

Abstract
Chronic pain is a life-altering condition affecting millions of people. Current treatments are inadequate and prolonged therapies come with severe side effects, especially dependence and addiction to opiates. Identification of non-narcotic analgesics is of paramount importance. Preclinical and clinical studies suggest that sphingolipid metabolism alterations contribute to neuropathic pain development. Functional S1P receptor 1 (S1PR1) antagonists, such as FTY720/Fingolimod, used clinically for non-pain conditions, are emerging as non-narcotic analgesics, supporting the repurposing of Fingolimod for chronic pain treatment and energizing drug discovery focused on S1P signalling. Here we summarize the role of sphingosine-1-phosphate (S1P) in pain to highlight the potential of targeting the S1P axis towards development of non-narcotic therapeutics which in turn will hopefully help lessen misuse of opioid pain medications and address the ongoing opioid epidemic.
Keywords: Sphingosine-1-phosphate (S1P), S1P receptors, neuropathic pain, neuroinflammation, non-narcotic treatments
Sphingosine-1-phosphate axis in pain
Chronic pain is prevalent worldwide and extremely difficult to treat, representing a huge economic burden on the healthcare system. Pain treatments have not really improved for decades and, even today, opioids are the main prescribed drugs for chronic pain management. However, long-term side effects such as tolerance, addiction and paradoxical hyperalgesia make research and development of novel non-narcotic analgesics imperative [1].
In the last few years, increasing evidence has implicated the sphingosine-1-phosphate (S1P) axis as an important modulator of pain pathways. Ceramide, the bioactive precursor of S1P, is generated both by enzymatic hydrolysis of sphingomyelin by sphingomyelinases (SMases, SM pathway) and by de novo biosynthesis (Figure 1) [2]. Ceramide biosynthesis begins with condensation of serine with a long chain saturated fatty acid, typically palmitate, mediated by serine palmitoyltransferase (SPT). The 3-keto-dihydrosphingosine intermediate is rapidly reduced to dihydrosphingosine, followed by N-acylation catalyzed by ceramide synthases (CerS) to dihydroceramide. Only then is the double bond introduced to form ceramide. The steady-state level of ceramide is further regulated by ceramidases that convert ceramide to sphingosine, which can be phosphorylated by sphingosine kinases (SphK1 and SphK2) [3] to S1P (Figure 1). Although SphK1 and SphK2 share overall homology and both produce S1P, they have different catalytic properties, subcellular locations, tissue distribution, and temporal expression patterns during development that are responsible for their unique and specific functions [3].
Figure 1. Sphingolipid biosynthetic pathway leading to ceramide and sphingosine 1-phosphate (S1P) formation.
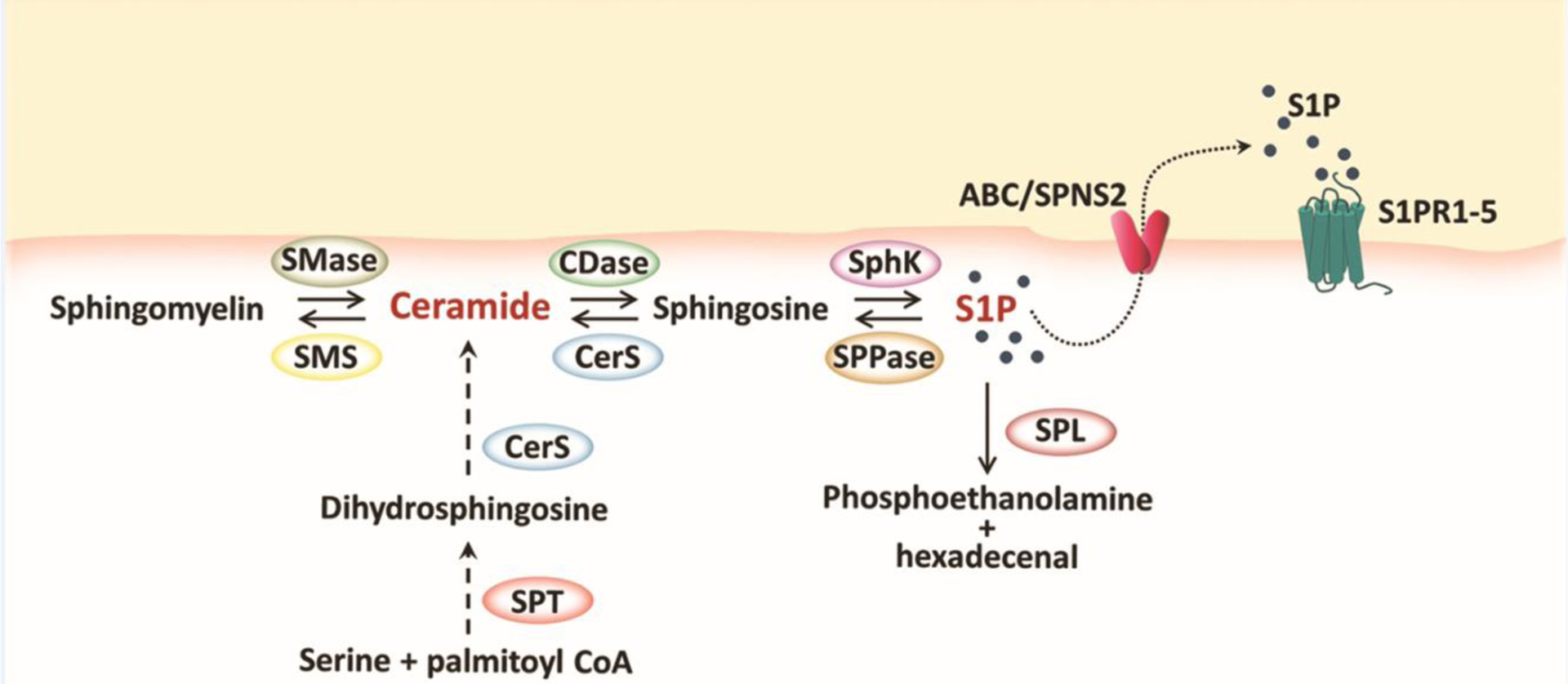
Ceramide can be formed either by a catabolic pathway, from the hydrolysis of membrane sphingomyelin, or by de novo biosynthesis, starting from the condensation of serine with palmitate in the endoplasmic reticulum. Ceramide and its bioactive metabolite S1P mediate several inflammatory, proapoptotic and nociceptive signalling cascades. S1P can cross the plasma membrane through either non-specific ABC transporters or through the recently discovered Spinster 2 (SPNS2) transporter, which is specific for S1P. S1P signalling is mediated by five G-protein coupled receptors (S1PR1-5). CDase, ceramidase; CerS, ceramide synthase; SMase, sphingomyelinase; SMS, sphingomyelin synthase; SphK, sphingosine kinases; SPL, S1P lyase; SPPase, sphingosine phosphate phosphatase; SPT, serine palmitoyltransferase.
S1P can be dephosphorylated back to sphingosine either by non-specific phosphatases or by S1P-specific phosphatases (SPP1 and SPP2) localized in the endoplasmic reticulum [4]. Sphingosine can then be recycled back to ceramide by CerS (Figure 1). S1P can also be irreversibly degraded by S1P lyase to phosphoethanolamine and hexadecenal [4]. Several direct intracellular S1P targets has been identified [5]; however, most of S1P cellular functions are mediated via its cell surface receptors [6]. S1P is transported outside the cell where it acts in a paracrine or autocrine manner by the process known as “inside-out signalling” [7]. Several non-specific S1P transporters belonging to the ABC family of transporters have been identified [7]. The passive transporter belonging to the spinster family, SPNS2, has more recently been recognized as an important and specific S1P transporter and its activity increases proportionally to the intracellular S1P concentration (Figure 1) [8].
S1P is the ligand of five G protein-coupled receptors (S1PR1-5) that are broadly expressed throughout the nervous system. In the peripheral nervous system (PNS), adult sensory neurons express S1PR1, S1PR2 and S1PR3. S1PR1 and S1PR2 are found mainly on nociceptors (see Glossary) and proprioceptive neurons, respectively, whereas S1PR3 is the predominant subtype within the dorsal root ganglion (DRG), where only a minor fraction of neurons also express S1PR2 [9, 10]. In the mature central nervous system (CNS), all S1PR subtypes except S1PR4 are expressed [11]. S1PR1 and S1PR3 are the most abundant subtypes in both astrocytes and microglia, and their expression increases following glial activation [12, 13]. S1PR5 is dominantly expressed in the white matter of the CNS and has been found in all developmental stages of oligodendrocytes [14]. S1PR4 is highly found in hematopoietic cells and lymphoid organs and is implicated in S1P-mediated immune responses [15].
S1P, together with its precursor ceramide, act as soluble signalling molecules sharing potent inflammatory and nociceptive actions [16]. S1P also has a role in pathogenesis of bladder pain syndrome/interstitial cystitis where S1P levels in serum has been proposed as a diagnostic marker [17]. Moreover, altered sphingolipid metabolism has been linked to clinically evident neuropathic pain [18, 19]. Mutations in the C1 and C2 subunits of SPT, the rate limiting enzyme in ceramide biosynthesis, have been associated with the development of hereditary sensory and autonomic neuropathy type 1 (HSAN1), which in the majority of patients is characterized by intense neuropathic pain [19, 20]. These mutations modify SPT specificity, resulting in the condensation of palmitoyl-CoA with alanine or glycine instead of with serine, leading to accumulation of neurotoxic 1-deoxysphingolipids, as they are not degraded in the canonical catabolic pathway [21]. Plasma levels of 1-deoxysphingolipids are also increased in chronic idiopathic axonal polyneuropathy (CIAP) patients [22] and in patients with chemotherapy-induced neuropathic pain (CINP) [23] where formation of 1-deoxysphingolipids correlates with the incidence and severity of the disease [23]. Moreover, 1-deoxysphingolipids are also associated with the development of neuropathy in type 1 diabetes patients, suggesting that these metabolites could be useful diagnostic and prognostic markers [24].
Below we first briefly discuss the different therapeutics that target S1P receptors (S1PRs) and then we summarize the most recent discoveries on the roles of the S1P axis in pain in both PNS and CNS in different in vitro and in vivo studies. S1P-based drugs are already approved for several clinical indications and could be repurposed also for the treatment of chronic pain states.
