

Abstract
Acute liver failure (ALF) causes severe liver dysfunction that can lead to multi-organ failure and death. Previous studies suggest that sphingosine kinase 1 (SphK1) protects against hepatocyte injury, yet not much is still known about its involvement in ALF. This study examines the role of SphK1 in D-galactosamine (GalN)/lipopolysaccharide (LPS)-induced ALF, which is a well-established experimental mouse model that mimics the fulminant hepatitis. Here we report that deletion of SphK1, but not SphK2, dramatically decreased GalN/LPS-induced liver damage, hepatic apoptosis, serum alanine aminotransferase levels, and mortality rate compared to wild-iype mice. Whereas GalN/LPS treatment-induced hepatic activation of NF-κB and JNK in wild-type and SphK2−/− mice, these signaling pathways were reduced in SphK1−/− mice. Moreover, repression of ALF in SphK1−/− mice correlated with decreased expression of the pro-inflammatory cytokine TNFα. Adoptive transfer experiments indicated that SphK1 in bone marrow-derived infiltrating immune cells but not in host liver-resident cells, contribute to the development of ALF. Interestingly, LPS-induced TNFα production was drastically suppressed in SphK1-deleted macrophages, whereas IL-10 expression was markedly enhanced, suggesting a switch to the anti-inflammatory phenotype. Finally, treatment with a specific SphK1 inhibitor ameliorated inflammation and protected mice from ALF. Our findings suggest that SphK1 regulates TNFα secretion from macrophages and inhibition or deletion of SphK1 mitigated ALF. Thus, a potent inhibitor of SphK1 could potentially be a therapeutic agent for fulminant hepatitis.
Keywords: acute liver failure, apoptosis, cytokines, sphingosine kinase
1 ∣. INTRODUCTION
Acute liver failure (ALF) is a critical medical condition defined as rapid development of hepatic dysfunction associated with increased levels of liver enzymes, hepatic encephalopathy, severe coagulopathy, systemic inflammatory response syndrome, and ultimately multi-organ failure.1-3 Although much progress has been made in the management of ALF, it remains an important cause of liver-related mortality and liver transplantation is the only proven therapy for those who develop severe ALF.2 Better understanding of the molecular mechanisms of disease progression and the development of multi-organ failure as well as new therapeutic approaches are still needed.
Bacterial infections are common complications in ALF that could lead to mortality.4 Lipopolysaccharide (LPS), a major component of Gram-negative bacteria that enters from altered gut barrier function, causes uncontrolled production of pro-inflammatory cytokines that are central to the pathogenesis of ALF.5 Patients with ALF have higher circulating concentrations of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNFα), interleukin1β (IL1β), and IL62,6 that further injure damaged liver hepatocytes. It is widely accepted that the mortality in ALF is a consequence of profound activation of systemic inflammatory responses and its attendant complications of recurrent sepsis and extra-hepatic organ dysfunction.2,7
Lipopolysaccharide (LPS) and D-galactosamine (GalN)-induced acute hepatic damage in mice has been extensively used as an experimental animal model that imitates the pathological hallmarks of human ALF and is relevant for exploring potential therapeutics.8 GalN, an amino sugar and hepatocyte-specific transcriptional inhibitor, increases LPS-induced hepatotoxicity.9 LPS stimulates the production of pro-inflammatory cytokines, including TNFα that triggers massive hepatocyte apoptosis and liver injury.10,11 Activation of liver-resident macrophages, Kupffer cells, by pathogens leads to the recruitment of bone marrow-derived monocytes that differentiate into macrophages expanding the macrophage pools that can promote tissue destruction or modify recovery from injury.12-14 While Kupffer cells are critical for homeostasis, secretion of TNFα by monocyte-derived macrophages predominate in acute or chronic liver injury.12-14 Indeed, depletion of macrophages and inhibition of TNFα production protected against GalN/LPS-induced hepatic injury.15 Furthermore, neutralizing anti-TNF antibodies protected mice against liver injury, supporting its critical roles in ALF.11
The bioactive sphingolipid metabolite sphingosine-1-phosphate (S1P) and the enzymes that produce it, sphingosine kinases (SphKs), have multiple effects on various components of the cellular immune response.16 Numerous pro-inflammatory stimuli, including LPS and TNFα, activate SphK1 leading to the formation of S1P and blockade of SphK1 inhibits pro-inflammatory responses triggered by these stimuli.16 Many of the actions of S1P are mediated by its binding to five G protein-coupled receptors, named S1PR1-5.16 S1P and its receptors are involved in liver pathophysiology and contribute to the development of liver diseases.17,18 Previous studies suggest that SphK1 protects against hepatocyte injury, yet not much is still known about the role of S1P and SphKs in ALF. Intriguingly, although in most studies S1P seems to contribute to liver damage, in others it seems to have a protective role. For example, S1P is important for recruitment and activation of liver-damaging immune cells, but in vitro studies indicate that S1P protects hepatocytes from apoptotic insults.19-21 Moreover, elevation of serum S1P attenuates impaired cardiac function in experimental sepsis22 and protects against LPS-induced death and organ injuries.23 In contrast, several reports indicated that SphK1 inhibitors including a poorly characterized inhibitor,24 a non-isozyme specific inhibitor,25 and a potent SphK1 inhibitor26 improved ALF in mice. Conversely, an inhibitor of SphK2 improved survival after hepatic ischemia-reperfusion and suppressed cell death in cultured hepatocytes.27 In the present study, we examined the roles of SphK1 and SphK2 in the well-characterized mouse model of GalN/LPS-induced ALF. We found that deletion or inhibition of SphK1, but not deletion of SphK2, markedly protected from fulminant hepatic failure by suppressing TNFα production by bone marrow-derived macrophages. Our findings support the notion that inhibition of SphK1 might be a potential therapeutic strategy for ALF by promoting a switch in macrophage function to a pro-resolution phenotype.
2 ∣. MATERIALS AND METHODS
2.1 ∣. Animal model of ALF and treatments
SphK1−/− and SphK2−/− and wild-type littermates were originally obtained from R. Proia (NIH, NIDDK) and bred at VCU for more than 20 generations. Male mice aged 8-10 weeks were used for all experiments. All animal procedures were performed as approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University. Acute liver failure was induced in mice by intraperitoneal injection of GalN (600 mg/kg; Sigma-Aldrich, St. Louis, MO) and LPS (10 μg/kg; Sigma-Aldrich). In some experiments, to inhibit SphK1 activity in vivo, mice were intraperitoneally injected 0.5 hour prior to the onset of ALF with 10 mg/kg of SK1-I (ENZO, Farmingdale, NY), a specific SphK1 inhibitor.
