

Abstract
Individuals with diabetes are exposed to a higher risk of perioperative stroke than non-diabetics mainly due to persistent hyperglycemia. LncRNA Meg3 has been considered as an important mediator in regulating ischemic stroke. However, the functional and regulatory roles of Meg3 in diabetic brain ischemic injury remain unclear. In this study, rat brain microvascular endothelial cells (RBMVECs) were exposed to 6 h of oxygen and glucose deprivation (OGD), and subsequent reperfusion via incubating cells with glucose of various high concentrations for 24 h to imitate in vitro diabetic brain ischemic injury. It was shown that the marker events of ferroptosis and increased Meg3 expression occurred after the injury induced by OGD combined with hyperglycemia. However, all ferroptotic events were reversed with the treatment of Meg3-siRNA. Moreover, in this in vitro model, p53 was also characterized as a downstream target of Meg3. Furthermore, p53 knockdown protected RBMVECs against OGD + hyperglycemic reperfusion-induced ferroptosis, while the overexpression of p53 exerted opposite effects, implying that p53 served as a positive regulator of ferroptosis. Additionally, the overexpression or knockdown of p53 significantly modulated GPX4 expression in RBMVECs exposed to the injury induced by OGD combined with hyperglycemic treatment. Furthermore, GPX4 expression was suppressed again after the reintroduction of p53 into cells silenced by Meg3. Finally, chromatin immunoprecipitation assay uncovered that p53 was bound to GPX4 promoter. Altogether, these data revealed that, by modulating GPX4 transcription and expression, the Meg3-p53 signaling pathway mediated the ferroptosis of RBMVECs upon injury induced by OGD combined with hyperglycemic reperfusion.
Key words: MEG3, p53, ferroptosis, ischemia, GPX4, OGD, hyperglycemia
Introduction
With the increasing trend of population aging in the Chinese mainland, the incidence of perioperative stroke is gradually increased. Perioperative stroke can cause significant morbidity and mortality, thus bringing heavy economic burden to the society and the families of patients.1,2 Epidemiological studies have identified diabetes mellitus (DM) as an independent factor in the increased risk of perioperative stroke in diabetic patients.2-4 Therefore, it is of a prime interest to develop new therapeutic strategies for the management of diabetic perioperative stroke.
Cerebral ischemic-reperfusion injury leads to the activation of cell death programs, including ferroptosis. Defined as cell death triggered by generation of excessive lipid reactive oxygen species (ROS) dependent on iron, ferroptosis is different from other classical modes of programmed cell death.5 Iron accumulation was previously observed in stroke patients,6 while iron inhibitors and chelators such as ferrostatin-1 and deferoxamine presented significant protective benefits against focal cerebral ischemia-reperfusion injury in mice.7
As a class of non-coding RNAs with a length of larger than 200 nucleotides, long non-coding RNAs (lncRNAs) are involved in many critical physiological and pathophysiological processes, such as cell differentiation, cell proliferation, immunity, inflammation and apoptosis, through modulating their target genes.8,9 Accumulative experimental evidence has revealed that dysregulation of lncRNAs is closely associated with neurologic functional disorders upon brain stroke.10-12 In addition, several studies have demonstrated that lncRNAs participated in the pathogenesis of DM and its complications.13,14 Therefore, it is very important to explore the relevance between lncRNAs and diabetic brain stroke.
Maternally expressed gene 3 (Meg3), is a lncRNA, which located in human chromosome 14q32. Previous studies have shown that Meg3 acted as a tumour suppressor in cancer initiation, progression, metastasis and chemo-resistance.15,16 More strikingly, it is now believed that Meg3 inhibition confers robust neuroprotection against cerebral ischemic reperfusion injury both in vivo and in vitro.17-19 p53, once considered as a downstream target of Meg3, is involved in initiating cellular responses to endogenous or exogenous stress.17,20,21 The functions of p53 in mediating the cellular response to oxidative stress-induced ferroptosis have been well established.22,23 Mechanistically, it appears plausible that the Meg3-p53 signaling pathway mediates ferroptotic cell death.
Based on available data, brain microvascular endothelial cells are affected by hyperglycemia in the early stages of diabetes to cause functional impairment, which is one of the initiating factors of diabetic cerebrovascular diseases.24 In addition, brain microvascular endothelial cell damage can lead to micro-vascular occlusion and micro-thrombosis, which are closely related to the significantly increased incidence of perioperative stroke in diabetic patients. Therefore, in this current study, rat brain microvascular endothelial cells (RBMVECs) were exposed to oxygen and glucose deprivation (OGD), and subsequently incubated with various concentrations of glucose to simulate in vitro diabetic brain ischemic injury. Furthermore, the regulatory roles of the Meg3-p53 axis in ferroptosis were studied to identify potential therapeutic strategies for diabetic brain stroke.
Materials and Methods
Cell separation and treatment
As described previously,25 Sprague Dawley (SD) rats (n=60) aged three weeks were purchased from the Experimental Animal Center of Xiangya Hospital of Central South University. All rats were bred in a specific pathogen-free environment in 12-h lightdark cycle and fed with rodent diet and water. All rats were anaesthetized with inhaling isoflurane (2%, CAS NO. 64181101, Lunan Pharmaceutical Co., LTD. Shandong, China) and sacrificed by cervical dislocation. The whole brain was removed after opening the cranial cavity.
Tissue was homogenized and then centrifuged at 720 × g for 5 minutes at 4°C. The pellet re-suspended in phosphate-buffered saline, while the supernatant was discarded. Then, the pellet was layered over 15 mL 18% dextran and centrifuged at 4500 × g for 20 minutes at 4°C. It was re-suspended in 10 ml phosphatebuffered saline (containing 0.1% bovine serum albumin). 10 ml of the suspension were added to 100 μL collagenase (100 mg/mL), 40 μl DNase I (10 mg/mL) and 100 μl N-alpha-tosyl-L-lysine chloromethyl ketone hydrochloride (14.7 μg/mL) and digested for 1 h at 37°C. After centrifugation at 1000 × g for 5 min, the pellet was re-suspended in 2 mL phosphate-buffered saline containing 0.1% bovine serum albumin. After incubation for 10 min at 4°C, cells were isolated with a Dynabeads FlowComp Flexi Kit (Thermo Fisher). Cells, cultured in ordinary medium for 3 days, were used for the following experiments. The endothelial cell markers, rabbit anti-factor VIII heavy chain polyclonal antibody (H-140) (1:400; Santa Cruz Biotechnology, Shanghai, China), was used to identify RBMVECs by immunofluorescence.
