

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a lethal disease and highly resistant to all forms of therapy. PDAC cells reprogram their metabolism extensively to promote their survival and growth. Reflecting the vital role of altered metabolism, experimental and clinical trials targeting the rewired metabolism are currently underway. In this review, we summarize the vital role of metabolic reprogramming in the development of PDAC and the future of novel therapeutic applications.
Keywords: autophagy, macropinocytosis, metabolism, pancreatic cancer, tumor microenvironment
Various types of metabolic reprogramming occur in pancreatic cancer cells, including metabolism of glucose, amino acids, and nutrient acquisition such as autophagy. Increased knowledge of these features can lead to the development of novel therapeutics.
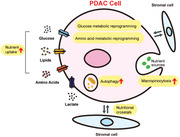
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers, characterized by early metastasis and resistance to all forms of treatment. 1 , 2 The extremely high mortality rate of patients with PDAC can be attributed to the lack of both an early diagnosis and appropriate targeted therapy. 3 Because the pancreas is located in a place difficult to observe, the early diagnosis of PDAC in routine examinations is nearly impossible. 4 Furthermore, current biomarkers are inadequate to detect PDAC efficiently, especially in the early stages. 5
In addition to the diagnostic shortcomings, effective therapeutic options are limited. Although improvements in chemotherapy have been achieved with the emergence of new combination chemotherapies, 6 , 7 the rapid development of chemoresistance usually leads to poor prognosis. Therefore, new treatment strategies are urgently required to improve the prognosis.
Recently, metabolic reprogramming, an emerging hallmark of cancer, 8 has generated renewed interest. Cancer cells rewire their metabolism to promote their growth and proliferation. 9 Based on recent evidence of metabolic adaptation in PDAC cells, 10 , 11 , 12 , 13 , 14 the metabolic features of PDAC could constitute attractive therapeutic opportunities.
In this review, we discuss how PDAC cells alter their metabolism to facilitate growth and how metabolism‐targeted therapies could be used to improve the prognosis of patients with PDAC.
2. GLUCOSE METABOLISM
Glucose is a major nutrient in cellular metabolism and biosynthesis. When used as a nutrient in normal cells, glucose is converted into carbon dioxide in mitochondria via oxidative phosphorylation to produce ATP. By contrast, cancer cells utilize more glucose carbon for anabolic reactions such as synthesis of ribose, amino acids, lipids, and glycosylation precursors. 15
In PDAC cells, oncogenic KRAS upregulates glucose transporter 1 (GLUT1), which increases glucose uptake 10 , 16 (Figure 1). Oncogenic KRAS also upregulates hexokinase 1/2 (HK1/2), phosphofructokinase 1, and lactate dehydrogenase A (LDHA) to promote glycolysis. 10 , 17 Furthermore, the hypoxic microenvironment and other mechanisms cooperate with oncogenic KRAS to increase the expression of glycolytic enzymes and maintain cytosolic ATP. 18 , 19 , 20 , 21 In addition to the transcriptional upregulation of glucose transporters and glycolytic enzymes, KRAS4A interacts with HK1 in mitochondria and regulates HK1 directly. 22
FIGURE 1.