2.2 ∣. Histological analysis
Mice were weighed and euthanized with inhaled isoflurane and cervical dislocation. Livers were removed and a portion sectioned, fixed in 4% formaldehyde, and embedded in paraffin for histological analysis. The remaining liver was snap-frozen in liquid nitrogen. Formalin fixed and paraffin-embedded liver slices were stained with hematoxylin and eosin (H&E). Apoptosis was measured by terminal uridine deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay using the ApopTag Peroxidase In Situ Apoptosis Detection Kit S7100 (Millipore). Formaldehyde fixed liver sections were also blocked with 2.5% of horse serum for 1 hour, and then, stained with anti-F4/80 (1:200, AbD Serotec). After two washes with PBS, sections were stained with Alexa Fluor 594-conjugated secondary antibodies (1:500, Invitrogen) for 30 minutes. Nuclei were counterstained with Hoechst 33432 (Invitrogen) for 5 minutes as previously described.28 Slides were examined with a Zeiss Axioimager A1 and images were captured with an AxioCam MRc camera (Zeiss, Thornwood).28 Stained sections were analyzed with ImageJ (NIH). Three fields were selected randomly from each section.
2.3 ∣. Caspase assays
Caspase 3/7 and 8 activities in livers were measured using the Caspase-Glo assay kit (Promega) as previously described.29 Briefly, livers were homogenized in 25 mM of HEPES buffer, pH7.5 containing 5 mM of MgCl2, 1 mM of EGTA, 1 mM of PMSF, and 1 μg/mL of each pepstatin, leupeptin, and aprotinin. Activities were measured by adding peptide substrates containing DEVD or LEHD sequences cleaved by caspase 3 and caspase 8, respectively, to cytosolic proteins in a white-walled 96-well plate. Luminescence was measured in a plate-reading luminometer (Perkin Elmer Victor X5 2030 multilabel plate reader).
2.4 ∣. Blood collection and analysis
Blood was collected from hepatic veins and serum obtained by centrifugation for 15 minutes at 1500g at 4°C. Serum samples were stored at −80°C until analysis. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured by Antech Diagnostics (Morrisville, NC). Cytokine levels were measured using an anti-mouse ELISA kit (RD Systems, Minneapolis).
2.5 ∣. Quantitative PCR
RNA was extracted from liver tissues and cells with TRIzol Reagent and converted by the High-Capacity cDNA Reverse Transcription Kit to cDNA. Premixed primer-probe sets and TaqMan Universal PCR Master Mix (Applied Biosystems) were used to examine mRNA levels. cDNAs were diluted 10-fold (for the target genes) or 100-fold (for GAPDH) and amplified using the ABI7900HT cycler. Gene expression levels were calculated by the ΔΔCt method and normalized to GAPDH expression.30
2.6 ∣. Western blot analysis
Liver tissues were pulverized in liquid nitrogen and then, lysed in buffer containing 50 mM of HEPES, pH 7.4, 150 mM of NaCl, 100 mM of NaF, 10% of glycerol, 10 mM of sodium pyrophosphate, 200 μM of sodium vanadate, 10 mM of EDTA, and 1% of Triton X-100, supplemented with Halt Protease and Phosphatase Inhibitors as described.30 Proteins were separated on 10% SDS-PAGE and then, transferred to 0.45 μm nitrocellulose membranes. Membranes were blocked with 5% BSA-TBST and incubated overnight with primary antibodies and immunopositive bands were visualized by enhanced chemiluminescence.30
2.7 ∣. Bone marrow chimeras
Bone marrows from femurs and tibia were isolated from SphK1+/+ congenic mice (expressing CD45.1 leukocyte antigen) and SphK1−/− (expressing CD45.2 leukocyte antigen) donor mice, and cells were re-suspended in PBS after washing. The recipients were given acidified, antibiotic-treated drinking water for 2 weeks, irradiated, and then, injected iv with 5 x 106 donor cells. Bone marrow reconstitution efficiency was verified after 6 weeks by staining for CD45.1 and CD45.2 in blood cells using FITC-conjugated anti-CD45.1 and PE-conjugated anti-CD45.2 (Biolegend) as we described previously.31
2.8 ∣. Peritoneal macrophages
Mouse peritoneal macrophages were elicited by intraperitoneal injection of 4% thioglycollate.32 Cells were washed, suspended in DMEM with 10% of FBS, cultured for 4 hours, and non-adherent cells removed as previously described.32
2.9 ∣. Quantification of S1P and SK1-I by mass spectrometry
Lipids were extracted from blood and liver and S1P and SK1-I quantified by liquid chromatography electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) using 4000 QTRAP (AB Sciex) as previously described.28
2.10 ∣. Statistical analysis
For mouse studies, three to eight randomly chosen mice were used for each experimental group. All cell culture data were from biological triplicates. Statistical analyses were performed using unpaired two-tailed Student's t test for comparison of two groups and analysis of variance (ANOVA) followed by post hoc tests for multiple comparisons. P < .05 was considered significant.
3 ∣. RESULTS
3.1 ∣. SphK1 null mice, but not SphK2 null mice, are resistant to ALF
To examine the role of SphKs in ALF, we used a model of ALF that involves co-administration of D-galactosamine and lipopolysaccharide (GalN/LPS).9 Consistent with numerous previous studies,9,11,33-35 within 6 hours after GalN/LPS treatment, 90%-100% of wild-type mice were dead. In sharp contrast, all of the SphK1−/− mice remained viable and the survival rate was 100% (8/8) at 48 hours (Figure 1A). Liver histology revealed injury damaged hepatic lobules, hemorrhage, and severe inflammatory cell infiltration in the livers of the GalN/LPS-treated WT mice but not in SphK1−/− mice (Figure 1B). Likewise, increases in serum of the liver enzymes, aspartate transaminase (AST), alanine transaminase (ALT), and markers of liver injury, observed in WT mice 4 hours after GalN/LPS injections were significantly lower in SphK1−/− mice (Figure 1C). Because SphK1 and SphK2 have some overlapping and some distinct functions as well as subcellular localizations,16 it was of interest to compare the effects on GalN/LPS-induced ALF in SphK2−/− mice. However, there was no significant difference in mortality between WT and SphK2−/− mice following challenge with GalN/LPS (Figure 1A). Moreover, as with WT mice, extensive hepatic parenchymal damage, hemorrhage, and immune cell infiltration were observed in SphK2−/− mice (Figure 1B).
