads1 | 0.75 | p=0.014 | p=0.11 | p<0.001 | p=0.013 |
| Srebf1 | 0.62 | p=0.007 | p=0.069 | p=0.39 | p=0.89 |
| Mogat1 | 49.12 | p<0.001 | p<0.001 | p=0.95 | p=1.00 |
| Lipases/Lipid Droplet Proteins | |||||
| Cidec | 197.9 | p<0.001 | p<0.001 | p<0.001 | p=0.004 |
| Pnpla2 | 5.65 | p<0.001 | p<0.001 | p=0.35 | p=0.85 |
| Lipoprotein Uptake and Metabolism | |||||
| Angptl4 | 1.95 | p<0.001 | p=0.002 | p=0.43 | p=0.94 |
| Apoa1 | 0.48 | p<0.001 | p<0.001 | p<0.001 | p=0.005 |
| Apoc3 | 0.26 | p<0.001 | p<0.001 | p=0.007 | p=0.07 |
| Lipc | 0.27 | p<0.001 | p<0.001 | p=0.003 | p=0.03 |
| Pctp | 1.86 | p<0.001 | p<0.001 | p<0.001 | p<0.001 |
| Vldlr | 4.57 | p<0.001 | p<0.001 | p<0.001 | p<0.001 |
| Cholesterol/Bile Transport and Metabolism | |||||
| Abca1 | 1.37 | p<0.001 | p<0.001 | p=0.09 | p=0.39 |
| Abcb4 | 2.49 | p<0.001 | p<0.001 | p=0.24 | p=0.69 |
| Cyp7a1 | 0.14 | p<0.001 | p<0.001 | p=0.01 | p=0.09 |
| Cyp27a1 | 0.56 | p<0.001 | p<0.001 | p=0.25 | p=0.70 |
| Nr1h3/Lxrα | 0.83 | p=0.008 | p=0.08 | p=0.41 | p=0.91 |
| Scarb2 | 1.29 | p<0.001 | p=0.003 | p=0.14 | p=0.52 |
| Slc10a2 | 0.05 | p<0.001 | p<0.001 | p=0.34 | p=0.84 |
Discussion
Epidemiological studies focusing on several model PFAS have found exposure to be associated with increased risk of numerous chronic diseases related to lipid and sterol metabolism, including cardiometabolic disorders and steatosis (Chen et al. 2020; Gaston et al. 2020; Jin et al. 2020; Wang et al. 2017). Studies across species have also reinforced the association between PFAS exposure and diseases such as steatosis and dyslipidemia. Numerous studies in rodents have found PFAS exposure to be associated with liver injury, including hypertrophy, apoptosis, and increased steatosis (Butenhoff et al. 2012; Han et al. 2018; Martin et al. 2007; Nakagawa et al. 2012; Qazi et al. 2010). However, in contrast to the hypercholesteremia demonstrated in human studies, PFAS exposure in animal studies, historically performed at high doses, have often produced decreased circulating cholesterol (i.e. hypocholesteremia) (Butenhoff et al. 2012; Han et al. 2018; Martin et al. 2007; Qazi et al. 2010; Seacat et al. 2002). Various reasons for these discrepancies have been offered, including interspecies genetic differences (Fragki et al. 2021). For example, one well-established PFAS signaling mechanism is through PPARα (Intrasuksri et al. 1998; Rosen et al. 2008; Takacs and Abbott 2007). A study conducted by Nakamura et al. revealed that human PPARα was much less responsive than mouse PPARα to PFAS exposure (Nakamura et al. 2009). This is further supported by findings that PPARα expression in humans is approximately 1/10 that expressed in mice (Palmer et al. 1998). However, a recent study using mice with liver-specific humanized PPARα demonstrated PFOA exposure to be positively associated with increased serum cholesterol levels in males (Schlezinger et al. 2020). It should also be noted, though, that PPARα agonists are widely used in humans clinically to produce cholesterol lowering effects (Staels et al. 1998). As PFAS are known activators of PPARα, the association of serum PFAS levels with increased serum cholesterol creates appears at odds with a PPARα-dependent mode of action. Studies investigating the in vitro activation of human PPARα by PFAS using reporter gene assays showed that PFOA, PFOS, PFHxS and GenX were all capable of PPARα activation. However, while PFOA and GenX demonstrated the strongest effect on PPARα activation, they were only able to produce 60-90% activation compared to the positive control GW7647 even at concentrations 250 times greater (Behr et al. 2020b). Therefore, although PFAS can activate PPARα, PPARα activation may have only a minor role in the observed PFAS-mediated adverse health effects. Several studies have also demonstrated that PPARα is not required for the induction of PFAS-mediated health effects in mice, including lipid accumulation with steatosis (Filgo et al. 2015; Wang et al. 2014).
For studies that focus on cardiometabolic disease, it is important to acknowledge that lipoprotein metabolism differs between species and may also contribute to the discrepancies between mouse and human data. For example, there is complete absence of cholesteryl ester transfer protein in rodents and a faster clearance of Apolipoprotein B from the liver in rodents compared to humans, resulting in a higher ratio of HDL-C to LDL-C in rodents (Princen et al. 2016). Additional explanations for the initial apparent discrepancies between animal and human PFAS studies include single chemical exposures versus real world complex mixtures, chronic versus acute exposures, and interactions with other stressors such as diet. The vast majority of the early rodent PFAS studies were conducted using a standard rodent chow diet, which contains low amounts of fat and negligible cholesterol. Thus, some more recent studies have investigated the effects of PFAS in conjunction with diets containing high amounts of fat and/or cholesterol. However, male mice exposed to PFOS or PFOA in conjunction with a high-fat diet also exhibited reduced serum cholesterol levels (Bijland et al. 2011; Pouwer et al. 2019; Wang et al. 2014). However, a study by Rebholz et al. found that mice fed a western diet and exposed to dietary PFOA resulted in elevated serum cholesterol levels in both male and female mice (Rebholz et al. 2016). While the majority of these studies suggest that dietary fat does not modify PFAS-induced serum cholesterol alterations, further studies are warranted to better understand the interactions of diet and PFAS toxicity.
However, the majority of mechanistic studies rely on single chemical exposures instead of utilizing PFAS mixtures when examining PFAS toxicity. Therefore, we wanted to investigate mechanisms linking exposure to a PFAS mixture, while on an atherogenic diet, with alterations in lipid metabolism. Here, we utilize unbiased transcriptomics and metabolomics to begin to characterize the toxicity of exposure to a PFAS mixture that incorporates the use of an atherogenic rodent diet and results in liver injury and increased circulating cholesterol even with concurrently strong PPARα activation. We examined the effect of a 12-week exposure to a mixture of five PFAS of differing chain length that have been identified in the blood of humans and that represent both legacy and emerging PFAS of concern (PFOA, PFOS, PFNA, PFHxS, and GenX) on liver injury and alterations in lipid metabolism in C57BL/6J wild-type male and female mice. PFAS caused liver injury and inflammation through mechanisms that likely involve increased lipid utilization and oxidative stress, especially in females. In addition, PFAS exposure led to higher levels of circulating cholesterol, bile acids and other measure of fatty acid metabolism implicating critical PFAS-mediated impacts on the enterohepatic circulation and energy metabolism. We believe this is a useful model to study mechanisms linking PFAS exposure to increased risk of cardiometabolic outcomes. It will be important to follow-up these studies using hyperlipidemic mouse models, such as LDL receptor-deficient mice, to determine the effectiveness of this model in helping to understand the progression of cardiometabolic disorders that result in atherosclerosis.
It is well established that PFAS exposure is associated with liver injury, both in humans and in animal models, often manifesting as increased steatosis and TAFLD (Butenhoff et al. 2012; Cave et al. 2016; Jain and Ducatman 2019a; Jin et al. 2020; Nakagawa et al. 2012; Schwingel et al. 2011). Our study reveals that, after 12 weeks of exposure, PFAS results in increased injury, which may be through a mechanism related to oxidative stress and that varies depending on sex. The PFAS-exposed mice exhibited increased plasma levels of ALT (Fig. 1H), increased liver weight (Fig. 1B), an enlargement of hepatocytes and lighter eosin staining, together illustrating a greater degree of hepatic injury and toxicity resulting from PFAS exposure. The genes Fabp4 and Fgf21, known to be associated with the progression of NAFLD, are also upregulated in our PFAS-exposed mice (Coilly et al. 2019; Rusli et al. 2016). Inflammation is also an important mediator of liver injury, and migration of inflammatory cells to the area of injury is an important indicator of level of injury (Del Campo et al. 2018). However, studies conducted over the past 20 years have shown that PFAS exposure generally produces a net anti-inflammatory effect, although the specific effect varies by exposure route, animal model, and dose (DeWitt et al. 2012). Additionally, the specific effect on inflammatory cytokine production can vary depending on the PFAS compound, dose, and the involvement of signaling through PPARα (DeWitt et al. 2012). Histological analysis of the mouse livers reveals that the PFAS-exposed mice exhibited a greater number of inflammatory foci, particularly portal and lobular, as well as increased migration of eosinophils to the liver, which are well known for activation of various proinflammatory and immunoregulatory pathways (Fig. 1C) (Marichal et al. 2017). In addition, the PFAS-exposed mice showed upregulated expression of several oxidative stress markers, including Sod2 and the Nrf2-target genes Gstm1 and Nqo1 (Ma 2013). Similar PFAS-induced oxidative stress has been demonstrated in recent studies, in which exposure to PFOS and PFHxS, in conjunction with a high-fat diet, led to increased hepatic expression of genes involved in oxidative stress (Pfohl et al. 2020). Oxidative stress is further exemplified through the increased hepatic lipid levels of 5-HETE in PFAS-exposed mice, which is an eicosanoid derived from arachidonic acid, shown to be upregulated during oxidative stress and that can be oxidized to 5-oxo-ETE, a potent eosinophil chemoattractant (Powell et al. 1995). The increased 5-HETE may at least in part, account for the increased inflammatory response especially in female mice. It is also possible that the increased 5-HETE in PFAS-exposed mice may be attributed to the elevated biotransformation of arachidonic acid, rather than its β-oxidation, especially given the reduced hepatic arachidoylcarnitine levels observed in female mice exposed to PFAS.