Targeting S1P receptors
The identification of myriocin, a fungal metabolite with potent immunosuppressive activity, as an inhibitor of sphingolipid biosynthesis was the first step for the development of S1P-based drugs. The need to reduce myriocin toxicity and improve solubility stimulated the synthesis of derivatives among which FTY720 (clinically known as Fingolimod) turned out to have even higher immunomodulatory activity in vivo [25]. Its mechanism of action on the immune system remained unclear until seminal work by Mandala et al. [26] showed that FTY720 is a sphingosine analog. FTY720 acts as a prodrug that is phosphorylated by SphK to its active counterpart, FTY720-P. FTY720-P acts as an agonist at all S1P receptors, except S1PR2, towards which it has no activity [26, 27]. It is now established that FTY720-P acts as a S1PR1 functional antagonist [28]. FTY720-P when bound to S1PR1 causes the internalization of the receptor, but it does not allow S1PR1 to recycle back to the plasma membrane. Indeed, FTY270-P leads to sustained depletion of S1PR1 from the cell surface by inducing its irreversible proteasomal degradation. In contrast, S1PR1 agonists, such as endogenous S1P and SEW2871, allow S1PR1 to recycle back to the plasma membrane after its internalization and thus do not functionally antagonize S1PR1 [27, 29].
Loss of S1PR1 on lymphocytes impairs their trafficking and leads to their accumulation in secondary lymphoid organs instead of being released into the bloodstream or lymphatic circulation [27]. In this way, attack by pathogenic lymphocytes is prevented, a quality that allowed Fingolimod to receive Food and Drug Administration (FDA) approval in 2010 as the first orally available drug for treatment of relapsing-remitting multiple sclerosis (MS), a demyelinating disease in which neuropathic pain is one of the most frequent symptoms [27]. The most common side effect of FTY720 is transient bradycardia, which occurs in about 1% of patients only after the first dose and requires heart rate monitoring [30].
The therapeutic success of Fingolimod led to the development of second generation functional S1PR1 antagonists including siponimod, ponesimod, ceralifimod and KRP-203 that are in advanced clinical trials for MS, autoimmune diseases and for transplant rejection [29] (Table 1). Moreover, the functional S1PR1 antagonist RPC1063 (clinically known as ozanimod) which has improved receptor selectivity, pharmacokinetic profiles and a more desirable clinical safety profile than Fingolimod [31], is also in advanced clinical trials for ulcerative colitis [29] and was approved recently for MS [32].
Table 1.
S1P receptors agonists and antagonists
| Drug | Structure | Major clinical studies | Ref |
|---|---|---|---|
| S1PR1 agonists | |||
| SEW2871 |
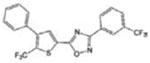
|
[29] | |
| S1PR1 functional antagonists | |||
| Fingolimod (FTY720) |
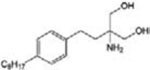
|
MS (FDA approved) Autoimmune diseases CINP | [25–27] |
| Siponimod |
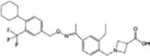
|
MS (FDA approved) Autoimmune diseases | [29] |
| Ponesimod |
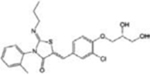
|
MS Autoimmune diseases Transplant rejection | [29] |
| Ceralifimod |
|
MS | [29] |
| KRP-203 |
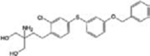
|
Autoimmune diseases Ulcerative colitis | [29] |
| Ozanimod (RPC1063) |
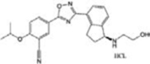
|
MS (FDA approved) Ulcerative colitis | [29, 32] |
| S1PR1 competitive antagonists | |||
| NIBR14/15 |
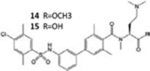
|
[33] | |
| TASP0277308 |
|
[33] | |
| W146 |
|
[33] | |
| S1PR2 agonists | |||
| CYM-5478 |
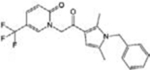
|
[34] | |
| S1PR2 antagonists | |||
| AB1 |
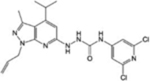
|
[35] | |
| S1PR3 agonists | |||
| SPM-354 |
|
[36] | |
| TY5–2156 |
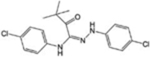
|
[36] | |
| S1PR5 agonists | |||
| A-971432 |
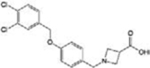
|
[40] | |
| S1PR5 antagonists | |||
| BIO027223 | Structure not available for publication | [41] | |
A number of competitive S1PR1 antagonists have also been developed and are in advanced preclinical evaluation for several indications. These include the methyl ester prodrug NIBR14, which is hydrolysed in vivo to the carboxylic acid NIBR15, TASP0277308 and W146; their efficacy has been documented in animal models of MS, colitis, rheumatoid arthritis and cancer (Table 1) [33].
The knowledge of S1PR2-3-4 functions is still rudimentary, and so far, selective modulators of these receptors have been poorly investigated in vivo (Table 1). Among them, the potent S1PR2 agonist CYM-5478 has recently been examined in vivo in the context of pain [34] (discussed below). The S1PR2 antagonist, AB1, shows remarkable antitumor activity and exhibits a good efficacy, potency and stability in vivo, but more studies are necessary to better define its selectivity [35]. Selective S1PR3 antagonists such as SPM-354 and TY-52156 have been found to modulate electric conduction of heart and neurons [36], respectively. Selective compounds targeting S1PR4 are also now available [37–39]: to date, neither the agonists (CYM50260, ML178, ML248 as well as a benzo-thiophene analog) nor the antagonists (CYM50358 and a series of compounds based on a 5-aryl furan-2-arylcarboxamide scaffold) have been tested in vivo, but they will surely help to elucidate the significance of S1PR4 signalling in physiological and pathological settings.
Targeting S1PR5 with the highly selective agonist A-971432 [40] has been suggested as a possible treatment for neurodegenerative disorders owing to its ability to revert age-related cognitive decline in rats and mice. Furthermore, the novel S1PR5 selective antagonist, BIO027223 [41], has been characterized in vivo, and found able to prevent the egress of natural killer cells from the bone marrow and secondary lymphoid organs, thus anticipating its possible application in autoimmune and inflammatory pathologies.
S1PRs have already been targeted successfully in several diseases associated with inflammatory conditions. Below we discuss the involvement of S1PRs and associated players in the S1P axis in peripheral and central pain mechanisms, providing evidence on how these can be also targeted to develop non-narcotic pain therapeutics.
S1P axis in peripheral sensitization
In chronic pain, immune cells recruited to damaged tissues release several mediators such as neuropeptides and neurotransmitters that act on specific receptors expressed on free nerve endings (nociceptors) that, in turn, regulate immune responses, and this neuro-immune crosstalk gives rise to inflammation. Inflammation lowers the sensory threshold, a phenomenon known as peripheral sensitization, so that innocuous mechanical or thermal stimuli induce pain (allodynia) and pain sensation associated with noxious stimulation, leading to drastically increased pain (hyperalgesia)i. One of the main mediators released at the inflammatory site is nerve growth factor (NGF), whose role in initiation and maintenance of both mechanical and thermal hypersensitivity is well established [42]. NGF along with p75 neurotrophin receptor (p75NTR) mediates mechanical allodynia also through neutral SMase activation, leading to increases in ceramide and S1P production [43] as well as S1PR1 stimulation [44] (Figure 2).
Figure 2. Schematic illustration of ceramide and S1P-induced peripheral sensitization.
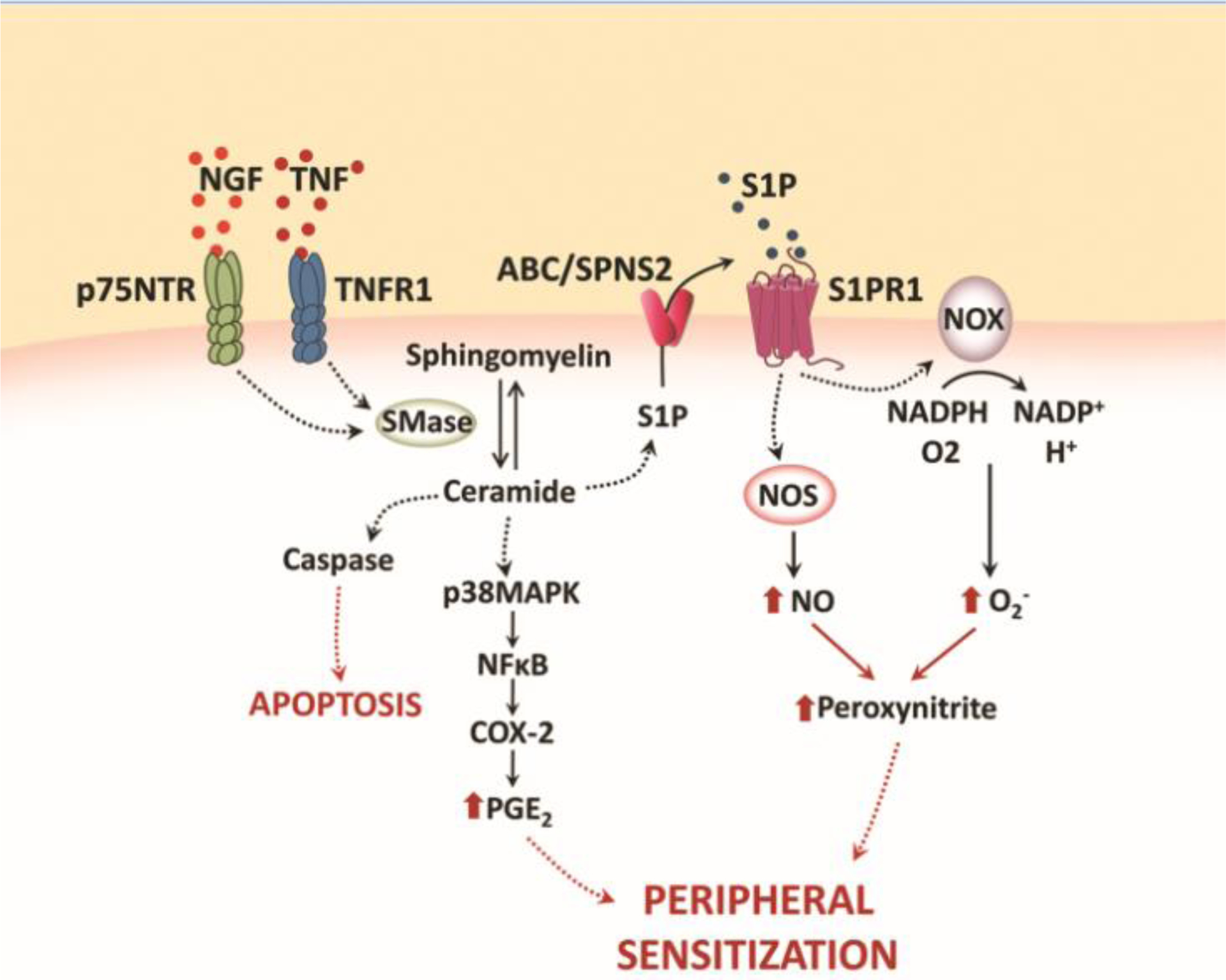
The illustration describes some inflammatory pathways triggered by ceramide and its bioactive downstream metabolite, S1P. Both NGF and TNF can induce the production of ceramide through the activation of sphingomyelinase (SMase). Once formed, ceramide promotes caspase-dependent apoptosis and stimulates cyclooxygenase-2 (COX-2) activation and prostaglandin E2 (PGE2) accumulation through a p38 MAPK-dependent NF-κB pathway. Ceramide-derived S1P acting on S1PR1 induces 1) assembly and activation of NADPH oxidase (NOX) holoenzyme and subsequent formation of superoxide (O2−), and 2) nitric oxide (NO) production from nitric oxide synthase (NOS) activation. Superoxide combines with nitric oxide to form peroxynitrite, leading to development of peripheral sensitization and hyperalgesia.