RBMVECs were transferred into an anaerobic incubator configured to an atmosphere of 5% CO2, 94% N2, 1% O2, 98% humidity and 37°C to induce OGD for 6 h. Then, the cells were reperfused by returning them to the incubation medium containing different concentrations of glucose (5, 10, 30, 45 and 75 mM) for 24 h of culture. The RBMVECs cultured in a medium with 5 mM glucose served as the control. In addition, 25 mM of mannitol were added into the 5 mM glucose medium to produce the same osmotic pressure as that produced by 30 mM of glucose, and the medium was used to treat RBMVECs for 24 h. In addition, to test whether RBMVECs are sensitive to ferroptotic cell death. Erastin and RSL3, two important ferroptosis activators (purchased from Sigma-Aldrich, St. Louis, MO, USA), were dissolved into DMSO with different concentrations. Erastin (2, 5, 10 μM) and RSL3 (60, 120, 180 μm) were used to treat RBMVECs in vitro for 48 h to induce ferroptosis.
All experiments were approved by Institutional Animal Care and Use committee in Central South University. The protocol for the use of rats followed the guidelines of the Care and Use of Laboratory animals.
Cell transfection
Meg3-siRNA, p53-siRNA and their negative controls were constructed by Genepharm Co., (Shanghai, China). In addition, the sequence of p53 was inserted into a pcDNA3.1 vector to generate the recombinant plasmid of p53. Meg3-siRNA (20 μM), p53- siRNA (25 μM) and recombinant plasmid of p53 (50 μM) were transiently transfected into RBMVECs with the help of Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The expression of Meg3 and p53 was detected 48 h after transfection.
Lactate dehydrogenase (LDH) and cell viability assay
Using a commercial CCK-8 (cell counting kit-8; CK04, provided by Dojindo, Tokyo, Japan), cell viability was assessed. Briefly, after the indicated experiments, a CCK-8 working solution was added to each well and incubated for 2 h at 37°C. The absorbance values were determined at 450 nm using a microplate spectrophotometer (Bio-Tek, Winooski, VT, USA). A LDH detection kit (purchased from Roche, Basel, Switzerland) was used as previously described,26,27 and OD (optical density) values were recorded at 450 nm. Cell survival rates and the values of LDH release were standardized to the values in the control group and expressed as percentages.
Real-time quantitative polymerase chain reaction (PCR)
After the indicated experiments, the total RNA was extracted by using Trizol (provided by Invitrogen, Carlsbad, CA, USA) according to the product instructions. PCR was performed with specific primers and a SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The 2-ΔΔCt method was used to calculate the relative levels of Meg3, p53, FTH1, GPX4 and ACSL4.28 The primer sequences are shown in Table 1.
Western blot
As described previously,29 cells were lysed by a lysis buffer. SDS-PAGE (12%, w/v) was used to separate the proteins, which were then transferred onto PVDF membranes (provided by Thermo Fisher, Waltham, MA, USA). Primary antibodies (monoclonal) against GPX4 (cat. no. ab125066, dilution 1:1000, Abcam, Cambridge, UK), p53 (cat. no. ab131442, dilution 1:1000, Abcam), FTH1 (cat. no. ab240277, dilution 1:1000, Abcam), ACSL4 (cat. no. ab227256, dilution 1:1000, Abcam UK) and β- actin (cat. no. ab179467, dilution 1:2000, Abcam), along with peroxidase- conjugated rabbit anti-IgG secondary antibodies (cat. no. A2074, dilution 1:4000, Sigma-Aldrich), were used in the Western blot analysis. The immunoreactive signals were visualized using enhanced chemiluminescence detection. To quantify protein levels, X-ray films were scanned and analyzed using Image J 1.47i software (National Institutes of Health, Bethesda, MD, USA).
Iron concentration assay
After indicated treatments, RBMVECs (≥1×105 for each well) were immediately homogenized in PBS (phosphate-buffered saline). The supernatant was collected after centrifugation. The iron concentration was measured by using an Iron Assay Kit (ab83366, Abcam) according to the product instructions. Iron concentrations were expressed as increments compared with the values of the control group (100%).
Lipid reactive oxygen species (ROS) measurement
After experiments, RBMVECs were incubated with C11- BODIPY 581/591 (D3861, Invitrogen). Then, the transplanted RBMVECs were cultured with the reagent at a working concentration of 2.5 μM for 30 min in an incubator. Collecting RBMVECs in centrifuge tube followed by washing twice with PBS, the pellets were re-suspended in 500 μL PBS. Using flow cytometry (provided by Becton Dickinson FACS Canto TM, USA), cellular fluorescence intensity was analyzed and cell images were acquired through a fluorescence microscope (IX81; Olympus). Lipid ROS production was expressed as increments compared with the values of the control group (100%).
Assessment of myeloperoxidase and glutathione expressions
RBMVECs (≥1×105 for each well) were homogenized and the supernatant was collected for the analysis of myeloperoxidase (MPO) and glutathione (GSH) expressions by using a MPO assay kit (A044, Jiancheng Bioengineering Institute, Nanjing, China) and a total GSH/oxidized GSH assay kit (A06, Jiancheng Bioengineering Institute), respectively. The ratio of GSH/GSSG was calculated. The levels of MPO and GSH were standardized to the values of the control group and were expressed as percentages.
Detection of cell apoptosis
The apoptosis of RBMVECs was detected using an Annexin VFITC/ PI apoptosis detection kit (Sigma-Aldrich Trading Co., Shanghai, China) following the manufacturer’s instructions. Apoptotic cells were quantified using a FACSCalibur cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Chromatin immunoprecipitation assay
Sonicated nuclear lysates were purified, and using a chromatin immunoprecipitation assay kit (P2078, Beyotime), immunoprecipitation was performed according to the method described in a previous study of the authors in this research.30
Statistical analysis
All data were presented as mean ± SD. One-way analysis of variance was applied with a Bonferroni post hoc test or Student’s t-test. At least three independent experiments were performed to confirm the results. Data were analyzed with SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). A p-value <0.05 indicated significant difference.