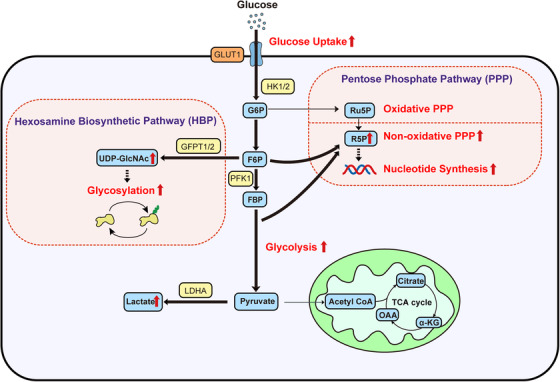
Glucose metabolism in PDAC cells. PDAC cells increase glucose uptake through oncogenic KRAS‐mediated upregulation of GLUT1. Oncogenic KRAS also upregulates other glycolytic enzymes, resulting in increased glycolytic flux. Glucose carbon is also important for anabolic metabolism in the HBP and the nonoxidative PPP phase. Bold arrows indicate accelerated metabolic pathways. α‐KG, α‐ketoglutarate; FBP, fructose 1,6‐bisphosphate; F6P, fructose 6‐phosphate; G6P, glucose 6‐phosphate; GLUT1, glucose transporter 1; GFPT1/2, glutamine fructose 6‐phosphate transamidase 1/2; HBP, hexosamine biosynthetic pathway; HK1/2, hexokinase 1/2; LDHA, lactate dehydrogenase A; OAA, oxaloacetate; PDAC, pancreatic ductal adenocarcinoma; PFK1, phosphofructokinase 1; R5P, ribose 5‐phosphate; Ru5P, ribulose 5‐phosphate; UDP‐GlcNAc, UDP‐N‐acetylglucosamine
Glucose also plays a crucial role in the anabolic pathway. Oncogenic KRAS activates the hexosamine biosynthetic pathway (HBP), 10 , 23 producing uridine diphosphate‐N‐acetylglucosamine, which has numerous functions, including intracellular signaling and posttranslational modification. 24 Oncogenic KRAS increases the HBP flux via the transcriptional upregulation of glutamine fructose 6‐phosphate transamidase 1 (GFPT1). 10 HBP flux is also increased through GFPT2, which is induced by hypoxia. 25
The pentose phosphate pathway (PPP) is another anabolic pathway through which oncogenic KRAS increases the glucose flux. This pathway is important for producing nucleotide synthesis intermediates and is subdivided into the oxidative and nonoxidative phases. In the oxidative phase, glucose 6‐phospate is converted into ribulose 5‐phosphate and two molecules of NADPH are produced simultaneously. Then, NADPH is used for redox control and fatty acid synthesis. The nonoxidative PPP phase consists of reactions that produce ribose 5‐phophate (R5P) for nucleotide synthesis. In a previous study, oncogenic KRAS in PDAC cells became dependent on the nonoxidative PPP phase 10 through MYC upregulation. 26 MUC1 also helps induce anabolic glucose metabolism by stabilizing HIF1α. 27 , 28 , 29 , 30 Because normal cells produce R5P in the oxidative phase, this differential reliance on the nonoxidative phase could be a metabolic vulnerability of pancreatic cancer. These glycolytic changes begin at the time of precancerous lesions and are maintained during tumor progression. 31
3. AMINO ACID METABOLISM
PDAC cells also reprogram amino acid metabolism. Various amino acid transporters are upregulated markedly in PDAC cells to meet the metabolic demand. 32 , 33 The amino acid glutamine (Gln) is important for tumor cell as a major source of carbon and nitrogen, contributing to biosynthesis of macromolecules 34 (Figure 2).
FIGURE 2.
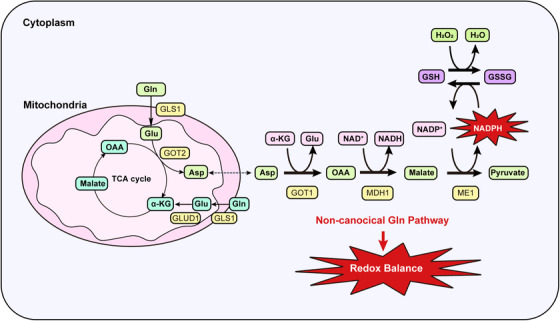
Glutamine metabolism in PDAC cells. PDAC cells depend on the noncanonical Gln pathway for redox balance. Gln‐derived Glu is metabolized to Asp by GOT2. This Asp is transported into the cytoplasm and then metabolized to OAA by GOT1. This OAA is metabolized to malate and then pyruvate, increasing the NADPH/NADP+ ratio. This sustains the reduced GSH levels needed for redox balance. α‐KG, α‐ketoglutarate; Asp, aspartate; GOT1, aspartate aminotransferase 1; GOT2, aspartate aminotransferase 2; Glu, glutamate; GLUD1, glutamate dehydrogenase 1; GLS1, glutaminase 1; Gln, glutamine; GSH, glutathione; MDH1, malate dehydrogenase 1; ME1, malic enzyme 1; OAA, oxaloacetate; GSSG, oxidized glutathione; PDAC, pancreatic ductal adenocarcinoma
Gln‐derived carbon continuously replenishes the tricarboxylic acid (TCA) cycle to produce reducing equivalents and intermediates for macromolecular synthesis. In the canonical Gln pathway, Gln‐derived glutamate (Glu) is converted into α‐ketoglutarate (α‐KG), replenishing the TCA cycle. In this process, NADH and the precursors of macromolecules and lipids are produced. Gln is also an important source of purines and pyrimidines. 35 , 36 Gln itself and Gln‐derived aspartate (Asp) can be used as substrates for nucleotide synthesis. 37
In addition, PDAC cells utilize Gln to maintain redox homeostasis. 11 Gln plays two roles in this process. First, Gln is a resource for the synthesis of glutathione, a tripeptide (composed of Glu, cysteine, and glycine) that protects cells from free radical damage by acting as an antioxidant. Second, oncogenic KRAS promotes the production of reducing equivalents in the form of NADPH via a noncanonical Gln metabolism pathway. Gln‐derived Glu is converted into Asp by aspartate aminotransferase (GOT2) in mitochondria. This Asp is transported into the cytoplasm and then metabolized by cytosolic aspartate aminotransferase (GOT1), malate dehydrogenase, and malic enzyme (ME1), which results in the production of reducing potential in the form of NADPH. GOT1 also plays an important role in an acidic tumor microenvironment. 38 PDAC cells can maintain the redox balance under acidosis stress by increasing anaplerotic Gln metabolism.