FIGURE 1.

Deletion of SphK1 improves survival, attenuates liver enzyme release, and tissue damage in acute liver failure in mice. (A-C) WT, SphK1−/−, and SphK2−/− mice were treated without or with GalN/LPS. n = 7-9 mice/group. A, Survival rates were determined by Kaplan-Meier analysis. P = .004, F = 8.276, log rank test for SphK1−/− compared to WT mice. B, Representative H&E-stained liver sections of mice at 4 hours after GalN/LPS administration. Magnification × 20. C, Serum ALT and AST levels were measured 4 hours after GalN/LPS administration. Data are means ± SEM. * P < .01 compared to WT
3.2 ∣. Deletion of SphK1 suppresses GalN/LPS-induced hepatocyte apoptosis
Massive hepatic apoptosis is a prominent pathogenic process that leads to ALF.10,36 We next examined the effect of SphK1 deletion on hepatocyte apoptosis determined by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. As expected, the number of apoptotic hepatocytes was greatly increased in the GalN/LPS-treated WT mice (Figure 2A,B). Consistent with the enhanced survival of SphK1−/−-treated mice (Figure 1A), apoptosis was not increased in these mice by GalN/LPS challenge (Figure 2A,B). In contrast, apoptotic hepatocyes in SphK2−/− mice were increased to a similar extent observed in liver sections from WT mice. Consistent with this, activation of caspase 3, the final executionary step preceding apoptosis, as measured by increased levels of cleaved caspase 3 in immunoblots was observed after GalN/LPS challenge only in WT and SphK2−/− but not in SphK1−/− mice (Figure 2C), suggesting SphK1 deletion protects the liver from hepatocyte apoptosis.
FIGURE 2.

SphK1 deletion protects from GalN/LPS-induced apoptosis. A-C, WT, SphK1−/−, and SphK2−/− mice were treated without or with GalN/LPS. (A,B) After 4 hours, liver sections were TUNEL stained to visualize apoptosis and quantified as described in Materials and Methods. Data are expressed as arbitrary units (AU) and are means ± SEM. n = 3. * P < .01 compared to WT. C, Proteins in liver lysates were separated by SDS-PAGE and analyzed by immunoblotting with antibody for cleaved caspase 3. Blots were re-probed with anti-GAPDH to show equal loading and transfer
3.3 ∣. Effect of deletion of SphK1 on signaling pathways induced by GalN/LPS
To elucidate the role of SphK1 in apoptosis and liver injury, we examined the signaling pathways involved in GalN/LPS-induced ALF. LPS binds to a receptor complex of Toll-like receptor 4 (TLR4), cluster of differentiation 14 (CD14), and MD2. Recruitment of the adaptor protein myeloid differentiation factor-88 (MyD88) leads to activation of the transcription factor-kB (NF-kB) that regulates the expression of many pro-inflammatory cytokines and the MAPK kinase JNK1/2, known to trigger hepatocyte apoptosis.37 In WT and SphK2−/− mice, GalN/LPS treatment induced a significant decrease in hepatic levels of IκB, a regulatory protein that inhibits NF-κB (Figure 3A,B). In contrast, IκB levels were only slightly decreased in SphK1−/− mice (Figure 3A), demonstrating reduction of active NF-κB signaling in these livers. Moreover, levels of active, phosphorylated p46, and p54 JNK were markedly elevated in GalN/LPS-treated WT and SphK2−/− mice (Figure 3B), whereas activation of JNK was markedly decreased in the absence of SphK1 expression (Figure 3A). Since it was previously suggested that another transcription factor, STAT3, mediates survival signaling in GalN/LPS-induced liver injury,38 we also examined the effect of deletion of SphK1 on STAT3 activation. Nevertheless, GalN/LPS induced similar increases in p-STAT3 levels in both WT and SphK1−/− mice (Figure 3A), suggesting that deletion of SphK1 has differential effects on signaling pathways involved in liver injury.
FIGURE 3.
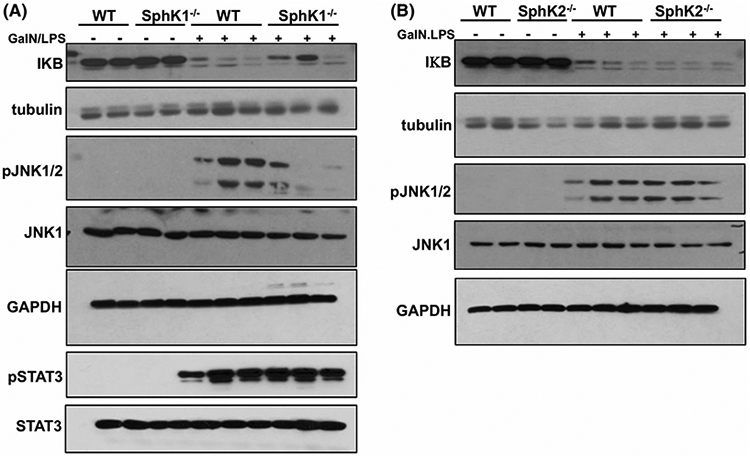
Effect of deletion of SphK1 on signaling pathways involved in GalN/LPS-induced acute liver failure. A and B, WT, SphK1−/−, and SphK2−/− mice were treated without or with GalN/LPS. n = 2-3 mice per group. One hour later, proteins in liver lysates were separated by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies. Tubulin and GAPDH were used as loading controls
3.4 ∣. Reduction of TNFα correlates with repression of ALF in SphK1−/− mice
GalN/LPS-induced hepatotoxicity and subsequent multi-organ failure depends on cytokine responses, particularly those of the pro-inflammatory cytokines TNFα and IL1β,10,36 and we, therefore, measured their hepatic levels. Consistent with previous studies,10,36 mRNA expressions of TNFα and IL1β in the livers were markedly increased after GalN/LPS challenge of WT mice (Figure 4A). Both of these were significantly reduced by 73% and 38%, respectively, in SphK1−/− livers. In contrast, hepatic mRNA expression of these cytokines in SphK2−/− livers were elevated to similar extents as in WT mice. Because GalN/LPS-induced ALF is mainly dependent on secreted TNFα signaling,36 we also measured the serum levels of TNFα. Serum TNFα increased rapidly and markedly in GalN/LPS-treated WT mice, reaching a maximal value within 1 hour after GalN/LPS administration.33,39 Consistent with the mRNA findings (Figure 4A), GalN/LPS failed to increase serum TNFα levels in SphK1−/− mice (Figure 4B). In sharp contrast, TNFα levels in serum from SphK2−/− mice were increased slightly more than in WT mice. Taken together these results suggest that the resistance of SphK1 deleted mice to ALF is due to a blockage in TNFα production.