Another indicator of oxidative stress, specifically in the mitochondria, is the high levels of acylcarnitines and ketone bodies measured in the PFAS-exposed mice. PPAR agonists have been found to produce reactive oxygen species in a PPARα-dependent manner (Kim and Yang 2013). As known agonists of PPARα, PFAS have been shown to upregulate mechanisms of fatty acid beta-oxidation both in humans and mice, including higher levels of acylcarnitines related to β-oxidation (Chen et al. 2020; Lu et al. 2019; Wang et al. 2020). Fatty acid β-oxidation, the process by which fatty acids are broken down to generate energy, involves the addition of a CoA group to a fatty acid followed by binding of free carnitine to produce acylcarnitines. Acyl carnitines have an essential role in the carnitine shuttle, which aids in the transportation of fatty acids into the mitochondria for beta oxidation (Knottnerus et al. 2018). These previous studies showing PFAS-induced upregulation of fatty acid β-oxidation pathways supports our data, which demonstrates an increase in the relative hepatic levels acylcarnitines in the PFAS-exposed mice. Furthermore, circulating levels of ketone bodies, which are produced via hepatic fatty acid oxidation and circulate to other organs during gluconeogenesis, were increased in PFAS-exposed mice in both the liver and plasma, indicating that these mice experience a shift in energy metabolism that is not limited to the liver (Lee et al. 2016). However, it is possible another mechanism responsible for the high hepatic levels of acylcarnitines could be PFAS-induced oxidative stress in the mitochondria, leading to increased energy production via acylcarnitines and ketone bodies. Short-, medium-, and long-chain fatty acid metabolism in the liver is increased, while very long-chain fatty acid metabolism is decreased, suggesting a differential regulation of oxidation of long to very long-chain fatty acids when exposed to PFAS. Interestingly, increased levels of corticosterone were observed in both the male and female PFAS-exposed mice, which could also be a reason for the reduction in fat observed in the PFAS-exposed mice (Supplementary Table 1 and Supplementary Figure 1).
Epidemiological studies conducted over the past several decades have revealed that humans exposed to both high and lower “background” PFAS exposure levels in the general U.S. population are positively associated with increased cholesterol (Geiger et al. 2014; Mora et al. 2018; Nelson et al. 2010; Sakr et al. 2007; Steenland et al. 2009). Furthermore, recent human and experimental data report on the emerging role of altered lipid metabolism, especially impaired hepatic cholesterol homeostasis, in the pathogenesis of NASH and NAFLD (Bashiri et al. 2013; Musso et al. 2013; Perla et al. 2017). Hepatic accumulation of free cholesterol has been shown to promote pathogenesis severity of NAFLD and NASH (Caballero et al. 2009; Puri et al. 2007; Van Rooyen et al. 2011; Wouters et al. 2010). In addition, studies aiming to reduce the accumulation of hepatic free cholesterol suggest improved disease severity (Deushi et al. 2007; Park et al. 2010; Yoneda et al. 2010; Zheng et al. 2008). However, the mechanisms involved in PFAS-induced lipid alterations remain poorly understood. Our study revealed that mice exposed to a mixture of PFAS while on a low-fat atherogenic diet results in increased relative plasma cholesterol levels in both PFAS-exposed females and PFAS-exposed males compared to control at 12 weeks (Table 3). However, total cholesterol levels were also measured at 3 and 5 weeks of PFAS exposure (Supplementary Figure 1H&I). Between 3 and 5 weeks, total cholesterol levels in the control mice decreased from 130 mg/dl to 54 mg/dl, while the PFAS-exposed mice only decreased from 126 mg/dl to 97 mg/dl. It is commonly seen in studies utilizing hyperlipidemic diets, that there is an initial increase in cholesterol levels, followed by a decrease and normalization over time (Petriello et al. 2018). These data suggest that the relatively elevated cholesterol levels in the PFAS-exposed mice may result from PFAS exposure preventing a general decrease in cholesterol levels, such as those seen in the control mice. Thus, PFAS could instead be thought of as causing a disruption in cholesterol rebalancing, as opposed to stimulating a general surge in overall cholesterol levels. Additionally, in conjunction with the significantly reduced body fat observed in our PFAS-exposed mice, the changes observed in circulating cholesterol levels could indicate increased reverse transportation of cholesterol under PFAS exposure (Marques et al. 2018). Furthermore, given the lower hepatic bile acid levels in our PFAS-exposed mice, it is possible that the cholesterol to bile acid metabolism is impaired, and the increased Vldlr expression could indicate an increased secretion of cholesterol into the blood. Interestingly, the relative hepatic and plasma levels of 4-cholesten-3-one, a cholestanol precursor, were increased in both PFAS-exposed males and females (Salen et al. 1984). It is possible that, due to PFAS exposure in the presence of dietary cholesterol, that cholesterol is being metabolized to a greater extent through oxysterol production of cholestanol unrelated to bile acid production. Another cholesterol metabolism pathway involves sulfonation of cholesterol to cholesterol sulfate, which is the C3β sulfate ester derived from cholesterol and an endogenous steroid that has roles in stabilizing cell membranes, supporting platelet adhesion, and regulating signal transduction pathways involving blood clotting, fibrinolysis, and epidermal cell adhesion (Strott and Higashi 2003). Our data reveals that hepatic levels of cholesterol sulfate are decreased in PFAS-exposed mice, while plasma levels are unchanged (Table 1). Although the reason for the hepatic decreases are unknown, it could be related to the source of hepatic cholesterol in the mice. Another cholesterol metabolite, 7-alpha-hydroxy-3-oxo-4-cholestenoate (7-Hoca), is a monohydroxy intermediate in the synthesis of primary bile acids that is produced primarily in liver mitochondria or extrahepatically (Axelson et al. 1992; Meaney et al. 2003; Shoda et al. 1993). Our data reveals that hepatic levels of 7-Hoca were decreased in PFAS-exposed females, while plasma levels of 7-Hoca were increased in PFAS-exposed males. These trends mirror those generally seen in our mice for bile acids levels in the liver and plasma, and so may be due to similar mechanisms.
The main route of cholesterol metabolism takes place through its conversion to bile acids, which is the primary method of cholesterol turnover (Luo et al. 2020). Bile acids are synthesized in the liver, secreted into the bile, reabsorbed and modified in the intestines, and circulated back to the liver via the portal vein, making up what is known as the enterohepatic circulation (Gonzalez 2012). Cholesterol is oxidized through multiple pathways into primary bile acids, two of the major primary bile acids being cholic acid and chenodeoxycholic acid in both mice and humans (Russell 2003). These primary bile acids can then be conjugated with taurine or glycine and secreted into the bile. In the intestines, primary bile acids can be deconjugated and converted by bacterial action to secondary bile acids. Previous studies have demonstrated the ability of PFAS exposure to impair bile acid metabolism and transport (Fenton et al. 2021). A recent study in humans has also found several PFAS to be highly associated with metabolites from predominantly lipid pathways (Salihovic et al. 2019). In vitro experiments exposing human cells to various PFAS report strong alterations in the expression of numerous genes involved in cholesterol and bile acid synthesis, metabolism, and transport, notably the strong decrease in Cyp7a1 expression (Behr et al. 2020a; Rowan-Carroll et al. 2021). Studies in mice have suggest that PFAS exposure impairs fatty acid transportation and bile acid synthesis from cholesterol, leading to steatosis via pathways that are PPARα-independent (Das et al. 2017; Filgo et al. 2015; Wang et al. 2014). Our study reveals that mice exposed to the PFAS mixture for 12 weeks have decreased levels of most hepatic primary bile acids in both males and females (Table 1). At the same time, PFAS-exposed mice exhibit increased circulating levels of bile acids, including many secondary bile acids, which may indicate an altered gut microbiome (Ridlon et al. 2014). An important factor affecting these bile acid levels involves downregulation of the bile acid synthesis pathways. High circulating levels of bile acids trigger feedback inhibition of cholesterol synthesis, bile acid synthesis, and bile acid transport (Eloranta and Kullak-Ublick 2008; Slijepcevic et al. 2017). Bile acids are a direct ligand for the nuclear receptor FXR (Ding et al. 2015; Hanafi et al. 2018). When bile acid levels are high, they can bind to and activate FXR, which induces expression of genes such as SHP and FGF-19 in order to protect against bile acid toxicity (Panzitt and Wagner 2021; Slijepcevic et al. 2017). FXR is enriched in the liver and ileum to allow for sensing and regulating bile acids in the enterohepatic circulation (Panzitt and Wagner 2021). Bile acids can bind to and activate FXR, which induces expression of target genes such as the inhibitory factor SHP1 (Goodwin et al. 2000). SHP1 inhibits transcription of CYP7A1 and CYP8B1 induced by liver receptor homolog 1 (LRH-1) (Goodwin et al. 2000). In the intestines, activation of FXR on ileal enterocytes leads to upregulation of fibroblast growth factor (FGF) 15/19, which can also act on hepatocytes to repress bile acid synthesis (Kliewer and Mangelsdorf 2015). Our RNASeq data supports activation of these negative feedback pathways, as Cyp7a1 and Cyp27a1 are both downregulated due to PFAS exposure, as well as other members of their downstream bile acid metabolism pathways (Figure 3). These pathways are likely activated by the high levels of circulating bile acids. It has also been shown that treatment with FXR agonists can downregulate cholesterol synthesis genes in mice (Gardès et al. 2013). In Supplementary Table 4, PFAS downregulation of cholesterol biosynthesis gene expression are reported. Only Hmgcs2, a direct PPARα target gene, was not downregulated. PFAS are also known agonists of PPARα (Wang et al. 2020; Wolf et al. 2012), which is widely known downregulate cholesterol synthesis genes when activated (Knight et al. 2005; König et al. 2007; Li and Chiang 2009). It is possible that the initial exposure to PFAS leads to downregulation of endogenous cholesterol synthesis. However, although endogenous cholesterol synthesis is repressed, our data shows that relative hepatic cholesterol levels do not significantly change in the PFAS-exposed mice. In our model, mice always had access to cholesterol via their diet throughout their exposure to PFAS. Thus, these results may better reflect the adaptation to changes in cholesterol source from endogenous to dietary. However, although hepatic cholesterol levels did not change significantly with PFAS exposure, hepatic bile acid levels saw an overall reduction, indicating a reduction in the cholesterol to bile acid conversion. Further studies are needed to determine the contributions from different sources of cholesterol to total cholesterol levels.
