

Abstract
The bromodomain and extra terminal (BET) protein family recognizes acetylated lysines within histones and transcription factors using two N-terminal bromodomains, D1 and D2. The protein–protein interactions between BET bromodomains, acetylated histones, and transcription factors are therapeutic targets for BET-related diseases, including inflammatory disease and cancer. Prior work demonstrated that methylated-1,2,3-triazoles are suitable N-acetyl lysine mimetics for BET inhibition. Here we describe a structure–activity relationship study of triazole-based inhibitors that improve affinity, D1 selectivity, and microsomal stability. These outcomes were accomplished by targeting a nonconserved residue, Asp144 and a conserved residue, Met149, on BRD4 D1. The lead inhibitors DW34 and 26 have a BRD4 D1 Kd of 12 and 6.4 nM, respectively. Cellular activity was demonstrated through suppression of c-Myc expression in MM.1S cells and downregulation of IL-8 in TNF-α-stimulated A549 cells. These data indicate that DW34 and 26 are new leads to investigate the anticancer and anti-inflammatory activity of BET proteins.
Graphical Abstract
INTRODUCTION
Epigenetic regulation is a reversible and dynamic process which involves chemical modification of DNA, RNA, and histone proteins.1 Post-translational modifications (PTMs) help regulate gene expression through alteration of the chromatin structure and recruitment of transcriptional complexes.2 Recognition of PTMs involves protein structural motifs that are known as epigenetic reader domains. Bromodomains are reader domains that bind to N-ε-acylated lysine residues on histones and recruit transcriptional complexes to promoter and enhancer regions on DNA.3,4 Enhancer binding of specific bromodomain-containing proteins, such as BRD4, regulates oncogene expression and inflammatory response genes that control cytokines and chemokine expression.4–7 As such, inhibiting protein–protein interactions between bromodomains and acetylated proteins is an emerging approach for anticancer and anti-inflammatory therapy.8
The 61 human bromodomains are classified into eight families based on structural and sequence similarities.9 The bromodomain and extra terminal (BET) family of proteins, BRD2, BRD3, BRD4, and BRDT, all contain two tandem bromodomains (D1 and D2) and an extra-terminal domain.9 Given their high sequence similarity, attaining selective inhibition within the BET family is challenging. As such, pan-BET inhibitors, such as (+)-JQ1 and iBET-151, have been widely used to investigate the function of BET proteins by simultaneously inhibiting multiple bromodomains.10–13 Although efficacious, the use of pan-BET inhibitors obscures the function of individual bromodomains and, in some cases, has led to incorrect attribution of biological activity to a specific BET bromodomain.8
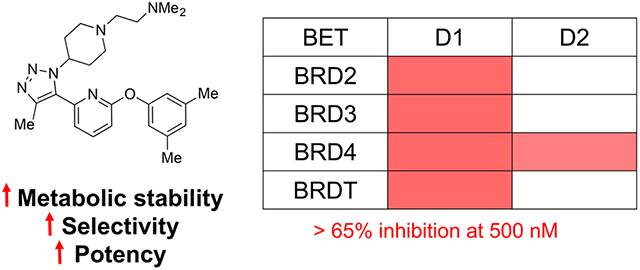
To improve our understanding of the role of individual bromodomains, selective inhibitors for specific BET bromodomains have been developed. Olinone (Figure 1A) was the first pan-D1 BET inhibitor with moderate affinity (Kd = 3.4 μM). The addition of olinone to oligodendrocyte progenitor cells demonstrated biological effects divergent from pan-BET inhibition.14 Whereas a pan-BET inhibitor impeded differentiation of oligodendrocytes, olinone induced this process.14 GSK778, another pan-D1-selective inhibitor (Figure 1A), was recently reported.15 Gilan et al. reported that pan-D1 inhibition was as efficient as pan-BET (D1 and D2) inhibition on the growth and viability of human cancer cells, including breast cancer and acute myeloid leukemia cells.15 Alternatively, ABBV-744 (Figure 1A) is a pan-D2 inhibitor with nanomolar affinity. ABBV-744 can displace BET bromodomains from androgen-receptor-enriched super-enhancer regions in prostate cancer cells.16 Notably, pan-D2 inhibition did not result in the characteristic dose-limiting toxicity observed for pan-BET inhibitors.16 More recently, Liu and co-workers developed ZL0516 as a BRD4 D1-selective inhibitor with significant anti-inflammatory effects in vitro (Figure 1A).17
Figure 1.

(A) Representative BET inhibitors with affinities against BRD4. (B) Exploiting D2/D1 differences to improve selectivity.
We also reported on UMN627, featuring an imidazole ring as the acetylated histone mimetic (Figure 1A), as a selective BRD4 D1 inhibitor.18,19 UMN627 engaged BRD4 at submicromolar concentrations, but, unexpectedly, it did not inhibit c-Myc expression at these concentrations.18 However, UMN627 effectively suppressed chemokine expression resulting from inflammation in the liver in vitro and in vivo.19 Given the sometimes unanticipated biological effects of pan-BET inhibition, new design strategies are needed to develop selective bromodomain inhibitors. More selective inhibitors could be used to illuminate the biological mechanisms of specific bromodomains and potentially be used as therapeutic agents. However, the long synthetic route needed to synthesize UMN627 precluded rapid assessment of the structure–activity relationships (SARs) to improve affinity and selectivity. To address this challenge, we developed a similar inhibitor based on a triazole scaffold using a click reaction.12,18 Starting from our previously reported inhibitor, 1,12 this report describes a systematic SAR study using the triazole scaffold that led to compounds with improved BRD4 D1 affinity, D1 vs D2 selectivity, and metabolic stability (Figure 1B).
RESULTS AND DISCUSSION
To improve the inhibitor’s affinity and selectivity, we focused on several key differences between the BRD4 D1 and BRD4 D2 bromodomains. One residue of difference is Asp144 in D1 instead of His437 in BRD4 D2 on the BC loop. In a cocrystal structure, the ethyl amide in ABBV-744 has a binding interaction with a unique channel in D2 of BRD2 formed by His433, Tyr386, and Pro430.20 Conversely, Wellaway et al. reported that a pan-D1 BET inhibitor formed a hydrogen bond to Asp144 through an intervening bridging water molecule.21 A second residue differing in the BC loop is an Ile in D1 and Val in D2, which was targeted for selective inhibitor development.16 In ABBV-744, the 2,6-dimethylphenyl ring is well tolerated by Val435 on BRD2 D2. On BRD2 D1, this residue corresponds to a larger Ile. The steric repulsion between the larger Ile and the 2,6-dimethylphenyl ring results in a lower D1 affinity and enhanced D2 selectivity. An overlay of the ABBV-744/BRD2 D1 cocrystal structure and triazole 27/BRD4 D112 cocrystal structure showed that the 2,6-dimethylphenyl motif of ABBV-744 was close to the 3,5-dimethylphenyl ring of triazole 27 (Figure 2B). Another dihydro-methyl-quinazolinone scaffold was also developed as a BET inhibitor by Fish et al., leading to PFI-1.22 A 10-fold affinity difference was found with different aryl groups in the PFI-1 series (Figure S12 in green). In the cocrystal structure, the aryl group of PFI-1 overlays similarly to the dimethylphenyl motif on triazole 27 and ABBV-744. Based on the SAR data reported for the ABBV-744 and PFI-1 series, it was hypothesized that altering the aryl group on the triazole scaffold could enable enhanced BRD4 D1 affinity and alter selectivity between D1 and D2.
Figure 2.

(A) Structure of ABBV-744 and previously reported triazole 27. (B) Overlay of the cocrystal structure of ABBV-744 with BRD2 D1 (PDB: 6ONY) and triazole 27 with BRD4 D1 (PDB: 7M16). The 2,6-dimethyl phenyl group of ABBV-744 and Ile163 is shown in green, and the 3,5-dimethylphenyl group of triazole 27 and Ile146 is shown in red.
Recent advances in synthetic chemistry have rapidly accelerated the optimization of inhibitors and have allowed access to new chemical spaces. In particular, cross-coupling reactions have become staples in medical chemistry.23–25 To efficiently synthesize the desired analogues, we employed two divergent steps. The phenolic ether linkage was constructed via nucleophilic aromatic substitution using a range of phenols and 2-fluoro-pyridine c (scheme not shown). This method is convenient when the necessary phenol is readily available. However, this study required phenols that are not commercially available and would likely require multiple steps to synthesize. To circumvent this issue, a second cross-coupling step was developed to facilitate late-stage divergence. A mono- or di- brominated arene was installed and subjected to a subsequent Kumada or Negishi cross-coupling reaction (Scheme 1). This approach allowed incorporation of many alkyl, cycloalkyl, and aryl groups. Azide a and propyne were combined in a copper-catalyzed azide–alkyne cycloaddition. Triazole b was deprotonated with n-BuLi, and the addition of ZnCl2 facilitated a Negishi coupling to form intermediate c. An SNAr reaction between fluoride c and 3-bromo-5-methylphenol generated the divergent intermediate d. Several alkyl groups were incorporated via Kumada or Negishi coupling conditions (compounds e–j). Subsequent deprotection afforded compounds 2–7, featuring varied groups to test for D2/D1 selectivity against BRD4.
Scheme 1.
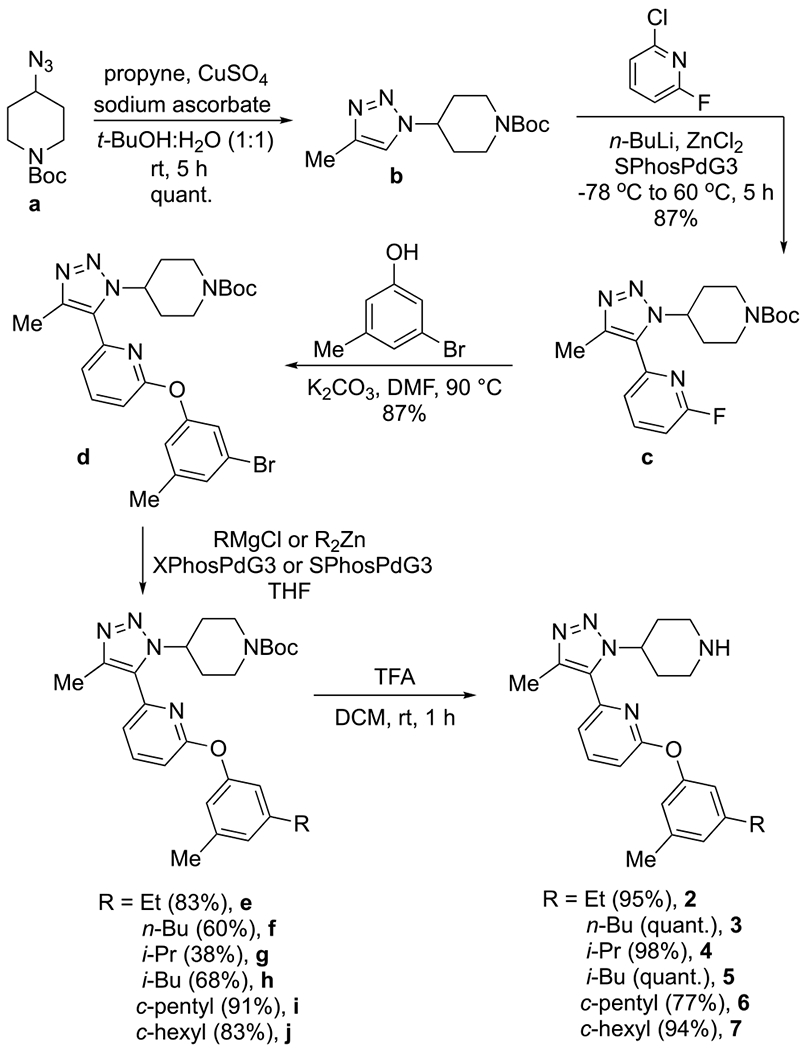
Synthesis of Triazole Analogues
The affinity and apparent selectivity of these compounds were examined by fluorescence anisotropy (FA, Table 1). Contrary to predictions drawing analogy to the ABBV-744 model, larger groups, such as c-hexyl and phenyl, improved the apparent D1 vs D2 selectivity. The size difference in the alkyl group appeared to have little effect on BRD4 D1 affinity. In the triazole 27 cocrystal structure (Figure 2B), only one edge of the phenolic ring points toward Ile146. The other edge is solvent exposed. Based on the similar affinity data, it was proposed that the phenolic ring rotates to leave the larger alkyl group solvent exposed. This would explain the minimal difference in the binding affinity and selectivity of compounds 2—7.
Table 1.
Affinity and Selectivity for Analogues with Unsymmetrical Benzene Ringsa

| ||||
|---|---|---|---|---|
| compound | R | BRD4 D1 IC50 (μM) | BRD4 D2 IC50 (μM) | D2/D1 selectivity |
| (+)-JQ1 | – | 0.0770 ± 0.006 | 0.0720 ± 0.02 | – |
| 1 | Me | 2.88 ± 0.3 | 4.93 ± 0.6 | 1.7 |
| 2 | Et | 1.57 ± 0.1 | 4.51 ± 1 | 2.8 |
| 3 | n-Bu | 2.40 ± 0.3 | 13.0 ± 3 | 5.4 |
| 4 | i-Pr | 3.23 ± 0.5 | 12.5 ± 2 | 1.5 |
| 5 | i-Bu | 3.21 ± 0.3 | 13.5 ± 1 | 2.3 |
| 6 | c-pentyl | 3.55 ± 0.7 | 13.3 ± 1 | 3.7 |
| 7 | c-hexyl | 4.75 ± 0.4 | 23.3 ± 3 | 4.9 |
| 8 | Ph | 2.78 ± 0.3 | 12.9 ± 2 | 4.6 |
| 9 | Br | 1.43 ± 0.2 | 2.54 ± 0.1 | 1.8 |
IC50 values were determined by FA. Data represent the mean and standard deviation of three experimental replicates.
A second series of inhibitors was synthesized through an analogous route utilizing 3,5-dibromophenol for the SNAr reaction. A double Kumada or Negishi cross-coupling formed compounds with the same R group at both the 3 and 5 positions (see SI for details). With these analogues, a more dramatic change in BRD4 D1 affinity was observed (Table 2). The BRD4 D1 selectivity was enhanced with slightly larger R groups (e.g., Et and n-Pr, and c-Pr). However, both affinity and selectivity were eroded with larger groups (e.g., n-Bu or c-pentyl). Inhibitor 13, featuring a bis-c-propyl phenolic ring, led to improved BRD4 D1 affinity and decreased BRD4 D2 affinity, which resulted in the highest apparent 7.7-fold selectivity toward D1.
Table 2.
Affinity and Selectivity for Analogues with Symmetrical Benzene Ringsa

| ||||
|---|---|---|---|---|
| compound | R | BRD4 D1 IC50 (μM) | BRD4 D2 IC50 (μM) | D2/D1 selectivity |
| (+)-JQ1 | – | 0.0770 ± 0.006 | 0.0720 ± 0.02 | – |
| 1 | Me | 2.88 ± 0.3 | 4.93 ± 0.6 | 1.7 |
| 10 | Et | 1.50 ± 0.2 | 6.12 ± 0.2 | 4.1 |
| 11 | n-Pr | 2.60 ± 0.1 | 12.6 ± 2 | 4.8 |
| 12 | n-Bu | 12.5 ± 1 | 33.5 ± 3 | 2.7 |
| 13 | c-propyl | 1.42 ± 0.1 | 10.9 ± 2 | 7.7 |
| 14 | c-pentyl | 21.8 ± 3 | 53.5 ± 0.7 | 2.5 |
| 15 | c-hexyl | 75.8 ± 13 | >100 | – |
| 16 | Cl | 2.40 ± 0.2 | 5.2 ± 0.2 | 2.2 |
| 17 | Br | 0.980 ± 0.1 | 2.43 ± 0.4 | 2.0 |
| 18 | CF3 | 14.3 ± 2 | 23.7 ± 2 | 3.2 |
| 19 | CH2OH | 40.9 ± 6 | >100 | – |
IC50 values were determined by FA. Data represent the mean and standard deviation of three experimental replicates.