Tumor necrosis factor (TNF) is another well-known inflammatory mediator. By binding its receptor, TNFR1, TNF stimulates SMase activation and production of ceramide, which, in turns, promote pain and caspase-dependent apoptosis. [45]. Furthermore, ceramide given intradermally has been shown to induce dose-dependent thermal and mechanical hyperalgesia in rats, driven by the S1P/S1PR1 axis [46] where S1PR1 stimulation triggers local activation of NADPH oxidase and nitric oxide synthase with the subsequent formation of peroxynitrite, a potent nitroxidative species implicated in many pain states (Figure 2) [47]. Moreover, ceramide-induced thermal hyperalgesia is also known to be mediated by nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB)- and p38 mitogen-activated protein kinase (MAPK)-dependent cyclooxygenase-2 (COX-2) induction and increased prostaglandin E2 (PGE2) production [48] (Figure 2). A recent study demonstrated also the involvement of nod-like receptor family, pyrin domain containing 2 (NLRP2) signalling in ceramide-induced hypersensitivity [49]: intraplantar injection of ceramide in mice increases the expression of NLRP2 and inflammatory cytokine IL-1β in DRG neurons.
S1P/S1PR1 has further been implicated in inflammation as studies with S1PR1 antagonists such as W146 have shown to ameliorate carrageenan-induced thermal hyperalgesia in a dose-dependent manner by reducing neutrophil infiltration at inflammatory sites [50]. Other studies have confirmed the S1P/S1PR1 axis contribution to peripheral hypersensitivity through modulation of immune cell trafficking and survival [51] and also by promoting persistence of activated CD4+ T cells at inflammatory sites [52]. Moreover, it has been shown that S1P/S1PR1 signalling in macrophages induces IL-6 production, which in turn promotes S1PR1 expression on cell membranes in a feed-forward cycle leading to the establishment of chronic inflammation and pain [53].
Early studies have also suggested involvement of additional S1PRs in S1P-mediated peripheral sensitization [54, 55]. Treating isolated sensory neurons with specific short interfering RNAs (siRNAs) targeted to individual S1PRs demonstrated that activation of S1PR2, S1PR4, or S1PR5 is not sufficient to elicit significant neuronal firing. Therefore, apart from S1PR1, attention has been directed to the role of S1PR3 [55]. Both of these S1PRs are expressed on sensory neurons and DRG [56].
In particular, S1PR3 has emerged as a key contributor to S1P-induced peripheral sensitization [9]. It was recently reported that S1PR3 knock-out mice show severe loss of mechanical sensitivity [36]. Likewise, intraplantar injection of the S1PR3-selective antagonist TY-52156 in mice causes dramatically decreased responsiveness to noxious stimulation [36]. The molecular mechanism of this S1P/S1PR3-mediated nociception was unravelled recently where it was shown that endogenous S1P activates S1PR3 on sensory neurons which in turn inhibits potassium channels KCNQ2/3, thus blocking potassium currents, another well-known mechanism that regulates neuronal excitability (Figure 3) [57].
Figure 3. Schematic illustration S1P-induced sensory neurons excitability.
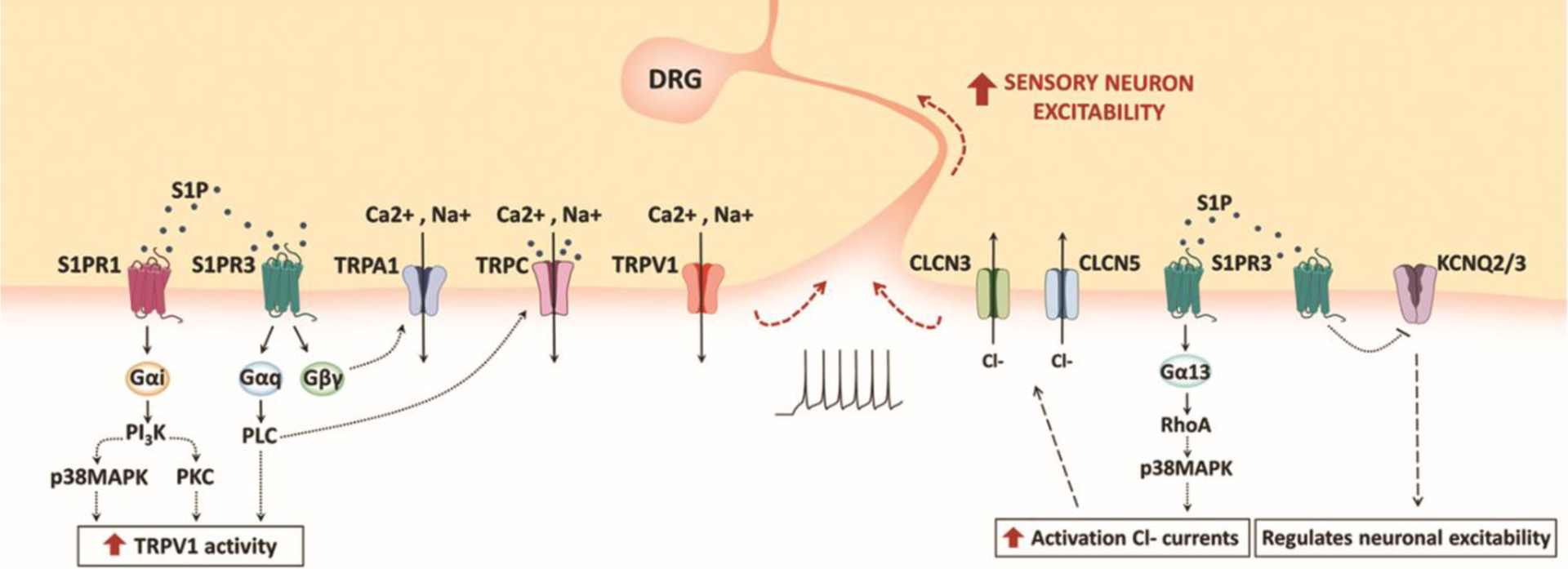
Both S1PR1 and S1PR3 are expressed in sensory neurons and DRG, and their stimulation lead to neuronal depolarization through both the potentiation of TRPV1 channels and the activation of chloride currents. However, the underlying molecular mechanisms seem to be independent of each other. S1PR3 stimulation also activate TRPC and TRPA1 channels through distinct G-protein coupled pathways. Furthermore, S1P regulates nociception by modulating potassium currents through the blockade of KCNQ2/3 channels.
It is known that different S1P concentrations at injury sites have opposite effects on the same population of sensory neurons based on the receptor subtype that is the most activated. For example, S1PR3 mediates growth cone collapse and neurite retraction through Ras homolog gene family, member A (RhoA) activation whereas S1PR1 stimulation results in elongation of neuronal processes [58]. Similarly, binding of S1P to S1PR3 evokes different neuronal responses in a concentration-dependent manner, activating ligand-gated ion channels, transient receptor potential ankyrin 1 (TRPA1) or transient receptor potential vanilloid 1 (TRPV1) through distinct G-protein pathways. Both TRPA1 and TRPV1 are expressed on nociceptors and are involved in transducing painful stimuli [59] and first observations reported that S1P-induced heat sensitization is strongly attenuated in mice lacking TRPV1 channels [10]. Indeed, it has been shown that there exist two distinct populations of S1P-responding sensory neurons that exhibit different expression patterns of TRPA1 [60]: TRPA1+/TRPV1+ sensory neurons respond to low, constitutive concentrations of S1P and mediate itch sensation through S1PR3-dependent TRPA1 activation, which requires Gβγ; TRPA1−/TRPV1+ neurons, instead, respond to high concentrations of S1P and are responsible for perception of both acute pain and heat hypersensitivity. In TRPA1−/TRPV1+ neurons, S1P trigger S1PR3-Gαq signalling, leading to phospholipase C (PLC) phosphorylation and activation of TRPV1 receptors (Figure 3) [60]. A recent study, even if mostly in accordance with the above-mentioned results, reported a major involvement of TRPV1 over TRPA1 in S1P-mediated pain and itch [61].
Similarly to TRPV1 and TRPA1, also canonical transient receptor potential (TRPC1-7) channels mediate Ca2+ and Na+ cell influx [62]. TRPC channels have been shown to be expressed in sensory neurons and their contribution to nociception and pathological pain is well established [62]. It has been suggested that S1P directly activates TRPC1 and TRPC5 in neurons [9] and an indirect activation pathway through S1PR3- Gαq-PLC has also been proposed [9, 63, 64] (Figure 3). Hence, S1P could contribute to sensory neurons excitability also through TRPC channel activation.