FIGURE 4.
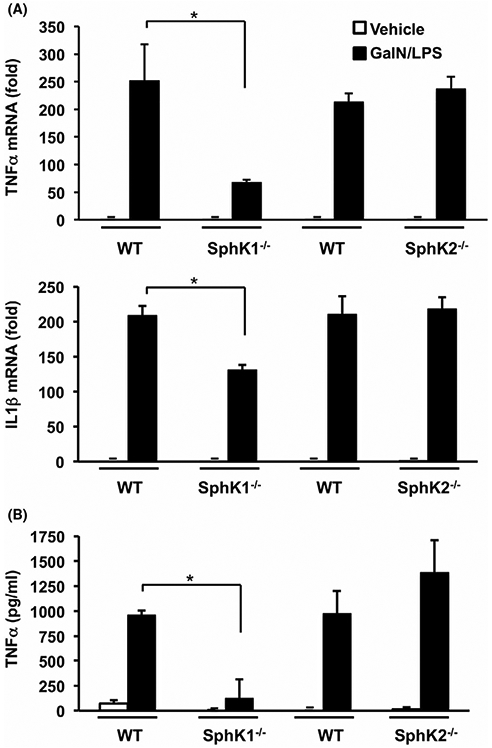
Lack of SphK1 but not SphK2, suppresses pro-inflammatory TNFα production triggered by GalN/LPS. (A,B) WT, SphK1−/−, and SphK2−/− mice were treated with vehicle or with GalN/LPS for 1 hour. n = 4-6. A, TNFα and IL1β mRNA expression in liver was determined by QPCR and normalized to GAPDH expression. B, TNFα levels in serum were measured by ELISA. Data are mean ± SD. * P < .05 compared to treated WT
3.5 ∣. SphK1 in infiltrating immune cells mediates GalN/LPS-induced liver damage
To examine whether GalN/LPS hepatotoxicity depends on expression of SphK1 in resident hepatocytes, stellate cells, or Kupffer cells, or by infiltrating immune cells, we generated reciprocal bone marrow chimeras by adoptively transferring bone marrow into lethally irradiated WT and SphK1 knockout recipient mice. Intriguingly, H&E and TUNEL staining after GalN/LPS challenge showed that SphK1−/− mice engrafted with WT bone marrow cells had increased liver damage (Figure 5A) and apoptosis (Figure 5B), compared to engraftment with SphK1−/− bone marrow cells. Moreover, engraftment of WT mice with SphK1−/− bone marrow greatly dampened GalN/LPS-induced liver damage and apoptosis (Figure 5A,B). Similarly, much higher AST serum levels were found in SphK1−/− mice when engrafted with WT but not with SphK1−/− bone marrow cells (Figure 5C).
FIGURE 5.

SphK1 in bone marrow-derived hematopoietic cells is critical for GalN/LPS-induced acute liver failure. A-C, Adoptive transfer of bone marrow from WT or SphK1−/− mice to the indicated recipient mice was carried out as described in Materials and Methods. After 6 weeks, mice were treated without or with GalN/LPS for 1 or 4 hours. n = 2 for untreated mice and n = 4 for treated mice. A, Liver damage 4 hours after the onset of ALF was visualized by H&E staining. B, After 4 hours, liver sections were TUNEL stained to visualize apoptosis. Magnification × 20. C, AST activity was measured in blood 1 hour after treatment with GalN/LPS. Data are mean ± SD. * P < .05
Hepatic expression of the pro-inflammatory cytokines TNFα, IL1β, and IL6 after GalN/LPS challenge, were significantly reduced in livers of SphK1−/− compared to WT mice (Figure 6A). Moreover, these cytokines were also increased in SphK1−/− chimeric mice. Conversely, WT chimeras had markedly reduced levels of these pro-inflammatory cytokines (Figure 6A). Importantly, after GalN/LPS challenge, expression of the anti-inflammatory cytokine, IL10, was enhanced in SphK1−/− compared to WT mice (Figure 6A). In addition, engraftment of SphK1−/− mice with WT bone marrow cells greatly reduced IL10 expression, whereas engraftment of WT mice with bone marrow cells from SphK1−/− mice enhanced it (Figure 6A). Together, these results indicate that SphK1 in bone marrow-derived cells, not SphK1 present in host liver-resident cells, contribute to the development of acute liver injury.
FIGURE 6.

Deletion of SphK1 in bone marrow-derived hematopoietic cells and macrophages suppresses TNFα and enhances IL10 expression. A, ALF was induced by GalN/LPS treatment of WT, SphK1−/− mice, or in bone marrow chimeric mice generated as described in Materials and Methods. One hour after treatment, TNFα, IL6, IL1β, and IL10 mRNA expression was determined in liver by QPCR and normalized to GAPDH expression. n = 2 for untreated mice and n = 3 for treated mice. Data are expressed as mean ± SD. ** P < .01, * P < .05. B and C, Peritoneal macrophages isolated from either WT or SphK1−/− mice were treated with LPS (100 ng/mL) for the indicated times. B, TNFα and IL10 mRNA levels were determined by QPCR and normalized to GAPDH expression. C, TNFα levels from peritoneal macrophages were measured by ELISA. n = 4. Data are mean ± SD. ** P < .01, * P < .05 compared to WT
3.6 ∣. Effect of SphK1 deletion on macrophage polarization
Because GalN/LPS-induced liver injury is critically dependent on macrophage-derived pro-inflammatory cytokines,14,15,40 it was of interest to examine whether deletion of SphK1 influences the activation and/or polarization of macrophages. To this end, peritoneal macrophages isolated from WT or SphK−/− mice were activated with LPS. As expected, TNFα mRNA expression and protein levels, a typical M1 phenotype marker, were rapidly increased after treatment of WT macrophages with LPS, reaching a peak at 2 hours (Figure 6B,C). In contrast, TNFα production was significantly suppressed in SphK1 deleted macrophages (Figure 6B,C). Moreover, production of IL10 was significantly enhanced in SphK1 deleted macrophages compared to WT (Figure 6B), suggesting a switch to the anti-inflammatory M2 phenotype.