Additionally, the 3,5-dichloro- (16) and 3,5-dibromo- (17) were tested to evaluate if the electron density of the phenolic ring is important to binding affinity. Analogue 17 demonstrated a 2.9-fold enhancement in D1 affinity relative to inhibitor 1, which was the first inhibitor with submicromolar affinity in this series. Alternatively, 3,5-bis-trifluoromethyl analogue 18 had a 15-fold weaker D1 affinity relative to inhibitor 17. This is likely due to a weakened edge-to-face π–π interaction with Trp81 in the WPF shelf.
A third set of analogues with different aryl and alkyl groups were synthesized and tested (Table 3). Inhibitors 20 and 21 have saturated side chains in place of the phenyl ring. Both inhibitors demonstrated inferior affinity. Utilizing the 2,6-dimethyl group present in ABBV-744 (22) fully eroded the D2/D1 selectivity as may be expected based on the SAR for ABBV-744. Inhibitor 23 with 2,5-dibromo substitution was the most potent inhibitor assayed with an IC50 of 790 nM, while the 2,5-disubstituted inhibitor 24 was less potent and less selective.
Table 3.
Affinity and Selectivity for Analogues with Alkyl Ethers and Substituted Benzene Ringsa
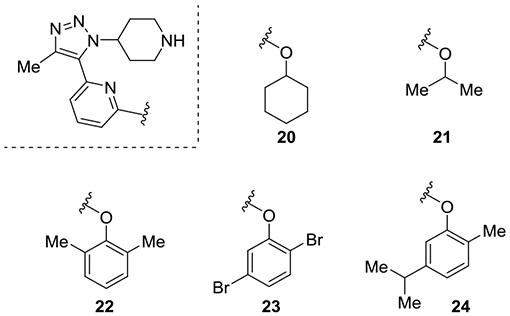
| |||
|---|---|---|---|
| compound | BRD4 D1 IC50 (μM) | BRD4 D2 IC50 (μM) | D2/D1 selectivity |
| (+)-JQ1 | 0.0770 ± 0.006 | 0.0720 ± 0.02 | – |
| 20 | 8.96 ± 0.8 | 22.5 ± 2 | 2.5 |
| 21 | >100 | >100 | – |
| 22 | 22.2 ± 4 | 20.2 ± 3 | 0.90 |
| 23 | 0.790 ± 0.03 | 3.14 ± 0.06 | 4.0 |
| 24 | 5.97 ± 2 | 12.2 ± 3 | 2.0 |
IC50 values were determined by FA. Data represents the mean and standard deviation of three experimental replicates.
Further insights into the inhibitor binding were gleaned through crystallography. Previously, an ethylamino group was used in UMN627 to target a nonconserved amino acid residue, Asp144, in BRD4 D1. In this case, a 2.1-fold affinity increase was observed; however, inspection of the resulting cocrystal structure indicated the basic amino group was oriented in an unfavorable direction, positioned 10.4 Å (N to O=C) away.18 To compare the triazole scaffold, a cocrystal structure of inhibitor 23 with BRD4 D1 was obtained (Figure 3A). A structural analysis indicated a more favorable orientation of the piperidyl ring pointing toward Asp 144 (Figure S14). This interaction indicated that attaching a basic group might be particularly advantageous. The distance between the piperidyl nitrogen atom on inhibitor 23 was 6.3 Å away from a carboxylate oxygen in Asp144 (Figure S14). With this information, an N,N-dimethylethylamino group was installed on inhibitors 1 and 23 to yield two new inhibitors 25, now referred to as DW34, and 26. By the FA assay, these inhibitors were more potent with IC50 values of 0.137 ± 0.02 μM (DW34) and 0.110 ± 0.02 μM (26), respectively (Figure 4A). This represents a potency increase of >10-fold and makes inhibitors DW34 and 26 similar in potency to (+)-JQ1 in the same FA assay. Furthermore, the apparent D2/D1 selectivity increased to 7.4- and 7.8-fold, respectively.
Figure 3.
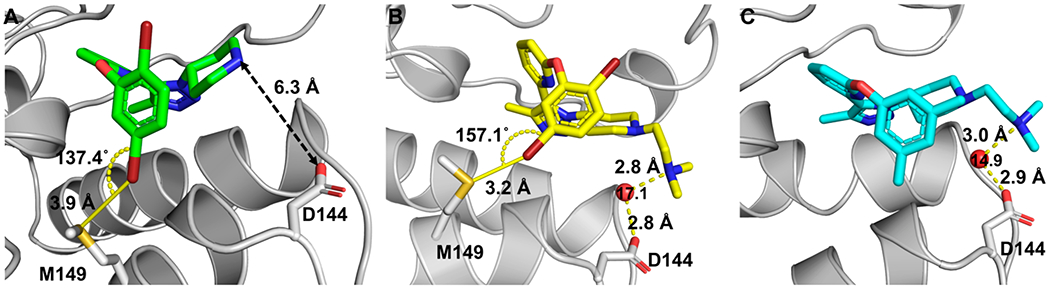
(A) Cocrystal structure of 23 with BRD4 D1. The distance between the piperidyl group of 23 and Asp144 is indicated. The distance and bond angle between bromide on the aryl group and Met149 are shown. PDB ID: 7MLS. (B) Cocrystal structure of 26 with BRD4 D1. The dimethylethylamino group has a hydrogen-bonding interaction with Asp144 via a structured water (red sphere). PDB ID: 7MLQ. The bromide forms a halogen bond with Met149. (C) Cocrystal structure of DW34 with BRD4 D1. The dimethylethylamino group has a hydrogen-bonding interaction with Asp144 via a structured water (red sphere). PDB ID: 7MLR. B-factors are labeled on the structured water molecules.
Figure 4.

(A) Affinity and selectivity of leading molecules 25 (DW34) and 26. (B) Selectivity of DW34 screened at 500 nM against a panel of 32 bromodomains by BROMOscan. Residual activity based on percent control is labeled in red circles. Sizes and cutoffs have been determined by Eurofins. Bromodomains not tested are shown in light gray.
Cocrystal structures of DW34 and 26 were obtained to better understand the molecular basis for the affinity (Figure 3B,C). The 1,2,3-triazole motif is the N-acetyl lysine mimic and forms a direct hydrogen-bonding interaction with Asn140 (Figure S15). Unexpectedly, in the case of 26, the side chain of Met149 moved toward the aryl ring of 26 and formed a halogen-bonding interaction with the bromine atom (Figure 3B). Halogen-bonding interactions between small molecules and bromodomains have only been reported recently. These interactions have been found with a different site in the histone binding pocket.26 In addition, the dimethylethylamino group formed a water-bridging hydrogen-bonding interaction with Asp144 via a structured water molecule. Although halogen-bonding interactions were not possible for DW34, a cocrystal structure also showed a water bridging hydrogen-bonding interaction with Asp144 via the structured water molecule (Figure 3C). This type of water bridging hydrogen-bonding interaction with Asp 144 was also found in GSK778 against BRD4 D1 (Figure S16).15 Together these results support targeting a nonconserved residue Asp144 and a conserved residue Met149, for improving selectivity and affinity, respectively.
Due to the large increase in potency demonstrated by compounds DW34 and 26, the affinity and selectivity were further characterized using a commercial BROMOscan assay from Eurofins/DiscoverX (Figure 4, Figure S17, and Table S2). The D1 Kd values were 12 nM and 6.4 nM for inhibitors DW34 and 26, respectively. The BRD4 D2/D1 selectivities were 6.2- and 4.2-fold, respectively. Based on its affinity, and metabolic stability (vide infra), DW34 was further screened against 32 bromodomains at 500 nM (approximately 50-fold above the Kd) to further evaluate the selectivity. DW34 has strong affinities against all BET D1s and BRD4 D2 (94–100% inhibition, Table S2). Weaker affinities were observed against the other three BET D2s consistent with targeting a nonconserved Asp residue. Minimal inhibitory activity was observed against non-BET bromodomains (0–35% inhibition). Kd determination confirmed this with BET D1 affinities ranging from 12 to 22 nM, while BRD2, BRD3, and BRDT D2 affinities were >16-fold weaker relative to BRD4 D1 (Kd = 200–310 nM, Figure 4A, Figure S17). The reason for the higher affinity toward BRD4 D2 versus the other D2 domains is unclear. While most inhibitors targeting BETs are either pan-BET, pan-D1, or pan-D2, this selectivity profile is unique. These data support DW34 as a potent and selective inhibitor for BET bromodomain-containing proteins.
Given the significant improvement in affinity and selectivity of inhibitors DW34 and 26, relative to inhibitor 1, a preliminary screen of metabolic stability was conducted. As an initial benchmark, human liver microsomes pooled from 150 donors were used to determine the microsomal metabolic half-life of UMN627 to be 54 min (Table 4, Table S3). Prior inhibitor 1 had an even shorter half-life of 47 min. In an effort to understand a liability, compounds 13, 16, and 17 were tested. Cyclopropyl groups are widely used to enhance the metabolic stability,27 and compound 13 had an 81 min half-life, which was the longest of the initially screened inhibitors. The two more advanced inhibitors (DW34 and 26) had the longest half-lives studied here of 117 and 83 min, respectively. A direct comparison of inhibitor 1 and DW34 indicates that this change can increase the half-life by approximately 2.5-fold.
Table 4.
Microsomal Stabilities of Selected Molecules
| compound | k | t1/2 (min) | CLint (μL/min/mg protein) | fu (fraction unbound) |
|---|---|---|---|---|
| UMN627 | 0.013 ± 0.001 | 54.2 ± 6.0 | 25.6 ± 2.5 | 0.080 ± 0.01 |
| 1 | 0.015 ± 0.002 | 46.7 ± 5.4 | 29.7 ± 3.1 | 0.13 ± 0.01 |
| 13 | 0.0090 ± 0.001 | 81.1 ± 10 | 17.1 ± 1.9 | 0.12 ± 0.02 |
| 16 | 0.020 ± 0.002 | 34.2 ± 3.0 | 40.5 ± 3.3 | 0.11 ± 0.03 |
| 17 | 0.020 ± 0.001 | 35.0 ± 2.4 | 39.5 ± 2.5 | 0.090 ± 0.02 |
| DW34 | 0.0060 ± 0.001 | 117 ± 15 | 11.9 ± 1.4 | 0.14 ± 0.01 |
| 26 | 0.0083 ± 0.001 | 83.5 ± 6.6 | 16.6 ± 1.2 | 0.13 ± 0.01 |
With the increased potency of these new inhibitors relative to parent compound 1, a preliminary assessment of cellular activity was conducted. The multiple myeloma cell line, MM.1S, is highly sensitive to pan-BET inhibitors due to its strong dependence on c-Myc expression. Encouragingly, the new inhibitors DW34 and 26 were highly effective at reducing c-Myc expression with EC50 values below 100 nM. Compound 26 was slightly more efficacious than DW34, which is consistent with the greater in vitro potency (Figure 5, Figure S18). Alternatively, the BRD4 D1-selective inhibitor UMN627 was unable to completely inhibit c-Myc expression at submicromolar concentrations (Figure S18). DW34 and 26 also reduced cell viability of MM.1S cells to comparable levels as (+)-JQ1 for 26, and 4.5-fold weaker for DW34 (Figure S19). BET inhibition can also lead to downregulation of cytokines, such as IL-8.28 Using non-small cell lung cancer A549 cells, we measured IL-8 levels to evaluate the anti-inflammatory effects of our inhibitors (Figure S21). At 1 μM concentration, DW34 and 26 reduced IL-8 levels to 14% and 17%, respectively without affecting the overall cell viability (Figures S20 and S21). Together, these results demonstrate high in vitro potency, improved metabolic stability, and anticancer and anti-inflammatory activity using two model cells lines. The good drug-like properties (Table 5), potency, and improved microsomal stability make compounds DW34 and 26 suitable candidates for future in vivo studies of BET-dependent disease.29
Figure 5.

Western blot measuring expression of c-Myc upon treatment of MM.1S cells with (+)-JQ1, DW34, and 26. Image is representative of three replicates (see Figure S16 for uncropped image and additional replicates).
Table 5.
Drug-like Properties of DW34 and UMN627
| compound | DW34 | UMN627 |
|---|---|---|
| MW | 434.59 | 535.62 |
| clogP | 3.78 | 3.32 |
| no. aromatic rings | 3 | 4 |
| LEa | 0.331 | 0.189 |
| LLEb | 4.14 | 2.17 |
Ligand efficiency (LE) = ΔG/number of heavy atoms, reported in kcal/mol per heavy atom.
Lipophilic ligand efficiency (LLE) = calculated as pKd – clogP.
CONCLUSION
In conclusion, we describe the development of new BET bromodomain inhibitors through a systematic SAR study. These inhibitors are based on a readily synthesized triazole inhibitor scaffold with a facile five-step synthesis with 28–66% overall yields. Furthermore, these compounds exhibit nano-molar affinities for BRD4 D1, a novel BET selectivity profile, and improved half-life relative to inhibitor 1 to guide future in vivo studies. Our preliminary cellular data demonstrated DW34 and 26 effectively reduced c-Myc expression at low nanomolar concentrations in a multiple myeloma cell line and downregulated cytokine IL-8 in a non-small lung cancer cell line. In addition, these design approaches can be applied to related scaffolds including our previously reported imidazole inhibitor, UMN627. With their high affinities and selectivity profile against BET bromodomains, improved metabolic stabilities, and high cellular efficacy, they will serve as useful tool compounds to study BET bromodomain function.
EXPERIMENTAL SECTION
General Procedures.
All reactions were conducted at elevated temperature using aluminum heating blocks with magnetic stirring (200 rpm). Reported temperatures were based on an external thermal couple. All commercially available chemicals were used without further purification. Dry tetrahydrofuran (THF), dimethylformamide (DMF), and dimethoxyethane (DME) were obtained from a commercial solvent system utilizing activated alumina columns under a positive pressure of argon. Thin-layer chromatography (TLC) was used for monitoring reaction progress. Visualization was conducted by using UV light, KMnO4, or PMA stains. Organic solutions were concentrated using a rotary evaporator under reduced pressure at or below 50 °C. Flash chromatography was performed on a Teledyne Isco CombiFlash Rf system utilizing normal phase precolumn load cartridges and gold high-performance columns. All proton (1H) nuclear magnetic resonance spectra were recorded at 400 or 500 MHz. All carbon (13C) nuclear magnetic resonance spectra were recorded at 101 or 126 MHz. The fluorine (19F) nuclear magnetic resonance spectra were recorded at 471 MHz without proton decoupling. Chemical shifts are expressed in parts per million and are referenced to residual solvent (CDCl3: 7.26 ppm for 1H NMR and CDCl3: 77.2 ppm for the central carbon in the 13C NMR. CD3OD: 3.31 ppm for 1H NMR and CD3OD: 49.0 ppm for 13C NMR. Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, and m = multiplet), integration, and coupling constant in Hertz (Hz). Infrared (IR) spectra were taken in a Nicolet Nexus 670 FT-IR with salt plates. IR spectra were reported in cm−1.