Moreover, pharmacological and genetic evidence indicate that apart from S1P/S1PR3, the S1P/S1PR1 axis is also a critical contributor to DRG and nociceptors hyperexcitability [65, 66] It has been shown that S1PR1 activation in DRG neurons enhances activity of TRPV1 via a Gαi-phosphoinositide-3-kinase (PI3K)-Protein kinase C (PKC)-p38 signalling cascade (Figure 3) [67]. Hence, both S1PR1 and S1PR3 downstream pathways are involved in TRPV1 sensitization and nociceptor responsiveness.
Nociceptor stimulation due to inflammation associated peripheral sensitization triggers phosphorylation and/or gating of specific ion channels on cell membranes that transduce the external stimulus into action potentials. Nociceptive inputs are then sent to the cell bodies in the DRG and relayed to the spinal cord and brain to be processed [68]. Besides the aforementioned sodium and calcium currents, in DRG sensory neurons, also chloride currents have been implicated in the transmission of peripheral nociceptive stimulation [69] where two voltage-gated chloride channels (CLCN), CLCN3 and CLCN5, directly regulate excitatory currents triggered by S1P stimulation [70]. While the downstream signalling events are not yet entirely understood, the proposed mechanism involves S1PR3-Gα13-dependent regulation of RhoA (Figure 3). Since phosphorylation is one regulatory mechanism that controls chloride channel activity [69] and RhoA can modulate p38-MAPK activity [71], p38-MAPK signalling may regulate S1P-induced Cl− efflux in DRG neurons [70]. However, S1P-induced Cl- currents and potentiation of TRPV1 functions mediated by S1P in sensory neurons described above, seem to be independent molecular mechanisms [70].
S1P axis in central sensitization
Apart from peripheral sensitization, intense, repeated and sustained noxious stimulation gives rise to specific molecular changes in the CNS, generating a persistent state of high neuronal reactivity. The main excitatory neurotransmitter in the CNS is glutamate, which has a leading role in pain transmission from the periphery to the brain [72]. Alteration in glutamatergic homeostasis associated with sustained neuroinflammation contributes to the establishment and maintenance of central sensitization [72]. In neuropathic pain states, endogenous IL-1β contributes to central sensitization by enhancing presynaptic glutamate release in the dorsal horn of the spinal cord (DHSC) and by increasing glutamate N-methyl-D-aspartate receptor (NMDAR) activity (Figure 4a) [73].
Figure 4. Schematic illustration of S1P-driven central sensitization.
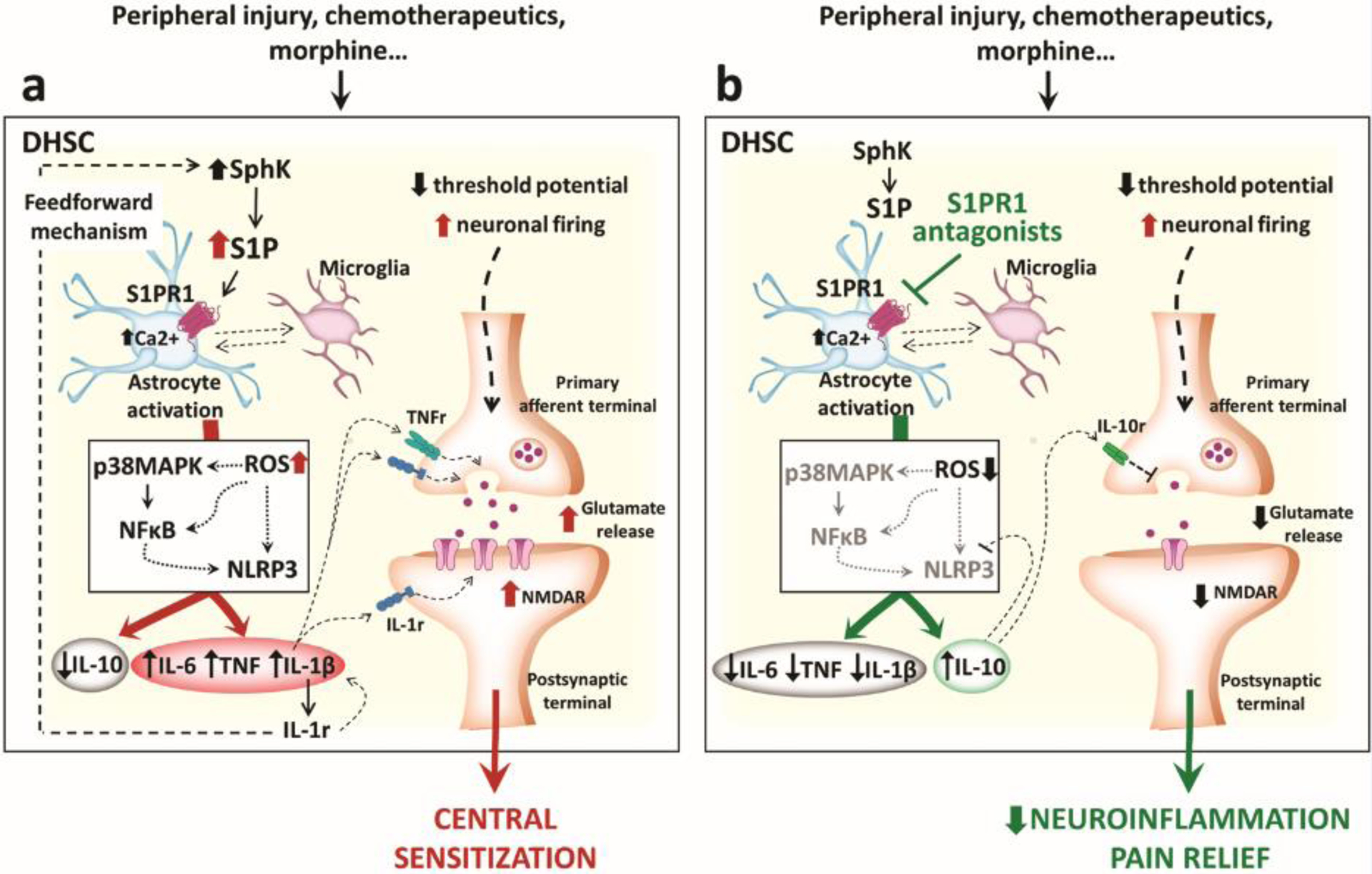
(a) The illustration shows how the dysregulation of sphingolipid signalling in the dorsal horn of the spinal cord (DHSC) leads to the establishment of central sensitization in neuropathic pain arising from different etiologies. Stimulation of S1PR1 expressed on spinal astrocytes activates NLRP3 inflammasomes, increasing the endogenous production and release of inflammatory cytokines, which in turn alter glutamatergic homeostasis and boost neuronal excitability. Moreover, activation of the IL-1β receptor (IL-1R) establishes a feedforward loop which increases inflammatory cytokine production and SphK activity, thus further increasing S1P. (b) Blocking S1PR1 signalling in astrocytes is sufficient to dampen inflammatory pathways and promote the release of anti-inflammatory IL-10, which restores glutamate homeostasis and promotes a neuroprotective state in the spinal cord.
The neuropharmacology of S1P signalling in the CNS has been largely investigated using different models of chronic pain, and important evidence for its role has emerged. It has been shown in mouse primary cortical neurons, that 1-deoxysphinganine and 1-deoxyceramide, neurotoxic sphingolipid metabolites, compromise cytoskeletal stability and membrane current properties, altering neuronal excitability, likely through allosteric modulation of NMDAR [74]. Other investigations have reported that S1P increases glutamate release, enhancing long-term potentiation in rat hippocampal neurons [75]. Moreover, inhibition of SMase attenuates the development of central sensitization in carrageenan-induced orofacial nociceptionii. Modifications of S1P levels in the spinal cord due to the absence of SphK2 also results in altered nociception: SphK2−/− mice show an anticipated licking response during the late phase of formalin-induced acute inflammation and develop bilateral mechano-hypersensitivity following unilateral complete Freund’s adjuvant intraplantar administration. SphK2 deletion decreases expression of brain-derived neurotrophic factor (BDNF), P2X4 receptor and inducible NOS in the DHSC and also impairs microglia and astrocyte activation [76].
Early studies from our group and others have also demonstrated the involvement of S1P signalling in the development of spinal sensitization through enhanced glial cell activation and increased production of inflammatory/neuroexcitatory cytokines and superoxide-derived peroxynitrite, both involved in the modulation of glutamate neurotransmission [77, 78]. For example, morphine-induced hyperalgesia correlates with increased production of both ceramide and S1P in the spinal cord which is associated with release of glia-derived TNF, IL-1β and IL-6 [77]. IL-1β can enhance SphK1 activity further promoting S1P production [79] and also inhibit the formation of the anti-inflammatory and neuroprotective IL-10 [80]. These events depend on S1PR1-mediated p38-MAPK pathway activation and contribute to the development of morphine-induced antinociceptive tolerance (Figure 4a) [81].
Intrathecal administration of the selective S1PR1 agonist, SEW2871, in rodents activates the nod-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome pathway leading to the release of IL-1β that evokes thermal and mechanical hyperalgesia [79]. These effects are abrogated in mice lacking the S1PR1 receptor on astrocytes, implying that the NLRP3 pathway is activated downstream of S1PR1 (Figure 4a). The role of astrocyte-mediated S1PR1 neuroinflammation has also been documented in models of traumatic nerve injury [82]. Peripheral nerve damage induces increased S1P production and S1PR1 expression in the dorsal horn of the spinal cord, leading to central sensitization triggered by NLRP3 inflammasome-derived IL-1β. Beneficial effects observed following S1PR1 antagonism are IL-10-dependent [82] (Figure 4b).
Besides astrocytes, also microglia, and the inflammatory factors they release, promote the transition from acute to pathological chronic pain states [72]. Hence, studying the molecular mechanisms through which S1P mediates central sensitization, a more complex scenario involving also a dynamic astrocyte-microglia interaction can be hypothesized (Figure 4a). Microglia, indeed, express both S1PR1 and NLRP3 [83, 84], release IL-1β under exogenous S1P stimulation [85] and are also able to produce IL-10. Moreover, activation of the microglial NLRP3 inflammasome has recently been linked to pathogenic neuroinflammation [84, 86]. Inhibition of S1PR1 signalling has no effect on microglia, which maintain an inflammatory phenotype [87]. However, it is possible that S1PR1 antagonism in astrocytes also impacts microglia functions. Indeed, it has been shown that activation of IL-10 signalling in astrocytes attenuates microglia activation under inflammatory conditions [88].