In addition to the role of KRAS in metabolic changes, p53 also plays an important role in rewiring Gln and glucose metabolism to accumulate α‐KG. 39 Accumulated α‐KG undergoes chromatin modification and exerts a p53‐mediated tumor suppressor effect.
The metabolism of other amino acids is reprogrammed in PDAC cells. Proline derived from the extracellular matrix (ECM) promotes PDAC cell survival under nutrient‐limited conditions. 40 Through macropinocytosis‐dependent and macropinocytosis‐independent mechanisms, PDAC cells take up ECM collagens to replenish TCA cycle when other fuels are limited. Cysteine is also important for supporting PDAC cell survival through the maintenance of nutritional and oxidative homeostasis. 41 The cystine/glutamate exchanger xCT is important for maintaining cysteine balance and may be a promising therapeutic target. Furthermore, elevation of plasma branched‐chain amino acids is associated with a future diagnosis of PDAC, which might be due to increased breakdown of tissue protein. 42 , 43
4. LIPID METABOLISM
Lipid metabolism is also important for PDAC progression. 44 In PDAC cells, cholesterol uptake and many enzymes associated with fatty acids and cholesterol synthesis are significantly upregulated. 45 , 46 A recent study showed that sterol O‐acyltransferase 1 (SOAT1) promotes the mevalonate pathway, preventing cholesterol feedback inhibition by unesterified cholesterol in PDAC cells with a p53 mutation and loss of heterozygosity. 47 By contrast, inhibition of SOAT1 does not affect the growth of normal cells with wild‐type p53, suggesting a potential therapeutic window.
5. NUTRIENT ACQUISITION
PDAC cells are poorly vascularized and in a state of nutrient deprivation. 48 , 49 Thus, PDAC cells have alternative mechanisms by which they acquire nutrients needed to survive and grow. To acquire sufficient fuel, PDAC cells activate mechanisms, such as autophagy, macropinocytosis, and metabolic interaction with surrounding noncancerous cells within the tumor microenvironment (Figure 3).
FIGURE 3.
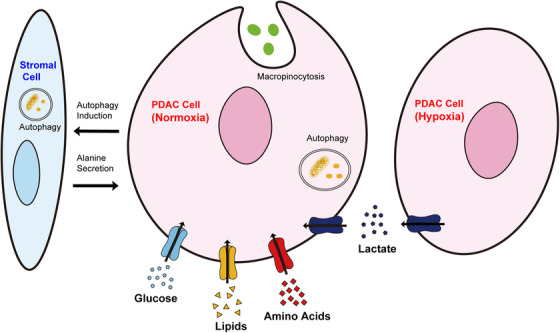
Nutrient acquisition strategies utilized by PDAC cells. PDAC cells acquire nutrients in various ways. The uptake of glucose, amino acids, and lipids is enhanced in PDAC cells. Macropinocytosis and autophagy are also promoted. PDAC cells undergo metabolic crosstalk with stromal cells. PDAC cells stimulate autophagy in stromal cells, inducing alanine secretion. Metabolic crosstalk also occurs among cancer cells. Lactate secreted by PDAC cells in the hypoxic region is taken up by PDAC cells in the normoxic region. These mechanisms cooperate to promote PDAC growth. PDAC, pancreatic ductal adenocarcinoma
5.1. Autophagy
Autophagy is a degradative process involving the formation of autophagosomes that swallow intracellular components for delivery to lysosomes. 50 The engulfed components are degraded by fusion of the autophagosome and lysosome, and the digested biomolecules are recycled as cellular nutrients 51 (Figure 3). Autophagy is regulated by highly controlled signaling events occurring at a basal level and is triggered by diverse signals. 52
In the progression of PDAC, autophagy plays critical but opposing roles. 53 At the tumor initiation stage, autophagy can be tumor suppressive via cellular quality control mechanism. In an established tumor, autophagy can be tumor promoting via an intracellular recycling mechanism, indicating that autophagy is essential for PDAC tumor growth. 54 The inhibition of autophagy in genetically engineered mouse models suppressed the progression of PDAC. 55 , 56 , 57