Purity Analysis.
Purities of 1–26 were checked by reverse-phase high-performance liquid chromatography (RP-HPLC) with a C-18 column and 10–60% 0.1% TFA water and acetonitrile over 60 min. HPLC traces are shown in Figures S1–S3. All lead compounds used were >95% pure by RP-HPLC except nonlead compounds 5 and 6 (92% and 91%). These two compounds were not evaluated in cells.
tert-Butyl 4-(4-Methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (b).
Compound b was prepared via a known literature procedure.12
tert-Butyl 4-(5-(6-Fluoropyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (c).
To a solution of triazole b (77.6 mg, 0.292 mmol) in DME (2 mL) cooled in a dry ice/acetone bath was added n-BuLi (0.13 mL, 2.5 M in hexanes, 0.32 mmol). After 15 min, a freshly prepared solution of ZnCl2 (67.1 mg, 0.493 mmol) in THF (2 mL) was added, and the reaction was warmed to rt. After 10 min, a solution of 2-chloro-6-fluoropyridine (77.5 mg, 0.589 mmol) and SPhosPdG3 (9.3 mg, 12 μmol) in DME (2 mL) was added, and the reaction was heated to 60 °C. After 4 h, the reaction was cooled to rt and quenched by the addition of saturated aqueous NH4Cl. The reaction mixture was extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (40–80% EtOAc in Hexanes) afforded compound c (91.8 mg, 87%). 1H NMR (500 MHz, CDCl3) δ 7.98 (apparent q, J = 7.9 Hz, 1H), 7.37 (d, J = 7.4 Hz, 1H), 7.01 (dd, J = 8.4, 2.8 Hz, 1H), 4.98 (tt, J = 11.3, 4.1 Hz, 1H), 4.26 (br, 2H), 2.89 (br, 2H), 2.47 (s, 3H), 2.24 (br, 2H), 2.11 (apparent d, J = 12.1 Hz, 2H), 1.48 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.1 (d, JC–F = 241.9 Hz), 154.6, 146.1 (d, JC–F = 14.4 Hz), 142.3, 142.1 (d, JC–F = 8.0 Hz), 130.7, 121.5 (d, JC–F = 4.2 Hz), 109.2 (d, JC–F = 36.7 Hz), 79.8, 57.3, 43.5 (br), 32.1, 28.4, 11.8. 19F NMR (471 MHz, CDCl3) δ −64.83 (d, J = 8.1 Hz). IR (NaCl, thin film, cm−1): 2976, 2932, 2864, 1692, 1601, 1575, 1474, 1425, 1366, 1332, 1246, 1166. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C18H24F5NaO2+ 384.1806, found 384.1819.
tert-Butyl 4-(5-(6-(3-Bromo-5-methylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (d).
General procedure I: To a solution of pyridine c (80.9 mg, 0.224 mmol) and 3-bromo-5-methylphenol (54.2 mg, 0.290 mmol) in DMF (1.1 mL) was added solid K2CO3 (55.0 mg, 0.399 mmol). The reaction was sealed under air and heated to 80 °C. After 24 h, the reaction was cooled to rt, quenched by addition of water, and extracted with EtOAc. The combined organic phases were washed with water, brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (0–60% EtOAc in hexanes) afforded bromide d (102 mg, 87%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.87 (apparent t, J = 7.9 Hz, 1H), 7.22–7.18 (m, 2H), 7.12 (s, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.90 (s, 1H), 4.65 (tt, J = 11.2, 4.1 Hz, 1H), 4.04 (br, 2H), 2.60–2.48 (m, 2H), 2.46 (s, 3H), 2.34 (s, 3H), 2.13–1.98 (m, 2H), 1.79–1.70 (m, 2H), 1.47 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.1, 154.6, 154.1, 145.4, 142.0, 141.5, 140.5, 131.3, 128.9, 122.2, 122.1, 121.3, 119.0, 111.3, 79.7, 56.8, 42.4 (br), 31.9, 28.4, 21.2, 12.0. IR (NaCl, thin film, cm−1): 2976, 2929, 2863, 1693, 1595, 1568, 1471, 1454, 1426, 1366, 1331, 1300, 1275, 1165, 1153. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C25H30BrN5NaO3+ 550.1424, found 550.1415.
tert-Butyl 4-(5-(6-(3-Ethyl-5-methylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (e).
To a solution of bromide d (52.9 mg, 0.100 mmol) and XPhosPdG3 (6.9 mg, 8.2 μmol) in THF (5 mL) at 0 °C was added Et2Zn (0.20 mL, 1 M in hexanes, 0.20 mmol) dropwise. The reaction was warmed to rt. After 1 h, the reaction was quenched by addition of saturated aqueous NH4Cl and extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (30–100% MTBE in hexanes) afforded compound e (39.8 mg, 83%). 1H NMR (500 MHz, CDCl3) δ 7.85 (apparent t, J = 7.8 Hz, 1H), 7.17 (d, J = 7.4 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 6.90 (s, 1H), 6.79 (s, 2H), 4.72 (tt, J = 11.1, 4.0 Hz, 1H), 4.00 (br, 2H), 2.62 (q, J = 7.6 Hz, 2H), 2.49 (s, 3H), 2.46–2.36 (m, 2H), 2.34 (s, 3H), 2.03 (apparent q, J = 12.5, 11.2 Hz, 2H), 1.77–1.68 (m, 2H), 1.48 (s, 9H), 1.22 (t, J = 7.6 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.6, 154.6, 153.6, 146.0, 145.4, 141.9, 140.2, 139.5, 131.4, 125.6, 119.5, 118.3, 118.2, 111.2, 79.6, 56.8, 43.0 (br), 31.9, 28.7, 28.4, 21.4, 15.4, 12.1. IR (NaCl, thin film, cm−1): 2966, 2932, 1692, 1572, 1425, 1302, 1245, 1164. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C27H35N5NaO3+ 500.2632, found 500.2641.
tert-Butyl 4-(5-(6-(3-Butyl-5-methylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (f).
To a solution of bromide d (50.9 mg, 96.4 μmol) and XPhosPdG3 (5.6 mg, 6.6 μmol) in THF (5 mL) cooled in a brine bath was added n-butylmagnesium chloride (0.10 mL, 2.0 M in THF, 0.19 mmol) dropwise. After 30 min, the reaction was quenched by addition of H2O and extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (10–50% IPA in hexanes) afforded compound f (29.3 mg, 60%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.84 (apparent t, J = 7.8 Hz, 1H), 7.17 (d, J = 7.4 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 6.88 (s, 1H), 6.78 (s, 1H), 6.77 (s, 1H), 4.71 (tt, J = 11.3, 4.1 Hz, 1H), 4.01 (br, 2H), 2.57 (t, J = 7.8 Hz, 2H), 2.48 (s, 3H), 2.42 (br, 2H), 2.33 (s, 3H), 2.11–1.98 (m, 2H), 1.72 (apparent d, J = 11.4 Hz, 2H), 1.57 (pent, J = 7.7 Hz, 2H), 1.48 (s, 9H), 1.34 (hept J = 7.4 Hz, 2H), 0.91 (t, J = 7.3 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.6, 154.5, 153.5, 145.3, 144.7, 141.9, 140.2, 139.4, 131.4, 126.1, 119.4, 118.7, 118.3, 111.2, 79.6, 56.8, 43.1 (br), 35.4, 33.4, 31.9, 28.4, 22.3, 21.4, 13.9, 12.1. IR (NaCl, thin film, cm−1): 2959, 2929, 2860, 1692, 1589, 1572, 1468, 1453, 1426, 1365, 1301, 1246, 1166, 1152. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C29H39N5NaO3+ 528.2945, found 528.2943.
tert-Butyl 4-(5-(6-(3-Isopropyl-5-methylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (g).
To a solution of bromide d (52.7 mg, 99.8 μmol) and SPhosPdG3 (4.8 mg, 6.2 μmol) in THF (5 mL) cooled in an ice bath was added isopropylmagnesium chloride (0.10 mL, 2.0 M in THF, 0.20 mmol) dropwise. After 3 h, the reaction was quenched by addition of H2O and extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (10–40% EtOAc in hexanes) afforded product g (18.8 mg, 38%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.85 (apparent t, J = 8.0 Hz, 1H), 7.18 (d, J = 7.3 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 6.93 (s, 1H), 6.82 (s, 1H), 6.79 (s, 1H), 4.74 (tt, J = 11.3, 4.1 Hz, 1H), 4.01 (br, 2H), 2.88 (hept, J = 6.9 Hz, 1H), 2.49 (s, 3H), 2.45–2.36 (m, 2H), 2.34 (s, 3H), 2.03 (apparent qd, J = 12.0, 3.4 Hz, 2H), 1.77–1.71 (m, 2H), 1.48 (s, 9H), 1.23 (d, J = 6.9 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.5, 154.6, 153.6, 150.8, 145.4, 141.9, 140.2, 139.4, 131.3, 124.1, 119.6, 118.3, 116.8, 111.1, 79.6, 56.8, 42.7 (br), 34.0, 31.9, 28.4, 23.9, 21.5, 12.2. IR (NaCl, thin film, cm−1): 2962, 2929, 2867, 1693, 1589, 1572, 1453, 1426, 1365, 1301, 1246, 1166, 1152. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C28H37N5NaO3+ 514.2789, found 514.2801.
tert-Butyl 4-(5-(6-(3-Isobutyl-5-methylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (h).
To a solution of ZnCl2 (76.4 mg, 0.562 mmol) in THF (2 mL) in an ice bath was added t-BuLi. After 10 min, a solution of bromide d (58.9 mg, 0.112 mmol) and SPhosPdG3 (4.8 mg, 6.2 μmol) in DME (2 mL) was added. The reaction was removed from the ice bath and heated to 60 °C. After 1 h, the reaction was quenched by addition of saturated aqueous NH4Cl. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (20–60% EtOAc in hexanes) afforded compound h (38.6 mg, 68%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.84 (apparent t, J = 7.8 Hz, 1H), 7.16 (d, J = 7.4 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 6.84 (s, 1H), 6.79 (s, 1H), 6.73 (s, 1H), 4.71 (tt, J = 11.3, 4.0 Hz, 1H), 4.01 (br, 2H), 2.51–2.37 (m, 2H), 2.48 (s, 3H), 2.43 (d, J = 7.1 Hz, 2H), 2.34 (s, 3H), 2.10–1.96 (m, 2H), 1.83 (hept, J = 6.7 Hz, 1H), 1.73 (d, J = 11.3 Hz, 2H), 1.48 (s, 9H), 0.89 (d, J = 6.7 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.6, 154.6, 153.4, 145.4, 143.5, 141.9, 140.2, 139.2, 131.4, 126.8, 119.4, 119.4, 118.3, 111.1, 79.6, 56.8, 45.2, 42.4 (br), 31.9, 30.1, 28.4, 22.3, 21.4, 12.0. IR (NaCl, thin film, cm−1): 2956, 2928, 2867, 1693, 1590, 1572, 1453, 1425, 1365, 1301, 1246, 1165, 1152. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C29H39N5NaO3 + 528.2945, found 528.2951.
tert-Butyl 4-(5-(6-(3-Cyclopentyl-5-methylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (i).
To a solution of bromide d (32.8 mg, 62.1 μmol) and XPhosPdG3 (2.0 mg, 2.4 μmol) in THF (0.60 mL) at rt was added cyclopentylmagnesium bromide (60 μL, 2.0 M in Et2O, 120 μmol). The reaction was heated to 60 °C. After 1 h, the reaction was cooled to rt and quenched by addition of saturated aqueous NH4Cl. The reaction mixture was extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (30–90% EtOAc in hexanes) afforded product i (29.1 mg, 91%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.84 (apparent t, J = 7.8 Hz, 1H), 7.18 (d, J = 7.4 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 6.94 (s, 1H), 6.83 (s, 1H), 6.78 (s, 1H), 4.74 (tt, J = 11.1, 4.0 Hz, 1H), 4.01 (br, 2H), 2.95 (p, J = 8.7 Hz, 1H), 2.49 (s, 3H), 2.44–2.35 (m, 2H), 2.34 (s, 3H), 2.09–1.96 (m, 4H), 1.84–1.76 (m, 2H), 1.75–1.65 (m, 4H), 1.62–1.52 (m, 2H), 1.48 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.5, 154.5, 153.5, 148.5, 145.4, 141.9, 140.2, 139.3, 131.3, 124.8, 119.6, 118.3, 117.5, 111.1, 79.6, 56.8, 45.8, 43.0 (br), 34.6, 31.9, 28.4, 25.4, 21.4, 12.2. IR (NaCl, thin film, cm−1): 2954, 2867, 1693, 1590, 1572, 1471, 1453, 1426, 1302, 1246, 1166, 1153. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C30H39N5NaO3 + 540.2945, found 540.2956.
tert-Butyl 4-(5-(6-(3-Cyclohexyl-5-methylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (j).
To a solution of bromide d (49.3 mg, 93.4 μmol) and XPhosPdG3 (4.3 mg, 5.1 μmol) in THF (5 mL) cooled in an ice bath was added cyclohexylmagnesium chloride (0.19 mL, 1.0 M in 2-methyltetrahydrofuran, 0.19 mmol). After 30 min, the reaction was quenched by addition of saturated aqueous NH4Cl and extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (0–50% IPA in hexanes) afforded product j (41.1 mg, 83%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.84 (apparent t, J = 7.8 Hz, 1H), 7.17 (d, J = 7.4 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 6.91 (s, 1H), 6.80 (s, 1H), 6.78 (s, 1H), 4.74 (tt, J = 11.3, 4.0 Hz, 1H), 4.00 (br, 2H), 2.50 (s, 3H), 2.48–2.43 (m, 1H), 2.42–2.35 (m, 2H), 2.33 (s, 3H), 2.02 (apparent qd, J = 12.0, 4.2 Hz, 2H), 1.87−1.80 (m, 4H), 1.76–1.68 (m, 3H), 1.48 (s, 9H), 1.42–1.34 (m, 4H), 1.29–1.17 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.5, 154.5, 153.5, 150.0, 145.3, 141.9, 140.2, 139.3, 131.3, 124.5, 119.6, 118.3, 117.2, 111.2, 79.6, 56.8, 44.4, 43.0 (br), 34.3, 31.9, 28.4, 26.8, 26.1, 21.4, 12.2. IR (NaCl, thin film, cm−1): 2974, 2927, 2852, 1693, 1589, 1573, 1470, 1452, 1426, 1365, 1331, 1303, 1276, 1246, 1166, 1152. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C31H41N5NaO3+ 554.3102, found 554.3112.
2-(3-Ethyl-5-methylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (2).