Consistent with these findings, GalN/LPS exposure increased F4/80+ macrophages in hepatic tissues in WT compared to SphK1−/− mice (Figure 7A). Moreover, in addition to M1 gene signatures including TNFα, IL1β, and IL6 (Figure 6), mRNA of CD86, a M1 type macrophage marker,41 was also significantly upregulated by GalN/LPS only in WT and SphK2−/− mice (Figure 7A). In support of a switch to the anti-inflammatory M2 phenotype in SphK1−/− mice (Figure 6B), the M2 signature gene CD20641 was upregulated only in ALF of SphK1−/− mice (Figure 7A).
FIGURE 7.

Effect of deletion and inhibition of SphK1 on GalN/LPS-induced macrophages infiltration. A, WT, SphK1−/−, and SphK2−/− mice were treated without or with GalN/LPS for 1 hour. Expression of F4/80, CD86, and CD206 mRNA were determined by QPCR and normalized to GAPDH. n = 2 for untreated mice and n = 3 for treated mice. Data are expressed as fold increases over the untreated group. B and C, WT mice were treated with the SphK1-specific inhibitor SKI-1 (10 mg/kg) or PBS (Vehicle) prior to GalN/LPS administration. After 4 hours liver sections were stained with anti-F4/80 antibody (red) and counterstained with Hoechst (blue). B, Representative confocal fluorescent images. C, Data are expressed as arbitrary fluorescence units (AFU) and are means ± SD
3.7 ∣. Inhibition of SphK1 protects from ALF
Next, we sought to examine the effects of the isotype-specific SphK1 inhibitor SK1-I42 on GalN/LPS-induced hepatic injury. Whereas vehicle treated mice began to die 4 hours after GalN/LPS injection, and none survived at 6 hours, treatment with SKI-1 dramatically increased the survival rate and 25% of the treated mice remained alive even at 48 hours (Figure 8A). Histological analysis of H&E sections and TUNEL staining confirmed attenuated liver injury and apoptosis induced by GalN/LPS in SK1-I-treated mice (Figure 8B,C). In agreement, SK1-I treatment markedly suppressed activation of caspase 3/7 and caspase 8 (Figure 8D). Immunohistochemical analysis of F4/80-stained liver cells, a marker for Kupffer cells and monocyte-derived macrophages, showed that macrophages infiltrated into liver tissues after GalN/LPS challenge, and SKI-1 pretreatment significantly prevented the infiltration (Figure 7B,C). Moreover, TNFα and IL1β expression, which were remarkably enhanced at 4 hours after GalN/LPS administration, were significantly reduced by SK1-I (Figure 8E), suggesting that it also ameliorated inflammation. Even TNFα levels in serum induced by GalN/LPS did not increase in SK1-I-treated mice (Figure 8F). Together, these results demonstrate that inhibition of SphK1 provides significant protection from ALF.
FIGURE 8.

Inhibition of SphK1 improves survival, attenuates apoptosis and pro-inflammatory cytokine production in GalN/LPS-induced liver damage. A-D, WT mice were treated with the SphK1-specific inhibitor SKI-1 (10 mg/kg) or PBS before GalN/LPS administration n = 8. A, Survival rate was determined by Kaplan-Meier analysis. (P = .0091). B, Representative H&E-stained liver sections 4 hours after GalN/LPS administration. Magnification × 20. C, After 4 hours of GalN/LPS treatment, liver sections were TUNEL stained to visualize apoptosis. D, Caspase 3/7 and caspase 8 activities measured 4 hours after the onset of ALF. E, After 4 hours of GalN/LPS treatment, mRNA levels of TNFα and IL1β in liver were determined by QPCR and normalized to GAPDH expression. F, TNFα levels in serum were measured by ELISA. Data are mean ± SD. *P < .05 compared to vehicle
As was previously reported 43,44 deletion or inhibition of SphK1 in mice reduced circulating S1P. GalN/LPS treatment decreased S1P levels in their blood (Figure 9A). This is consistent with the observation that S1P levels are decreased in serum from patients with ALF45 due to decrease of the S1P carrier ApoM46 and subsequent vascular leakage.47 Like acetaminophen-induced ALF,26 GalN/LPS treatment also increased hepatic S1P levels, which were blunted in SphK1−/− mice or in WT mice treated with SK1-I (Figure 9B). Surprisingly, however, SK1-I levels in the blood and liver were greater in GalN/LPS treated than in non-treated mice (Figure 9C,D), even though the amount of SK1-I administered was identical, suggesting that either GalN/LPS administration or the injury itself increased retention or uptake of SK1-I.
FIGURE 9.