The autophagic reliance of PDAC was recently shown to be partly mediated by MiT/TFE family of transcriptional factors. 58 In PDAC, microphthalmia/transcription factor E (MiT/TFE) proteins can activate basal autophagy independently of mechanistic target of rapamycin (mTOR) activity and are necessary for the maintenance of amino acid pools. Furthermore, an ULK1 phosphatase (PP2A‐B55α complex), which activates autophagy, was also shown to be crucial for high levels of basal autophagy in PDAC. 59
5.2. Macropinocytosis
Although autophagy plays an important role in metabolic scavenging, it cannot create a net increase in biomass. PDAC cells are also dependent on another pathway to satisfy their metabolic needs. Macropinocytosis is an endocytic process that involves the nonspecific uptake of extracellular material through large, heterogeneous vesicles known as macropinosomes. 60 The internalized molecules undergo lysosomal degradation, yielding precursor molecules that can be used for macromolecular biosynthesis.
In PDAC cells, oncogenic KRAS promotes macropinocytosis to acquire protein sources to survive in nutrient‐poor conditions by replenishing amino acid pools 48 , 49 , 61 , 62 , 63 , 64 (Figure 3). Enhanced macropinocytosis might enhance the delivery of nanomedicines, such as nanoparticle albumin‐bound (nab)‐paclitaxel, and partly explain the efficacy of the drug. 7 , 65
5.3. Metabolic crosstalk with the microenvironment
The molecular and cellular heterogeneity of PDAC is well characterized. 66 , 67 , 68 , 69 Metabolic heterogeneity also exists in cancer cells, 9 , 70 , 71 , 72 which depends on differences in cell state. For instance, pancreatic cancer stem cells are mainly dependent on oxidative phosphorylation, but have metabolic plasticity leading to resistance of mitochondrial inhibition. 73 MYC and PGC‐1α cooperatively determine such plasticity. In addition, environmental factors, such as local nutrient and oxygen status, affect metabolic heterogeneity.
Accordingly, metabolically different cell populations utilize cross‐feeding mechanisms in which one population can use metabolites from another (Figure 3). A recent study showed that cancer cells were shown to use lactate as a substrate for the TCA cycle. 74 Lactate secreted by cancer cells in the hypoxic region is captured by cancer cells in the normoxic region and feeds cancer cells. 25 PDAC cells upregulate the expression of monocarboxylate transporter 1 (MCT1) and MCT4 to promote lactate exchange. 75 , 76 Lactate secreted by cancer cells is also captured by mesenchymal stem cells and converted into α‐KG, causing extensive epigenetic changes and promoting cancer‐associated fibroblast differentiation. 77
Metabolites derived from stromal cells also feed cancer cells. A recent study showed that stroma‐associated pancreatic stellate cells (PSCs) had an important role in PDAC proliferation via the release of free amino acids, especially alanine. 78 PDAC cells activate autophagy in PSCs and selectively utilize the released alanine, fueling the TCA cycle.
5.4. Preclinical and clinical trials targeting cancer metabolism and conclusion
Although metabolism‐targeted therapy is not yet standard therapy for many cancers, several experimental and clinical trials targeting altered metabolism are currently underway.