Results
A mimetic model of diabetic brain ischemic reperfusion injury was successfully established in vitro
To induce diabetic brain ischemic reperfusion injury in vitro, RBMVECs were exposed to 6 h of in vitro ischemia induced by OGD in an anaerobic incubator, followed by subsequent reperfusion via returning the RBMVECs to normoxic conditions, in which the RBMVECs were cultured for 24 h in culture media containing different concentrations of glucose (5, 10, 30, 45 and 75 mM). To elevate the in vitro insult to RBMVECs, a survival rate of approximately 50-60% was considered appropriate. The data demonstrated that as compared to 5 mM glucose (served as control), the exposure to higher glucose concentrations (≥30 mM) during the reperfusion period caused a significant reduction in cell viability and a marked increase of LDH release in a dose-dependent manner after exposure to OGD (p<0.05). However, higher concentrations of glucose (45 or 75 mM) caused too excessive damage to RBMVECs (with a survival rate of less than 50%). Hence, continuous incubation at a relatively high concentration of glucose (30 mM) was chosen for subsequent experiments. Additionally, there was no change in cell viability and LDH leakage between 5 mM and mannitol groups (p>0.05), which suggested that the impact of osmolality on cell survival was minimal. Thus, these findings indicated that an in vitro mimetic model of diabetic brain ischemic reperfusion injury was successfully established (Figure 1 A,B).
Table 1.
Primer sequence.
| Name | Sequence (5'-3') |
|---|---|
| MEG3 | GTGAAGGTCGGAGTGAACG |
| CTCGCTCCTGGAAGATGGTG | |
| p53 | GAGATGTTCCGAGAGCTGA |
| TCAGCTCTCGGAACATCTC | |
| FTH1 | GGCAGTGACCTTTTCTGAGC |
| TCATTGTCAAAGGGTGCAAA | |
| GPX4 | TCAGCAAGATCTGCGTGAAC |
| GGGGCAGGTCCTTCTCTATC | |
| ACSL4 | AATGCAGCCAAATGGAAAAG |
| CACAGAAGATGGCAATGGTG | |
| ACSL4 | AACGGATTTGGTCGTATTGGG |
| CCTGGAAGATGGTGATGGGAT |
Meg3 mediated the cellular damage induced by exposure to OGD + high glucose
As depicted in Figure 2A, after 6 h of exposure to OGD and subsequent reperfusion in normal (5 mM) media for 24 h, OGD injury significantly induced the up-regulation of Meg3 expression, compare with control group (P<0.05). Additionally, the Meg3 level in OGD combined with high glucose media (30 mM) was higher than that in OGD in the cultures exposed to normal glucose (5 mM). Mannitol did not affect Meg3 expression (p>0.05). These findings suggest that continuous culture at a moderately high concentration of glucose (30 mM) significantly elevated Meg3 expression of RBMVECs treated with OGD.
si-Meg3 (Meg3 siRNA) was transfected into RBMVECs and the results showed that Meg3 expression was markedly down-regulated in RBMVECs transfected with si-Meg3, which confirmed the successful transfection of RBMVECs with si-Meg3 (p<0.05, Figure 2B). Next, we evaluated the effect of si-Meg3 in OGD and OGD combined with hyperglycemic condition and found that si- Meg3 also significantly inhibited Meg3 elevation (p<0.05, Figure 2C). Moreover, to assess the effects of Meg3 knockdown on the survival of RBMVECs, cell viability and LDH leakage were detected under OGD + HG conditions following transfection with either si-Meg3 or NC-siRNA. The results indicated that Meg3 knockdown promoted cell survival and reduced the release of LDH in RBMVECs (p<0.05, Figure 2 D,E). Moreover, it was found that the exposure to moderately high glucose (30 mM) induced the apoptosis of RBMVECs after their exposure to the injury induced by OGD + hyperglycemic treatment (20.54±0.81% in OGD+HG group vs 5.03±0.12% in Control group). Similarly, Meg3 knockdown remarkably alleviated apoptosis triggered by OGD + HG exposure (14.23±0.65% in OGD+HG+MEG3-KD group vs 19.74±0.77% in OGD+HG+MEG3-NC group, p<0.05, Figure 2 F,G).
Inhibition of Meg3 conferred protection to RBMVECs against OGD + HG-induced ferroptosis
Some pivotal proteins, such as ferritin heavy chain 1 (FTH1), Acyl-CoA synthetase long-chain family member 4 (ACSL4) and glutathione peroxidase 4 (GPX4), are considered as core regulators of ferroptosis. Therefore, the expression levels of these proteins were measured under OGD + HG conditions with or without si- Meg3 transfection. The ACSL4 (a positive regulator) level was notably increased, while the levels of GPX4 and FTH1 (two negative regulators) were reduced in RBMVECs exposed to OGD + HG (Figure 3 A-D). Interestingly, the silencing of Meg3 by siRNA not only obviously inhibited ACSL4 expression, but also reversed the levels of FTH1 and GPX4.
Additionally, cellular iron concentration, generation of lipid ROS, the product of lipid peroxidation, such as MPO and GSH, and GSH/GSSG ratio were assayed after exposure to OGD + HG. Similar to the above results, OGD + HG significantly elevated intracellular iron concentration, lipid ROS generation and MPO content, while markedly reduced the level of GSH and the GSH/GSSG ratio. Similarly, the effects of OGD + hyperglycemic treatment on these ferroptosis-associated indictors were all abolished by si-Meg3 (p<0.05, Figure 3 E-J).
To gain more solid evidence, it was tested whether RBMVECs undergo ferroptosis in response to erastin and RSL3, direct inhibitors of system XC- and GPX4, respectively. At first, different concentrations of erastin and RSL3 were used to induce the ferroptosis of RBMVECs to determine an optimum concentration. Depicted in Figure 4 A,B, erastin suppressed cell viability and enhanced LDH leakage in a dose-dependent manner, and the optimum concentration was 5 μM. However, when 60, 120, and 180 μM of RSL3 were used, RSL3 triggered cytotoxicity in a doseindependent manner and the optimum concentration was 60 μM (p<0.05, Figure 4 C,D). The cytotoxicity induced by both erastin and RSL3 was alleviated upon treatment with si-Meg3 (p<0.05, Figure 4 E-H).
Figure 1.
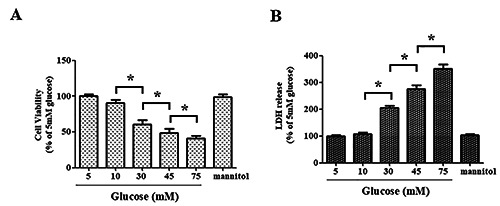
Establishment of an in vitro model of diabetic brain ischemia in adult rat brain microvascular endothelial cells (RBMVECs). RBMVECs were exposed to OGD for 6 h before they were returned to cultured media containing various concentrations of glucose for an additional 24 h of incubation. A) The survival rate of RBMVECs following in vitro ischemia. B) LDH leakage was measured using LDH assays. mannitol: RBMVECs was treated for 24 h with 5 mM glucose + 25 mM mannitol. CCK-8 and LDH assays were used to rule out the influence of osmolality on cell survival. Data were expressed as the mean ± SD. The experiments were conducted in triplicate; *p<0.05.