General procedure IV: To a solution of compound e (14.8 mg, 31.0 μmol) in DCM (0.50 mL) was added TFA (0.5 mL) at rt. After 1.5 h, the reaction was concentrated under reduced pressure which afforded product 2 (16.8 mg, 95%) as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.02 (apparent t, J = 7.9 Hz, 1H), 7.39 (d, J = 7.4 Hz, 1H), 7.12 (d, J = 8.3 Hz, 1H), 6.96 (s, 1H), 6.84 (s, 2H), 4.94–4.84 (m, 1H), 3.38–3.33 (m, 2H), 2.70 (apparent t, J = 11.6 Hz, 2H), 2.64 (q, J = 7.9 Hz, 2H), 2.46 (s, 3H), 2.36 (s, 3H), 2.32–2.21 (m, 2H), 2.03 (apparent dd, J = 14.2, 4.0 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H). 13C{1H} NMR (126 MHz, MeOD) δ 163.7, 154.0, 146.1, 144.2, 141.5, 140.9, 139.7, 132.1, 125.1, 119.2, 118.9, 117.9, 111.7, 53.6, 42.7, 28.6, 28.2, 20.0, 14.7, 10.2. IR (NaCl, thin film, cm−1): 2969, 2929, 2852, 1688, 1678, 1590, 1573, 1454, 1432, 1330, 1298, 1248, 1202, 1135. HRMS (ESI-TOF) m/z [M + H]+ calcd for C22H28N5O+ 378.2288, found 378.2281. HPLC purity is >99%.
2-(3-Butyl-5-methylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (3).
General procedure IV was used and product 3 (27.7 mg, 96%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.02 (apparent t, J = 7.9 Hz, 1H), 7.38 (d, J = 7.4 Hz, 1H), 7.12 (d, J = 8.3 Hz, 1H), 6.93 (s, 1H), 6.85 (s, 1H), 6.79 (s, 1H), 4.90–4.83 (m, 1H), 3.38–3.33 (m, 2H), 2.72 (apparent td, J = 12.6, 3.1 Hz, 2H), 2.60 (t, J = 7.7 Hz, 2H), 2.46 (s, 3H), 2.36 (s, 3H), 2.31–2.20 (m, 2H), 2.06–1.98 (m, 2H), 1.57 (pent, J = 7.6 Hz, 2H), 1.32 (hex, J = 7.5 Hz, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C{1H} NMR (126 MHz, MeOD) δ 163.8, 154.0, 144.7, 144.1, 141.4, 140.9, 139.6, 132.2, 125.7, 119.1, 118.9, 118.6, 111.7, 53.6, 42.7, 34.9, 33.4, 28.6, 21.8, 20.0, 12.8, 10.1. IR (NaCl, thin film, cm−1): 2959, 2930, 2744, 2529, 1678, 1591, 1573, 1454, 1431, 1311, 1298, 1248, 1202, 1137. HRMS (ESI-TOF) m/z [M + H]+ calcd for C24H32N5O+ 406.2601, found 406.2616. HPLC purity is 98%.
2-(3-Isopropyl-5-methylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (4).
General procedure IV was used, and product 4 (21.9 mg, 93%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.02 (apparent t, J = 7.9 Hz, 1H), 7.39 (d, J = 7.4 Hz, 1H), 7.11 (d, J = 8.3 Hz, 1H), 6.99 (s, 1H), 6.85 (apparent s, 2H), 4.90–4.84 (m, 1H), 3.39–3.34 (m, 2H), 2.89 (hept, J = 6.9 Hz, 1H), 2.71 (apparent t, J = 11.5 Hz, 2H), 2.46 (s, 3H), 2.38 (s, 3H), 2.32–2.21 (m, 2H), 2.05 (apparent dd, J = 14.2, 4.0 Hz, 2H), 1.23 (d, J = 6.9 Hz, 6H). 13C{1H} NMR (126 MHz, MeOD) δ 163.7, 154.0, 150.9, 144.2, 141.5, 140.9, 139.6, 132.2, 123.6, 119.1, 118.9, 116.6, 111.6, 53.5, 42.7, 33.8, 28.6, 22.9, 20.1, 10.1. IR (NaCl, thin film, cm−1): 2962, 2929, 2858, 1677, 1591, 1573, 1460, 1431, 1200, 1176, 1141. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C23H29N5NaO+ 414.2264, found 414.2252. HPLC purity is 97%.
2-(3-Isobutyl-5-methylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (5).
General procedure IV was used, and product 5 (48.6 mg, quant.) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.01 (apparent t, J = 7.8 Hz, 1H), 7.38 (d, J = 7.3 Hz, 1H), 7.11 (d, J = 8.2 Hz, 1H), 6.90 (s, 1H), 6.86 (s, 1H), 6.76 (s, 1H), 4.86 (tt, J = 10.8, 4.1 Hz, 1H), 3.39–3.34 (m, 2H), 2.75 (apparent td, J = 12.6, 3.1 Hz, 2H), 2.46 (d, J = 7.2 Hz, 2H), 2.45 (s, 3H), 2.36 (s, 3H), 2.32–2.20 (m, 2H), 2.06–1.98 (m, 2H), 1.82 (nonet, J = 6.8 Hz, 1H), 0.87 (d, J = 6.9 Hz, 6H). 13C{1H} NMR (126 MHz, MeOD) δ 163.8, 153.9, 144.1, 143.5, 141.4, 140.9, 139.4, 132.2, 126.4, 119.3, 119.2, 118.9, 111.6, 53.6, 44.7, 42.7, 30.0, 28.5, 21.2, 20.0, 10.1. IR (NaCl, thin film, cm−1): 2962, 2927, 2850, 1677, 1591, 1573, 1454, 1431, 1299, 1249, 1202, 1177, 1137. HRMS (ESI-TOF) m/z [M + H]+ calcd for C24H32N5O+ 406.2601, found 406.2595. HPLC purity is 92%.
2-(3-Cyclopentyl-5-methylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (6).
General procedure IV was used, and product 6 (21.6 mg, 73%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.02 (apparent t, J = 7.8 Hz, 1H), 7.39 (d, J = 7.4 Hz, 1H), 7.11 (d, J = 8.3 Hz, 1H), 7.00 (s, 1H), 6.86 (s, 1H), 6.84 (s, 1H), 4.90–4.85 (m, 1H), 3.37–3.33 (m, 2H), 2.99 (pent, J = 8.7, 8.1 Hz, 1H), 2.69 (apparent td, J = 12.7, 3.1 Hz, 2H), 2.46 (s, 3H), 2.37 (s, 3H), 2.31–2.22 (m, 2H), 2.09–2.00 (m, 4H), 1.86–1.77 (m, 2H), 1.75–1.66 (m, 2H), 1.61–1.50 (m, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 163.7, 153.9, 148.4, 144.1, 141.5, 140.9, 139.5, 132.1, 124.3, 119.1, 118.9, 117.2, 111.6, 53.6, 45.7, 42.7, 34.3, 28.6, 25.0, 20.1, 10.2. IR (NaCl, thin film, cm−1): 2956, 2868, 1678, 1591, 1573, 1454, 1431, 1298, 1249, 1202, 1137. HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H32N5O+ 418.2601, found 418.2609. HPLC purity is 91%.
2-(3-Cyclohexyl-5-methylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (7).
General procedure IV was used, and product 7 (42.5 mg, 94%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.01 (apparent t, J = 7.8 Hz, 1H), 7.38 (d, J = 7.4 Hz, 1H), 7.10 (d, J = 8.3 Hz, 1H), 6.96 (s, 1H), 6.84 (s, 1H), 6.82 (s, 1H), 4.88 (tt, J = 10.6, 4.1 Hz, 1H), 3.37–3.33 (m, 2H), 2.69 (apparent td, J = 12.6, 3.1 Hz, 2H), 2.53–2.47 (m, 1H), 2.46 (s, 3H), 2.36 (s, 3H), 2.31–2.21 (m, 2H), 2.04 (apparent dd, J = 14.2, 4.0 Hz, 2H),1.82 (apparent d, J = 6.7 Hz, 4H), 1.74 (d, J = 12.5 Hz, 1H), 1.41 (t, J = 10.1 Hz, 4H), 1.33–1.20 (m, 1H). 13C{1H} NMR (126 MHz, MeOD) δ 163.7, 153.9, 150.0, 144.1, 141.5, 140.9, 139.5, 132.2, 124.1, 119.1, 118.9, 117.0, 111.7, 53.6, 44.3, 42.6, 34.2, 28.6, 26.5, 25.7, 20.1, 10.2. IR (NaCl, thin film, cm−1): 2927, 2852, 2746, 1675, 1613, 1590, 1573, 1470, 1453, 1430, 1298, 1248, 1201, 1139. HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H34N5O+ 432.2758, found 432.2759. HPLC purity is >99%.
2-(4-Methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)-6-((5-methyl-[1,1′-biphenyl]-3-yl)oxy)pyridine (8).
General procedure IV was used, and product 8 (53.7 mg, quant.) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.04 (apparent t, J = 7.9 Hz, 1H), 7.62 (d, J = 7.7 Hz, 2H), 7.44 (apparent t, J = 7.6 Hz, 2H), 7.40 (d, J = 7.5 Hz, 1H), 7.39 (s, 1H), 7.35 (t, J = 7.3 Hz, 1H), 7.28 (s, 1H), 7.18 (d, J = 8.3 Hz, 1H), 7.02 (s, 1H), 4.88 (tt, J = 10.7, 4.2 Hz, 1H), 3.27 (apparent d, J = 13.1 Hz, 2H), 2.63 (apparent td, J = 12.6, 3.1 Hz, 2H), 2.44 (apparent s, 6H), 2.29–2.18 (m, 2H), 2.00 (apparent dd, J = 14.2, 4.0 Hz, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 163.6, 154.5, 144.2, 142.7, 141.5, 141.0, 140.5, 139.8, 132.2, 128.8, 127.6, 126.5, 124.0, 120.9, 119.1, 117.0, 111.8, 53.5, 42.5, 28.5, 20.1, 10.1. IR (NaCl, thin film, cm−1): 2977, 2850, 2743, 1678, 1592, 1572, 1455, 1431, 1319, 1296, 1247, 1201, 1142. HRMS (ESI-TOF) m/z [M + H]+ calcd for C26H28N5O+ 426.2288, found 426.2292. HPLC purity is >99%.
2-(3-Bromo-5-methylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (9).
General procedure IV was used, and product 9 (50.2 mg, 94%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.05 (apparent t, J = 7.9 Hz, 1H), 7.42 (d, J = 7.4 Hz, 1H), 7.30 (s, 1H), 7.24 (s, 1H), 7.17 (d, J = 8.3 Hz, 1H), 7.03 (s, 1H), 4.87 (tt, J = 10.4, 4.0 Hz, 1H), 3.42 (apparent dt, J = 13.2, 4.1 Hz, 2H), 2.81 (apparent td, J = 12.6, 3.2 Hz, 2H), 2.45 (s, 3H), 2.36 (s, 3H), 2.35–2.26 (m, 2H), 2.05 (apparent dd, J = 14.2, 4.0 Hz, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 163.2, 154.5, 144.1, 142.1, 141.6, 141.2, 132.1, 128.5, 121.9, 121.7, 121.3, 119.5, 111.9, 53.5, 42.7, 28.6, 19.7, 10.0. IR (NaCl, thin film, cm−1): 2990, 2743, 1678, 1596, 1568, 1455, 1430, 1416, 1298, 1257, 1201, 1143. HRMS (ESI-TOF) m/z [M + H]+ calcd for C20H23BrN5O+ 428.1080, found 450.0900. HPLC purity is >99%.
2-(3,5-Diethylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (10).
General procedure IV was used, and product 10 (17.9 mg, quant.) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.02 (dd, J = 8.3, 7.4 Hz, 1H), 7.39 (dd, J = 7.5, 0.7 Hz, 1H), 7.12 (dd, J = 8.3, 0.7 Hz, 1H), 6.98 (t, J = 1.5 Hz, 1H), 6.86 (d, J = 1.5 Hz, 2H), 4.93–4.83 (m, 1H), 3.37–3.33 (m, 2H), 2.71 (apparent dd, J = 12.7, 3.1 Hz, 2H), 2.66 (q, J = 7.6 Hz, 4H), 2.46 (s, 3H), 2.32–2.20 (m, 2H), 2.04 (apparent dd, J = 14.1, 3.3 Hz, 2H), 1.24 (t, J = 7.6 Hz, 6H). 13C{1H} NMR (126 MHz, MeOD) δ 163.7, 154.1, 146.2, 144.2, 141.5, 140.9, 132.1, 123.9, 118.9, 118.0, 111.6, 53.5, 42.7, 28.6, 28.3, 14.7, 10.2. IR (NaCl, thin film, cm−1): 2966, 2930, 2854, 1683, 1590, 1573, 1456, 1431, 1298, 1248, 1202, 1178, 1135. HRMS (ESI-TOF) m/z [M + H]+ calcd for C23H30N5O+ 392.2445, found 392.2432. HPLC purity is >99%.
2-(3,5-Dipropylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (11).
General procedure IV was used, and product 11 (35.4 mg, 95%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.02 (dd, J = 8.4, 7.4 Hz, 1H), 7.38 (dd, J = 7.5, 0.7 Hz, 1H), 7.11 (dd, J = 8.4, 0.7 Hz, 1H), 6.94 (t, J = 1.5 Hz, 1H), 6.84 (d, J = 1.5 Hz, 2H), 4.87 (tt, J = 10.7, 4.1 Hz, 1H), 3.38–3.33 (m, 2H), 2.74 (apparent td, J = 12.7, 3.2 Hz, 2H), 2.60 (t, J = 7.4 Hz, 4H), 2.45 (s, 3H), 2.31–2.20 (m, 2H), 2.03 (apparent dd, J = 14.1, 3.6 Hz, 2H), 1.63 (hex, J = 7.4 Hz, 4H), 0.94 (t, J = 7.4 Hz, 6H). 13C{1H} NMR (126 MHz, MeOD) δ 163.8, 153.9, 144.5, 144.2, 141.5, 140.9, 132.2, 125.2, 118.9, 118.7, 111.6, 53.6, 42.7, 37.4, 28.6, 24.3, 12.7, 10.1. IR (NaCl, thin film, cm−1): 2961, 2931, 2871, 1677, 1591, 1573, 1453, 1431, 1298, 1248, 1202, 1177, 1136. HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H34N5O+ 420.2758, found 420.2767. HPLC purity is >99%.
2-(3,5-Dibutylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (12).
General procedure IV was used, and product 12 (44.0 mg, quant.) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (400 MHz, MeOD) δ 8.02 (apparent t, J = 7.9 Hz, 1H), 7.38 (d, J = 7.4 Hz, 1H), 7.11 (d, J = 8.3 Hz, 1H), 6.93 (s, 1H), 6.82 (s, 2H), 4.85 (tt, J = 10.6, 4.1 Hz, 1H), 3.40–3.33 (m, 2H), 2.75 (apparent td, J = 12.6, 3.1 Hz, 2H), 2.62 (t, J = 7.6 Hz, 4H), 2.45 (s, 3H), 2.33–2.18 (m, 2H), 2.08–1.97 (m, 2H), 1.59 (pent, J = 7.5 Hz, 4H), 1.34 (hex, J = 7.3 Hz, 4H), 0.91 (t, J = 7.3 Hz, 6H). 13C{1H} NMR (126 MHz, MeOD) δ 163.8, 154.0, 144.7, 144.1, 141.4, 140.9, 132.2, 125.1, 118.9, 118.6, 111.6, 53.6, 42.7, 35.0, 33.4, 28.6, 21.9, 12.8, 10.1. IR (NaCl, thin film, cm−1): 2958, 2931, 2859, 2741, 1677, 1591, 1573, 1455, 1431, 1298, 1249, 1202, 1177, 1136. HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H38N5O+ 448.3071, found 448.3081. HPLC purity is 99%.