Other than S1PR1, recently also S1PR2 and S1PR3 involvement has been proposed in central pain mechanisms. In particular, in a model of chronic constriction injury in rats, S1PR2 activation in the spinal cord resulted in anti-inflammatory effects [89], whereas an in vitro study showed that activation of S1PR3 on astrocytes promotes the release of cytotoxic IL-6, COX-2 and vascular endothelial growth factor A (VEGFa) through RhoA activation [90]. Moreover, S1PR2/S1PR3-induced TRPC6-mediated Ca2+ influx and C-X-C Motif Chemokine Ligand 1 (CXCL1) release in astrocytes has been recently proposed as a contributor of central neuroinflammation [91].
Increased S1P levels at spinal cord injury sites [92] are known to attract microglia and macrophages, whose activation exacerbates the inflammatory response. Both inhibition of ceramide biosynthesis [93] and FTY720 administration shortly after spinal contusion ameliorate injury outcomes and promote functional recovery [94]. Recently, a study revealed that FTY720 reduces spinal cord injury-induced neuropathic pain by dampening neuroinflammation and inhibiting glial scar formation [95].
Collectively, the dysregulation of sphingolipid metabolism in the DHSC cord has been linked to the development of persistent neuroinflammation and nociceptive behaviors in several chronic pain models [77, 82, 96–98] with most recent data support the predominant role of S1PR1 signalling in astrocytes as the driving force of central sensitization.
In contrast with the increasing data available in the spinal cord, the role of S1P signalling in higher pain centers is still largely unknown. On hippocampal slices, it has been shown that S1PR1 signalling influences NMDAR properties [99]. Similarly, ablation of S1PR3 impairs neuronal hippocampal excitability in vitro [100] and reduces inflammatory processes in medial prefrontal cortex in mice [101], both areas involved in pain processing [102]. However, more in-depth studies on S1P signalling need to be addressed also towards other brain structures responsible for pain modulation such as cingulate cortex, thalamus, periaqueductal grey and rostroventral medulla.
Exploiting S1P signalling towards managing chronic pain
As mentioned above, high plasma levels of 1-deoxysphingolipids have been found in patients with CINP [23]. CINP is a major dose-limiting side effect of widely used chemotherapeutics with distinct antitumor modes of action including taxanes, platinum-based drugs, vinka alkaloids and proteasome inhibitors [103]. Recent investigations following docetaxel administration in mice, revealed a strong increase of neurotoxic 1-deoxysphingolipids in DRG, which could contribute to nerve endings degeneration and neuropathy onset [104].
Sphingolipidomic analysis of spinal cord tissues have revealed that CNS alterations in S1P also contribute to the development of CINP via S1PR1 [96, 97]. Ceramide is known to promote activation of several anti-proliferative and tumor-suppressive pathways. In contrast, S1P positively regulates pro-survival and anti-apoptotic responses [105]. Hence it has been hypothesized that the ceramide/S1P rheostat plays a critical role in regulating cancer cell fate with elevated levels of ceramide inducing cell death and elevated levels of S1P leading to survival and proliferation [105]. Thus, therapeutic strategies aimed at reducing S1P bioavailability or signalling are attracting attention as novel anticancer agents [105]. Numerous studies have demonstrated that FTY720 has potent antitumor activity [106] and can synergize with anticancer agents. For example, FTY720 synergizes with bortezomib to target multiple myeloma cells in vitro and multiple myeloma xenografts in vivo [107]. Thus, drugs, such as FTY720, may enhance the anticancer effects of chemotherapeutic agents while minimizing CINP, one of their major side effects.
Further investigations using paclitaxel, oxaliplatin, and bortezomib have revealed how activation of S1PR1 signalling at the spinal cord level acts in chemotherapy-induced hyperalgesia. As mentioned earlier, S1PR1 mediated activation of glial cells [108], NFκB and p38-MAPK [96] increase production and release of TNF and IL-1β [108] and enhance presynaptic glutamate release (Figure 4a) [97, 108]. Blocking S1PR1 with functional and competitive antagonists attenuate these neuroinflammatory processes and block CINP without interfering with anticancer effects [96, 97] (Figure 4b). Moreover, mice with specific deletion of S1PR1 on astrocytes do not develop CINP, underscoring the importance of an astrocyte-based S1PR1 signalling pathway in these conditions [97]. Importantly, sex differences in drug responses to bortezomib-induced neuropathic pain (BINP) have been reported [109]. In particular, S1PR1 antagonists, which alleviates paclitaxel and oxaliplatin CINP in both sexes, failed to prevent and reverse BINP in females rodents [109]. These data are particularly relevant and have to be considered in the clinical translation of these compounds since S1PR1-based therapies could result ineffective in the treatment of CINP in women who receive bortezomib chemotherapy.
Interestingly, a recent study suggested the possibility of targeting also S1PR2 in CINP [34]. Intraperitoneal administration of the S1PR2 agonist CYM-5478 in rats blocked the development of cisplatin-induced neuropathic pain by inhibiting peripheral gliosis and myelin disruption in DRG neurons [34].
Results obtained from CINP studies also correlate with central mechanisms observed in MS where neuropathic pain is one of the most frequent symptoms where blocking S1PR1 suppresses pathogenic astrocyte activation and prevents disease progression, decreasing proinflammatory markers of central sensitization and up-regulating IL-10 expression [87]. Furthermore, conditional astrocyte depletion of S1PR1 mimics the effects observed following S1PR1 antagonist administration in an animal model of MS [110]. Moreover, in MS patients, increased CNS levels of N,N-dimethylsphingosine [111] has been observed, a metabolite that is known to induce neuropathic pain in rats when injected intrathecally [112]. Oligodendrocytes were identified as the source of N,N-dimethylsphingosine and its production increases after exposure to white matter-damaging stimuli, confirming the inflammatory demyelinating aspect of MS [111].
Ultimately, even if analgesic effects have been attributed to S1PR1 agonism [113, 114], or combined S1PR1/S1PR3 agonism [115], recent in-depth investigation through multidisciplinary and pharmacological approaches [82] have revealed S1PR1 inhibition, and not activation, as beneficial in neuropathic pain. Thus, the disruption of S1P-S1PR1 pathway is a promising pharmacological strategy to be pursued to block and reverse persistent pain states (Figure 4b).
Concluding remarks and future perspective
It has become apparent that alterations in S1P signalling and associated bioactive sphingolipid metabolites such as S1P play critical roles in the development of several pain states (Table 2). In particular, S1PR1 has emerged as a viable target for therapeutic intervention with functional and competitive S1PR1 antagonists. These compounds do not induce tolerance with prolonged use and exert their beneficial effects without engaging endogenous opioid circuits and can therefore be considered as non-narcotic analgesics. Moreover, preclinical and clinical data indicate that alterations in ceramide/S1P may be implicated in anxiety, depression and cognitive dysfunction [116, 117], which are common comorbidities associated with chronic pain states. These findings highlight the need to explore repurposing drugs such as FTY720 for the treatment of pain of diverse etiologies. Noteworthy, clinical trials for proof-of-concept in patients with breast cancer have begun with FTY720 to explore efficacy in preventing and/or treating CINP (Clinical Trial Numberiii NCT03941743; NCT03943498). In support of these ongoing clinical evaluations, a recent pharmacogenomic study revealed that patients with CINP have single nucleotide polymorphisms (SNPs) in a genomic region that regulate S1PR1 gene expression. These findings on genetic variations validate pharmacological studies and could explain patient susceptibility and drive mechanisms underlying CINP development [118]. Validation of the next generation of S1PR modulators requires preclinical and clinical trials (see Outstanding Questions). For example, a few studies have suggested that S1PR2 agonists may be effective [34, 89]. However, several therapeutic agents that lack activity at S1PR2 (i.e. FTY720) show beneficial results in models of neuropathic pain states, suggesting a non-critical involvement of this receptor [82, 96, 97]. Emerging evidence also points to interesting contributions of S1PR3 [36, 60, 70] and development of highly selective S1PR3 antagonists could provide valuable tools for in-depth exploration of their effects for pain treatment. There is no doubt that the S1P/S1PR1 axis represents a fertile area for therapeutic intervention.
Table 2.