Figure 2.
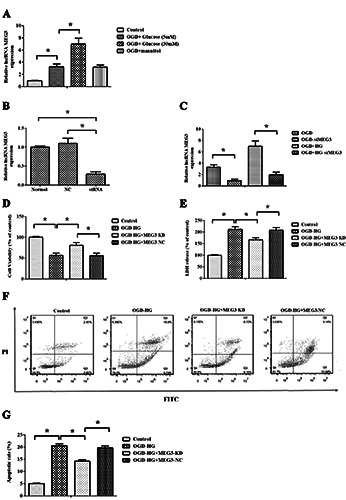
Meg3 mediated OGD + hyperglycemic (HG) reperfusion injury in RBMVECs. A) RT-qPCR analysis of Meg3 expression in RBMVECs undergoing OGD + HG treatment; mannitol did not affect Meg3 expression. B) Meg3 was inhibited by siRNA transfection, which was verified by RT-qPCR. C) si-Meg3 also successfully abated the elevation of Meg3 expression caused by OGD and hyperglycemic reperfusion. D-E) The CCK-8 and LDH assays were used to detect the survival of RBMVECs, and showed that OGD + HG caused RBMVEC damage, while the inhibition of Meg3 protected RBMVECs. F-G) The cell apoptosis was enhanced in RBMVECs elicited by OGD+HG, while it was decreased by inhibited Meg3. Data were expressed as the mean ± SD. The experiments were carried out for three times; *p<0.05.
Figure 3.
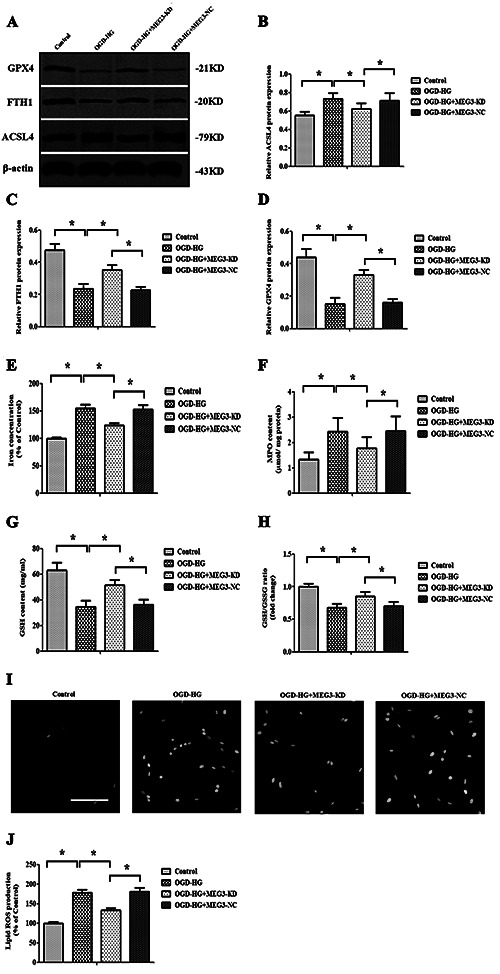
Inhibition of Meg3 protected RBMVECs against ferroptosis elicited by OGD+HG. Knockdown of Meg3 altered the expression of FTH1, GPX4 and ACSL4 induced by OGD + HG. A) The content of ferroptosis-related proteins was determined by quantitative analyses of FTH1 (B), GPX4 (C) and ACSL4 (D) expression and Western blotting. E-H) si-Meg3 attenuates OGD + HG induced-lipid peroxidation; the levels of iron (E), MPO (F) and GSH (G) were determined using commercial kits. H) The ratio of GSH/GSSG after OGD + HG exposure with or without si-Meg3 treatment. I-J) Lipid ROS production was detected by BODIPY 581/591 C11 staining using fluorescence microscopy; scale bar:100 μm. Data were expressed as the mean ± SD. The experiments were carried out for three times; *p<0.05.
Taken together, all these findings proved that the inhibition of Meg3 neutralized the ferroptosis induced by OGD+ hyperglycemic treatment.
Figure 4.
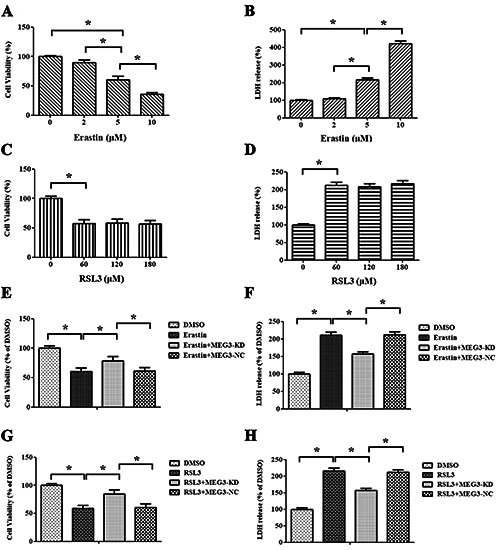
Ferroptosis inducer reversed the protective effect of si-Meg3 on RBMVECs following OGD + HG stimulation. A,B) Different doses of erastin induced ferroptosis in RBMVECs, as shown by cell viability and LDH leakage. C,D) Various concentrations of RSL3 triggered ferroptosis, as evidenced by cell survival and LDH release. E-H) Meg3 knockdown using siRNA rescued RBMVECs from erastin- and RSL3-induced lethality. Data were expressed as the mean ± SD. The experiments were carried out for three times; *p<0.05.
Figure 5.