The observed decrease in the hepatic levels of bile acids is also Supplementary by the decrease in hepatic expression of transporters responsible for bile acid uptake, including Na+-taurocholate cotransporting polypeptide (NTCP), OATPs, and mEH (Kosters and Karpen 2008). NTCP, encoded by the gene Slc10a1, is a high-affinity Na+-dependent transporter located solely on the basolateral membrane of hepatocytes and known to transport bile salts, sulfated steroid hormones, and thyroid hormones as well (Döring et al. 2012). NTCP accounts for ~90% of bile acid uptake and its major bile substrates include all the main taurine- and glycine-conjugated bile acids (Hagenbuch and Meier 1994; Hata et al. 2003; Platte et al. 1996), while unconjugated bile acids are only moderate substrates (Hata et al. 2003; Mita et al. 2006). Our data demonstrates a significant downregulation of Slc10a1 is PFAS-exposed mice of both sexes. Another major uptake transporter for bile acids into the liver is the organic anion transporting polypeptide 1 (OATP1). OATP1, encoded by the gene Slco1a1, is a Na+-independent transporter located on the basolateral membrane of hepatocytes and most efficiently transports unconjugated or sulfated bile acids (Hata et al. 2003; Meier et al. 1997). Our data demonstrates a significant downregulation of Slco1a1 in PFAS-exposed mice. A recent study examining the effects of PFAS administration in the presence of a high-fat diet reported a similar reduction in expression of hepatic PFAS uptake transporters, including OATPs (Pfohl et al. 2021). However, unlike the NTCP and OATP1 transporters, the microsomal epoxide hydrolase (mEH) expression was shown to be upregulated in PFAS-exposed mice. mEH, encoded by the gene Ephx1 and located on the basolateral membrane of hepatocytes (Von Dippe et al. 1993; von Dippe et al. 2003), was originally associated with the conversion of epoxides to trans-dihydrodiols (Oesch et al. 1971). Although much about the preferred substrates for mEH remain unknown, it has been reported that mEH can transport cholate and taurocholate (von Dippe et al. 1996). The increase in Ephx1 could be due to its transcriptional regulation by GATA-4 (Zhu et al. 2004) or the HNF-4α/CAR/RXR/PSF complex (Peng et al. 2013), as opposed to the FXR pathway that regulates Ntcp and Oatps (Peng et al. 2015). In addition, mEH is a bifunctional enzyme that can be induced by Nrf2 via the antioxidant response pathway and plays an important role in oxidative stress (Ma et al. 2006). It is possible that the increased expression of Ephx1 seen in our data is due to mEH’s role in detoxification and oxidative stress. It has also been previously shown that some of these bile acid transporters, such as NTCP, OATPs, and ASBT to transport PFAS as well as bile acids (Zhao et al. 2015; Zhao et al. 2017), which could compete for binding with bile acids and contribute to the decreased hepatic bile acid levels observed.
The low hepatic levels of bile acids and high plasma levels of bile acids can also be regulated by transporters responsible for bile acid efflux from the liver and bile acid influx to the intestines. The hepatic bile acid efflux transporters include the bile salt export pump (Bsep), encoded by the gene Abcb11. Bsep is an ATP-dependent transporter located on the hepatocyte canalicular membrane and is the major efflux transporter in the liver, efficiently transporting conjugated bile acids and driving the flow of bile (Byrne et al. 2002; Stieger et al. 2007). Previous reporter gene studies have demonstrated the ability of FXR to directly bind the promoter region and positively control transcription of Abcb11 (Ananthanarayanan et al. 2001; Matsubara et al. 2013; Plass et al. 2002). Studies involving human patients have revealed that Bsep deficiencies are associated with intrahepatic cholestasis (Stieger et al. 2007; Strautnieks et al. 1998). Lacking or having a defective form of the Bsep protein results in an impaired ability to release bile from liver cells, leading to a build-up of bile in the liver and diminished liver function (Sohail et al. 2021). Surprisingly, our data reveal that Abcb11 is downregulated ~0.3-fold in PFAS-exposed mice compared to vehicle mice. Therefore, the low hepatic bile acid levels in the PFAS-exposed mice may occur via a mechanism unrelated to Bsep efflux. It is also possible Bsep is being downregulated by inflammatory injury, as has been previously described (Wagner et al. 2010), or that Bsep is being directly regulated by PFAS by an unknown mechanism. In the intestine, bile acid reabsorption is largely controlled by the apical sodium dependent transporter (ASBT), which is located on the apical membrane of ileal enterocytes and encoded by the gene Slc10a2 (Balakrishnan and Polli 2006). Studies have shown FXR to negatively regulate ASBT expression in the intestines of mice (Chen et al. 2003; Matsubara et al. 2013). This is in agreement with our results, which show Slc10a2 to be downregulated and high levels of circulating bile acids. Patients with ASBT deficiency have been shown to display substantial bile acid malabsorption (Oelkers et al. 1997). However, it is possible that hepatic gene expression levels do not reflect intestinal expression levels. Further analysis examining the intestine specifically are required in the future. Although our metabolomics data show a decrease in the majority of secondary bile acids in PFAS-exposed mice, some specific exceptions do appear. For example, relative hepatic levels of taurohyocholic acid significantly increased in PFAS-exposed males compared to vehicle, while levels in female mice did not change with PFAS exposure, which could possibly be due to sex-dependent differences in hepatic uptake of secondary bile acids or differences in hepatic taurine content.
Throughout this study, sex has been a significant variable impacting the direction and degree of our results. For example, our study reveals that exposure to PFAS for 12 weeks results in increased injury, inflammation, and oxidative stress that varies depending on sex. Interestingly, the increase in liver weight and upregulation of TALFD-related gene Fgf21 was significantly increased in PFAS-exposed females compared to PFAS-exposed males, indicating a higher propensity for females to develop PFAS-induced hepatic toxicity (Fig. 1B&E). PFAS-exposed females also displayed greater lobular and portal inflammation (Fig. 1C). In addition, the upregulation of markers of oxidative stress Gstm1, Nqo1, and Sod2 by PFAS exposure, as well as hepatic levels of the metabolite 5-HETE, all demonstrated a significant interaction between PFAS exposure and sex, with greater expression in females (Fig.1 F&G). It is possible that the greater propensity for liver injury in PFAS-exposed females compared to males derives from the greater activation of these inflammatory and oxidative stress pathways in females. Furthermore, although the females did exhibit increasing water intake compared to males, only hepatic and plasma levels of PFOA were found to be increased in PFAS-exposed females compared to males (Supplementary Figure 1B and Table 3). A unique aspect of our work is the ability to see relative serum and hepatic PFAS levels in exposed male and female mice. Although there are examples of serum PFAS concentrations after PFAS exposure (Butenhoff et al. 2012; Loveless et al. 2006), sex-stratified direct liver-to-blood comparisons are not readily presented. It is possible that the sex-dependent progression of liver injury derives from toxicity induced specifically by PFOA, which was found at greater levels in both the plasma and livers of PFAS-exposed females (Table 3). Similar to our results, a recent study in which female and male mice exposed to PFOA and an American diet exhibited increased serum cholesterol in male mice only (Schlezinger et al. 2020). When examining human epidemiological data, in the general U.S. adult population, total cholesterol levels are higher in females than males, although this can largely be attributed to a higher HDL fraction in females than males and varied depending on age and race (Carroll et al. 2012; Jain and Ducatman 2019b; Swiger et al. 2014). This trend can be seen in data from NHANES surveys conducted from 1959-2008 ((CDC) 2009). In terms of age, younger women are largely protected from cardiovascular disease due to estrogens (Meyer and Barton 2016), while after menopause the risk factors for cardiovascular disease increase(Barrett-Connor 2013). In one study of primary care patients either at-risk for cardiovascular disease or having cardiovascular disease, women had higher total cholesterol and higher LDL-C than, but also received less lipid-lowering treatment (Rachamin et al. 2021). In a study of PFAS-exposed populations, it was found that men had higher total cholesterol than women, although the trend varied with age (Sakr et al. 2007).