2-(3,5-Dicyclopropylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (13).
General procedure IV was used, and compound 13 (14.4 mg, 86%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a mono-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.02 (dd, J = 8.3, 7.5 Hz, 1H), 7.39 (d, J = 7.4 Hz, 1H), 7.11 (d, J = 8.2 Hz, 1H), 6.72 (t, J = 1.7 Hz, 1H), 6.65 (d, J = 1.6 Hz, 2H), 4.94–4.90 (m, 1H), 3.42–3.35 (m, 2H), 2.72 (td, J = 12.8, 3.2 Hz, 2H), 2.47 (s, 3H), 2.32–2.23 (m, 2H), 2.08–2.01 (m, 2H), 1.90 (tt, J = 8.4, 5.1 Hz, 2H), 1.02–0.97 (m, 4H), 0.71–0.65 (m, 4H). 13C{1H} NMR (126 MHz, MeOD) δ 163.6, 154.3, 146.3, 144.2, 141.6, 140.9, 132.1, 119.1, 118.8, 115.3, 111.7, 53.6, 42.8, 28.6, 14.7, 10.3, 8.7. IR (NaCl, thin film, cm−1): 2999, 2726, 1678, 1589, 1572, 1470, 1456, 1430, 1297, 1247, 1202, 1136. HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H30N5O+ 416.2445, found 416.2456. HPLC purity is 96%.
2-(3,5-Dicyclopentylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (14).
General procedure IV was used, and product 14 (17.6 mg, quant.) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a tris-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.02 (dd, J = 8.3, 7.4 Hz, 1H), 7.39 (dd, J = 7.4, 0.7 Hz, 1H), 7.10 (dd, J = 8.2, 0.7 Hz, 1H), 7.06 (t, J = 1.7 Hz, 1H), 6.89 (d, J = 1.5 Hz, 2H), 4.90–4.83 (m, 1H), 3.37–3.33 (m, 2H), 3.02 (tt, J = 9.9, 7.5 Hz, 2H), 2.70 (apparent td, J = 12.7, 3.2 Hz, 2H), 2.46 (s, 3H), 2.32–2.21 (m, 2H), 2.12–2.02 (m, 6H), 1.87–1.77 (m, 4H), 1.76–1.66 (m, 4H), 1.62–1.53 (m, 4H). 13C{1H} NMR (126 MHz, MeOD) δ 163.7, 153.8, 148.3, 144.1, 141.5, 140.9, 132.2, 122.5, 118.9, 117.2, 111.6, 53.6, 45.8, 42.7, 34.3, 28.7, 25.0, 10.1. IR (NaCl, thin film, cm−1): 2953, 2866, 1677, 1590, 1572, 1455, 1430, 1298, 1248, 1201, 1176, 1137. HRMS (ESI-TOF) m/z [M + H]+ calcd for C29H38N5O+ 472.3071, found 472.3059. HPLC purity is >99%.
2-(3,5-Dicyclohexylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (15).
General procedure IV was used, and product 15 (28.1 mg, 81%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a tris-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.01 (dd, J = 8.3, 7.4 Hz, 1H), 7.39 (d, J = 7.1 Hz, 1H), 7.08 (d, J = 8.3 Hz, 1H), 6.99 (t, J = 1.6 Hz, 1H), 6.86 (d, J = 1.5 Hz, 2H), 4.87 (tt, J = 10.6, 4.2 Hz, 1H), 3.36–3.30 (m, 2H), 2.72 (apparent td, J = 12.8, 3.0 Hz, 2H), 2.58–2.50 (m, 2H), 2.46 (s, 3H), 2.32–2.22 (m, 2H), 2.10 (apparent dd, J = 14.1, 3.2 Hz, 2H), 1.85 (apparent d, J = 8.8 Hz, 8H), 1.75 (d, J = 12.8 Hz, 2H), 1.51–1.37 (m, 8H), 1.36–1.25 (m, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 163.6, 153.8, 149.9, 144.2, 141.5, 140.9, 132.3, 121.8, 119.0, 116.9, 111.6, 53.6, 44.4, 42.6, 34.3, 28.7, 26.5, 25.7, 10.1. IR (NaCl, thin film, cm−1): 2926, 2852, 1677, 1590, 1572, 1449, 1430, 1310, 1248, 1201, 1180, 1139. HRMS (ESI-TOF) m/z [M + H]+ calcd for C31H42N5O+ 500.3384, found 500.3393. HPLC purity is >99%.
2-(3,5-Dichlorophenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (16).
General procedure IV was used, and compound 16 (15.7 mg, 76%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a mono-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.09 (dd, J = 8.3, 7.4 Hz, 1H), 7.46 (d, J = 7.2 Hz, 1H), 7.40 (t, J = 1.8 Hz, 1H), 7.31 (d, J = 1.9 Hz, 2H), 7.23 (d, J = 8.1 Hz, 1H), 4.89–4.83 (m, 1H), 3.46 (dt, J = 13.1, 3.7 Hz, 2H), 2.95–2.84 (m, 2H), 2.44 (s, 3H), 2.39–2.27 (m, 2H), 2.11–2.02 (m, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 162.8, 155.1, 144.2, 141.7, 141.5, 135.3, 132.1, 124.9, 120.9, 120.2, 112.0, 53.3, 42.6, 28.6, 9.9. IR (NaCl, thin film, cm−1): 2968, 2730, 1680, 1577, 1425, 1247, 1202, 1134. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C19H20Cl2N5O+ 404.1039, found 404.1046. HPLC purity is >99%.
2-(3,5-Dibromophenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (17).
General procedure IV was used, and product 17 (33.0 mg, quant.) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.09 (dd, J = 8.3, 7.4 Hz, 1H), 7.68 (t, J = 1.7 Hz, 1H), 7.48 (d, J = 1.7 Hz, 2H), 7.46 (dd, J = 7.5, 0.7 Hz, 1H), 7.22 (dd, J = 8.4, 0.7 Hz, 1H), 4.89–4.83 (m, 1H), 3.47 (apparent dt, J = 13.1, 3.6 Hz, 2H), 2.90 (apparent td, J = 12.8, 3.0 Hz, 2H), 2.44 (s, 3H), 2.40–2.28 (m, 2H), 2.08 (apparent dd, J = 14.5, 4.0 Hz, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 162.8, 155.1, 144.2, 141.8, 141.5, 132.1, 130.4, 124.2, 122.7, 120.2, 112.0, 53.3, 42.6, 28.6, 10.0. IR (NaCl, thin film, cm−1): 2923, 2851, 1676, 1568, 1419, 1246, 1203, 1135. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C19H19Br2N5NaO+ 513.9849, found 513.9851. HPLC purity is >99%.
2-(3,5-Bis(trifluoromethyl)phenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (18).
General procedure IV was used, and product 18 (25.5 mg, quant.) was isolated as a white solid. Integration of the 19F NMR indicated formation of a bis-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.13 (dd, J = 8.3, 7.4 Hz, 1H), 7.91 (apparent s, 3H), 7.49 (dd, J = 7.5, 0.7 Hz, 1H), 7.31 (dd, J = 8.4, 0.7 Hz, 1H), 4.80 (tt, J = 10.3, 4.2 Hz, 1H), 3.39 (apparent dt, J = 13.8, 4.4 Hz, 2H), 2.85 (apparent td, J = 12.9, 3.1 Hz, 2H), 2.41 (s, 3H), 2.36–2.22 (m, 2H), 2.06–1.94 (m, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 162.6, 154.8, 144.2, 141.8, 141.7, 132.8 (q, JC–F = 33.9 Hz), 132.1, 124.1 (q, JC–F = 273.4 Hz), 122.6 (q, JC–F = 4.0 Hz), 120.6, 118.22 (hept, JC–F = 3.9 Hz), 112.2, 53.0, 42.2, 28.5, 9.8. 19F NMR (471 MHz, MeOD) δ −64.2 (s, 6F, ArCF3), −77.5 (s, 6F, TFA). IR (NaCl, thin film, cm−1): 2990, 2850, 1676, 1597, 1577, 1370, 1280, 1175, 1135. HRMS (ESI-TOF) m/z [M + H]+ calcd for C21H20F6N5O+ 472.1567, found 472.1572. HPLC purity is >99%.
(5-((6-(4-Methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridin-2-yl)oxy)-1,3-phenylene)dimethanol (19).
To a solution of alcohol S17 in DCM (1.0 mL) was added TFA (0.2 mL). The reaction was sealed under air at rt. After 20 min, the reaction was concentrated under reduced pressure. Final purification by column chromatography (5–50% MeOH in EtOAc with 1% TEA) afforded product 19 (9.9 mg, 54%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.01 (dd, J = 8.3, 7.4 Hz, 1H), 7.35 (d, J = 7.3 Hz, 1H), 7.22 (s, 1H), 7.14 (d, J = 8.3 Hz, 1H), 7.09 (s, 2H), 4.71 (tt, J = 11.4, 4.0 Hz, 1H), 4.65 (s, 4H), 2.96 (dt, J = 13.5, 3.6 Hz, 2H), 2.42 (s, 3H), 2.29 (td, J = 12.7, 2.7 Hz, 2H), 1.98 (apparent qd, J = 12.4, 4.2 Hz, 2H), 1.81ȓ1.74 (m, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 163.6, 154.2, 144.6, 143.8, 141.3, 140.8, 131.7, 121.0, 119.0, 118.2, 111.5, 63.1, 56.6, 44.3, 32.1, 10.1. IR (NaCl, thin film, cm−1): 3294, 2923, 2852, 1641, 1590, 1572, 1451, 1429, 1298, 1246, 1191. HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H30N5O+ 416.2445, found 416.2456. HPLC purity is 95%.
2-(Cyclohexyloxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (20).
General procedure IV was used, and compound 20 (32.3 mg, 84%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a mono-TFA salt. 1H NMR (500 MHz, MeOD) δ 7.85 (dd, J = 8.4, 7.3 Hz, 1H), 7.18 (dd, J = 7.3, 0.8 Hz, 1H), 6.86 (dd, J = 8.4, 0.8 Hz, 1H), 5.17 (tt, J = 10.2, 4.2 Hz, 1H), 5.01 (tt, J = 8.9, 3.8 Hz, 1H), 3.65 (dt, J = 12.9, 3.9 Hz, 2H), 3.22 (ddd, J = 13.1, 11.5, 3.4 Hz, 2H), 2.60–2.47 (m, 2H), 2.46–2.40 (m, 2H), 2.41 (s, 3H), 2.09ȓ1.98 (m, 2H), 1.90–1.81 (m, 2H), 1.68–1.54 (m, 3H), 1.48–1.35 (m, 3H). 13C{1H} NMR (126 MHz, MeOD) δ 163.6, 143.6, 141.3, 139.8, 133.2, 117.6, 112.0, 73.3, 53.0, 42.6, 31.4, 28.8, 25.2, 23.6, 9.7. IR (NaCl, thin film, cm−1): 2939, 2859, 1678, 1574, 1439, 1203, 1133. HRMS (ESI-TOF) m/z [M + H]+ calcd for C19H28N5O+ 342.2288, found 342.2287. HPLC purity is >99%.
2-Isopropoxy-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (21).
General procedure IV was used, and compound 21 (41.0 mg, 85%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a mono-TFA salt. 1H NMR (500 MHz, MeOD) δ 7.85 (dd, J = 8.4, 7.3 Hz, 1H), 7.18 (d, J = 7.2 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 5.29 (hept, J = 6.2 Hz, 1H), 5.23 (tt, J = 10.2, 4.3 Hz, 1H), 3.64 (dt, J = 13.2, 3.9 Hz, 2H), 3.21 (td, J = 13.0, 3.3 Hz, 2H), 2.59–2.49 (m, 2H), 2.46–2.39 (m, 2H), 2.42 (s, 3H), 1.39 (d, J = 6.2 Hz, 6H). 13C{1H} NMR (126 MHz, MeOD) δ 163.7, 143.6, 141.4, 139.8, 133.1, 117.6, 112.0, 68.2, 53.1, 42.6, 28.8, 21.0, 9.7. IR (NaCl, thin film, cm−1): 2982, 2837, 2740, 2527, 1677, 1599, 1574, 1439, 1314, 1257, 1203, 1140. HRMS (ESI-TOF) m/z [M + H]+ calcd for C16H24N5O+ 302.1975, found 302.1969. HPLC purity is >99%.
2-(2,6-Dimethylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (22).
General procedure IV was used, and compound 22 (34.8 mg, 73%) was isolated as a yellow solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a mono-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.05 (dd, J = 8.4, 7.4 Hz, 1H), 7.37 (dd, J = 7.4, 0.7 Hz, 1H), 7.20 (dd, J = 8.4, 0.4 Hz, 1H), 7.19–7.16 (m, 2H), 7.15–7.11 (m, 1H), 4.80 (tt, J = 10.0, 4.2 Hz, 1H), 3.40 (dt, J = 13.5, 4.3 Hz, 2H), 2.84–2.68 (m, 2H), 2.43 (s, 3H), 2.26–2.16 (m, 2H), 2.15 (s, 6H), 1.92–1.84 (m, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 162.9, 150.5, 144.2, 141.7, 141.1, 132.0, 131.0, 128.6, 125.3, 118.6, 110.8, 53.0, 42.5, 28.1, 15.4, 10.2. IR (NaCl, thin film, cm−1): 2925, 2851, 1679, 1595, 1575, 1470, 1456, 1429, 1298, 1268, 1246, 1201, 1178, 1132. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C21H26N5O+ 364.2132, found 364.2143. HPLC purity is >99%.
2-(2,5-Dibromophenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (23).
General procedure IV was used, and compound 23 (24.1 mg, 80%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a mono-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.10 (dd, J = 8.3, 7.4 Hz, 1H), 7.67 (d, J = 8.6 Hz, 1H), 7.60 (d, J = 2.3 Hz, 1H), 7.45–7.41 (m, 2H), 7.26 (dd, J = 8.4, 0.7 Hz, 1H), 4.86–4.80 (m, 1H), 3.50 (dt, J = 13.5, 4.0 Hz, 2H), 3.01–2.91 (m, 2H), 2.41 (s, 3H), 2.35–2.25 (m, 2H), 2.03–1.97 (m, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 162.6, 151.7, 144.0, 141.8, 141.5, 134.6, 132.1, 130.0, 127.5, 121.1, 119.9, 115.7, 111.6, 52.9, 42.6, 28.4, 9.8. IR (NaCl, thin film, cm−1): 2985, 2848, 2740, 1676, 1567, 1466, 1428, 1388, 1203. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C19H20Br2N5O+ 492.0029, found 492.0019. HPLC purity is >99%.
2-(5-Isopropyl-2-methylphenoxy)-6-(4-methyl-1-(piperidin-4-yl)-1H-1,2,3-triazol-5-yl)pyridine (24).