Chronological studies on bioactive sphingolipid metabolites in pain
| Hereditary sensory neuropathy (HSAN1) in humans is linked to mutations in serine palmitoyltransferase (SPT) and increased SPT enzymatic activity | 2001 | [19] |
| Hindpaw injection of ceramide causes hyperalgesia in rats | 2004 | [45] |
| Ceramide/S1P enhance the excitability of rat sensory neurons | 2006 | [54] |
| FTY720 blocks neuropathic pain in spared nerve injury model in rats | 2008 | [114] |
| Spinal injection of the S1PR1 agonist, SEW2871, has no effect on formalin phase 1/2 response in rats | 2008 | [114] |
| Chronic morphine administration in rats upregulates ceramide and S1P in the spinal cord resulting in the development of antinociceptive tolerance and central sensitization | 2009–2010 | [77, 78] |
| Inhibition of sphingomyelinase attenuates carrageenan-induced orofacial mechanical allodynia in mice | 2009 | ii |
| Inhibition of ceramide biosynthesis ameliorates neuropathic pain and promote functional recovery after spinal cord injury in mice. Similar results are obtained with oral FTY720 administration shortly after spinal contusion | 2009–2012 | [93, 94] |
| S1PR1 is implicated in S1P-mediated enhancement of isolated rat sensory neuron excitability | 2010 | [65] |
| S1P enhances BV2 microglia-induced inflammation and production of nitroxidative species | 2010 | [85] |
| S1P increases glutamate release and enhances long-term potentiation in rat hippocampal neurons | 2010 | [75] |
| Administration of S1PR1 agonist, SEW2871, in mice and rats recapitulates pain behavior phenotypes | 2011–2020 | [10, 47, 50, 79, 82, 96] |
| Ceramide-induced pain behaviors in rats requires bioconversion of ceramide to S1P and S1PR1-mediated activation of NF-kB- and p38-dependent COX2/PGE2 pathways | 2011 | [48] |
| S1P-mediated activation of S1PR1 induces pain behaviors in rats through peroxynitrite formation | 2011 | [47] |
| Mass spectrometry-based metabolomics implicates altered sphingolipid signalling in neuropathic pain states in rats | 2012 | [112] |
| S1P induces spontaneous pain in mice via S1P3-dependent activation of an excitatory Cl- conductance | 2013 | [9] |
| Spinal injection of the S1PR1 agonist, SEW2871, in mice and rats does not block traumatic nerve injury-induced pain | 2014–2019 | [82, 115] |
| Chemotherapy-induced increase in S1P in spinal cord contributes to neuropathic pain in rats and mice. Functional and competitive S1PR1 antagonists block paclitaxel, oxaliplatin and bortezomib induced neuropathic pain without altering their anticancer activity. The S1PR1 agonist, SEW2871, does not block chemotherapy-induced pain | 2014–2018 | [96, 97] |
| S1P-S1PR1 binding enhances the activity of TRPV1 via Gαi-PI3K-PKC-p38 signalling in mouse DRG neurons | 2014 | [67] |
| Sphk2 activity in the spinal cord is involved in the facilitation of nociceptive circuits leading to pain behaviors in mice | 2015 | [76] |
| Neurotoxic 1-deoxysphinganine and 1-deoxyceramide enhance neuronal excitability through an allosteric modulation of NMDAR | 2016 | [74] |
| Sphingolipidomic analysis of spinal cord samples reveals that alterations in the S1P-S1PR1 axis underlie cancer-induced bone pain and neuroinflammation in mice. Functional and competitive S1PR1 antagonists block bone-cancer pain | 2017 | [98] |
| Fingolimod (FTY720) reduces neuropathic pain behaviors in an autoimmune mouse model of multiple sclerosis. Similar results are obtained with systemic administration of selective S1PR1 agonist, SEW2871 | 2018 | [113] |
| Both S1PR3 knock-out and pharmacological inhibition of S1PR3 cause dramatic decrease responsiveness to noxious stimulation in mice | 2018 | [36] |
| S1P through S1PR3 regulates neuronal excitability by inhibiting KCNQ2/3 potassium channels in mouse sensory neurons | 2018 | [36] |
| CLCN3 and CLCN5 mediate excitatory Cl- currents activated by S1P/S1PR3 in mouse sensory neurons | 2018 | [70] |
| S1P-to-S1PR3 signalling mediates pain behaviors in mice through the activation of TRPV1 in sensory neurons | 2018 | [60] |
| Sexual dymorphic responses are observed with bortezomib. Unlike paclitaxel and oxaliplatin, S1PR1-based therapies do not block bortezomib-induced neuropathic pain in female rats and mice | 2019 | [109] |
| Ceramide-induced pain behaviors in mice involve NLRP2 inflammasome activation in the DRG | 2019 | [49] |
| Activation of S1PR1 in spinal astrocytes, following intrathecal injection of SEW2871, leads to NLRP3 inflammasome activation, IL-1β production and pain behaviors in mice and rats | 2019 | [79] |
| Functional and competitive S1PR1 antagonists block traumatic nerve injury-induced neuropathic pain in mice and rats. S1PR1 expressed on astrocytes is identified as target for therapeutic intervention with S1PR1 antagonists | 2019 | [82] |
| S1PR2 activation in spinal cord attenuates neuropathic pain and neuroinflammation in a model of traumatic nerve injury in rats | 2019 | [89] |
| Clinical trials for proof of concept in patients with breast cancer have begun with Fingolimod to explore efficacy in preventing and/or treating chemo-induced neuropathic pain (NCTID: NCT03941743; NCT03943498) | 2019 | iii |
| FTY720 ameliorates spinal cord injury induced-neuropathic pain in rats by dampening neuroinflammation and inhibiting glial scar formation | 2020 | [95] |
| Role for S1PR1 in opioid-mediated adverse events is identified. Functional and competitive S1PR1 antagonists block morphine-induced hyperalgesia, tolerance and dependence | 2020 | [81] |
| Activation of S1PR2 following systemic administration of the selective S1PR2 agonist CYM-5478 attenuates cisplatin-induced neuropathic pain in rats | 2020 | [34] |
| Patients with CINP have single nucleotide polymorphisms (SNPs) in a genomic region that regulate S1PR1 gene expression. These genetic variations validate pharmacological studies and could explain patient susceptibility to CINP development | 2020 | [118] |
Outstanding Questions.
How is sphingolipid expression and signaling modified in higher pain centers (periaqueductal grey, rostroventral medulla, hippocampus, prefrontal cortex…) during chronic pain states?
What is the consequence of inhibiting S1PR1 signaling at supraspinal sites?
Limited evidence suggests that S1PR1 agonism and not antagonism contributes to the beneficial effects observed in models of autoimmune-dependent chronic pain such as MS; extensive work is needed to determine whether agonism or antagonism is required.
What are the interactions between opioids and sphingolipids in the context of chronic pain?
How do strategies of blocking S1PR3 and activating S1PR2 compare with S1PR1 inhibition?
What effects do S1PR agonists/antagonists have on depression, anxiety, and cognitive dysfunctions, comorbidities that are common in patients with chronic pain syndromes?
Highlights.
Alteration in sphingolipid metabolism and increased formation of ceramide and sphingosine-1-phosphate (S1P) in the nervous system are linked to the development of chronic pain states.
Functional and competitive S1P receptor 1 (S1PR1) antagonists block neuropathic pain states of diverse etiologies.
Inhibition of neuro-immune processes known to increase neuronal excitability are important pharmacological actions resulting from S1PR1 inhibition.
S1PR1 has emerged as a non-opioid target for therapeutic intervention with functional and competitive S1PR1 antagonists. Astrocytes are a key cellular hub for S1PR1 activity.
The functional S1PR antagonists, Fingolimod and Ozanimod, are approved for non-pain indications, suggesting repurposing these drugs for chronic pain indications.
ACKNOWLEDGEMENTS
We would like to acknowledge support from NIH grant R01GM043880 (SS) and RO1DA043543 (DS). We thank Dr. T. M. Doyle (Saint Louis University) for input during the preparation of the manuscript and Dr J. Eissenberg (Saint Louis University) for editing the manuscript.
GLOSSARY
- Dorsal root ganglion (DRG)
Cluster of neuronal cell bodies located in the dorsal root of a spinal nerve. Here, the peripheral information carried by sensory neurons is processed and sent to the central nervous system.
- Bradycardia
is a slower than normal heart rate. In adults, a resting heart beat that is less than 60 beats per minute.
- Functional antagonist
A compound whose binding to its receptor initiates effects that are functionally opposite those of an agonist. Conversely, a competitive antagonist binds at the same site as the agonist, preventing the activation of the receptor.
- Gating
Process that controls the opening and closing of cell membrane ion channels.
- Growth cone
Terminal portion of a neurite with a distinctive cytoskeletal organization which allows a high motility to explore the extracellular environment. The growth cone guides axonal extension, determining the direction of growth.
- Long-term potentiation (LTP)
long-lasting strengthening of synapses (it is considered the main form of synaptic plasticity) that occur with repeated and sustained neuronal stimulation. This mechanism facilitates neuronal communication and it is crucially involved in memory formation.
- Morphine-induced antinociceptive tolerance
Opioid tolerance is defined as a reduced responsiveness to an opioid analgesic. Thus, increasing doses of the drug are needed to obtain the same analgesic effect. Other severe side effects induced by prolonged opioid treatment include addiction, physical and psychological dependence and a paradoxical increased sensitivity to pain.
- Nociceptors
Sensory neurons activated by noxious stimuli that can potentially damage or threaten tissue integrity. They are classified according to their responses to mechanical, thermal and chemical stimuli.
- Peripheral sensitization
Increased sensitivity of peripheral sensory (afferent) neurons.
- Pharmacogenomic study
Study of how genes affect a person’s response to drugs. This relatively new field combines pharmacology and genomics to develop effective, safe medications and doses that will be tailored to a person’s genetic makeup.
- Prodrug
Biologically inactive compound that can be metabolized in the body to produce an active therapeutic drug.
- Ras homolog gene family, member A (RhoA)
Small GTPase, an enzyme able to bind and hydrolyze guanosine triphosphate (GTP). All GTPases belong to the Ras superfamily whose members can be divided into families and subfamilies based on their structure, sequence and function. The five main families are Ras, Rho, Ran, Rab, and Arf GTPases.
- Single nucleotide polymorphisms (SNPs)
Substitution of a single nucleotide within a gene or in a regulatory region. They can act as biological markers, helping scientists locate genes that are associated with disease.
- Small (short) interfering RNA (siRNA)
Double-stranded RNA molecules, 20–25 base pairs in length that interfere with the expression of specific genes (gene silencing) through repression of translation or targeted degradation of transcripts.
- Sphingolipidomics
Analysis of the all of the sphingolipids (sphingolipidome) in a biological sample.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLAIMER STATEMENT
Dr. Salvemini has patents submitted by Saint Louis University that cover some of the intellectual property described in this manuscript (U.S. patent number 8,747,844 and its divisional, U.S. patent number 8,945,549). All other authors declare no conflict of interest.