Our data also reveals significant interaction between PFAS exposure and sex for transcriptional regulation of the bile acid influx transporters Slc10a1, Slco1a1, and Ephx1. Slc10a1 had greater fold change repression in PFAS-exposed males compared to females. Slco1a1, which is highly expressed in male vehicle mice and expressed at low levels in female vehicle mice, was almost completely repressed in both sexes. Lastly, Ephx1 was upregulated to a significantly greater degree in PFAS-exposed females compared males. Previous studies have also observed sex-specific differences in the expression and abundance of bile acid transporters, such as the male-predominant Oatp1a1 and Oatp1a3 in mouse liver (Cheng et al. 2005; Gotoh et al. 2002; Isern et al. 2001; Melià et al. 1998). These differences have been shown to be the result, at least in part, of the abundance of sex-specific hormones, such as androgen (Cheng et al. 2006). These effects contribute to the gender differences observed in bile acid synthesis and bile acid pool composition (Li-Hawkins et al. 2002; Phelps et al. 2019). For example, males have been shown to have a much larger bile acid pools than females (Bennion et al. 1978; Turley et al. 1998; Xiang et al. 2012). Furthermore, gender differences in bile acid transporters could be having an effect on hepatic PFAS levels. Our data reveals male vehicle mice have significantly higher levels of PFOA, PFOS, and PFHxS in the liver compared to female vehicle mice. This could be due to the greater male expression of transporters such as OATP1a1, which also contribute to PFAS uptake into the liver. Our data also showed greater relative concentrations of hepatic PFOA in PFAS-exposed females compared to PFAS-exposed males, which could be due to gender-specific transporter abundance along with the specific affinity of PFOA for certain transporters. It is not likely that these differences were due to the observed increased intake of water in PFAS-exposed females, as the other PFAS measured did not follow the same sexually dimorphic pattern. Certain limitations are present throughout this study. The dosing regimen used mirrors blood levels of high occupational exposure to single PFAS, such as those found in workers at PFOA manufacturing plants, where studies have found average serum PFOA concentrations of 1.3-12.9ug/mL (Costa et al. 2009; Sakr et al. 2007). The average serum PFOA concentrations in our mice was 7.3 ug/mL in males and 23.4 ug/mL in females (Table 3B). The concentrations of other PFAS used in our model are unknown in these studies. Using NHANES as a guide for the PFAS serum levels in the general population, it is known that our measured concentrations of PFOA were 2000-6000-fold higher than the general population (Nelson et al. 2010). While average serum GenX concentrations in our mice were 0.10 ug/mL in males and 0.19 ug/mL in females (Table 3B), previous studies measuring serum levels of GenX in a population exposure to PFAS-contaminated water found GenX not to be detected above their limit of detection (2ng/mL) (Kotlarz et al. 2020). Our study is not attempting to mimic water exposures, but rather blood concentrations similar to highly exposed individuals. Investigation of the effects of lower PFAS dosage regimens will be valuable in future studies. The critical next steps would be to utilize a mixture of PFAS that more closely matches the observed water toxicity levels and that reflect the measured concentrations found in human blood. The body weights, food intake, and water intake were recorded weekly until around week 5-6, when facilities were shut down due to COVID-19 for all except emergency personnel. Mice continued to receive treatment via their water source during this time, but detailed monitoring could not be performed and additional blood and tissue samples could not be collected until study completion. Because of this, it is unknown what the pattern of weight gain or weight loss looks like over time, especially the definitive time the PFAS-exposed mice began losing weight. Future studies that utilize the same diet but with exposures of lower concentration are critical to limit the impact of weight loss as an additional variable. Similarly, although the PFAS-exposed female mice displaying a trend in increased water intake, the trend beyond week 5 is unknown. In addition, after 12 weeks of exposure to PFAS, the female mice displayed signs of wasting that could by itself obscure the specific pathways being mediated by PFAS exposure. Furthermore, the estrogenic influences on PFAS toxicity were not examined in this study, but are an interesting variable for further study. Moreover, although our study focused largely on PFAS exposure’s effect on sterols, there are numerous different species in the lipid milieu that could provide the setting for follow-up studies in the future (Quehenberger and Dennis 2011). Our untargeted metabolomics approach did not include measurement for metabolites involved in the cholesterol to bile acid metabolism pathway, which would help to highlight specific PFAS-induced modulations of these pathways leading to the overall effects. Furthermore, levels of cholesterol subsets, such as HDL/LDL, were not measured. Additionally, although we exposed mice to 5 distinct PFAS chemicals, this should still be considered a simple mixture model. Future studies investigating full factorial combinatorial mixtures may help tease out synergistic effects of these PFAS pollutants.
Studies of the effects of PFAS on human health have and continue to demonstrate an association with increased risk of chronic diseases such as steatosis and cardiometabolic disorders. Here, we begin to characterize the toxicity of exposure to a PFAS mixture in combination with an atherogenic diet that results in a pattern of liver injury and increased circulating cholesterol similar to that seen in humans. In our model, exposure to the PFAS mixture resulted in accelerated liver injury, especially in females, that likely occur through mechanisms involving increased oxidative stress and altered lipid metabolism. Furthermore, mice exposed to the PFAS mixture displayed increased circulating cholesterol levels and alterations in levels of bile acid and fatty acid metabolism that indicate impaired functions of the enterohepatic circulation and energy metabolism. Our results suggest that the positive associations observed in epidemiological studies between PFAS and circulating cholesterol may be more related to impacts on cholesterol metabolism and transport than on endogenous cholesterol formation.
Supplementary Material
Highlights.
PFAS exposures are associated with dysregulated lipid and sterol homeostasis
A mixture of 5 PFAS increased cholesterol and bile acids in mice
Female mice displayed increased liver injury
Circulating PFOA were increased in exposed females compared to males.
Acknowledgments:
We thank Katherine Gurdziel at the Wayne State University Genomics Core for support related to RNA sequencing and generation of heatmaps.
Funding:
This research was supported in part by the National Institute of Environmental Health Sciences [P30ES020957, R00ES028734] and the National Institute of Diabetes and Digestive and Kidney [R01DK106540] at the National Institutes of Health and the Office of the Vice President for Research at Wayne State University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that there are no competing financial interests.
References
- Center for Disease Control and Prevention (CDC) CfDCaP. 2005. Nhanes 2003–2004 public data general release file documentation.