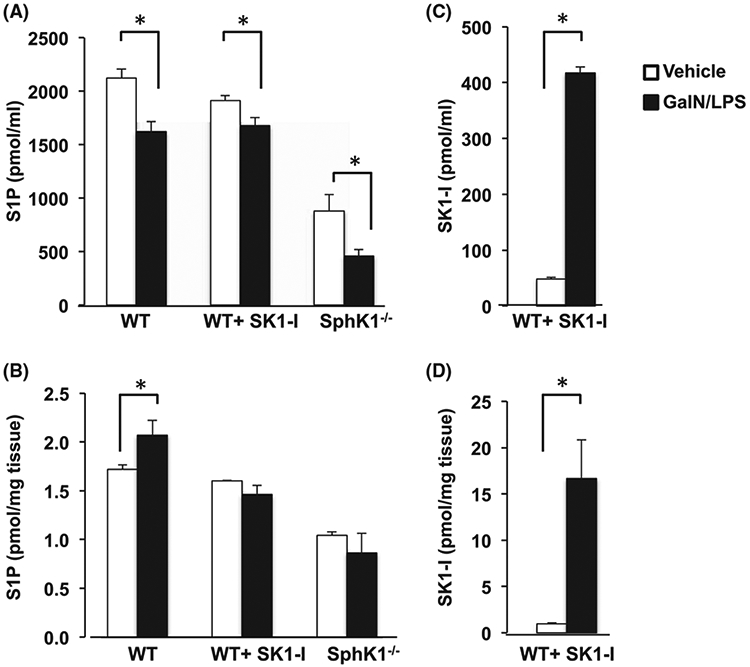
Effect of deletion or inhibition of SphK1 on blood and liver S1P levels. (A) WT mice treated without or with the SphK1-specific inhibitor SKI-1 (10 mg/kg) or SphK1−/− mice were challenged without (vehicle) or with GalN/LPS for 1 hour. Levels of S1P (A,B) and SK1-I (C,D) were determined by LC-ESI-MS/MS in blood (A,C) or liver (B,D). For A, n = 4-6; for B-D, n = 2 for untreated mice; n = 4 for GalN/LPS-treated mice. Data are mean ± SD. *P < .05 compared to vehicle
4 ∣. DISCUSSION
ALF is a severe consequence of abrupt hepatocyte injury that can lead to systemic inflammatory responses and multi-organ failure.1,2 GalN/LPS-induced ALF in mice is a widely used experimental model for better understanding of progression of the disease and for development of potential therapeutic agents.8,9,48 Here we found that deletion or inhibition of SphK1 protected mice against GalN/LPS-induced liver injury, hepatic apoptosis, and prolonged survival of the animals. Deletion of SphK1, but not SphK2, decreased the circulating levels of pro-inflammatory cytokines in mice injected with GalN/LPS and completely dampened the expression of TNFα induced by LPS in peritoneal macrophages, whereas the anti-inflammatory cytokine IL10 was greatly enhanced. Adoptive transfer of bone marrow cells into lethally irradiated recipient mice also demonstrated that SphK1 in bone marrow-derived inflammatory cells, and not in liver-resident Kupffer cells, hepatocytes, or stellate cells, is responsible for the elevated TNFα production and reduced IL10, and plays important roles in development of ALF. Consistent with these observations, previous studies showed that increased IL10 and decreased TNFα contribute to hepatoprotection.49,50 Our results fit with data indicating that SphK2 is a negative regulator of macrophage activation, as inhibition or deletion of SphK2 in mouse peritoneal macrophages increased LPS-induced inflammatory cytokine production.51
Although the sentinel functions of Kupffer cells normally dominate the hepatic macrophage pool in homeostasis, during injury, the liver macrophage pool is augmented by recruitment of bone marrow-derived monocytes which mature into macrophages and contribute to the development and/or resolution of hepatic inflammation and ALF. Our data suggest that SphK1 expression in bone marrow-derived macrophages is critical for the production of pro-inflammatory cytokines, including TNFα, and conversely, its deletion or inhibition switches them from “M1-like,” pro-inflammatory, to alternatively activated “M2-like,” anti-inflammatory phenotype with enhanced IL10 production that have been implicated in inflammation resolution and repair.52 Our data suggest that inhibition of SphK1 is a novel means to regulate the balance between pro- and anti-inflammatory cytokines and the switch from inflammatory to restorative macrophages that limit hepatic damage.
It was suggested that a predominance of NF-κB activation promotes M1 macrophage polarization, while in contrast, a predominance of STAT3 activation results in M2 macrophage polarization.53 In this regard, it is interesting to note that deletion of SphK1 reduced GalN/LPS-induced activation of NF-κB, whereas STAT3 activation was not affected. However, it has become apparent that liver macrophages cannot be described solely as M1 or M2 phenotypes as macrophages have substantial plasticity and these dichotomous phenotypes represent the extremes of different macrophage activation states.53
There is growing evidence that the SphK1/S1P axis is involved in inflammation and that targeting this axis could be a potential therapy for inflammatory disorders.16,54 Although surprisingly, SphK1 and S1P protect cultured hepatocytes from apoptotic insults in vitro,19-21 we found that SphK1 contributes to liver damage induced by GalN/LPS. Consistent with our findings, N,N-dimethylsphingosine, which inhibits both SphK1 and SphK2 but also has other activities,55 reduced mortality, liver inflammation, and serum levels of the pro-inflammatory cytokines TNFα, IL-1, and IL-625 by reduced activation of PKCδ.56 Another poorly characterized SphK1 inhibitor, SKI-5c, also reduced GalN/LPS-induced ALF. Yet, although in addition to hepatic SphK1, S1PR1, and S1PR3 levels were also elevated in ALF, antagonists of S1PR1 or S1PR3 were ineffective.24 However, treatment with the S1PR1 functional antagonists FTY720 or KRP203, suppressed Con A-induced hepatitis by targeting CD4+ peripheral blood T lymphocytes.57 Moreover, administration of S1PR2 or S1PR3 antagonists reduced bone marrow-derived monocyte/macrophage recruitment in cholestatic liver injury in mice and attenuated hepatic inflammation and fibrosis.58 However, in contrast to the studies implicating SphK1 in ALF,24-26 others demonstrated that treatment with a SphK2 inhibitor improved survival and liver function in acute liver damage induced by ischemia and reperfusion.27 To resolve these seemingly contradictory results regarding the role of SphK1 or SphK2 in ALF and to avoid problems associated with nonspecific inhibitors of SphK1 and/or SphK2, we induced ALF in SphK1 and SphK2 knockout mice. Deletion of SphK1, but not SphK2, and treatment with a very isozyme-specific inhibitor of SphK1 markedly decreased GalN/LPS-induced liver damage, hepatic apoptosis, and mortality rate compared to WT mice. These results clearly demonstrated that it is SphK1, not SphK2 that plays a critical role in ALF. In agreement, SphK1 deficient mice were protected from acetaminophen-induced liver damage.26 However, adoptive transfer experiments indicated that SphK1 in bone marrow-derived cells did not contribute to the development of acute inflammatory liver injury induced by acetaminophen.26 In this regard, we have found that SphK1 expressed in bone marrow-derived infiltrating immune cells, but not in host liver-resident cells, contribute to the development of ALF induced by GalN/LPS. These differences might be due to distinct mechanisms of hepatotoxicity or because adoptive transfer of bone marrow monocytes aggravates acute acetaminophen-induced liver injury in mice.59
We also observed that SphK1 inhibition attenuated liver enzyme release, reduced liver inflammation and hepatocyte apoptosis, and decreased production of the pro-inflammatory cytokine TNFα while increasing the anti-inflammatory cytokine IL10 that can restrain and limit inflammatory responses and liver damage. Taken together with previous reports using SphK1 inhibitors,24-26 our results also support the notion that targeting SphK1 represents a potential new strategy to ameliorate ALF. Interestingly, although the concentration of SK1-I in blood was lower than S1P, in the liver, SK1-I levels were 10-fold higher than those of S1P, suggesting that it might more potently inhibit SphK1 in liver than in the circulation. This might have important clinical implications, as it is not desirable to reduce S1P levels in the circulation that can cause vascular leakage.16 Nevertheless, the effects of treatment with SK1-I were less dramatic than SphK1 deletion possibly due to insufficient pharmacokinetic optimization. In this regard, a more potent SphK1 inhibitor PF-543 protected mice from acetaminophen-induced death to the same extent as SphK1 deletion.26 Further studies utilizing novel approaches to enhance the targeting efficiency of more potent inhibitors of SphK1 to the liver may pave the way for the development of a clinically applicable therapeutic strategy to control acute liver injury.