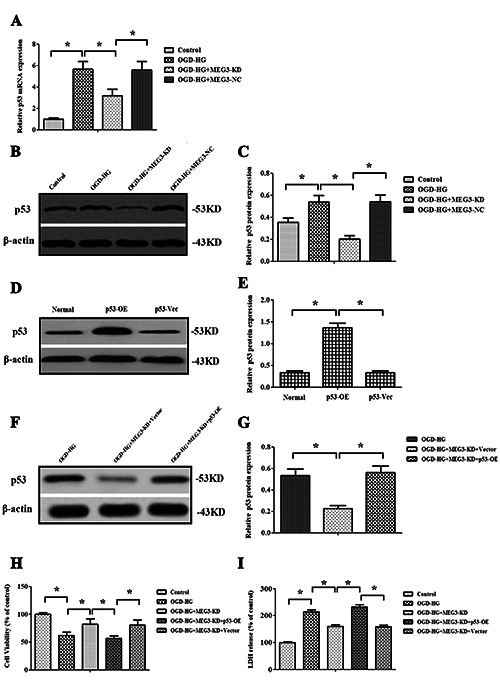
Meg3 regulated OGD + HG-induced injury by targeting p53. A) Relative expression of p53 gene was detected by RT-qPCR analysis. B,C) Western blot images and quantitative analysis of p53 protein expression in RBMVECs following OGD + HG exposure in the presence or absence of si-Meg3; RBMVECs transfected with a wt-p53-encoding plasmid or its negative vector, and the p53 level was determined by Western blotting (D) and quantitative analysis (E). F-G) Western blot images and quantitative analysis indicated that the transfection of pcDNA-p53 significantly restored the expression of p53 in si-Meg3-transfected RBMVECs; overexpression of p53 abrogated the protective role of si-Meg3. Cell viability was detected by CCK-8 (H) and LDH (I) assays. Data were expressed as the mean ± SD. The experiments were carried out for three times; *p<0.05.
The Meg3-p53 axis mediated OGD + HG treatmentinduced ferroptosis
Previous studies have reported that Meg3 could stimulate p53- mediated ischemic neuronal death.17,25 In this present study, the results also demonstrated that Meg3 knockdown notably inhibited the elevation of p53 gene (Figure 5A) and protein expression (Figure 5 B,C) induced by OGD + hyperglycemic reperfusion injury, suggesting that p53 might act as a downstream target of Meg3 (p<0.05). To further validate whether Meg3 modulated OGD + HG-induced injury through targeting p53, rescue experiments were performed. Firstly, RBMVECs were transfected with a wildtype p53 encoded specifically by plasmid DNA. Then, Western blotting indicated that p53 levels were dramatically increased by pcDNA-p53 in normal control cells (p<0.05, Figure 5 D,E). Secondly, it was shown that the transfection of pcDNA-p53 significantly restored the expression of p53 in si-Meg3 -transfected RBMVECs exposed to OGD + HG stimulation (p<0.05, Figure 5 F,G). Furthermore, the pcDNA-p53 treatment also reduced RBMVECs viability compared to that of RBMVECs transfected with si-Meg3 (p<0.05; Figure 5H). Finally, LDH leakage was decreased by the transfection with si-Meg3, which was abolished by pcDNA-p53 transfection in RBMVECs treated by OGD + HG (p<0.05, Figure 5I). The results showed that the overexpression of p53 significantly abrogated the protective effect of si-Meg3.
p53 participated in OGD + HG-induced ferroptosis through modulating GPX4
In vitro gain of function and loss of function analyses of p53 were also employed to assess its effect on ferroptosis elicited by OGD + HG and the associated signaling pathway by using siRNAs. As summarized in Figure 6 A,B, p53 knockdown was validated on the protein level.
To further investigate the biological function of p53 in regulating OGD + HG-induced injury, the effect of p53 on OGD + HG reperfusion-induced ferroptosis was evaluated. It was found that the inhibition of p53 significantly improved cell survival, as evidenced by increased cell viability and reduced LDH release (p<0.05, Figure 6 C,D). Moreover, p53 knockdown not only reduced cellular iron concentrations, lipid ROS production and MPO levels, but also increased GSH contents and GSH/GSSG ratios. However, the overexpression of p53 exerted exactly opposite effects (p<0.05, Figure 6 E-J).
With a desire to better elucidate the underlying mechanisms of p53 in modulating the ferroptosis induced by OGD + hyperglycemic reperfusion injury, the impact of p53 overexpression or inhibition on the expression of FTH1, GPX4 and ACSL4 was assessed. The findings revealed that neither the overexpression nor knockdown of p53 exerted any significant impact on the gene expression of FTH1 and ACSL4 (p>0.05, Figure 7 A, B). However, GPX4 gene expression was significantly down-regulated by p53 overexpression and remarkably up-regulated by p53 depletion in RBMVECs exposed to OGD + HG-induced injury (p<0.05, Figure 7C). Meanwhile, it was also observed that the GPX4 protein expression was regulated by the overexpression or knockdown of p53 (p<0.05, Figure 7 D,E). Furthermore, the overexpression of p53 also reversed the impact of si-Meg3 on the protein expression of GPX4 (p<0.05, Figure 7 F,G).
Using a chromatin-anti-p53 immunoprecipitation assay, it was shown that p53 was structurally bound to GPX4 promoter. Upon OGD + HG insult, p53 interacted with GPX4 promoter to suppress its transcription. However, p53 depletion inhibited the role of p53 in a way similar to the results obtained from RBMVECs silenced by Meg3. In contrast, p53 overexpression rescued the interaction between p53 and GPX4 promoter in RBMVECs silenced by Meg3 (p<0.05, Figure 7 H,I). Collectively, these results indicated that Meg3 regulated OGD + hyperglycemic reperfusion-induced ferroptosis at least partially through p53-mediated GPX4 transcription.
Discussion
Perioperative stroke is featured by the acute development of cerebral circulation disorders caused by cerebral ischemia or embolism during the perioperative period, and the morbidity and mortality of perioperative stroke remain high. Perioperative stroke is also one of the common serious complications observed during or after surgery and anesthesia.1,2 Over the past decade, numerous risk factors, including DM, have been well characterized for perioperative stroke.3,4 DM is a chronic, persistent, and severe metabolic health problem characterized by hyperglycemia. Individuals with DM have almost twice the risk of stroke compared to nondiabetics.
Moreover, approximately 30% of perioperative stroke patients have DM.3,4 Unfortunately, so far, few data are available to clarify the possible molecular mechanisms of diabetic brain stroke.
Long non-coding RNAs are recognized as a unique class of non-coding RNAs with a length of above 200 bp. Recent studies have pointed to the essential role of lncRNAs in brain stroke,10,11,17-19 and lncRNAs have been identified as pivotal biomarkers of diabetes mellitus.31,32 Maternally expressed gene 3 (Meg3), a newly discovered lncRNA, shows the strongest genetic susceptibility for neurovascular17-19 and neurodegenerative disease.33,34 Recently, a growing body of evidence indicates that up-regulated Meg3 expression is positively correlated with a poor prognosis of ischemic brain injury.17-19 Furthermore, previous studies reported that Meg3 is highly expressed in vascular endothelial cells.35 Nevertheless, up to now, almost no literature has discussed the correlation between Meg3 and diabetic brain stroke. In this study, RBMVECs were exposed to OGD injury followed by reperfusion in a high glucose medium to simulate an in vitro diabetic brain stroke model. The results suggested that a relatively high concentration of glucose (30 mM) significantly reduced the survival of RBMVECs. Moreover, it was also found that Meg3 was robustly activated in RBMVECs, whereas the silencing of Meg3 expression using small interfering RNA (siRNA) resulted in prominent inhibition of cell death after OGD + hyperglycemic reperfusion, suggesting that Meg3 might accelerate the pathologic process of diabetic brain stroke. Thus, it is urgent to explore the mechanisms underlying the role of Meg3 in diabetic brain stroke.