- (CDC) CfDCaP. 2009. Quickstats: Average total cholesterol level among men and women aged 20--74 years -- national health and nutrition examination survey, united states, 1959--1962 to 2007—2008. Morbidity and mortality weekly reports. [Google Scholar]
- Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. 2001. Human bile salt export pump promoter is transactivated by the farnesoid x receptor/bile acid receptor. J Biol Chem 276:28857–28865. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. Htseq--a python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelson M, Shoda J, Sjövall J, Toll A, Wikvall K. 1992. Cholesterol is converted to 7 alpha-hydroxy-3-oxo-4-cholestenoic acid in liver mitochondria. Evidence for a mitochondrial sterol 7 alpha-hydroxylase. J Biol Chem 267:1701–1704. [PubMed] [Google Scholar]
- Balakrishnan A, Polli JE. 2006. Apical sodium dependent bile acid transporter (asbt, slc10a2): A potential prodrug target. Mol Pharm 3:223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett-Connor E 2013. Menopause, atherosclerosis, and coronary artery disease. Curr Opin Pharmacol 13:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselli GA, Dongiovanni P, Rametta R, Meroni M, Pelusi S, Maggioni M, et al. 2020. Liver transcriptomics highlights interleukin-32 as novel nafld-related cytokine and candidate biomarker. Gut 69:1855–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashiri A, Tavallaee G, Li L, Ng DS. 2013. Emerging role of cellular cholesterol in the pathogenesis of nonalcoholic fatty liver disease. Curr Opin Lipidol 24:275–276. [DOI] [PubMed] [Google Scholar]
- Behr AC, Kwiatkowski A, Ståhlman M, Schmidt FF, Luckert C, Braeuning A, et al. 2020a. Impairment of bile acid metabolism by perfluorooctanoic acid (pfoa) and perfluorooctanesulfonic acid (pfos) in human heparg hepatoma cells. Arch Toxicol 94:1673–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr AC, Plinsch C, Braeuning A, Buhrke T. 2020b. Activation of human nuclear receptors by perfluoroalkylated substances (pfas). Toxicol In Vitro 62:104700. [DOI] [PubMed] [Google Scholar]
- Bennion LJ, Drobny E, Knowler WC, Ginsberg RL, Garnick MB, Adler RD, et al. 1978. Sex differences in the size of bile acid pools. Metabolism 27:961–969. [DOI] [PubMed] [Google Scholar]
- Bijland S, Rensen PC, Pieterman EJ, Maas AC, van der Hoorn JW, van Erk MJ, et al. 2011. Perfluoroalkyl sulfonates cause alkyl chain length-dependent hepatic steatosis and hypolipidemia mainly by impairing lipoprotein production in apoe*3-leiden cetp mice. Toxicol Sci 123:290–303. [DOI] [PubMed] [Google Scholar]
- Blomberg AJ, Shih YH, Messerlian C, Jørgensen LH, Weihe P, Grandjean P. 2021. Early-life associations between per- and polyfluoroalkyl substances and serum lipids in a longitudinal birth cohort. Environ Res 200:111400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brase RA, Mullin EJ, Spink DC. 2021. Legacy and emerging per- and polyfluoroalkyl substances: Analytical techniques, environmental fate, and health effects. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck RC, Franklin J, Berger U, Conder JM, Cousins IT, de Voogt P, et al. 2011. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integr Environ Assess Manag 7:513–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck RC, Korzeniowski SH, Laganis E, Adamsky F. 2021. Identification and classification of commercially relevant per- and poly-fluoroalkyl substances (pfas). Integr Environ Assess Manag. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butenhoff JL, Chang SC, Olsen GW, Thomford PJ. 2012. Chronic dietary toxicity and carcinogenicity study with potassium perfluorooctanesulfonate in sprague dawley rats. Toxicology 293:1–15. [DOI] [PubMed] [Google Scholar]
- Byrne JA, Strautnieks SS, Mieli-Vergani G, Higgins CF, Linton KJ, Thompson RJ. 2002. The human bile salt export pump: Characterization of substrate specificity and identification of inhibitors. Gastroenterology 123:1649–1658. [DOI] [PubMed] [Google Scholar]
- Caballero F, Fernández A, De Lacy AM, Fernández-Checa JC, Caballería J, García-Ruiz C. 2009. Enhanced free cholesterol, srebp-2 and star expression in human nash. J Hepatol 50:789–796. [DOI] [PubMed] [Google Scholar]
- Calafat AM, Wong LY, Kuklenyik Z, Reidy JA, Needham LL. 2007. Polyfluoroalkyl chemicals in the u.S. Population: Data from the national health and nutrition examination survey (nhanes) 2003-2004 and comparisons with nhanes 1999-2000. Environ Health Perspect 115:1596–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll MD, Kit BK, Lacher DA. 2012. Total and high-density lipoprotein cholesterol in adults: National health and nutrition examination survey, 2009-2010. NCHS Data Brief:1–8. [PubMed] [Google Scholar]
- Cave MC, Clair HB, Hardesty JE, Falkner KC, Feng W, Clark BJ, et al. 2016. Nuclear receptors and nonalcoholic fatty liver disease. Biochim Biophys Acta 1859:1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Ma L, Dawson PA, Sinal CJ, Sehayek E, Gonzalez FJ, et al. 2003. Liver receptor homologue-1 mediates species- and cell line-specific bile acid-dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J Biol Chem 278:19909–19916. [DOI] [PubMed] [Google Scholar]
- Chen Z, Yang T, Walker DI, Thomas DC, Qiu C, Chatzi L, et al. 2020. Dysregulated lipid and fatty acid metabolism link perfluoroalkyl substances exposure and impaired glucose metabolism in young adults. Environ Int 145:106091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Maher J, Chen C, Klaassen CD. 2005. Tissue distribution and ontogeny of mouse organic anion transporting polypeptides (oatps). Drug Metab Dispos 33:1062–1073. [DOI] [PubMed] [Google Scholar]
- Cheng X, Maher J, Lu H, Klaassen CD. 2006. Endocrine regulation of gender-divergent mouse organic anion-transporting polypeptide (oatp) expression. Mol Pharmacol 70:1291–1297. [DOI] [PubMed] [Google Scholar]
- Clara M, Scheffknecht C, Scharf S, Weiss S, Gans O. 2008. Emissions of perfluorinated alkylated substances (pfas) from point sources--identification of relevant branches. Water Sci Technol 58:59–66. [DOI] [PubMed] [Google Scholar]
- Coilly A, Desterke C, Guettier C, Samuel D, Chiappini F. 2019. Fabp4 and mmp9 levels identified as predictive factors for poor prognosis in patients with nonalcoholic fatty liver using data mining approaches and gene expression analysis. Sci Rep 9:19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa G, Sartori S, Consonni D. 2009. Thirty years of medical surveillance in perfluooctanoic acid production workers. J Occup Environ Med 51:364–372. [DOI] [PubMed] [Google Scholar]
- Darrow LA, Groth AC, Winquist A, Shin HM, Bartell SM, Steenland K. 2016. Modeled perfluorooctanoic acid (pfoa) exposure and liver function in a mid-ohio valley community. Environ Health Perspect 124:1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das KP, Wood CR, Lin MT, Starkov AA, Lau C, Wallace KB, et al. 2017. Perfluoroalkyl acids-induced liver steatosis: Effects on genes controlling lipid homeostasis. Toxicology 378:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva AO, Armitage JM, Bruton TA, Dassuncao C, Heiger-Bernays W, Hu XC, et al. 2021. Pfas exposure pathways for humans and wildlife: A synthesis of current knowledge and key gaps in understanding. Environ Toxicol Chem 40:631–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Campo JA, Gallego P, Grande L. 2018. Role of inflammatory response in liver diseases: Therapeutic strategies. World J Hepatol 10:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deushi M, Nomura M, Kawakami A, Haraguchi M, Ito M, Okazaki M, et al. 2007. Ezetimibe improves liver steatosis and insulin resistance in obese rat model of metabolic syndrome. FEBS Lett 581:5664–5670. [DOI] [PubMed] [Google Scholar]
- DeWitt JC, Peden-Adams MM, Keller JM, Germolec DR. 2012. Immunotoxicity of perfluorinated compounds: Recent developments. Toxicol Pathol 40:300–311. [DOI] [PubMed] [Google Scholar]
- di Gregorio MC, Cautela J, Galantini L. 2021. Physiology and physical chemistry of bile acids. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Yang L, Wang Z, Huang W. 2015. Bile acid nuclear receptor fxr and digestive system diseases. Acta Pharm Sin B 5:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. 2013. Star: Ultrafast universal rna-seq aligner. Bioinformatics 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds JN, Hopkins ZR, Knappe DRU, Baker ES. 2020. Rapid characterization of per- and polyfluoroalkyl substances (pfas) by ion mobility spectrometry-mass spectrometry (ims-ms). Anal Chem 92:4427–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo JL, Nadal M. 2017. Per- and polyfluoroalkyl substances (pfass) in food and human dietary intake: A review of the recent scientific literature. J Agric Food Chem 65:533–543. [DOI] [PubMed] [Google Scholar]
- Domingo JL, Nadal M. 2019. Human exposure to per- and polyfluoroalkyl substances (pfas) through drinking water: A review of the recent scientific literature. Environ Res 177:108648. [DOI] [PubMed] [Google Scholar]
- Döring B, Lütteke T, Geyer J, Petzinger E. 2012. The slc10 carrier family: Transport functions and molecular structure. Curr Top Membr 70:105–168. [DOI] [PubMed] [Google Scholar]
- Eloranta JJ, Kullak-Ublick GA. 2008. The role of fxr in disorders of bile acid homeostasis. Physiology (Bethesda) 23:286–295. [DOI] [PubMed] [Google Scholar]
- Fenton SE, Ducatman A, Boobis A, DeWitt JC, Lau C, Ng C, et al. 2021. Per- and polyfluoroalkyl substance toxicity and human health review: Current state of knowledge and strategies for informing future research. Environ Toxicol Chem 40:606–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filgo AJ, Quist EM, Hoenerhoff MJ, Brix AE, Kissling GE, Fenton SE. 2015. Perfluorooctanoic acid (pfoa)-induced liver lesions in two strains of mice following developmental exposures: Pparα is not required. Toxicol Pathol 43:558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. 1957. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226:497–509. [PubMed] [Google Scholar]
- Fragki S, Dirven H, Fletcher T, Grasl-Kraupp B, Bjerve Gützkow K, Hoogenboom R, et al. 2021. Systemic pfos and pfoa exposure and disturbed lipid homeostasis in humans: What do we know and what not? Crit Rev Toxicol 51:141–164. [DOI] [PubMed] [Google Scholar]
- Frisbee SJ, Shankar A, Knox SS, Steenland K, Savitz DA, Fletcher T, et al. 2010. Perfluorooctanoic acid, perfluorooctanesulfonate, and serum lipids in children and adolescents: Results from the c8 health project. Arch Pediatr Adolesc Med 164:860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Leonardi G, Genser B, Lopez-Espinosa MJ, Frisbee SJ, Karlsson L, et al. 2012. Serum perfluorooctanoate (pfoa) and perfluorooctane sulfonate (pfos) concentrations and liver function biomarkers in a population with elevated pfoa exposure. Environ Health Perspect 120:655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon SA, Fasano WJ, Mawn MP, Nabb DL, Buck RC, Buxton LW, et al. 2016. Absorption, distribution, metabolism, excretion, and kinetics of 2,3,3,3-tetrafluoro-2-(heptafluoropropoxy)propanoic acid ammonium salt following a single dose in rat, mouse, and cynomolgus monkey. Toxicology 340:1–9. [DOI] [PubMed] [Google Scholar]