ACKNOWLEDGMENTS
The authors thank Dr Proia for providing the founding mice colonies, Dr Mingxia Liu for helping with ALT and AST determinations, and the VCU Lipidomics and Microscopy Cores, which are supported, in part by funding from the National Cancer Institute Support Grant P30 CA016059.
Funding information
This work was supported by National Institutes of Health Grant RO1GM043880 (to SS)
Abbreviations:
- ALF
acute liver failure
- GalN
D-(+)-galactosamine
- LPS
lipopolysaccharide
- S1P
sphingosine-1-phosphate
- SphK
sphingosine kinase
- WT
wild type
Footnotes
CONFLICT OF INTEREST
Dr Spiegel is a co-inventor on patent number US 8,372,888 B2 titled “Sphingosine kinase type 1 inhibitors, compositions and processes for using same.” Dr Sanyal has served as a consultant to AbbVie, Astra Zeneca, Nitto Denko, Ardelyx, Conatus, Nimbus, Amarin, Salix, Tobira, Takeda, Fibrogen, Jannsen, Gilead, Boehringer, Lilly, Zafgen, Novartis, Novo Nordisk, and Pfizer. He has served as an unpaid consultant to Exhalenz, Intercept, Echosens, Immuron, Galectin, Fractyl, Northsea Pharma, Gencia, Syntlogic, Affimune, Chemomab, Nordic Bioscience Zydus, and Bristol Myers Squibb. His institution has received grant support from Gilead, Salix, Tobira, Bristol Myers, Shire, Intercept, Merck, Astra Zeneca, Mallinckrodt, Cumberland, and Novartis. He receives royalties from Elsevier and UptoDate. He is the President of Sanyal Biotechnology and owns stock in Genfit, Akarna, Tiziana, Indalo, and Durect. The other authors declare that they have no competing financial interests.
REFERENCES
- 1.Saliba F, Samuel D. Acute liver failure: current trends. J Hepatol. 2013;59:6–8. [DOI] [PubMed] [Google Scholar]
- 2.Bernal W, Lee WM, Wendon J, et al. Acute liver failure: a curable disease by 2024? J Hepatol. 2015;62:S112–S120. [DOI] [PubMed] [Google Scholar]
- 3.Stravitz RT, Lee WM. Acute liver failure. Lancet. 2019;394:869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology. 1990;11:49–53. [DOI] [PubMed] [Google Scholar]
- 5.Antoniades CG, Berry PA, Wendon JA, et al. The importance of immune dysfunction in determining outcome in acute liver failure. J Hepatol. 2008;49:845–861. [DOI] [PubMed] [Google Scholar]
- 6.Atillasoy E, Berk PD. Fulminant hepatic failure: pathophysiology, treatment, and survival. Annu Rev Med. 1995;46:181–191. [DOI] [PubMed] [Google Scholar]
- 7.Chan G, Taqi A, Marotta P, et al. Long-term outcomes of emergency liver transplantation for acute liver failure. Liver Transpl. 2009;15:1696–1702. [DOI] [PubMed] [Google Scholar]
- 8.Tuñón MJ, Alvarez M, Culebras JM, et al. An overview of animal models for investigating the pathogenesis and therapeutic strategies in acute hepatic failure. World J Gastroenterol. 2009;15:3086–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehmann V, Freudenberg MA, Galanos C. Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J Exp Med. 1987;165:657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leist M, Gantner F, Bohlinger I, et al. Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol. 1995;146:1220–1234. [PMC free article] [PubMed] [Google Scholar]
- 11.Olleros ML, Vesin D, Fotio AL, et al. Soluble TNF, but not membrane TNF, is critical in LPS-induced hepatitis. J Hepatol. 2010;53:1059–1068. [DOI] [PubMed] [Google Scholar]
- 12.Zimmermann HW, Trautwein C, Tacke F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol. 2012;3:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. 2014;193:344–353. [DOI] [PubMed] [Google Scholar]
- 14.Guillot A, Tacke F. Liver macrophages: old dogmas and new insights. Hepatol Commun. 2019;3:730–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang W, Sun R, Wei H, et al. Toll-like receptor 3 ligand attenuates LPS-induced liver injury by down-regulation of toll-like receptor 4 expression on macrophages. Proc Natl Acad Sci USA. 2005;102:17077–17082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleuser B Divergent role of sphingosine 1-phosphate in liver health and disease. Int J Mol Sci. 2018;19:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park WJ, Song JH, Kim GT, et al. Ceramide and sphingosine 1-phosphate in liver diseases. Mol Cells. 2020;43:419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osawa Y, Uchinami H, Bielawski J, et al. Roles for C16-ceramide and sphingosine 1-phosphate in regulating hepatocyte apoptosis in response to tumor necrosis factor-alpha. J Biol Chem. 2005;280:27879–27887. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Saiyan S, Men TY, et al. Hepatopoietin Cn reduces ethanol-induced hepatoxicity via sphingosine kinase 1 and sphingosine 1-phosphate receptors. J Pathol. 2013;230:365–376. [DOI] [PubMed] [Google Scholar]
- 21.Nowatari T, Murata S, Nakayama K, et al. Sphingosine 1-phosphate has anti-apoptotic effect on liver sinusoidal endothelial cells and proliferative effect on hepatocytes in a paracrine manner in human. Hepato Res. 2015;45:1136–1145. [DOI] [PubMed] [Google Scholar]
- 22.Coldewey SM, Benetti E, Collino M, et al. Elevation of serum sphingosine-1-phosphate attenuates impaired cardiac function in experimental sepsis. Sci Rep. 2016;6:27594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurano M, Tsuneyama K, Morimoto Y, et al. Apolipoprotein M protects lipopolysaccharide-treated mice from death and organ injury. Thromb Haemost. 2018;118:1021–1035. [DOI] [PubMed] [Google Scholar]
- 24.Tian T, Tian W, Yang F, et al. Sphingosine kinase 1 inhibition improves lipopolysaccharide/D-galactosamine-induced acute liver failure by inhibiting mitogen-activated protein kinases pathway. United Eur Gastroenterol J. 2016;4:677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lei YC, Yang LL, Li W, et al. Inhibition of sphingosine kinase 1 ameliorates acute liver failure by reducing high-mobility group box 1 cytoplasmic translocation in liver cells. World J. Gastroenterol 2015;21:13055–13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Wang H, Zhang J, et al. SPHK1 deficiency protects mice from acetaminophen-induced ER stress and mitochondrial permeability transition. Cell Death Differ. 2020;27:1924–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi Y, Rehman H, Ramshesh VK, et al. Sphingosine kinase-2 inhibition improves mitochondrial function and survival after hepatic ischemia-reperfusion. J Hepatol. 2012;56:137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagahashi M, Ramachandran S, Kim EY, et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangio-genesis. Cancer Res. 2012;72:726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu D, Li C, Chen Y, et al. Nuclear import of proinflammatory transcription factors is required for massive liver apoptosis induced by bacterial lipopolysaccharide. J Biol Chem. 2004;279:48434–48442. [DOI] [PubMed] [Google Scholar]