General procedure IV was used, and compound 24 (23.0 mg, 65%) was isolated as a white solid. Analysis by 19F NMR with 2,2,2-trifluoroethanol as the internal standard indicated formation of a mono-TFA salt. 1H NMR (500 MHz, MeOD) δ 8.03 (dd, J = 8.4, 7.4 Hz, 1H), 7.38 (dd, J = 7.4, 0.7 Hz, 1H), 7.27 (d, J = 7.8 Hz, 1H), 7.14 (dd, J = 8.4, 0.7 Hz, 1H), 7.11 (dd, J = 7.8, 1.8 Hz, 1H), 6.99 (d, J = 1.8 Hz, 1H), 4.86 (tt, J = 10.2, 4.2 Hz, 1H), 3.38 (dt, J = 13.0, 4.2 Hz, 2H), 2.90 (heptet, J = 6.9 Hz, 1H), 2.79–2.66 (m, 2H), 2.45 (s, 3H), 2.27–2.18 (m, 2H), 2.17 (s, 3H), 2.00–1.92 (m, 2H), 1.22 (d, J = 6.9 Hz, 6H). 13C{1H} NMR (126 MHz, MeOD) δ 163.6, 152.0, 148.5, 144.1, 141.7, 141.0, 132.0, 131.0, 127.6, 123.2, 120.3, 118.6, 111.1, 53.2, 42.5, 33.4, 28.3, 23.0, 14.8, 10.2. IR (NaCl, thin film, cm−1): 2979, 2850, 2737, 1681, 1575, 1202, 1178, 1132. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C23H30N5O+ 392.2445, found 392.2455. HPLC purity is >99%.
2-(4-(5-(6-(3,5-Dimethylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidin-1-yl)-N,N-dimethylethan-1-amine (DW34).
To a solution of compound 1 (352 mg, 0.738 mmol) in methyl ethyl ketone (3.0 mL) were added 2-chloro-N,N-dimethylethylamine hydrochloride (117 mg, 0.815 mmol), K2CO3 (310 mg, 2.25 mmol), and NaI (125 mg, 0.834 mmol). The reaction was sealed under air and heated to 40 °C. After 18 h, the reaction was cooled to rt and quenched by the addition of saturated aqueous K2CO3. The reaction mixture was extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), and concentrated under reduced pressure. Final purification by column chromatography (5–30% MeOH in DCM) afforded compound DW34 (22.7 mg, 7%) as a white solid. 1H NMR (500 MHz, MeOD) δ 7.99 (dd, J = 8.3, 7.4 Hz, 1H), 7.34 (d, J = 7.4 Hz, 1H), 7.09 (d, J = 8.3 Hz, 1H), 6.91 (s, 1H), 6.79 (s, 2H), 4.61 (tt, J = 11.7, 4.1 Hz, 1H), 2.85 (apparent d, J = 12.6 Hz, 2H), 2.56–2.46 (m, 4H), 2.43 (s, 3H), 2.34 (s, 6H), 2.33 (s, 6H), 2.13 (apparent qd, J = 12.3, 3.8 Hz, 2H), 1.78–1.70 (m, 4H). 13C{1H} NMR (126 MHz, MeOD) δ 163.6, 153.9, 144.6, 141.3, 140.7, 139.3, 131.9, 126.4, 119.1, 118.7, 111.5, 56.8, 56.1, 55.2, 52.5, 44.4, 31.4, 20.1, 10.1. IR (NaCl, thin film, cm−1): 3428, 2954, 2829, 1682, 1589, 1573, 1432, 1301, 1210, 1181, 1135. HRMS (ESI-TOF) m/z [M + H]+ calcd for C25H35N6O+ 435.2867, found 435.2884. HPLC purity is 98%.
2-(4-(5-(6-(2,5-Dibromophenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidin-1-yl)-N,N-dimethylethan-1-amine (26).
To a solution of compound 23 (183 mg, 0.302 mmol) in methyl ethyl ketone (6.0 mL) were added 2-chloro-N,N-dimethylethylamine (52.6 mg, 0.365 mmol), K2CO3 (136 mg, 0.986 mmol), and NaI (50.2 mg, 0.335 mmol). The reaction was sealed under air and heated to 40 °C. After 18 h, the reaction was cooled to rt and quenched by the addition of saturated aqueous K2CO3. The reaction mixture was extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), and concentrated under reduced pressure. Final purification by column chromatography (10–50% MeOH in DCM) afforded compound 26 (11.8 mg, 7%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.07 (apparent t, J = 7.9 Hz, 1H), 7.65 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 2.3 Hz, 1H), 7.41 (dd, J = 8.6, 2.3 Hz, 1H), 7.38 (d, J = 7.4 Hz, 1H), 7.23 (d, J = 8.3 Hz, 1H), 4.49 (tt, J = 11.5, 4.2 Hz, 1H), 3.00–2.90 (m, 2H), 2.60–2.53 (m, 4H), 2.39 (s, 3H), 2.34 (s, 6H), 2.16 (apparent qd, J = 12.2, 3.6 Hz, 2H), 1.96 (td, J = 12.2, 2.4 Hz, 2H), 1.74–1.67 (m, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 162.5, 151.6, 144.4, 141.5, 141.2, 134.7, 131.8, 129.9, 127.5, 121.1, 119.8, 115.7, 111.3, 56.5, 56.0, 55.2, 52.6, 44.4, 31.4, 9.8. IR (NaCl, thin film, cm−1): 2922, 2850, 2816, 1596, 1581, 1565, 1463, 1427, 1384, 1296, 1241. HRMS (ESI-TOF) m/z [M + H]+ calcd for C23H29Br2N6O+ 563.0764, found 563.0756. HPLC purity is 97%.
tert-Butyl-4-(5-(6-(3,5-dibromophenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S1).
To a solution of pyridine c (1.07 g, 2.97 mmol) and 3,5-dibromophenol (1.07 g, 4.23 mmol) in DMF (5 mL) was added solid K2CO3 (637 mg, 4.62 mmol) at rt. The reaction was sealed under air and heated to 90 °C. After 48 h, the reaction was cooled to rt, quenched by addition of H2O, and extracted with DCM. The combined organic phases were dried (Na2SO4), filtered, and concentrated under reduced pressure. Initial purification was by column chromatography (5–30% IPA in hexanes followed by 30% IPA in DCM). Final purification by recrystallization (DCM/hexanes, ca. 1:10) afforded bromide S1 (1.22 g, 69%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.91 (apparent t, J = 7.9 Hz, 1H), 7.55 (s, 1H), 7.29 (s, 2H), 7.24 (d, J = 7.4 Hz, 1H), 7.04 (d, J = 8.3 Hz, 1H), 4.63 (tt, J = 10.8, 4.0 Hz, 1H), 4.09 (br, 2H), 2.63–2.49 (m, 2H), 2.47 (s, 3H), 2.10 (apparent qd, J = 12.0, 4.3 Hz, 2H), 1.83–1.74 (m, 2H), 1.48 (s, 9H).13C{1H} NMR (126 MHz, CDCl3) δ 162.6, 154.6, 154.6, 145.5, 142.1, 140.9, 131.2, 130.8, 124.1, 123.1, 119.6, 111.5, 79.8, 56.9, 43.0 (br), 31.9, 28.5, 11.9. IR (NaCl, thin film, cm−1): 3073, 2929, 2862, 1691, 1566, 1566, 1418, 1298, 1244, 1165. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C24H27Br2N5NaO3+ 614.0373, found 614.0357.
tert-Butyl-4-(4-methyl-5-(6-((5-methyl-[1,1′-biphenyl]-3-yl)oxy)-pyridin-2-yl)-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S3).
To a solution of bromide d (51.9 mg, 98.2 μmol), phenyl boronic acid (31.1 mg, 255 μmol), and XPhosPdG3 (9.0 mg, 10.6 μmol) in THF (0.5 mL) was added K3PO4 (80.3 mg, 0.378 mmol). The reaction was heated to 80 °C. After 2 h, the reaction was cooled to rt, filtered through Celite with EtOAc, and concentrated under reduced pressure. Final purification by column chromatography (50–100% EtOAc in hexanes) afforded compound S3 (46.8 mg, 91%). 1H NMR (500 MHz, CDCl3) δ 7.87 (apparent t, J = 7.8 Hz, 1H), 7.56–7.53 (m, 2H), 7.43 (apparent t, J = 7.5 Hz, 2H), 7.35 (t, J = 7.3 Hz, 1H), 7.29 (s, 1H), 7.22–7.16 (m, 2H), 7.04 (d, J = 8.3 Hz, 1H), 6.97 (s, 1H), 4.72 (tt, J = 10.9, 4.0 Hz, 1H), 3.95 (br, 2H), 2.48 (s, 3H), 2.44 (s, 3H), 2.40–2.29 (m, 2H), 2.02 (apparent qd, J = 12.2, 4.2 Hz, 2H), 1.75–1.67 (m, 2H), 1.46 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.5, 154.5, 154.0, 145.4, 142.9, 142.0, 140.3, 140.2, 140.1, 131.3, 128.9, 127.7, 127.0, 124.8, 121.1, 118.6, 117.6, 111.3, 79.6, 56.8, 43.0 (br), 31.9, 28.4, 21.5, 12.1. IR (NaCl, thin film, cm−1): 2974, 2928, 2862, 1691, 1590, 1571, 1426, 1245, 1166, 1151. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C31H35N5NaO3+ 548.2632, found 548.2655.
tert-Butyl-4-(5-(6-(3,5-dichlorophenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S4).
General procedure I was used, and compound S4 (32.3 mg, 79%) was isolated as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.91 (dd, J = 8.2, 7.6 Hz, 1H), 7.25 (t, J = 1.8 Hz, 1H), 7.23 (dd, J = 7.5, 0.7 Hz, 1H), 7.09 (d, J = 1.8 Hz, 2H), 7.04 (dd, J = 8.4, 0.7 Hz, 1H), 4.62 (tt, J = 11.2, 4.0 Hz, 1H), 4.09 (br, 2H), 2.53 (br, 2H), 2.46 (s, 3H), 2.17–2.03 (m, 2H), 1.86–1.69 (m, 2H), 1.48 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 162.6, 154.5, 154.5, 145.5, 142.1, 140.9, 135.5, 131.2, 125.4, 120.8, 119.6, 111.6, 79.8, 56.9, 43.1 (br), 31.9, 28.4, 11.8. IR (NaCl, thin film, cm−1): 3070, 2975, 2929, 2861, 1692, 1589, 1578, 1423, 1246, 1165. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C24H27Cl2N5NaO3+ 526.1383, found 526.1387.
tert-Butyl-4-(5-(6-(3,5-bis(trifluoromethyl)phenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S5).
To a solution of pyridine c (33.2 mg, 92.0 μmol) and 3,5-bis-(trifluoromethyl)phenol (21 μL, 138 μmol) in DMF (0.5 mL) was added solid K2CO3 (36.4 mg, 264 μmol) at rt. The reaction was sealed under air and heated to 130 °C. After 18 h, the reaction was cooled to rt and quenched by addition of H2O. The reaction mixture was extracted with EtOAc. The combined organic phases were washed with H2O, brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (30–50% EtOAc in hexanes) afforded product S5 (32.3 mg, 61%). 1H NMR (500 MHz, CDCl3) δ 7.97 (dd, J = 8.3, 7.5 Hz, 1H), 7.77 (s, 1H), 7.67 (s, 2H), 7.28 (d, J = 7.5 Hz, 1H), 7.12 (d, J = 8.3 Hz, 1H), 4.52 (tt, J = 11.1, 4.0 Hz, 1H), 4.01 (br, 2H), 2.47 (s, 3H), 2.37 (ddd, J = 13.6, 12.0, 2.9 Hz, 2H), 2.07 (apparent qd, J = 12.2, 4.3 Hz, 2H), 1.69 (apparent d, J = 12.9 Hz, 2H), 1.47 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 162.2, 154.5, 154.2, 145.6, 142.3, 141.1, 133.3 (q, JC–F = 33.8 Hz), 130.9, 122.8 (q, JC–F = 273.5 Hz), 122.3 (d, JC–F = 4.0 Hz), 120.1, 118.7 (hept, JC–F = 4.0 Hz), 111.6, 79.8, 56.8, 42.6 (br), 31.8, 28.3, 11.9. 19F NMR (471 MHz, CDCl3) δ −62.8. IR (NaCl, thin film, cm−1): 2924, 2851, 1693, 1457, 1428, 1368, 1278, 1245, 1175, 1141. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C26H27F6N5NaO3+ 594.1910, found 594.1919.
tert-Butyl-4-(5-(6-(3,5-dipropylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S6).
General procedure II: To a solution of ZnCl2 (54.3 mg, 0.399 mmol) in THF (3 mL) at rt was added n-propylmagnesium chloride (0.10 mL, 2.0 M in Et2O, 0.20 mmol). After 10 min, a solution of Pd(PhCN)2Cl2 (1.6 mg, 4.1 μmol), dppf (3.3 mg, 6.0 μmol) and bromide S1 (52.3 mg, 88.2 μmol) in THF (4 mL) was added. The reaction was heated to 50 °C. After 20 h, the reaction was cooled to rt and quenched by addition of saturated aqueous NH4Cl. The reaction mixture was extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (40% EtOAc in hexanes) afforded product S6 (45.5 mg, 99%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.84 (dd, J = 8.3, 7.4 Hz, 1H), 7.17 (d, J = 7.4 Hz, 1H), 6.98 (d, J = 8.2 Hz, 1H), 6.88 (t, J = 1.5 Hz, 1H), 6.79 (d, J = 1.5 Hz, 2H), 4.73 (tt, J = 11.2, 4.0 Hz, 1H), 3.99 (br, 2H), 2.56 (t, 4H), 2.49 (s, 3H), 2.40 (br, 2H), 2.02 (apparent qd, J = 12.3, 4.2 Hz, 2H), 1.77–1.70 (m, 2H), 1.62 (hex, J = 7.4 Hz, 4H), 1.48 (s, 9H), 0.94 (t, J = 7.3 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.6, 154.5, 153.5, 145.4, 144.4, 141.9, 140.2, 131.3, 125.5, 118.9, 118.3, 111.1, 79.6, 56.8, 43.1 (br), 37.9, 31.9, 28.4, 24.4, 13.9, 12.1. IR (NaCl, thin film, cm−1): 2960, 2931, 2870, 1695, 1589, 1572, 1468, 1452, 1427, 1302, 1246, 1166, 1152. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C30H41N5NaO3+ 542.3102, found 542.3114.
tert-Butyl-4-(5-(6-(3,5-diethylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S7).
General procedure II was used, and product S7 (13.1 mg, 58%) was isolated as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.85 (dd, J = 8.3, 7.4 Hz, 1H), 7.18 (d, J = 7.4 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 6.93 (s, 1H), 6.81 (s, 2H), 4.74 (tt, J = 11.2, 4.0 Hz, 1H), 3.98 (br, 2H), 2.64 (q, J = 7.6 Hz, 4H), 2.49 (s, 3H), 2.38 (br, 2H), 2.02 (apparent qd, J = 12.3, 4.1 Hz, 2H), 1.77–1.69 (m, 2H), 1.48 (s, 9H), 1.23 (t, J = 7.6 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.5, 154.6, 153.6, 146.0, 145.4, 141.9, 140.2, 131.3, 124.4, 118.4, 118.3, 111.1, 79.6, 56.8, 43.0 (br), 31.9, 28.7, 28.4, 15.4, 12.2. IR (NaCl, thin film, cm−1): 2966, 2931, 2862, 1693, 1589, 1572, 1452, 1426, 1365, 1331, 1302, 1276, 1246, 1166, 1152. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C28H37N5NaO3+ 514.2789, found 514.2798.
tert-Butyl-4-(5-(6-(3,5-dibutylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S8).