- Gardès C, Chaput E, Staempfli A, Blum D, Richter H, Benson GM. 2013. Differential regulation of bile acid and cholesterol metabolism by the farnesoid x receptor in ldlr −/− mice versus hamsters. J Lipid Res 54:1283–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston SA, Birnbaum LS, Jackson CL. 2020. Synthetic chemicals and cardiometabolic health across the life course among vulnerable populations: A review of the literature from 2018 to 2019. Curr Environ Health Rep 7:30–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger SD, Xiao J, Ducatman A, Frisbee S, Innes K, Shankar A. 2014. The association between pfoa, pfos and serum lipid levels in adolescents. Chemosphere 98:78–83. [DOI] [PubMed] [Google Scholar]
- Giesy JP, Kannan K. 2001. Global distribution of perfluorooctane sulfonate in wildlife. Environ Sci Technol 35:1339–1342. [DOI] [PubMed] [Google Scholar]
- Gleason JA, Post GB, Fagliano JA. 2015. Associations of perfluorinated chemical serum concentrations and biomarkers of liver function and uric acid in the us population (nhanes), 2007-2010. Environ Res 136:8–14. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ. 2012. Nuclear receptor control of enterohepatic circulation. Compr Physiol 2:2811–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. 2000. A regulatory cascade of the nuclear receptors fxr, shp-1, and lrh-1 represses bile acid biosynthesis. Mol Cell 6:517–526. [DOI] [PubMed] [Google Scholar]
- Gotoh Y, Kato Y, Stieger B, Meier PJ, Sugiyama Y. 2002. Gender difference in the oatpl-mediated tubular reabsorption of estradiol 17beta-d-glucuronide in rats. Am J Physiol Endocrinol Metab 282:E1245–1254. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Meier PJ. 1994. Molecular cloning, chromosomal localization, and functional characterization of a human liver na+/bile acid cotransporter. J Clin Invest 93:1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han R, Hu M, Zhong Q, Wan C, Liu L, Li F, et al. 2018. Perfluorooctane sulphonate induces oxidative hepatic damage via mitochondria-dependent and nf-κb/tnf-α-mediated pathway. Chemosphere 191:1056–1064. [DOI] [PubMed] [Google Scholar]
- Hanafi NI, Mohamed AS, Sheikh Abdul Kadir SH, Othman MHD. 2018. Overview of bile acids signaling and perspective on the signal of ursodeoxycholic acid, the most hydrophilic bile acid, in the heart. Biomolecules 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata S, Wang P, Eftychiou N, Ananthanarayanan M, Batta A, Salen G, et al. 2003. Substrate specificities of rat oatp1 and ntcp: Implications for hepatic organic anion uptake. Am J Physiol Gastrointest Liver Physiol 285:G829–839. [DOI] [PubMed] [Google Scholar]
- He W, Huang C, Zhang X, Wang D, Chen Y, Zhao Y, et al. 2021. Identification of transcriptomic signatures and crucial pathways involved in non-alcoholic steatohepatitis. Endocrine. [DOI] [PubMed] [Google Scholar]
- Intrasuksri U, Rangwala SM, O’Brien M, Noonan DJ, Feller DR. 1998. Mechanisms of peroxisome proliferation by perfluorooctanoic acid and endogenous fatty acids. Gen Pharmacol 31:187–197. [DOI] [PubMed] [Google Scholar]
- Isern J, Hagenbuch B, Stieger B, Meier PJ, Meseguer A. 2001. Functional analysis and androgen-regulated expression of mouse organic anion transporting polypeptide 1 (oatp1) in the kidney. Biochim Biophys Acta 1518:73–78. [DOI] [PubMed] [Google Scholar]
- Jain RB, Ducatman A. 2019a. Selective associations of recent low concentrations of perfluoroalkyl substances with liver function biomarkers: Nhanes 2011 to 2014 data on us adults aged ≥20 years. J Occup Environ Med 61:293–302. [DOI] [PubMed] [Google Scholar]
- Jain RB, Ducatman A. 2019b. Roles of gender and obesity in defining correlations between perfluoroalkyl substances and lipid/lipoproteins. Sci Total Environ 653:74–81. [DOI] [PubMed] [Google Scholar]
- Jin R, McConnell R, Catherine C, Xu S, Walker Dl, Stratakis N, et al. 2020. Perfluoroalkyl substances and severity of nonalcoholic fatty liver in children: An untargeted metabolomics approach. Environ Int 134:105220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Yang Q. 2013. Peroxisome-proliferator-activated receptors regulate redox signaling in the cardiovascular system. World J Cardiol 5:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Mangelsdorf DJ. 2015. Bile acids as hormones: The fxr-fgf15/19 pathway. Dig Dis 33:327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight BL, Hebbachi A, Hauton D, Brown AM, Wiggins D, Patel DD, et al. 2005. A role for pparalpha in the control of srebp activity and lipid synthesis in the liver. Biochem J 389:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knottnerus SJG, Bleeker JC, Wüst RCI, Ferdinandusse S, L IJ, Wijburg FA, et al. 2018. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord 19:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König B, Koch A, Spielmann J, Hilgenfeld C, Stangl Gl, Eder K. 2007. Activation of pparalpha lowers synthesis and concentration of cholesterol by reduction of nuclear srebp-2. Biochem Pharmacol 73:574–585. [DOI] [PubMed] [Google Scholar]
- Kosters A, Karpen SJ. 2008. Bile acid transporters in health and disease. Xenobiotica 38:1043–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlarz N, McCord J, Collier D, Lea CS, Strynar M, Lindstrom AB, et al. 2020. Measurement of novel, drinking water-associated pfas in blood from adults and children in Wilmington, north Carolina. Environ Health Perspect 128:77005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Choi J, Scafidi S, Wolfgang MJ. 2016. Hepatic fatty acid oxidation restrains systemic catabolism during starvation. Cell Rep 16:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Hawkins J, Gåfvels M, Olin M, Lund EG, Andersson U, Schuster G, et al. 2002. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J Clin Invest 110:1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chiang JY. 2009. Regulation of bile acid and cholesterol metabolism by ppars. PPAR Res 2009:501739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Barregard L, Xu Y, Scott K, Pineda D, Lindh CH, et al. 2020. Associations between perfluoroalkyl substances and serum lipids in a Swedish adult population with contaminated drinking water. Environ Health 19:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Lin LY, Chiang CK, Wang WJ, Su YN, Hung KY, et al. 2010. Investigation of the associations between low-dose serum perfluorinated chemicals and liver enzymes in us adults. Am J Gastroenterol 105:1354–1363. [DOI] [PubMed] [Google Scholar]
- Lindstrom AB, Strynar MJ, Libelo EL. 2011. Polyfluorinated compounds: Past, present, and future. Environ Sci Technol 45:7954–7961. [DOI] [PubMed] [Google Scholar]
- Liu HS, Wen LL, Chu PL, Lin CY. 2018. Association among total serum isomers of perfluorinated chemicals, glucose homeostasis, lipid profiles, serum protein and metabolic syndrome in adults: Nhanes, 2013-2014. Environ Pollut 232:73–79. [DOI] [PubMed] [Google Scholar]
- Loveless SE, Finlay C, Everds NE, Frame SR, Gillies PJ, O’Connor JC, et al. 2006. Comparative responses of rats and mice exposed to linear/branched, linear, or branched ammonium perfluorooctanoate (apfo). Toxicology 220:203–217. [DOI] [PubMed] [Google Scholar]
- Lu Y, Gao K, Li X, Tang Z, Xiang L, Zhao H, et al. 2019. Mass spectrometry-based metabolomics reveals occupational exposure to per- and polyfluoroalkyl substances relates to oxidative stress, fatty acid β-oxidation disorder, and kidney injury in a manufactory in china. Environ Sci Technol 53:9800–9809. [DOI] [PubMed] [Google Scholar]
- Luo J, Yang H, Song BL. 2020. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol 21:225–245. [DOI] [PubMed] [Google Scholar]
- Ma Q, Battelli L, Hubbs AF. 2006. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor nrf2. Am J Pathol 168:1960–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q 2013. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marichal T, Mesnil C, Bureau F. 2017. Homeostatic eosinophils: Characteristics and functions. Front Med (Lausanne) 4:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques LR, Diniz TA, Antunes BM, Rossi FE, Caperuto EC, Lira FS, et al. 2018. Reverse cholesterol transport: Molecular mechanisms and the non-medical approach to enhance hdl cholesterol. Front Physiol 9:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MT, Brennan RJ, Hu W, Ayanoglu E, Lau C, Ren H, et al. 2007. Toxicogenomic study of triazole fungicides and perfluoroalkyl acids in rat livers predicts toxicity and categorizes chemicals based on mechanisms of toxicity. Toxicol Sci 97:595–613. [DOI] [PubMed] [Google Scholar]
- Matsubara T, Li F, Gonzalez FJ. 2013. Fxr signaling in the enterohepatic system. Mol Cell Endocrinol 368:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney S, Babiker A, Lütjohann D, Diczfalusy U, Axelson M, Björkhem I. 2003. On the origin of the cholestenoic acids in human circulation. Steroids 68:595–601. [DOI] [PubMed] [Google Scholar]
- Meier PJ, Eckhardt U, Schroeder A, Hagenbuch B, Stieger B. 1997. Substrate specificity of sinusoidal bile acid and organic anion uptake systems in rat and human liver. Hepatology 26:1667–1677. [DOI] [PubMed] [Google Scholar]
- Melià MJ, Bofill N, Hubank M, Meseguer A. 1998. Identification of androgen-regulated genes in mouse kidney by representational difference analysis and random arbitrarily primed polymerase chain reaction. Endocrinology 139:688–695. [DOI] [PubMed] [Google Scholar]
- Meyer MR, Barton M. 2016. Estrogens and coronary artery disease: New clinical perspectives. Adv Pharmacol 77:307–360. [DOI] [PubMed] [Google Scholar]
- Miller MJ, Kennedy AD, Eckhart AD, Burrage LC, Wulff JE, Miller LA, et al. 2015. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J Inherit Metab Dis 38:1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita S, Suzuki H, Akita H, Hayashi H, Onuki R, Hofmann AF, et al. 2006. Vectorial transport of unconjugated and conjugated bile salts by monolayers of llc-pk1 cells doubly transfected with human ntcp and bsep or with rat ntcp and bsep. Am J Physiol Gastrointest Liver Physiol 290:G550–556. [DOI] [PubMed] [Google Scholar]
- Mora AM, Fleisch AF, Rifas-Shiman SL, Woo Baidal JA, Pardo L, Webster TF, et al. 2018. Early life exposure to per- and polyfluoroalkyl substances and mid-childhood lipid and alanine aminotransferase levels. Environ Int 111:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso G, Gambino R, Cassader M. 2013. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog Lipid Res 52:175–191. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Ramdhan DH, Tanaka N, Naito H, Tamada H, Ito Y, et al. 2012. Modulation of ammonium perfluorooctanoate-induced hepatic damage by genetically different pparα in mice. Arch Toxicol 86:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Ito Y, Yanagiba Y, Ramdhan DH, Kono Y, Naito H, et al. 2009. Microgram-order ammonium perfluorooctanoate may activate mouse peroxisome proliferator-activated receptor alpha, but not human pparalpha. Toxicology 265:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JW, Hatch EE, Webster TF. 2010. Exposure to polyfluoroalkyl chemicals and cholesterol, body weight, and insulin resistance in the general u.S. Population. Environ Health Perspect 118:197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson A, Maizel AC, Kulkarni PR, Adamson DT, Kornuc JJ, Higgins CP. 2020. Enhanced extraction of afff-associated pfass from source zone soils. Environ Sci Technol. [DOI] [PubMed] [Google Scholar]
- OECD. 2018. (the organisation for economic co-operation and development)toward a new comprehensive global database of per- and polyfluoroalkyl substances (pfass): Summary report on updating the oecd 2007 list of per and polyfluoroalkyl substances (pfass).