As a new pathway of programmed cell death, ferroptosis is iron-dependent and activated by lipid peroxides. A study by Tuo et al. confirmed an elevated iron concentration in brain stroke injury and showed that ferroptotic inhibitors reduced the size of brain injury and improved neurological functions in a middle cerebral artery occlusion (MCAO) model,7 demonstrating the link between ferroptosis and ischemic brain injury. Since iron dysregulation and subsequent lipid peroxidation are indispensable for the induction of ferroptosis,5 iron concentrations, lipid ROS production, levels of MPO and GSH contents, and GSH/GSSG ratio were assayed in this study after OGD + hyperglycemic reperfusion. The results showed that OGD + HG led to significant cytoplasmic elevation of levels of iron, lipid ROS and MPO along with a remarkable reduction in the GSH content and GSH/GSSG ratio, implying ferroptosis. Furthermore, all events associated with ferroptosis were blocked by the treatment with si-Meg3. Moreover, to further verify the role of Meg3 in ferroptosis, erastin and RSL3 were used to induce ferroptosis in RBMVECs. The results showed a noticeably suppressive effect of si-Meg3 on the lethality of erastin and RSL3. Additionally, several reports have described that FTH1, ACSL4, and GPX4 act as essential factors in ferroptosis regulation.36-38 Therefore, to obtain more evidence and further investigate the molecular mechanisms underlying the role of OGD + HG in ferroptosis regulation, the expression levels of above proteins were evaluated and the results showed that the expression of the positive ferroptosis regulator ACSL4 was enhanced, while the expression of two negative ferroptosis regulators (GPX4 and FTH1) was suppressed under OGD + HG conditions. Similar to the above results, the administration of si-Meg3 reversed the changes in the expression of these proteins in ferroptosis. Thus, these results suggested that the knockdown of Meg3 could inhibit ferroptosis following OGD + HG insult.
Figure 6.
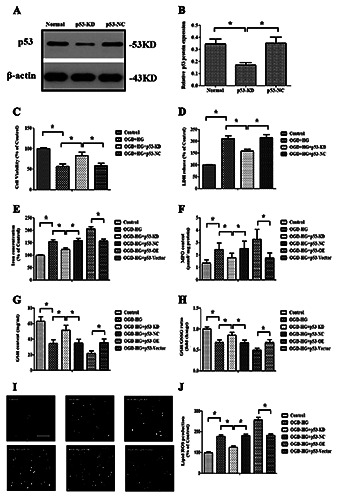
p53 modulated ferroptosis. A,B) Western blot images and quantitative analysis showed the effectiveness of p53-siRNA. C,D) The effect of p53-siRNA on cell survival and LDH leakage; the effect of p53 overexpression or inhibition on the OGD + HG inducedlipid peroxidation; the levels of iron (E), MPO (F) and GSH (G) were determined using commercial kits. H) GSH/GSSG ratios. Determination of the effect of p53 overexpression or inhibition on the production of lipid ROS (I-J) by a C11-BODIPY (581/591) probe. Data were expressed as the mean ± SD. The experiments were carried out for three times; *p<0.05.
Figure 7.
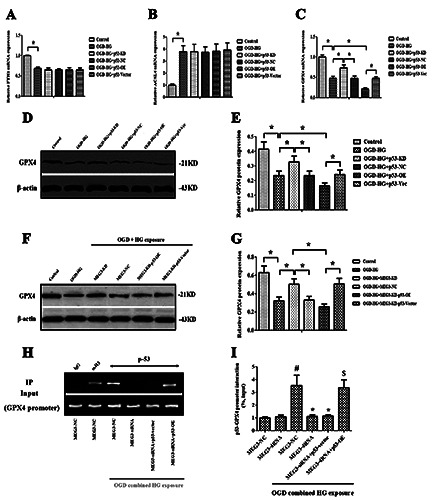
Meg3 regulated GPX4 via p53. A,B) RT-qPCR analysis showed that the overexpression and knockdown of p53 had no impact on FTH1 and ACSL4 expressions. C) The impact of p53 overexpression and knockdown on GPX4 expression was evaluated using RTqPCR analysis. D,E) Western blot images and quantitative analysis showed the effect of p53 overexpression and knockdown on GPX4 protein expression. F,G) Western blot images and quantitative analysis showed that the overexpression of p53 also reversed the impact of si-Meg3 on the protein expression of GPX4. H,I) Chip assay suggested that p53 significantly bound to GPX4 promoter in injury elicited by OGD + HG; anti-histone-3 (α-H3) served as a positive control, and IgG was used as a negative control. Data were expressed as the mean ± SD. The experiments were carried out for three times. *p<0.05 vs the OGD-HG+ Meg3-NC group; $p<0.05 vs the OGDHG+ Meg3-siRNA group; #p<0.05 vs the Meg3-NC group.
Additionally, it was found Meg3 was positively correlated with p53 expression in RBMVECs. The depletion of Meg3 caused the marked reduction of p53 expression. Notably, reintroduction of p53 reversed the protective effect of Meg3 inhibition against OGD + HG-induced injury. Furthermore, it was reported that Meg3 directly acted on p53 to regulate the expression of p53 and sensitize p53-mediated neuronal death in vivo and in vitro.17,18,20,21 Recently, Zhan and his colleagues also used an RNA-immunoprecipitation (RIP) assay to affirm that Meg3 could interact with p53 in RBMVECs.25 Hence, it seems plausible that p53 serves as a downstream target of Meg3 in brain stroke. p53 has been characterized as a positive regulator of ferroptosis associated with iron metabolism disorder and excessive lipid peroxidation in certain cancer cells.22,23 Moreover, the activation of p53 sensitizes cells to ferroptosis through transcriptional repression of TIGAR, GLS2 and SCO2.39 Nevertheless, tumor cells expressing p53 mutations are more vulnerable to ferroptosis.40 Considering these facts, it is reasonable to speculate that the Meg3-p53 signaling pathway mediates ferroptosis in the model of this study. To validate this hypothesis, in vitro gain- and loss-of-function analyses of p53 were performed to estimate the effect of p53 on ferroptosis. Lipid peroxidation, down-regulated GPX4 expression and GSH depletion are crucial events in the ferroptosis assay. The data showed that the inhibition of p53 not only significantly alleviated lipid peroxidation and elevated iron and MPO contents upon exposure to OGD + hyperglycemic reperfusion, but also reserved the expression of GPX4 and GSH, whereas the overexpression of p53 exerted opposite effects. Although both overexpression and inhibition of p53 had no impact on FTH1 and ACSL4 expressions, it could still be concluded that p53 activation invariably caused ferroptosis in OGD + hyperglycemic reperfusion injury.