General procedure III: To a solution of ZnCl2 (74.6 mg, 0.549 mmol) in THF (2 mL) was added n-butylmagnesium chloride (0.10 mL, 2.0 M in THF, 0.20 mmol) at rt. After 5 min, the reaction was cooled in an ice bath. A solution of bromide S1 (48.6 mg, 82.0 μmol) in THF (2 mL) was added, followed by Pd(dppf)Cl2 (10.4 mg, 12.7 μmol). The reaction was removed from the ice bath and heated to 50 °C. After 18 h, the reaction was cooled to rt and quenched by addition of saturated aqueous NH4Cl. The reaction mixture was extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (30% EtOAc in hexanes) afforded product S8 (40.3 mg, 90%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.84 (dd, J = 8.3, 7.4 Hz, 1H), 7.17 (dd, J = 7.4, 0.7 Hz, 1H), 6.98 (dd, J = 8.3, 0.7 Hz, 1H), 6.88 (t, J = 1.6 Hz, 1H), 6.79 (d, J = 1.5 Hz, 2H), 4.73 (tt, J = 11.3, 4.0 Hz, 1H), 4.01 (br, 2H), 2.58 (t, J = 7.8 Hz, 4H), 2.49 (s, 3H), 2.40 (br, 2H), 2.03 (apparent qd, J = 12.3, 11.7, 3.3 Hz, 2H), 1.77–1.70 (m, 2H), 1.58 (p, J = 7.6 Hz, 4H), 1.48 (s, 9H), 1.35 (hex, J = 7.4 Hz, 4H), 0.92 (t, J = 7.4 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.6, 154.5, 153.5, 145.4, 144.6, 141.9, 140.2, 131.3, 125.5, 118.8, 118.3, 111.1, 79.6, 56.8, 43.2 (br), 35.5, 33.5, 31.9, 28.4, 22.4, 13.9, 12.1. IR (NaCl, thin film, cm−1): 2957, 2930, 2859, 1695, 1589, 1572, 1468, 1453, 1426, 1302, 1247, 1166, 1152. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C32H45N5NaO3+ 570.3415, found 570.3410.
tert-Butyl-4-(5-(6-(3,5-dicyclopentylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S9).
General procedure III was used, and product S9 (35.3 mg, 82%) was isolated as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.85 (dd, J = 8.3, 7.4 Hz, 1H), 7.18 (dd, J = 7.5, 0.7 Hz, 1H), 7.01 (t, J = 1.6 Hz, 1H), 6.98 (dd, J = 8.4, 0.7 Hz, 1H), 6.85 (d, J = 1.6 Hz, 2H), 4.76 (tt, J = 11.2, 4.0 Hz, 1H), 4.18–3.75 (m, 2H), 2.96 (tt, J = 10.0, 7.4 Hz, 2H), 2.51 (s, 3H), 2.36–2.24 (m, 2H), 2.10–1.91 (m, 6H), 1.84–1.75 (m, 4H), 1.74–1.62 (m, 6H), 1.61–1.51 (m, 4H), 1.47 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.5, 154.5, 153.4, 148.2, 145.3, 142.0, 140.2, 131.1, 123.0, 118.2, 117.7, 111.1, 79.5, 56.8, 46.0, 42.9 (br), 34.6, 31.9, 28.4, 25.4, 12.3. IR (NaCl, thin film, cm−1): 2954, 2867, 1694, 1589, 1572, 1469, 1452, 1426, 1303, 1246, 1166, 1152. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C34H45N5NaO3+ 594.3415, found 594.3405.
tert-Butyl-4-(5-(6-(3,5-dicyclohexylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S10).
General procedure III was used, and product S10 (30.2 mg, 60%) was isolated as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.84 (dd, J = 8.3, 7.5 Hz, 1H), 7.18 (d, J = 7.4 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 6.95 (t, J = 1.6 Hz, 1H), 6.82 (d, J = 1.5 Hz, 2H), 4.76 (tt, J = 11.2, 4.0 Hz, 1H), 4.11–3.81 (m, 2H), 2.51 (s, 3H), 2.49–2.45 (m, 2H), 2.33–2.24 (m, 2H), 2.00 (apparent qd, J = 12.2, 4.1 Hz, 2H), 1.88–1.81 (m, 8H), 1.74 (apparent d, J = 12.5 Hz, 4H), 1.47 (s, 9H), 1.44–1.32 (m, 8H), 1.30–1.17 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.4, 154.4, 153.4, 149.8, 145.4, 141.9, 140.2, 131.2, 122.4, 118.2, 117.4, 111.1, 79.5, 56.9, 44.6, 42.9 (br), 34.4, 31.9, 28.4, 26.8, 26.1, 12.3. IR (NaCl, thin film, cm−1): 2926, 2851, 1694, 1589, 1572, 1449, 1426, 1304, 1246, 1166, 1152. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C36H49N5NaO3+ 622.3728, found 622.3719.
tert-Butyl-4-(5-(6-(3,5-dicyclopropylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S11).
General procedure III was used, and product S11 (16.3 mg, 35%) was isolated as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.85 (dd, J = 8.3, 7.4 Hz, 1H), 7.18 (d, J = 7.4 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 6.65–6.61 (m, 3H), 4.77 (tt, J = 11.2, 4.0 Hz, 1H), 4.02 (br, 2H), 2.52 (s, 3H), 2.47–2.34 (m, 2H), 2.09–1.96 (m, 2H), 1.88–1.81 (m, 2H), 1.77–1.70 (m, 2H), 1.49 (s, 9H), 0.99–0.94 (m, 4H), 0.68–0.63 (m, 4H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.6, 154.5, 153.8, 146.0, 145.0, 141.6, 140.2, 131.5, 119.6, 118.3, 116.1, 111.4, 79.6, 57.1, 42.2 (br), 31.9, 28.5, 15.4, 12.0, 9.45. IR (NaCl, thin film, cm–1): 3081, 2973, 2866, 1691, 1589, 1571, 1467, 1452, 1427, 1366, 1301, 1276, 1246, 1167. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C30H37N5NaO3+ 538.2789, found 538.2773.
tert-Butyl-4-(5-(6-(5-isopropyl-2-methylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S12).
General procedure I was used, and compound S12 (34.4 mg, 69%) was isolated as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.83 (dd, J = 8.3, 7.4 Hz, 1H), 7.18 (d, J = 7.8 Hz, 1H), 7.14 (d, J = 7.3 Hz, 1H), 7.04 (dd, J = 7.8, 1.7 Hz, 1H), 6.98 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 1.6 Hz, 1H), 4.66 (tt, J = 11.1, 4.1 Hz, 1H), 3.97 (br, 2H), 2.86 (heptet, J = 6.9 Hz, 1H), 2.46 (s, 3H), 2.42 (br, 2H), 2.12 (s, 3H), 1.99 (br, 2H), 1.64 (apparent d, J = 11.1 Hz, 2H), 1.46 (s, 9H), 1.20 (d, J = 6.9 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.5, 154.7, 151.9, 148.5, 145.5, 142.1, 140.4, 131.4, 131.3, 127.9, 123.7, 120.5, 118.2, 110.7, 79.8, 56.7, 42.1 (br), 33.7, 31.9, 28.6, 24.1, 16.4, 12.3. IR (NaCl, thin film, cm–1): 2963, 2928, 2869, 1693, 1426, 1301, 1247, 1166. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C28H37N5NaO3+ 514.2789, found 514.2799.
tert-Butyl-4-(5-(6-(2,5-dibromophenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S13).
General procedure I was used, and compound S13 (31.0 mg, 63%) was isolated as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.92 (dd, J = 8.3, 7.4 Hz, 1H), 7.53 (d, J = 8.5 Hz, 1H), 7.37 (d, J = 2.2 Hz, 1H), 7.30 (dd, J = 8.7, 2.2 Hz, 1H), 7.20 (d, J = 7.3 Hz, 1H), 7.12 (d, J = 8.3 Hz, 1H), 4.58 (tt, J = 11.2, 4.2 Hz, 1H), 4.08 (br, 2H), 2.63 (br, 2H), 2.43 (s, 3H), 2.08 (br, 2H), 1.72–1.60 (m, 2H), 1.49 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 162.4, 154.5, 151.4, 145.3, 142.0, 140.8, 134.4, 131.3, 129.9, 127.7, 121.4, 119.5, 115.7, 111.1, 79.8, 56.4, 43.2 (br), 31.9, 28.5, 11.7. IR (NaCl, thin film, cm−1):2975, 2930, 2860, 1691, 1565, 1463, 1426, 1297, 1244, 1165. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C24H27Br2N5NaO3+ 614.0373, found 614.0373.
tert-Butyl-4-(5-(6-(2,6-dimethylphenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S14).
General procedure I was used, and compound S14 (46.4 mg, 63%) was isolated as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.84 (dd, J = 8.3, 7.4 Hz, 1H), 7.11 (d, J = 7.5 Hz, 1H), 7.09–7.04 (m, 3H), 7.00 (d, J = 8.3 Hz, 1H), 4.56 (tt, J = 11.2, 4.2 Hz, 1H), 4.01 (br, 2H), 2.51 (br, 2H), 2.42 (s, 3H), 2.11 (s, 6H), 2.08–1.91 (m, 2H), 1.62–1.53 (m, 2H), 1.47 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 162.8, 154.5, 150.4, 145.4, 141.9, 140.4, 131.5, 130.9, 128.8, 125.4, 118.1, 110.2, 79.7, 56.4, 43.0, 31.7, 28.5, 16.7, 11.8. IR (NaCl, thin film, cm−1): 2975, 2929, 2866, 1693, 1574, 1425, 1301, 1245, 1166. HRMS (ESITOF) m/z [M + Na]+ calcd for C26H33N5NaO3+ 486.2476, found 486.2474.
tert-Butyl-4-(5-(6-(cyclohexyloxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S15).
To a solution of cyclohexanol (168 mg, 1.67 mmol) in MeCN (1.0 mL) was added NaOt-Bu (68.7 mg, 0.815 mmol) at rt. After 5 min, a solution of pyridine c (47.0 mg, 0.130 mmol) in MeCN (1.5 mL) was added. The reaction was sealed under air and heated to 80 °C. After 4 days, the reaction was cooled to rt and extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (10–50% EtOAc in hexanes) afforded product S15 (37.5 mg, 65%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.70 (dd, J = 8.4, 7.2 Hz, 1H), 6.98 (dd, J = 7.2, 0.8 Hz, 1H), 6.76 (dd, J = 8.4, 0.8 Hz, 1H), 5.02–4.90 (m, 2H), 4.28 (br, 2H), 2.82 (br, 2H), 2.43 (s, 3H), 2.36–2.22 (m, 2H), 2.14–1.93 (m, 4H), 1.88–1.76 (m, 2H), 1.67–1.53 (m, 3H), 1.49 (s, 9H), 1.40–1.31 (m, 3H).13C{1H} NMR (126 MHz, CDCl3) δ 163.4, 154.5, 144.7, 141.6, 139.3, 132.4, 117.2, 111.8, 79.9, 73.5, 56.6, 43.5 (br), 32.2, 31.9, 28.4, 25.5, 24.1, 11.5.IR (NaCl, thin film, cm−1): 2930, 2855, 1693, 1573, 1435, 1304, 1247. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C24H35N5NaO3+ 464.2632, found 464.2632.
tert-Butyl-4-(5-(6-isopropoxypyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S16).
To a solution of isopropanol (0.40 mL, 5.26 mmol) in MeCN (1.0 mL) was added NaOt-Bu (68.7 mg, 0.815 mmol) at rt. After 5 min, a solution of pyridine 3 (49.9 mg, 0.138 mmol) in MeCN (1.5 mL) was added. The reaction was sealed under air and heated to 80 °C. After 24 h, the reaction was cooled to rt and extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (20–80% EtOAc in hexanes) afforded product S16 (46.4 mg, 84%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.71 (dd, J = 8.4, 7.3 Hz, 1H), 6.99 (dd, J = 7.3, 0.8 Hz, 1H), 6.75 (dd, J = 8.4, 0.8 Hz, 1H), 5.28 (hept, J = 6.2 Hz, 1H), 5.00 (tt, J = 11.4, 4.1 Hz, 1H), 4.28 (br, 2H), 2.82 (br, 2H), 2.44 (s, 3H), 2.36–2.27 (m, 2H), 2.14–1.99 (m, 2H), 1.49 (s, 9H), 1.38 (d, J = 6.3 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 163.4, 154.5, 144.6, 141.6, 139.4, 132.3, 117.2, 111.8, 79.9, 68.2, 56.7, 43.1 (br), 32.2, 28.4, 22.2, 11.6. IR (NaCl, thin film, cm−1): 2977, 2931, 2866, 1695, 1574, 1471, 1435, 1308, 1249, 1166. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C21H31N5NaO3+424.2319, found 424.2323.
tert-Butyl-4-(5-(6-(3,5-bis(hydroxymethyl)phenoxy)pyridin-2-yl)-4-methyl-1H-1,2,3-triazol-1-yl)piperidine-1-carboxylate (S17).
To a solution of pyridine c (45.1 mg, 0.125 mmol) and 3,5-bis-(hydroxymethyl)phenol (32.7 mg, 0.212 mmol) in DMSO (2.0 mL) was added Cs2CO3 (62.3 mg, 0.191 mmol) at rt. The reaction was sealed under air and heated to 110 °C. After 24 h, the reaction was cooled to rt and extracted with EtOAc. The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (40–75% EtOAc in DCM) afforded product S17 (33.1 mg, 53%) as a white solid. 1H NMR (500 MHz, MeOD) δ 8.01 (dd, J = 8.3, 7.4 Hz, 1H), 7.36 (dd, J = 7.4, 0.7 Hz, 1H), 7.21 (s, 1H), 7.14 (dd, J = 8.3, 0.7 Hz, 1H), 7.09 (s, 2H), 4.73 (tt, J = 11.2, 4.1 Hz, 1H), 4.64 (s, 4H), 3.94 (dt, J = 13.7, 2.8 Hz, 2H), 2.61–2.47 (m, 2H), 2.42 (s, 3H), 1.89 (apparent qd, J = 12.2, 4.3 Hz, 2H), 1.791.70 (m, 2H), 1.49 (s, 9H). 13C NMR (126 MHz, MeOD) δ 163.7, 155.0, 154.2, 144.5, 143.8, 141.4, 140.9, 131.9, 121.0, 119.0, 118.2, 111.6, 79.9, 63.1, 56.7, 42.2 (br), 31.6, 27.3, 10.1. IR (NaCl, thin film, cm−1): 3392, 2975, 2930, 2866, 1691, 1673, 1591, 1573, 1452, 1428, 1367, 1302, 1277, 1247, 1166. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C26H33N5NaO5+ 518.2374, found 518.2390.
Fluorescence Anisotropy.
Procedures were performed according to our prior report.18
Crystallization Conditions and X-ray Data Collection Methods.
Compounds 23, DW34, and 26 (500 μM) were cocrystallized with BRD D1 (300 μM, in 10 mM HEPES, 100 mM NaCl, pH 7.4) in 100 mM Bis-Tris propane, 20% (v/v) PEG 3350, 10% (v/v) ethylene glycol at pH 8.5 using the hanging drop method. Harvestable crystals grew in 1–2 days at ambient temperature. Crystals were harvested, cryoprotected in ethylene glycol, and flash frozen. Data were collected at Advanced Photon Source with the NE-CAT 24-ID-C and E beamlines. Phaser was used to solve the structure via molecular replacement using PDB ID 3MXF as a search model.30 Phenix31 and Coot32 were used for structure refinement.
Cell Culture.