RESOURCES
Vardeh, D. and Naranjo, J.F. (2017) Peripheral and Central Sensitization. In Pain Medicine: An Essential Review (Yong, R.J. et al. eds), pp. 15–17, Springer International Publishing
REFERENCES
- 1.Montgomery LS (2020) Pain management with opioids in adults. J Neurosci Res. [DOI] [PubMed] [Google Scholar]
- 2.Kolesnick R (2002) The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J Clin Invest 110 (1), 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pyne S et al. (2018) Sphingosine Kinases as Druggable Targets. Handb Exp Pharmacol. [DOI] [PubMed] [Google Scholar]
- 4.Maceyka M and Spiegel S (2014) Sphingolipid metabolites in inflammatory disease. Nature 510 (7503), 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hait NC et al. (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325 (5945), 1254–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hannun YA and Obeid LM (2018) Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol 19 (3), 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagahashi M et al. (2014) Sphingosine-1-phosphate transporters as targets for cancer therapy. Biomed Res Int 2014, 651727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spiegel S et al. (2019) New insights into functions of the sphingosine-1-phosphate transporter SPNS2. J Lipid Res 60 (3), 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camprubi-Robles M et al. (2013) Sphingosine-1-phosphate-induced nociceptor excitation and ongoing pain behavior in mice and humans is largely mediated by S1P3 receptor. J Neurosci 33 (6), 2582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mair N et al. (2011) Genetic evidence for involvement of neuronally expressed S1P(1) receptor in nociceptor sensitization and inflammatory pain. PLoS One 6 (2), e17268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Healy LM and Antel JP (2016) Sphingosine-1-Phosphate Receptors in the Central Nervous and Immune Systems. Curr Drug Targets 17 (16), 1841–1850. [DOI] [PubMed] [Google Scholar]
- 12.Tham CS et al. (2003) Microglial activation state and lysophospholipid acid receptor expression. Int J Dev Neurosci 21 (8), 431–43. [DOI] [PubMed] [Google Scholar]
- 13.Van Doorn R et al. (2010) Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia 58 (12), 1465–76. [DOI] [PubMed] [Google Scholar]
- 14.Yu N et al. (2004) Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes. Glia 45 (1), 17–27. [DOI] [PubMed] [Google Scholar]
- 15.Olesch C et al. (2017) Beyond Immune Cell Migration: The Emerging Role of the Sphingosine-1-phosphate Receptor S1PR4 as a Modulator of Innate Immune Cell Activation. Mediators Inflamm 2017, 6059203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salvemini D et al. (2013) Therapeutic targeting of the ceramide-to-sphingosine 1-phosphate pathway in pain. Trends Pharmacol Sci 34 (2), 110–8. [DOI] [PubMed] [Google Scholar]
- 17.Asi T et al. (2019) Serum sphingosine-1-phosphate levels in bladder pain syndrome/interstitial cystitis patients: could it help in diagnosis? World J Urol. [DOI] [PubMed] [Google Scholar]
- 18.Yu FPS et al. (2018) Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J Rare Dis 13 (1), 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dawkins JL et al. (2001) Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet 27 (3), 309–12. [DOI] [PubMed] [Google Scholar]
- 20.Suriyanarayanan S et al. (2019) A Novel Variant (Asn177Asp) in SPTLC2 Causing Hereditary Sensory Autonomic Neuropathy Type 1C. Neuromolecular Med 21 (2), 182–191. [DOI] [PubMed] [Google Scholar]
- 21.Penno A et al. (2010) Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem 285 (15), 11178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hube L et al. (2017) Metabolic Syndrome, Neurotoxic 1-Deoxysphingolipids and Nervous Tissue Inflammation in Chronic Idiopathic Axonal Polyneuropathy (CIAP). PLoS One 12 (1), e0170583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kramer R et al. (2015) Neurotoxic 1-deoxysphingolipids and paclitaxel-induced peripheral neuropathy. Faseb j 29 (11), 4461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hammad SM et al. (2017) Increased Plasma Levels of Select Deoxy-ceramide and Ceramide Species are Associated with Increased Odds of Diabetic Neuropathy in Type 1 Diabetes: A Pilot Study. Neuromolecular Med 19 (1), 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adachi KK, T.; Nakao N; Arita M; Chiba K; Mishina T; Sasaki S; Fujita T (1995) Design, synthesis, and structure-activity relationships of 2-substituted-2-amino-1,3-propanediols: discovery of a novel immunosuppressant, FTY720. Bioorg. Med. Chem. Lett 5, 853–856. [Google Scholar]
- 26.Mandala S et al. (2002) Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 296 (5566), 346–9. [DOI] [PubMed] [Google Scholar]
- 27.Brinkmann V et al. (2010) Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov 9 (11), 883–97. [DOI] [PubMed] [Google Scholar]
- 28.Huwiler A and Zangemeister-Wittke U (2018) The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: Recent findings and new perspectives. Pharmacol Ther 185, 34–49. [DOI] [PubMed] [Google Scholar]
- 29.Park SJ and Im DS (2017) Sphingosine 1-Phosphate Receptor Modulators and Drug Discovery. Biomol Ther (Seoul) 25 (1), 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Camm J et al. (2014) Cardiac and vascular effects of fingolimod: mechanistic basis and clinical implications. Am Heart J 168 (5), 632–44. [DOI] [PubMed] [Google Scholar]
- 31.Swallow E et al. (2020) Comparative safety and efficacy of ozanimod versus fingolimod for relapsing multiple sclerosis. J Comp Eff Res 9 (4), 275–285. [DOI] [PubMed] [Google Scholar]
- 32.Lamb YN (2020) Ozanimod: First Approval. Drugs 80 (8), 841–848. [DOI] [PubMed] [Google Scholar]
- 33.Bigaud M et al. (2014) Second generation S1P pathway modulators: research strategies and clinical developments. Biochim Biophys Acta 1841 (5), 745–58. [DOI] [PubMed] [Google Scholar]
- 34.Wang W et al. (2020) Activation of sphingosine 1-phosphate receptor 2 attenuates chemotherapy-induced neuropathy. J Biol Chem 295 (4), 1143–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li MH et al. (2015) Antitumor Activity of a Novel Sphingosine-1-Phosphate 2 Antagonist, AB1, in Neuroblastoma. J Pharmacol Exp Ther 354 (3), 261–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill RZ et al. (2018) The signaling lipid sphingosine 1-phosphate regulates mechanical pain. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guerrero M et al. (2012) Discovery, design and synthesis of novel potent and selective sphingosine-1-phosphate 4 receptor (S1P(4)-R) agonists. Bioorg Med Chem Lett 22 (1), 537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urbano M et al. (2011) SAR analysis of innovative selective small molecule antagonists of sphingosine-1-phosphate 4 (S1P(4)) receptor. Bioorg Med Chem Lett 21 (18), 5470–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hur W et al. (2017) A benzo[b]thiophene-based selective type 4 S1P receptor agonist. Bioorg Med Chem Lett 27 (1), 1–5. [DOI] [PubMed] [Google Scholar]
- 40.Hobson AD et al. (2015) Discovery of A-971432, An Orally Bioavailable Selective Sphingosine-1-Phosphate Receptor 5 (S1P5) Agonist for the Potential Treatment of Neurodegenerative Disorders. J Med Chem 58 (23), 9154–70. [DOI] [PubMed] [Google Scholar]
- 41.Drouillard A et al. (2018) S1PR5 is essential for human natural killer cell migration toward sphingosine-1 phosphate. J Allergy Clin Immunol 141 (6), 2265–2268.e1. [DOI] [PubMed] [Google Scholar]
- 42.Denk F et al. (2017) Nerve Growth Factor and Pain Mechanisms. Annu Rev Neurosci 40, 307–325. [DOI] [PubMed] [Google Scholar]
- 43.Khodorova A et al. (2013) The p75NTR signaling cascade mediates mechanical hyperalgesia induced by nerve growth factor injected into the rat hind paw. Neuroscience 254, 312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khodorova A et al. (2017) The TrkA receptor mediates experimental thermal hyperalgesia produced by nerve growth factor: Modulation by the p75 neurotrophin receptor. Neuroscience 340, 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joseph EK and Levine JD (2004) Caspase signalling in neuropathic and inflammatory pain in the rat. Eur J Neurosci 20 (11), 2896–902. [DOI] [PubMed] [Google Scholar]
- 46.Doyle T et al. (2011) Sphingosine-1-phosphate acting via the S1P(1) receptor is a downstream signaling pathway in ceramide-induced hyperalgesia. Neurosci Lett 499 (1), 4–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doyle T et al. (2011) Role for peroxynitrite in sphingosine-1-phosphate-induced hyperalgesia in rats. Pain 152 (3), 643–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doyle T et al. (2011) Intraplantar-injected ceramide in rats induces hyperalgesia through an NF-kappaB- and p38 kinase-dependent cyclooxygenase 2/prostaglandin E2 pathway. Faseb j 25 (8), 2782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsuoka Y et al. (2019) NLRP2 inflammasome in dorsal root ganglion as a novel molecular platform that produces inflammatory pain hypersensitivity. Pain 160 (9), 2149–2160. [DOI] [PubMed] [Google Scholar]
- 50.Finley A et al. (2013) Sphingosine 1-phosphate mediates hyperalgesia via a neutrophil-dependent mechanism. PLoS One 8 (1), e55255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aoki M et al. (2016) Sphingosine-1-Phosphate Signaling in Immune Cells and Inflammation: Roles and Therapeutic Potential. Mediators Inflamm 2016, 8606878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jaigirdar SA et al. (2017) Sphingosine-1-Phosphate Promotes the Persistence of Activated CD4 T Cells in Inflamed Sites. Front Immunol 8, 1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao S et al. (2018) Sphingosine-1-phosphate receptor 1 mediates elevated IL-6 signaling to promote chronic inflammation and multitissue damage in sickle cell disease. Faseb j 32 (5), 2855–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang YH et al. (2006) Sphingosine-1-phosphate via activation of a G-protein-coupled receptor(s) enhances the excitability of rat sensory neurons. J Neurophysiol 96 (3), 1042–52. [DOI] [PubMed] [Google Scholar]
- 55.Li C et al. (2015) Sphingosine 1-phosphate enhances the excitability of rat sensory neurons through activation of sphingosine 1-phosphate receptors 1 and/or 3. J Neuroinflammation 12, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kays JS et al. (2012) Expression of sphingosine 1-phosphate receptors in the rat dorsal root ganglia and defined single isolated sensory neurons. Physiol Genomics 44 (18), 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Passmore GM et al. (2003) KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci 23 (18), 7227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quarta S et al. (2017) Sphingosine-1-Phosphate and the S1P3 Receptor Initiate Neuronal Retraction via RhoA/ROCK Associated with CRMP2 Phosphorylation. Front Mol Neurosci 10, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Basso L and Altier C (2017) Transient Receptor Potential Channels in neuropathic pain. Curr Opin Pharmacol 32, 9–15. [DOI] [PubMed] [Google Scholar]