Ferroptosis can be activated by the treatment with RSL3 and direct inactivation of GPX4, which block the conversion of reduced GSH to oxidized GSH and lipid hydroperoxides/hydrogen peroxides to their end products, such as alcohol and water. Ferroptosis was induced by knockdown of GPX4 in a ROS-, MEK-, and iron-dependent manner, while RSL3 resistance was caused by GPX4 overexpression.41 Furthermore, cerebral neuronal loss was found in GPX4 knockout (neuron-specific) mice, which was explained by induced ferroptosis in part.42 In this study, it was proven that p53 suppressed GPX4 expression at the pre- and posttranscriptional level. Mechanistically, the most pivotal issue to address is how p53 modulates GPX4 expression. SLC7A11 is a major component of System XC - and the latter is indispensable for importing cystine, which is used to synthesize antioxidant GSH.43 Previous studies have demonstrated that p53 directs the transcriptionally suppressed SLC7A11 expression,44,45 thereby suppressing system XC - activity to trigger ferroptosis. In a similar mechanism depicted in Figure 7H, under OGD + HG conditions, p53 directly inhibits GPX4 transcription by binding to a specific locus located on GPX4 promoter. Similarly, Meg3 knockdown blocked this interaction, while the reintroduction of p53 promoted this interaction in RBMVECs silenced by Meg3. Based on the findings of this study, it can be conceived that diabetic cerebral ischemic reperfusion injury significantly up-regulated Meg3 expression, resulting the activation of its downstream target p53 and subsequently suppressing GPX4 expression, eventually leading to lipid peroxidation- triggered plasma membrane rupture and ferroptosis. Although we elucidated that Meg3 acted as ferroptotic promoter to mediate OGD combined high glucose-induced death of RBMVECs, whether Meg3 has similar effects on the neurons and other types of glial cells should be further clarified. The findings achieved in the current study remain to be further confirmed in animal models of cerebral ischemia.
Figure 8.
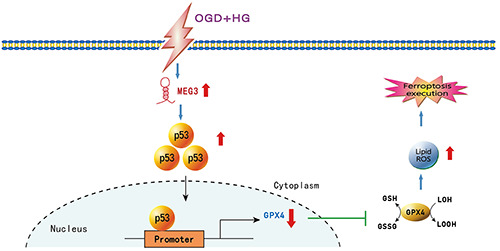
Schematic diagram of Meg3-p53-GPX4 axis regulating ferroptosis triggered by OGD + HG injury. OGD combined with hyperglycemic reperfusion promoted Meg3 expression and there was positive correlation between Meg3 and p53 expression in RBMVECs. Subsequently, p53 inhibited the activity of GPX4 by binding with its promoter. GPX4 inactivation led to the accumulation of lipid peroxides and final ferroptosis execution. Moreover, Meg3 depletion was conducive to protect RBMVECs against OGD + HGinduced ferroptosis.
Taken together, we found that the knockdown of Meg3 protected RBMVECs against the injury induced by OGD + hyperglycemic treatment. Most importantly, the present study also provided novel insights that Meg3 served as a ferroptotic promoter to mediate diabetic brain ischemic damage by functionally activating p53 to inhibit GPX4 activity. Therefore, Meg3 promoted ferroptosis in RBMVECs induced by OGD + HG through the p53-GPX4 axis (Figure 8). Such information may help to develop a novel strategy for the therapeutic intervention of diabetic brain ischemia.
Funding Statement
Funding: This work was supported by the National Natural Science Foundation of China (grant 81771422) and Hunan Provincial Natural Science Foundation (no. 2019JJ50931).
References
- 1.Vlisides P, Mashour GA. Perioperative stroke. Can J Anaesth 2016;63:193-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pai SL, Wang RD, Aniskevich S.Perioperative stroke: incidence, etiologic factors, and prevention. Minerva Anestesiol 2017;83:1178-89. [DOI] [PubMed] [Google Scholar]
- 3.Chen R, Ovbiagele B, Feng W.Diabetes and stroke: Epidemiology, pathophysiology, pharmaceuticals and outcomes. Am J Med Sci 2016;351:380-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alloubani A, Saleh A, Abdelhafiz I.Hypertension and diabetes mellitus as a predictive risk factors for stroke. Diabetes Metab Syndr 2018;12:577-84. [DOI] [PubMed] [Google Scholar]
- 5.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almutairi MMA, Xu G, Shi H.Iron pathophysiology in stroke. Adv Exp Med Biol 2019;1173:105-23. [DOI] [PubMed] [Google Scholar]
- 7.Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Li XL, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry 2017;22:1520-30. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez Calle A, Kawamura Y, Yamamoto Y, Takeshita F, Ochiya T.Emerging roles of long non-coding RNA in cancer. Cancer Sci 2018;109:2093-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 2016;17:47-62. [DOI] [PubMed] [Google Scholar]
- 10.Deng Y, Chen D, Wang L, Gao F, Jin B, Lv H, et al. Silencing of long noncoding RNA nespas aggravates microglial cell death and neuroinflammation in ischemic stroke. Stroke 2019;50:1850-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Zhu XL, Ji BY, Cao X, Yu LJ, Zhang Y, et al. LncRNA-1810034E14Rik reduces microglia activation in experimental ischemic stroke. J Neuroinflammation 2019;16:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang J, Wang Q, Li JQ, Guo T, Yu D.Long non-coding RNA MEG3 promotes cerebral ischemia-reperfusion injury through increasing pyroptosis by targeting miR-485/AIM2 axis. Exp Neurol 2020;325:113139. [DOI] [PubMed] [Google Scholar]
- 13.Zhang P, Sun Y, Peng R, Chen W, Fu X, Zhang L, et al. Long non-coding RNA Rpph1 promotes inflammation and proliferation of mesangial cells in diabetic nephropathy via an interaction with Gal-3. Cell Death Dis 2019;10:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdulle LE, Hao JL, Pant OP, Liu XF, Zhou DD, Gao Y, et al. MALAT1 as a diagnostic and therapeutic target in diabetesrelated complications: A promising long-noncoding RNA. Int J Med Sci 2019;16:548-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sommerkamp P, Renders S, Ladel L, Hotz-Wagenblatt A, Schonberger K, Zeisberger P, et al. The long non-coding RNA Meg3 is dispensable for hematopoietic stem cells. Sci Rep 2019;9:2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao H, Wang X, Feng X, Li X, Pan L, Liu J, et al. Long noncoding RNA MEG3 regulates proliferation, apoptosis, and autophagy and is associated with prognosis in glioma. J Neurooncol 2018;140:281-8. [DOI] [PubMed] [Google Scholar]