- Oelkers P, Kirby LC, Heubi JE, Dawson PA. 1997. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (slc10a2). J Clin Invest 99:1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesch F, Jerina DM, Daly JW. 1971. Substrate specificity of hepatic epoxide hydrase in microsomes and in a purified preparation: Evidence for homologous enzymes. Arch Biochem Biophys 144:253–261. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, et al. 2007. Half-life of serum elimination of perfluorooctanesulfonate,perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ Health Perspect 115:1298–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CN, Hsu MH, Griffin KJ, Raucy JL, Johnson EF. 1998. Peroxisome proliferator activated receptor-alpha expression in human liver. Mol Pharmacol 53:14–22. [PubMed] [Google Scholar]
- Panzitt K, Wagner M. 2021. Fxr in liver physiology: Multiple faces to regulate liver metabolism. Biochim Biophys Acta Mol Basis Dis:166133. [DOI] [PubMed] [Google Scholar]
- Park H, Hasegawa G, Shima T, Fukui M, Nakamura N, Yamaguchi K, et al. 2010. The fatty acid composition of plasma cholesteryl esters and estimated desaturase activities in patients with nonalcoholic fatty liver disease and the effect of long-term ezetimibe therapy on these levels. Clin Chim Acta 411:1735–1740. [DOI] [PubMed] [Google Scholar]
- Peng H, Zhu QS, Zhong S, Levy D. 2013. Transcription of the human microsomal epoxide hydrolase gene (ephx1) is regulated by an hnf-4α/car/rxr/psf complex. Biochim Biophys Acta 1829:1000–1009. [DOI] [PubMed] [Google Scholar]
- Peng H, Zhu QS, Zhong S, Levy D. 2015. Transcription of the human microsomal epoxide hydrolase gene (ephx1) is regulated by parp-1 and histone h1.2. Association with sodium-dependent bile acid transport. PLoS One 10:e0125318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perla FM, Prelati M, Lavorato M, Visicchio D, Anania C. 2017. The role of lipid and lipoprotein metabolism in non-alcoholic fatty liver disease. Children (Basel) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petriello MC, Brandon JA, Hoffman J, Wang C, Tripathi H, Abdel-Latif A, et al. 2018. Dioxin-like pcb 126 increases systemic inflammation and accelerates atherosclerosis in lean ldl receptor-deficient mice. Toxicol Sci 162:548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfohl M, Ingram L, Marques E, Auclair A, Barlock B, Jamwal R, et al. 2020. Perfluorooctanesulfonic acid and perfluorohexanesulfonic acid alter the blood lipidome and the hepatic proteome in a murine model of diet-induced obesity. Toxicol Sci 178:311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfohl M, Marques E, Auclair A, Barlock B, Jamwal R, Goedken M, et al. 2021. An ‘omics approach to unraveling the paradoxical effect of diet on perfluorooctanesulfonic acid (pfos) and perfluorononanoic acid (pfna)-induced hepatic steatosis. Toxicol Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps T, Snyder E, Rodriguez E, Child H, Harvey P. 2019. The influence of biological sex and sex hormones on bile acid synthesis and cholesterol homeostasis. Biol Sex Differ 10:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plass JR, Mol O, Heegsma J, Geuken M, Faber KN, Jansen PL, et al. 2002. Farnesoid x receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 35:589–596. [DOI] [PubMed] [Google Scholar]
- Platte HD, Honscha W, Schuh K, Petzinger E. 1996. Functional characterization of the hepatic sodium-dependent taurocholate transporter stably transfected into an immortalized liver-derived cell line and v79 fibroblasts. Eur J Cell Biol 70:54–60. [PubMed] [Google Scholar]
- Pouwer MG, Pieterman EJ, Chang SC, Olsen GW, Caspers MPM, Verschuren L, et al. 2019. Dose effects of ammonium perfluorooctanoate on lipoprotein metabolism in apoe*3-leiden.Cetp mice. Toxicol Sci 168:519–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell WS, Chung D, Gravel S. 1995. 5-oxo-6,8,11,14-eicosatetraenoic acid is a potent stimulator of human eosinophil migration. J Immunol 154:4123–4132. [PubMed] [Google Scholar]
- Princen HMG, Pouwer MG, Pieterman EJ. 2016. Comment on “hypercholesterolemia with consumption of pfoa-laced western diets is dependent on strain and sex of mice” by rebholz s.L. Et al. Toxicol. Rep. 2016 (3) 46-54. Toxicol Rep 3:306–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, et al. 2007. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46:1081–1090. [DOI] [PubMed] [Google Scholar]
- Qazi MR, Abedi MR, Nelson BD, DePierre JW, Abedi-Valugerdi M. 2010. Dietary exposure to perfluorooctanoate or perfluorooctane sulfonate induces hypertrophy in centrilobular hepatocytes and alters the hepatic immune status in mice. Int Immunopharmacol 10:1420–1427. [DOI] [PubMed] [Google Scholar]
- Quehenberger O, Dennis EA. 2011. The human plasma lipidome. N Engl J Med 365:1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachamin Y, Grischott T, Rosemann T, Meyer MR. 2021. Inferior control of low-density lipoprotein cholesterol in women is the primary sex difference in modifiable cardiovascular risk: A large-scale, cross-sectional study in primary care. Atherosclerosis. [DOI] [PubMed] [Google Scholar]
- Rebholz SL, Jones T, Herrick RL, Xie C, Calafat AM, Pinney SM, et al. 2016. Hypercholesterolemia with consumption of pfoa-laced western diets is dependent on strain and sex of mice. Toxicol Rep 3:46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. 2014. Bile acids and the gut microbiome. Curr Opin Gastroenterol 30:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MB, Lee JS, Ren H, Vallanat B, Liu J, Waalkes MP, et al. 2008. Toxicogenomic dissection of the perfluorooctanoic acid transcript profile in mouse liver: Evidence for the involvement of nuclear receptors ppar alpha and car. Toxicol Sci 103:46–56. [DOI] [PubMed] [Google Scholar]
- Rowan-Carroll A, Reardon A, Leingartner K, Gagné R, Williams A, Meier MJ, et al. 2021. High-throughput transcriptomic analysis of human primary hepatocyte spheroids exposed to per- and polyfluoroalkyl substances as a platform for relative potency characterization. Toxicol Sci 181:199–214. [DOI] [PubMed] [Google Scholar]
- Rusli F, Deelen J, Andriyani E, Boekschoten MV, Lute C, van den Akker EB, et al. 2016. Fibroblast growth factor 21 reflects liver fat accumulation and dysregulation of signalling pathways in the liver of c57bl/6j mice. Sci Rep 6:30484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DW. 2003. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72:137–174. [DOI] [PubMed] [Google Scholar]
- Sakr CJ, Leonard RC, Kreckmann KH, Slade MD, Cullen MR. 2007. Longitudinal study of serum lipids and liver enzymes in workers with occupational exposure to ammonium perfluorooctanoate. J Occup Environ Med 49:872–879. [DOI] [PubMed] [Google Scholar]
- Salen G, Shefer S, Tint GS. 1984. Transformation of 4-cholesten-3-one and 7 alpha-hydroxy-4-cholesten-3-one into cholestanol and bile acids in cerebrotendinous xanthomatosis. Gastroenterology 87:276–283. [PubMed] [Google Scholar]
- Salihovic S, Fall T, Ganna A, Broeckling CD, Prenni JE, Hyötyläinen T, et al. 2019. Identification of metabolic profiles associated with human exposure to perfluoroalkyl substances. J Expo Sci Environ Epidemiol 29:196–205. [DOI] [PubMed] [Google Scholar]
- Schlezinger JJ, Puckett H, Oliver J, Nielsen G, Heiger-Bernays W, Webster TF. 2020. Perfluorooctanoic acid activates multiple nuclear receptor pathways and skews expression of genes regulating cholesterol homeostasis in liver of humanized pparα mice fed an american diet. Toxicol Appl Pharmacol 405:115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwingel PA, Cotrim HP, Salles BR, Almeida CE, dos Santos CR Jr., Nachef B, et al. 2011. Anabolic-androgenic steroids: A possible new risk factor of toxicant-associated fatty liver disease. Liver Int 31:348–353. [DOI] [PubMed] [Google Scholar]