- 60.Hill RZ et al. (2018) S1PR3 Mediates Itch and Pain via Distinct TRP Channel-Dependent Pathways. J Neurosci 38 (36), 7833–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kittaka H et al. (2020) Differential contribution of sensory transient receptor potential channels in response to the bioactive lipid sphingosine-1-phosphate. Mol Pain 16, 1744806920903515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun ZC et al. (2020) Canonical Transient Receptor Potential (TRPC) Channels in Nociception and Pathological Pain. Neural Plast 2020, 3764193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lepannetier S et al. (2016) Sphingosine-1-phosphate-activated TRPC1 channel controls chemotaxis of glioblastoma cells. Cell Calcium 60 (6), 373–383. [DOI] [PubMed] [Google Scholar]
- 64.Pulli I et al. (2018) Sphingolipid-mediated calcium signaling and its pathological effects. Biochim Biophys Acta Mol Cell Res 1865 (11 Pt B), 1668–1677. [DOI] [PubMed] [Google Scholar]
- 65.Chi XX and Nicol GD (2010) The sphingosine 1-phosphate receptor, S1PR(1), plays a prominent but not exclusive role in enhancing the excitability of sensory neurons. J Neurophysiol 104 (5), 2741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xie W et al. (2012) Knockdown of the sphingosine-1-phosphate receptor S1PR1 reduces pain behaviors induced by local inflammation of the rat sensory ganglion. Neurosci Lett 515 (1), 61–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langeslag M et al. (2014) Sphingosine 1-phosphate to p38 signaling via S1P1 receptor and Galphai/o evokes augmentation of capsaicin-induced ionic currents in mouse sensory neurons. Mol Pain 10, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pinho-Ribeiro FA et al. (2017) Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol 38 (1), 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Funk K et al. (2008) Modulation of chloride homeostasis by inflammatory mediators in dorsal root ganglion neurons. Mol Pain 4, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qi Y et al. (2018) Identification of Chloride Channels CLCN3 and CLCN5 Mediating the Excitatory Cl(−) Currents Activated by Sphingosine-1-Phosphate in Sensory Neurons. Front Mol Neurosci 11, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shatanawi A et al. (2011) Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am J Physiol Cell Physiol 300 (5), C1181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ji RR et al. (2018) Neuroinflammation and Central Sensitization in Chronic and Widespread Pain. Anesthesiology 129 (2), 343–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yan X and Weng HR (2013) Endogenous interleukin-1beta in neuropathic rats enhances glutamate release from the primary afferents in the spinal dorsal horn through coupling with presynaptic N-methyl-D-aspartic acid receptors. J Biol Chem 288 (42), 30544–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guntert T et al. (2016) 1-Deoxysphingolipid-induced neurotoxicity involves N-methyl-d-aspartate receptor signaling. Neuropharmacology 110 (Pt A), 211–222. [DOI] [PubMed] [Google Scholar]
- 75.Kanno T et al. (2010) Regulation of synaptic strength by sphingosine 1-phosphate in the hippocampus. Neuroscience 171 (4), 973–80. [DOI] [PubMed] [Google Scholar]
- 76.Canlas J et al. (2015) Sphingosine kinase 2-deficiency mediated changes in spinal pain processing. Front Mol Neurosci 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muscoli C et al. (2010) Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J Neurosci 30 (46), 15400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ndengele MM et al. (2009) Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J Pharmacol Exp Ther 329 (1), 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Doyle TM et al. (2019) Activation of Sphingosine-1-Phosphate Receptor 1 in the Spinal Cord Produces Mechanohypersensitivity Through the Activation of Inflammasome and IL-1beta Pathway. J Pain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zielinski CE et al. (2012) Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature 484 (7395), 514–8. [DOI] [PubMed] [Google Scholar]
- 81.Doyle TM et al. (2020) Activation of sphingosine-1-phosphate receptor subtype 1 in the central nervous system contributes to morphine-induced hyperalgesia and antinociceptive tolerance in rodents. PAIN Articles in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen Z et al. (2019) Sphingosine-1-phosphate receptor 1 activation in astrocytes contributes to neuropathic pain. Proc Natl Acad Sci U S A 116 (21), 10557–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Y et al. (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34 (36), 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.He W et al. (2019) Microglial NLRP3 inflammasome activation mediates IL-1beta release and contributes to central sensitization in a recurrent nitroglycerin-induced migraine model. J Neuroinflammation 16 (1), 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nayak D et al. (2010) Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience 166 (1), 132–44. [DOI] [PubMed] [Google Scholar]
- 86.Deora V et al. (2020) The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 68 (2), 407–421. [DOI] [PubMed] [Google Scholar]
- 87.Rothhammer V et al. (2017) Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc Natl Acad Sci U S A 114 (8), 2012–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Norden DM et al. (2014) TGFbeta produced by IL-10 redirected astrocytes attenuates microglial activation. Glia 62 (6), 881–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li Y et al. (2019) Sphingosine-1-phosphate receptor 2 modulates pain sensitivity by suppressing the ROS-RUNX3 pathway in a rat model of neuropathy. J Cell Physiol. [DOI] [PubMed] [Google Scholar]
- 90.Dusaban SS et al. (2017) Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. Journal of Neuroinflammation 14 (1), 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shirakawa H et al. (2017) Sphingosine-1-phosphate induces Ca(2+) signaling and CXCL1 release via TRPC6 channel in astrocytes. Glia 65 (6), 1005–1016. [DOI] [PubMed] [Google Scholar]
- 92.Kimura A et al. (2007) Essential roles of sphingosine 1-phosphate/S1P1 receptor axis in the migration of neural stem cells toward a site of spinal cord injury. Stem Cells 25 (1), 115–24. [DOI] [PubMed] [Google Scholar]
- 93.Cuzzocrea S et al. (2009) Inhibition of ceramide biosynthesis ameliorates pathological consequences of spinal cord injury. Shock 31 (6), 634–44. [DOI] [PubMed] [Google Scholar]
- 94.Norimatsu Y et al. (2012) FTY720 improves functional recovery after spinal cord injury by primarily nonimmunomodulatory mechanisms. Am J Pathol 180 (4), 1625–35. [DOI] [PubMed] [Google Scholar]
- 95.Yamazaki K et al. (2020) FTY720 attenuates neuropathic pain after spinal cord injury by decreasing systemic and local inflammation in a rat spinal cord compression model. J Neurotrauma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Janes K et al. (2014) The development and maintenance of paclitaxel-induced neuropathic pain require activation of the sphingosine 1-phosphate receptor subtype 1. J Biol Chem 289 (30), 21082–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stockstill K et al. (2018) Dysregulation of sphingolipid metabolism contributes to bortezomib-induced neuropathic pain. J Exp Med 215 (5), 1301–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grenald SA et al. (2017) Targeting the S1P/S1PR1 axis mitigates cancer-induced bone pain and neuroinflammation. Pain 158 (9), 1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Attiori Essis S et al. (2015) GluN2B-containing NMDA receptors are upregulated in plasma membranes by the sphingosine-1-phosphate analog FTY720P. Brain Res 1624, 349–358. [DOI] [PubMed] [Google Scholar]
- 100.Weth-Malsch D et al. (2016) Ablation of Sphingosine 1-Phosphate Receptor Subtype 3 Impairs Hippocampal Neuron Excitability In vitro and Spatial Working Memory In vivo. Front Cell Neurosci 10, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Corbett BF et al. (2019) Sphingosine-1-phosphate receptor 3 in the medial prefrontal cortex promotes stress resilience by reducing inflammatory processes. Nat Commun 10 (1), 3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang S and Chang MC (2019) Chronic Pain: Structural and Functional Changes in Brain Structures and Associated Negative Affective States. Int J Mol Sci 20 (13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zajaczkowska R et al. (2019) Mechanisms of Chemotherapy-Induced Peripheral Neuropathy. Int J Mol Sci 20 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Becker KA et al. (2020) Role of 1-Deoxysphingolipids in docetaxel neurotoxicity. J Neurochem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ogretmen B and Hannun YA (2004) Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 4 (8), 604–16. [DOI] [PubMed] [Google Scholar]
- 106.White C et al. (2016) The emerging role of FTY720 (Fingolimod) in cancer treatment. Oncotarget 7 (17), 23106–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Beider K et al. (2017) The Sphingosine-1-Phosphate Modulator FTY720 Targets Multiple Myeloma via the CXCR4/CXCL12 Pathway. Clin Cancer Res 23 (7), 1733–1747. [DOI] [PubMed] [Google Scholar]
- 108.Doyle T et al. (2012) Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J Neurosci 32 (18), 6149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stockstill K et al. (2019) Sexually dimorphic therapeutic response in bortezomib-induced neuropathic pain reveals altered pain physiology in female rodents. Pain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Choi JW et al. (2011) FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A 108 (2), 751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen YJ et al. (2014) Inflammation triggers production of dimethylsphingosine from oligodendrocytes. Neuroscience 279, 113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Patti GJ et al. (2012) Metabolomics implicates altered sphingolipids in chronic pain of neuropathic origin. Nat Chem Biol 8 (3), 232–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Doolen S et al. (2018) Fingolimod reduces neuropathic pain behaviors in a mouse model of multiple sclerosis by a sphingosine-1 phosphate receptor 1-dependent inhibition of central sensitization in the dorsal horn. Pain 159 (2), 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Coste O et al. (2008) Antinociceptive activity of the S1P-receptor agonist FTY720. J Cell Mol Med 12 (3), 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang DD et al. (2015) Antinociceptive effects of FTY720 during trauma-induced neuropathic pain are mediated by spinal S1P receptors. Biol Chem 396 (6–7), 783–94. [DOI] [PubMed] [Google Scholar]
- 116.Schneider M et al. (2017) Lipids in psychiatric disorders and preventive medicine. Neurosci Biobehav Rev 76 (Pt B), 336–362. [DOI] [PubMed] [Google Scholar]
- 117.Eakin KA et al. (2019) Plasma Sphingolipids Mediate a Relationship Between Type 2 Diabetes and Memory Outcomes in Patients with Coronary Artery Disease Undertaking Exercise. J Alzheimers Dis 69 (3), 717–727. [DOI] [PubMed] [Google Scholar]
- 118.Chua KC et al. (2020) Genomewide Meta-Analysis Validates a Role for S1PR1 in Microtubule Targeting Agent-Induced Sensory Peripheral Neuropathy. Clin Pharmacol Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]