- 17.Yan H, Yuan J, Gao L, Rao J, Hu J.Long noncoding RNA MEG3 activation of p53 mediates ischemic neuronal death in stroke. Neuroscience 2016;337:191-9. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Li Q, Zhang KS, Hu B, Niu X, Zhou SM, et al. Downregulation of the long non-coding RNA meg3 promotes angiogenesis after ischemic brain injury by activating notch signaling. Mol Neurobiol 2017;54:8179-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan H, Rao J, Yuan J, Gao L, Huang W, Zhao L, et al. Long non-coding RNA MEG3 functions as a competing endogenous RNA to regulate ischemic neuronal death by targeting miR- 21/PDCD4 signaling pathway. Cell Death Dis 2017;8:3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyu Y, Lou J, Yang Y, Feng J, Hao Y, Huang S, et al. Dysfunction of the WT1-MEG3 signaling promotes AML leukemogenesis via p53-dependent and -independent pathways. Leukemia 2017;31:2543-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uroda T, Anastasakou E, Rossi A, Teulon JM, Pellequer JL, Annibale P, et al. Conserved pseudoknots in lncRNA MEG3 are essential for stimulation of the p53 pathway. Mol Cell 2019;75:982-95.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mao C, Wang X, Liu Y, Wang M, Yan B, Jiang Y, et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res 2018;78:3484-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol 2019;21:579-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, Chen Z, Yan M, He P, Chen Z, Dai H.The protective role of isorhamnetin on human brain microvascular endothelial cells from cytotoxicity induced by methylglyoxal and oxygenglucose deprivation. J Neurochem 2016;136:651-9. [DOI] [PubMed] [Google Scholar]
- 25.Zhan R, Xu K, Pan J, Xu Q, Xu S, Shen J.Long noncoding RNA MEG3 mediated angiogenesis after cerebral infarction through regulating p53/NOX4 axis. Biochem Biophys Res Commun 2017;490:700-6. [DOI] [PubMed] [Google Scholar]
- 26.Pan Y, Wang N, Xia P, Wang E, Guo Q, Ye Z.Inhibition of Rac1 ameliorates neuronal oxidative stress damage via reducing Bcl-2/Rac1 complex formation in mitochondria through PI3K/Akt/mTOR pathway. Exp Neurol 2018;300:149-66. [DOI] [PubMed] [Google Scholar]
- 27.Xia P, Pan Y, Zhang F, Wang N, Wang E, Guo Q, et al. Pioglitazone confers neuroprotection against ischemia-induced pyroptosis due to its inhibitory effects on HMGB-1/RAGE and Rac1/ROS pathway by activating PPAR. Cell Physiol Biochem 2018;45:2351-68. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z, Ye Z, Huang G, Wang N, Wang E, Guo Q.Sevoflurane post-conditioning enhanced hippocampal neuron resistance to global cerebral ischemia induced by cardiac arrest in rats through PI3K/Akt survival pathway. Front Cell Neurosci 2016;10:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye Z, Li Q, Guo Q, Xiong Y, Guo D, Yang H, et al. Ketamine induces hippocampal apoptosis through a mechanism associated with the caspase-1 dependent pyroptosis. Neuropharmacology 2018;128:63-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia P, Zhang F, Yuan Y, Chen C, Huang Y, Li L, et al. ALDH 2 conferred neuroprotection on cerebral ischemic injury by alleviating mitochondria-related apoptosis through JNK/caspase-3 signing pathway. Int J Biol Sci 2020;16:1303-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiu GZ, Tian W, Fu HT, Li CP, Liu B.Long noncoding RNAMEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem Biophys Res Commun 2016;471:135-41. [DOI] [PubMed] [Google Scholar]
- 32.Tong P, Peng QH, Gu LM, Xie WW, Li WJ. LncRNA-MEG3 alleviates high glucose induced inflammation and apoptosis of retina epithelial cells via regulating miR-34a/SIRT1 axis. Exp Mol Pathol 2019;107:102-9. [DOI] [PubMed] [Google Scholar]
- 33.Chanda K, Das S, Chakraborty J, Bucha S, Maitra A, Chatterjee R, et al. Altered levels of long NcRNAs meg3 and neat1 in cell and animal models of Huntington's disease. RNA Biol 2018;15:1348-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson R. Long non-coding RNAs in Huntington's disease neurodegeneration. Neurobiol Dis 2012;46:245-54. [DOI] [PubMed] [Google Scholar]
- 35.He C, Yang W, Yang J, Ding J, Li S, Wu H, et al. Long noncoding RNA MEG3 negatively regulates proliferation and angiogenesis in vascular endothelial cells. DNA Cell Biol 2017;36:475-81. [DOI] [PubMed] [Google Scholar]
- 36.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ 2016;23:369-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Angeli JPF, Shah R, Pratt DA, Conrad M.Ferroptosis inhibition: Mechanisms and opportunities. Trends Pharmacol Sci 2017;38:489-98. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, et al. Ischemiainduced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ 2019;26:2284-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W, Gai C, Ding D, Wang F, Li W.Targeted p53 on small-molecules-induced ferroptosis in cancers. Front Oncol 2018;8:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015;520:57-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156:317-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Hambright WS, Na R, Ran Q.Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem 2015;290:28097-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu T, Jiang L, Tavana O, Gu W.The Deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res 2019;79:1913-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye J, Jiang X, Dong Z, Hu S, Xiao M.Low-concentration PTX And RSL3 inhibits tumor cell growth synergistically by inducing ferroptosis in mutant p53 hypopharyngeal squamous carcinoma. Cancer Manag Res 2019;11:9783-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang C, Yang M, Deng J, Li P, Su W, Jiang R.Upregulation and activation of p53 by erastininduced reactive oxygen species contribute to cytotoxic and cytostatic effects in A549 lung cancer cells. Oncol Rep 2018;40:2363-70. [DOI] [PubMed] [Google Scholar]