- 30.Hait NC, Allegood J, Maceyka M, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang J, Nagahashi M, Kim E, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Avni D, Philosoph A, Meijler MM, et al. The ceramide-1-phosphate analogue PCERA-1 modulates tumour necrosis factor-alpha and interleukin-10 production in macrophages via the cAMP-PKA-CREB pathway in a GTP-dependent manner. Immunology. 2010;129:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakama T, Hirono S, Moriuchi A, et al. Etoposide prevents apoptosis in mouse liver with D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure resulting in reduction of lethality. Hepatology. 2001;33:1441–1450. [DOI] [PubMed] [Google Scholar]
- 34.Ali M, Fritsch J, Zigdon H, et al. Altering the sphingolipid acyl chain composition prevents LPS/GLN-mediated hepatic failure in mice by disrupting TNFR1 internalization. Cell Death Dis. 2013;4:e929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ilyas G, Zhao E, Liu K, et al. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1beta. J Hepatol. 2016;64:118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowak M, Gaines GC, Rosenberg J, et al. LPS-induced liver injury in D-galactosamine-sensitized mice requires secreted TNF-alpha and the TNF-p55 receptor. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1202–R1209. [DOI] [PubMed] [Google Scholar]
- 37.Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol. 2014;26:237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang K, Damjanov I, Wan Y-J. The protective role of pregnane X receptor in lipopolysaccharide/D-galactosamine-induced acute liver injury. Lab Invest. 2010;90:257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Singh R, Lefkowitch JH, et al. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong X, Liu J, Xu Y, et al. Role of macrophages in experimental liver injury and repair in mice. Exp Ther Med. 2019;17:3835–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porta C, Riboldi E, Ippolito A, et al. Molecular and epigenetic basis of macrophage polarized activation. Semin Immunol. 2015;27:237–248. [DOI] [PubMed] [Google Scholar]
- 42.Paugh SW, Paugh BS, Rahmani M, et al. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112:1382–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allende ML, Sasaki T, Kawai H, et al. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004;279:52487–52492. [DOI] [PubMed] [Google Scholar]
- 44.Price MM, Oskeritzian CA, Falanga YT, et al. A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent murine model of allergic asthma. J Allergy Clin Immunol. 2013;131:501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mücke VT, Maria Schwarzkopf K, Thomas D, et al. Serum sphingosine-1-phosphate is decreased in patients with acute-on-chronic liver failure and predicts early mortality. Hepatol Commun. 2020;4:1477–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winkler MS, Märtz KB, Nierhaus A, et al. Loss of sphingosine 1-phosphate (S1P) in septic shock is predominantly caused by decreased levels of high-density lipoproteins (HDL). J Intensive Care. 2019;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christensen PM, Liu CH, Swendeman SL, et al. Impaired endothelial barrier function in apolipoprotein M-deficient mice is dependent on sphingosine-1-phosphate receptor 1. FASEB J. 2016;30:2351–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silverstein R D-galactosamine lethality model: scope and limitations. J Endotoxin Res. 2004;10:147–162. [DOI] [PubMed] [Google Scholar]
- 49.Santucci L, Fiorucci S, Chiorean M, et al. Interleukin 10 reduces lethality and hepatic injury induced by lipopolysaccharide in galactosamine-sensitized mice. Gastroenterology. 1996;111:736–744. [DOI] [PubMed] [Google Scholar]
- 50.Louis H, Le Moine O, Peny M, et al. Production and role of interleukin-10 in concanavalin A-induced hepatitis in mice. Hepatology. 1997;25:1382–1389. [DOI] [PubMed] [Google Scholar]
- 51.Weigert A, von Knethen A, Thomas D, et al. Sphingosine kinase 2 is a negative regulator of inflammatory macrophage activation. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:1235–1246. [DOI] [PubMed] [Google Scholar]
- 52.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59:2034–2042. [DOI] [PubMed] [Google Scholar]
- 54.Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol. 2005;5:560–570. [DOI] [PubMed] [Google Scholar]
- 55.Megidish T, Cooper J, Zhang L, et al. A novel sphingosine-dependent protein kinase (SDK1) specifically phosphorylates certain isoforms of 14-3-3 protein. J Biol Chem. 1998;273:21834–21845. [DOI] [PubMed] [Google Scholar]
- 56.Lei YC, Yang LL, Li W, et al. Sphingosine kinase 1 dependent protein kinase C-delta activation plays an important role in acute liver failure in mice. World J. Gastroenterol 2015;21:13438–13446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaneko T, Murakami T, Kawana H, et al. Sphingosine-1-phosphate receptor agonists suppress concanavalin A-induced hepatic injury in mice. Biochem Biophys Res Commun. 2006;345:85–92. [DOI] [PubMed] [Google Scholar]
- 58.Yang L, Han Z, Tian L, et al. Sphingosine 1-phosphate receptor 2 and 3 mediate bone marrow-derived monocyte/macrophage motility in cholestatic liver injury in mice. Sci Rep. 2015;5:13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mossanen JC, Krenkel O, Ergen C, et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology. 2016;64:1667–1682. [DOI] [PubMed] [Google Scholar]