MM.1S cells and A549 cells were grown in a humidified 5% CO2 environment at 37 °C. Cells were cultured in RPMI 1640 media (Corning) supplemented with 10% fetal-bovine serum (FBS, Gibco), and penicillin/streptomycin (50 μ/mL each, Gibco)18,28.
MM.1S Cells.
The mixed suspension/adherent cells were subcultured at a 1:10 dilution by decanting suspended cells and dissociating adherent cells from plates in 0.25% trypsin/EDTA (Gibco) with 2 min incubation times. Cell-line authenticity was verified using the short-tandem-repeat (STR)-profiling service provided by ATCC.
A549 Cells.
The adherent cells were subcultured at a 1:10 dilution by dissociating adherent cells from the plates in 0.25% trypsin/EDTA (Gibco) with 2 min incubation times. Cell-line authenticity was verified using the STR-profiling service provided by ATCC.
Western Blotting.
Procedures were performed according to our prior report.18
IL-8 ELISA.
Procedures were performed according to our prior report.28
Viability Assays.
Procedures were performed according to our prior report.18,28
Human Liver Microsomal Binding Assay.
Triplicate 100 μL samples were prepared with 1.0 mg/mL HLM150 and 1 nM test compound in 100 mM potassium phosphate buffer (pH 7.4) in microcentrifuge tubes. Negative controls without HLM150 were prepared for comparison. Samples were centrifuged at 200,000 × g and 25 °C for 24 h in a Beckman TI-100 ultracentrifuge (Beckman Coulter, Brea, CA). Following centrifugation, 50 μL of the middle fraction was removed and mixed in a full-volume microplate with a 2-fold volume of ice-cold methanol containing 10 μM internal standard dansylamide. The samples were then evaporated to dryness using an Organomation Microvap Nitrogen Evaporator System (Organomation Associates, Inc., Berlin, MA). Dried wells were resuspended in mobile phase (1:4 acetonitrile and water with 0.1% formic acid) for mass spectroscopic analysis.
Assessment of Compound Metabolic Stability.
We assessed metabolic stability over time using human liver microsomes 150 (HLM150) as a model for the average adult liver. All substrate stocks were prepared at 10 mM in DMSO. Reactions contained final concentrations of 1.0 mg/mL HLM150 and 1 μM substrate in 100 mM potassium phosphate buffer, pH 7.4. Reactions were initiated upon addition of a NADPH regenerating system (0.4 U/μL glucose-6-phosphate dehydrogenase, 10 mM glucose 6-phosphate, 2 mM MgCl2, 500 μM NADP+) and incubated at 37 °C with shaking at 350 rpm using a BMG Labtech THERMOstar incubator (Ortenberg, Germany). Samples without addition of the NADPH regenerating system or without substrate served as negative controls. At 0, 5, 10, 15, 20, 30, 45, and 60 min, reaction aliquots were quenched by adding a 2-fold volume of ice-cold acetonitrile containing 5 μM internal standard dansylamide and incubated on ice for 10 min to optimize precipitation of proteins and phosphate buffer. After 1800 × g centrifugation at 4 °C for 15 min using a Sorvall ST 16R Centrifuge (Thermo Scientific; Waltham, MA), the supernatant was transferred to a 96-well half-volume microplate and evaporated to dryness using an Organomation Microvap Nitrogen Evaporator System (Organomation Associates, Inc., Berlin MA). Dried wells were then resuspended in mobile phase (20:80 water:acetonitrile + 0.01% formic acid) for LC-MS analysis. Each set of reactions was performed in triplicate. Samples were separated by a 4.6 × 150 mm Waters XSelect HSS C18 3.5 μm column using a Waters quaternary solvent manager-R liquid chromatograph, and parent masses were detected using a Waters Acquity QDa mass spectrometer. Mobile phase consisted of solvents A (0.1% formic acid in deionized water) and B (0.1% formic acid in acetonitrile). The gradient method started with 40% solvent A, decreased to 10% over 3 min, held stable for 1 min, increased to 40% over 1 min, and held for the remainder of the run. The total flow rate was 1 mL/min, and total run time per sample was 6 min. The ESI source was operated in positive-ion mode with a cone voltage of 15 V, and ion spectra were acquired in full scan mode monitoring the m/z range of 100–1000 amu. Analyte responses were normalized to internal standard dansylamide. Calculation of metabolic parameters was performed using GraphPad Prism 7.4 from GraphPad Software, Inc. (San Diego, CA).
Supplementary Material
ACKNOWLEDGMENTS
DW34 has been selected as a compound name in acknowledgement of the significant cancer awareness advocacy of DeAngelo Williams.
Funding
This research was supported by the National Institutes of Health’s National Center for Advancing Translational Sciences, grant UL1TR002494. A.C. was supported by the Wayland E. Noland Fellowship and UMN Doctoral Dissertation Fellowship. This research was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under award numbers R35GM124718 (J.J.T.) and R35GM140837 (W.C.K.P) . We also acknowledge NIH Shared Instrumentation grant number S10OD011952. Further support for this research was from the NIH (R35-GM118047 to H.A.), and A.D. and C.R.B. were supported by the NIH chemistry–biology interface training grant (T32-GM132029–02) A.D. was also supported by a University of Minnesota Doctoral Dissertation Fellowship. H.Z. was supported by a IEM doctoral fellowship. M.S. was supported by the NIGMS Systems Pharmacology and Toxicology Training grant (T32-GM106999). M.S. and G.M. were supported by Winthrop P. Rockefeller Cancer Institute Team Science Award. D.H. acknowledges funding from the Masonic Cancer Center at the University of Minnesota with resources from Minnesota Masonic Charities. This work is based on research conducted at the NE-CAT beamlines at the Advanced Photon Source, which is supported by the NIH (P30-GM124165). The Pilatus 6 M detector on 24-ID-C beamline is funded by a NIH-ORIP HEI grant (S10-RR029205). We thank staff at the NE-CAT beamlines for assistance in data collection.
ABBREVIATIONS USED
- BET
bromodomain and extra terminal
- BRD4
bromodomain-containing protein 4
- BRD2
bromodomain-containing protein 2
- BRD3
bromodomain-containing protein 3
- BRDT
bromodomain testis-specific protein
- c-Myc
Myc protooncogene protein
- D1
N-terminal BET bromodomain
- D2
C-terminal BET bromodomain
- FA
fluorescence anisotropy
- TLC
thin-layer chromatography
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00933.
Molecular-string files for the final compounds (CSV)
All corresponding spectra along with HPLC traces and all relevant biophysical data and additional crystallographic information. Full Western blots are also provided (PDF)
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health’s National Center for Advancing Translational Sciences. Authors will release the new atomic coordinates and experimental data upon article publication. The authors declare no competing financial interest.
Contributor Information
Huarui Cui, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Angela S. Carlson, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Mary A. Schleiff, Department of Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, Arkansas 72205, United States
Anand Divakaran, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Jorden A. Johnson, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Caroline R. Buchholz, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Huda Zahid, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Nora R. Vail, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Ke Shi, Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Hideki Aihara, Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Daniel A. Harki, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States; Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Grover P. Miller, Department of Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, Arkansas 72205, United States
Joseph J. Topczewski, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
William C. K. Pomerantz, Department of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States; Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States.
REFERENCES
- (1).Jenuwein T; Allis CD Translating the Histone Code. Science 2001, 293 (5532), 1074–1080. [DOI] [PubMed] [Google Scholar]
- (2).Allis CD; Muir TW Spreading Chromatin into Chemical Biology. ChemBioChem 2011, 12 (2), 264–279. [DOI] [PubMed] [Google Scholar]
- (3).Filippakopoulos P; Picaud S; Mangos M; Keates T; Lambert JP; Barsyte-Lovejoy D; Felletar I; Volkmer R; Müller S; Pawson T; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149 (1), 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lovén J; Hoke HA; Lin CY; Lau A; Orlando DA; Vakoc CR; Bradner JE; Lee TI; Young RA Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153 (2), 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Brown JD; Lin CY; Duan Q; Griffin G; Federation AJ; Paranal RM; Bair S; Newton G; Lichtman AH; Kung AL; et al. Nf-Kb Directs Dynamic Super Enhancer Formation in Inflammation and Atherogenesis. Mol. Cell 2014, 56 (2), 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Delmore JE; Issa GC; Lemieux ME; Rahl PB; Shi J; Jacobs HM; Kastritis E; Gilpatrick T; Paranal RM; Qi J; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell 2011, 146 (6), 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Huang B; Yang X-D; Zhou M-M; Ozato K; Chen L-F Brd4 Coactivates Transcriptional Activation of NF- B via Specific Binding to Acetylated RelA. Mol. Cell. Biol 2009, 29 (5), 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Andrieu G; Belkina AC; Denis GV Clinical Trials for BET Inhibitors Run Ahead of the Science. Drug Discovery Today: Technol. 2016, 19, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Filippakopoulos P; Knapp S The Bromodomain Interaction Module. FEBS Lett. 2012, 586 (17), 2692–2704. [DOI] [PubMed] [Google Scholar]
- (10).Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468 (7327), 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ciceri P; Müller S; O’Mahony A; Fedorov O; Filippakopoulos P; Hunt JP; Lasater EA; Pallares G; Picaud S; Wells C; et al. Dual Kinase-Bromodomain Inhibitors for Rationally Designed Polypharmacology. Nat. Chem. Biol 2014, 10 (4), 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Carlson AS; Cui H; Divakaran A; Johnson JA; Brunner RM; Pomerantz WCK; Topczewski JJ Systematically Mitigating the P38α Activity of Triazole-Based BET Inhibitors. ACS Med. Chem. Lett 2019, 10 (9), 1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Seal J; Lamotte Y; Donche F; Bouillot A; Mirguet O; Gellibert F; Nicodeme E; Krysa G; Kirilovsky J; Beinke S; et al. Identification of a Novel Series of BET Family Bromodomain Inhibitors: Binding Mode and Profile of I-BET151 (GSK1210151A). Bioorg. Med. Chem. Lett 2012, 22 (8), 2968–2972. [DOI] [PubMed] [Google Scholar]
- (14).Gacias M; Gerona-Navarro G; Plotnikov AN; Zhang G; Zeng L; Kaur J; Moy G; Rusinova E; Rodriguez Y; Matikainen B; et al. Selective Chemical Modulation of Gene Transcription Favors Oligodendrocyte Lineage Progression. Chem. Biol 2014, 21 (7), 841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gilan O; Rioja I; Knezevic K; Bell MJ; Yeung MM; Harker NR; Lam EYN; Chung C; Bamborough P; Petretich M Selective Targeting of BD1 and BD2 of the BET Proteins in Cancer and Immuno-Inflammation. Science 2020, 368 (6489), 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Faivre EJ; McDaniel KF; Albert DH; Mantena SR; Plotnik JP; Wilcox D; Zhang L; Bui MH; Sheppard GS; Wang L; et al. Selective Inhibition of the BD2 Bromodomain of BET Proteins in Prostate Cancer. Nature 2020, 578 (7794), 306–310. [DOI] [PubMed] [Google Scholar]
- (17).Liu Z; Chen H; Wang P; Li Y; Wold EA; Leonard PG; Joseph S; Brasier AR; Tian B; Tian B; et al. Discovery of Orally Bioavailable Chromone Derivatives as Potent and Selective BRD4 Inhibitors: Scaffold Hopping, Optimization, and Pharmacological Evaluation. J. Med. Chem 2020, 63 (10), 5242–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cui H; Divakaran A; Pandey AK; Johnson JA; Zahid H; Hoell ZJ; Ellingson MO; Shi K; Aihara H; Harki DA; et al. Selective N-Terminal BET Bromodomain Inhibitors by Targeting Non-Conserved Residues and Structured Water Displacement**. Angew. Chem., Int. Ed 2021, 60, 1220–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gao J; Wei B; Liu M; Hirsova P; Sehrawat TS; Cao S; Hu X; Xue F; Yaqoob U; Kang N; et al. Endothelial P300 Promotes Portal Hypertension and Hepatic Fibrosis through C-C Motif Chemokine Ligand 2-mediated Angiocrine Signaling. Hepatology 2021, 73 (6), 2468–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sheppard GS; Wang L; Fidanze SD; Hasvold LA; Liu D; Pratt JK; Park CH; Longenecker K; Qiu W; Torrent M; et al. Discovery of N-Ethyl-4-[2-(4-Fluoro-2,6-Dimethyl-Phenoxy)-5-(1-Hydroxy-1-Methyl-Ethyl)Phenyl]-6-Methyl-7-Oxo-1 H-Pyrrolo-[2,3-c]Pyridine-2-Carboxamide (ABBV-744), a BET Bromodomain Inhibitor with Selectivity for the Second Bromodomain. J. Med. Chem 2020, 63 (10), 5585–5623. [DOI] [PubMed] [Google Scholar]
- (21).Wellaway CR; Bamborough P; Bernard SG; Chung CW; Craggs PD; Cutler L; Demont EH; Evans JP; Gordon L; Karamshi B; et al. Structure-Based Design of a Bromodomain and Extraterminal Domain (BET) Inhibitor Selective for the N-Terminal Bromodomains That Retains an Anti-Inflammatory and Antiproliferative Phenotype. J. Med. Chem 2020, 63, 9020. [DOI] [PubMed] [Google Scholar]
- (22).Fish PV; Filippakopoulos P; Bish G; Brennan PE; Bunnage ME; Cook AS; Federov O; Gerstenberger BS; Jones H; Knapp S; et al. Identification of a Chemical Probe for Bromo and Extra C-Terminal Bromodomain Inhibition through Optimization of a Fragment-Derived Hit. J. Med. Chem 2012, 55 (22), 9831–9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dombrowski AW; Gesmundo NJ; Aguirre AL; Sarris KA; Young JM; Bogdan AR; Martin MC; Gedeon S; Wang Y Expanding the Medicinal Chemist Toolbox: Comparing Seven C(Sp2)-C(Sp3) Cross-Coupling Methods by Library Synthesis. ACS Med. Chem. Lett 2020, 11 (4), 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Boström J; Brown DG; Young RJ; Keserü GM Expanding the Medicinal Chemistry Synthetic Toolbox. Nat. Rev. Drug Discovery 2018, 17 (10), 709–727. [DOI] [PubMed] [Google Scholar]
- (25).Brown DG; Boström J Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem 2016, 59 (10), 4443–4458. [DOI] [PubMed] [Google Scholar]
- (26).Lucas SCC; Atkinson SJ; Bamborough P; Barnett H; Chung CW; Gordon L; Mitchell DJ; Phillipou A; Prinjha RK; Sheppard RJ; et al. Optimization of Potent ATAD2 and CECR2 Bromodomain Inhibitors with an Atypical Binding Mode. J. Med. Chem 2020, 63 (10), 5212–5241. [DOI] [PubMed] [Google Scholar]
- (27).Talele TT The “Cyclopropyl Fragment” Is a Versatile Player That Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem 2016, 59 (19), 8712–8756. [DOI] [PubMed] [Google Scholar]
- (28).Divakaran A; Talluri SK; Ayoub AM; Mishra N; Cui H; Widen JC; Berndt N; Zhu J-Y; Carlson AS; Topczewski JJ; et al. Molecular Basis for the N-Terminal Bromodomain and Extra Terminal (BET) Family Selectivity of a Dual Kinase-Bromodomain Inhibitor. J. Med. Chem 2018, 61, 9316–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev 2012, 64, 4–17. [DOI] [PubMed] [Google Scholar]
- (30).Mccoy AJ; Grosse-kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Research Papers Phaser Crystallographic Software Research Papers. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.