- Seacat AM, Thomford PJ, Hansen KJ, Olsen GW, Case MT, Butenhoff JL. 2002. Subchronic toxicity studies on perfluorooctanesulfonate potassium salt in cynomolgus monkeys. Toxicol Sci 68:249–264. [DOI] [PubMed] [Google Scholar]
- Shoda J, Toll A, Axelson M, Pieper F, Wikvall K, Sjövall J. 1993. Formation of 7 alpha- and 7 beta-hydroxylated bile acid precursors from 27-hydroxycholesterol in human liver microsomes and mitochondria. Hepatology 17:395–403. [PubMed] [Google Scholar]
- Slijepcevic D, Roscam Abbing RLP, Katafuchi T, Blank A, Donkers JM, van Hoppe S, et al. 2017. Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology 66:1631–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail MI, Dönmez-Cakil Y, Szöllősi D, Stockner T, Chiba P. 2021. The bile salt export pump: Molecular structure, study models and small-molecule drugs for the treatment of inherited bsep deficiencies. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. 1998. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 98:2088–2093. [DOI] [PubMed] [Google Scholar]
- Steenland K, Tinker S, Frisbee S, Ducatman A, Vaccarino V. 2009. Association of perfluorooctanoic acid and perfluorooctane sulfonate with serum lipids among adults living near a chemical plant. Am J Epidemiol 170:1268–1278. [DOI] [PubMed] [Google Scholar]
- Stieger B, Meier Y, Meier PJ. 2007. The bile salt export pump. Pflugers Arch 453:611–620. [DOI] [PubMed] [Google Scholar]
- Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, et al. 1998. A gene encoding a liver-specific abc transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 20:233–238. [DOI] [PubMed] [Google Scholar]
- Strott CA, Higashi Y. 2003. Cholesterol sulfate in human physiology: What’s it all about? J Lipid Res 44:1268–1278. [DOI] [PubMed] [Google Scholar]
- Swiger KJ, Martin SS, Blaha MJ, Toth PP, Nasir K, Michos ED, et al. 2014. Narrowing sex differences in lipoprotein cholesterol subclasses following mid-life: The very large database of lipids (vldl-10b). J Am Heart Assoc 3:e000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs ML, Abbott BD. 2007. Activation of mouse and human peroxisome proliferator-activated receptors (alpha, beta/delta, gamma) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol Sci 95:108–117. [DOI] [PubMed] [Google Scholar]
- Touchstone JC. 1995. Thin-layer chromatographic procedures for lipid separation. J Chromatogr B Biomed Appl 671:169–195. [DOI] [PubMed] [Google Scholar]
- Trier X, Granby K, Christensen JH. 2011. Polyfluorinated surfactants (pfs) in paper and board coatings for food packaging. Environ Sci Pollut Res Int 18:1108–1120. [DOI] [PubMed] [Google Scholar]
- Turley SD, Schwarz M, Spady DK, Dietschy JM. 1998. Gender-related differences in bile acid and sterol metabolism in outbred cd-1 mice fed low- and high-cholesterol diets. Hepatology 28:1088–1094. [DOI] [PubMed] [Google Scholar]
- Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, loannou G, Kuver R, et al. 2011. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 141:1393–1403, 1403.e1391-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Dippe P, Amoui M, Alves C, Levy D. 1993. Na(+)-dependent bile acid transport by hepatocytes is mediated by a protein similar to microsomal epoxide hydrolase. Am J Physiol 264:G528–534. [DOI] [PubMed] [Google Scholar]
- von Dippe P, Amoui M, Stellwagen RH, Levy D. 1996. The functional expression of sodium-dependent bile acid transport in madin-darby canine kidney cells transfected with the cdna for microsomal epoxide hydrolase. J Biol Chem 271:18176–18180. [DOI] [PubMed] [Google Scholar]
- von Dippe P, Zhu QS, Levy D. 2003. Cell surface expression and bile acid transport function of one topological form of m-epoxide hydrolase. Biochem Biophys Res Commun 309:804–809. [DOI] [PubMed] [Google Scholar]
- Wagner M, Zollner G, Trauner M. 2010. Nuclear receptor regulation of the adaptive response of bile acid transporters in cholestasis. Semin Liver Dis 30:160–177. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang Y, Liang Y, Li J, Liu Y, Zhang J, et al. 2014. Pfos induced lipid metabolism disturbances in balb/c mice through inhibition of low density lipoproteins excretion. Sci Rep 4:4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Liu L, Zhang W, Zhang J, Du X, Huang Q, et al. 2017. Serum metabolome biomarkers associate low-level environmental perfluorinated compound exposure with oxidative /nitrosative stress in humans. Environ Pollut 229:168–176. [DOI] [PubMed] [Google Scholar]
- Wang Y, Nakajima T, Gonzalez FJ, Tanaka N. 2020. Ppars as metabolic regulators in the liver: Lessons from liver-specific ppar-null mice. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Cousins IT, Scheringer M, Hungerbuehler K. 2015. Hazard assessment of fluorinated alternatives to long-chain perfluoroalkyl acids (pfaas) and their precursors: Status quo, ongoing challenges and possible solutions. Environ Int 75:172–179. [DOI] [PubMed] [Google Scholar]
- Winquist A, Steenland K. 2014. Modeled pfoa exposure and coronary artery disease, hypertension, and high cholesterol in community and worker cohorts. Environ Health Perspect 122:1299–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf CJ, Schmid JE, Lau C, Abbott BD. 2012. Activation of mouse and human peroxisome proliferator-activated receptor-alpha (pparalpha) by perfluoroalkyl acids (pfaas): Further investigation of c4-c12 compounds. Reprod Toxicol 33:546–551. [DOI] [PubMed] [Google Scholar]
- Wouters K, van Bilsen M, van Gorp PJ, Bieghs V, Lütjohann D, Kerksiek A, et al. 2010. Intrahepatic cholesterol influences progression, inhibition and reversal of non-alcoholic steatohepatitis in hyperlipidemic mice. FEBS Lett 584:1001–1005. [DOI] [PubMed] [Google Scholar]
- Xiang X, Backman JT, Neuvonen PJ, Niemi M. 2012. Gender, but not cyp7a1 or slco1b1 polymorphism, affects the fasting plasma concentrations of bile acids in human beings. Basic Clin Pharmacol Toxicol 110:245–252. [DOI] [PubMed] [Google Scholar]
- Yoneda M, Fujita K, Nozaki Y, Endo H, Takahashi H, Hosono K, et al. 2010. Efficacy of ezetimibe for the treatment of non-alcoholic steatohepatitis: An open-label, pilot study. Hepatol Res 40:566–573. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zitzow JD, Ehresman DJ, Chang SC, Butenhoff JL, Forster J, et al. 2015. Na+/taurocholate cotransporting polypeptide and apical sodium-dependent bile acid transporter are involved in the disposition of perfluoroalkyl sulfonates in humans and rats. Toxicol Sci 146:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zitzow JD, Weaver Y, Ehresman DJ, Chang SC, Butenhoff JL, et al. 2017. Organic anion transporting polypeptides contribute to the disposition of perfluoroalkyl acids in humans and rats. Toxicol Sci 156:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Hoos L, Cook J, Tetzloff G, Davis H Jr., van Heek M, et al. 2008. Ezetimibe improves high fat and cholesterol diet-induced non-alcoholic fatty liver disease in mice. Eur J Pharmacol 584:118–124. [DOI] [PubMed] [Google Scholar]
- Zhu QS, Qian B, Levy D. 2004. Regulation of human microsomal epoxide hydrolase gene (ephx1) expression by the transcription factor gata-4. Biochim Biophys Acta 1676:251–260. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. 2014. Mitochondrial reactive oxygen species (ros) and ros-induced ros release. Physiol Rev 94:909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.