In vitro and in preclinical mouse models, WZB117, a specific GLUT1 inhibitor, was shown to be a promising agent targeting cancer stem cells. 79 In preclinical mouse models, FX11, a LDHA inhibitor, was also shown to suppress tumor growth. 80 , 81 In a phase I clinical trial, 2‐deoxy‐D‐glucose in combination with docetaxel was shown to be feasible with clinical benefit (NCT00096707). 82 For mitochondrial metabolism, the mitochondrial inhibitor phenformin showed antitumor effects in PDAC xenograft models. 83 For altered Gln metabolism, GLS1 inhibitors and β‐lapachone (ARQ761) selectively induced PDAC cell death in vivo. 84 A phase I clinical trial using ARQ761 in combination with gemcitabine/nab‐paclitaxel in PDAC is currently underway (NCT02514031). For lipid metabolism, SB‐204990, an ATP citrate lyase inhibitor, reduced tumor growth in vivo. 85
In addition, the autophagy inhibitor hydroxychloroquine is under clinical evaluation. Hydroxychloroquine alone has shown limited efficacy, 86 , 87 but was clinically beneficial in combination with gemcitabine/nab‐paclitaxel (NCT01978184). In a recent study, dual inhibition of the RAF‐MEK‐ERK pathway and autophagy synergistically impaired PDAC proliferation. 88 , 89 Because direct RAS inhibitors, previously thought to be undruggable, and more specific autophagy inhibitors are under development; their use could potentially enhance the therapeutic effect. 90 , 91
Although attempts to treat PDAC by targeting tumor metabolism have succeeded to some extent, the development of resistance is a concern when designing metabolism‐targeted therapy. Metabolic networks are very plastic and reportedly can be rewired to avoid targeted therapies. 92 , 93 In addition, PDAC cells have heterogeneous metabolic subtypes. 70 , 71 Different metabolic subtypes have different prognoses and metabolic vulnerability. 94 , 95 Therefore, these factors should be considered when designing effective metabolism‐targeted therapy. In the near future, novel treatment strategies based on the findings of PDAC metabolism are expected to improve the prognosis of patients with PDAC.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
TS, MO, and KK wrote the manuscript. TS, TI, and TK prepared the figures.
ACKNOWLEDGMENTS
This work was supported by Grants‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (#19H03430) (to MO), by JST CREST (to MO, #JPMJCR19H5), and by the Practical Research for Innovative Cancer Control Program from AMED (to MO, #JP20ck0106557).
Suzuki T, Otsuka M, Seimiya T, Iwata T, Kishikawa T, Koike K. The biological role of metabolic reprogramming in pancreatic cancer. MedComm. 2020;1:302–310. 10.1002/mco2.37
REFERENCES
- 1. Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Notta F, Chan‐Seng‐Yue M, Lemire M, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature. 2016;538:378‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7‐34. [DOI] [PubMed] [Google Scholar]
- 4. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039‐1049. [DOI] [PubMed] [Google Scholar]
- 5. Singhi AD, Koay EJ, Chari ST, Maitra A. Early detection of pancreatic cancer: opportunities and challenges. Gastroenterology. 2019;156:2024‐2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817‐1825. [DOI] [PubMed] [Google Scholar]
- 7. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab‐paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691‐1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 9. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368:eaaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS‐regulated metabolic pathway. Nature. 2013;496:101‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qin C, Yang G, Yang J, et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol Cancer. 2020;19:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biancur DE, Kimmelman AC. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochim Biophys Acta Rev Cancer. 2018;1870:67‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Halbrook CJ, Lyssiotis CA. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell. 2017;31:5‐19. [DOI] [PubMed] [Google Scholar]
- 15. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441‐464. [DOI] [PubMed] [Google Scholar]
- 16. Zhang C, Liu J, Liang Y, et al. Tumour‐associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gaglio D, Metallo CM, Gameiro PA, et al. Oncogenic K‐Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chaika NV, Gebregiworgis T, Lewallen ME, et al. MUC1 mucin stabilizes and activates hypoxia‐inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci USA. 2012;109:13787‐13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baek G, Tse YF, Hu Z, et al. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014;9:2233‐2249. [DOI] [PubMed] [Google Scholar]
- 20. Shi M, Cui J, Du J, et al. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin Cancer Res. 2014;20:4370‐4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cui J, Shi M, Xie D, et al. FOXM1 promotes the Warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin Cancer Res. 2014;20:2595‐2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Amendola CR, Mahaffey JP, Parker SJ, et al. KRAS4A directly regulates hexokinase 1. Nature. 2019;576:482‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ma Z, Vocadlo DJ, Vosseller K. Hyper‐O‐GlcNAcylation is anti‐apoptotic and maintains constitutive NF‐kappaB activity in pancreatic cancer cells. J Biol Chem. 2013;288:15121‐15130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Slawson C, Hart GW. O‐GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer. 2011;11:678‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guillaumond F, Leca J, Olivares O, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 2013;110:3919‐3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Santana‐Codina N, Roeth AA, Zhang Y, et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun. 2018;9:4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gunda V, Souchek J, Abrego J, et al. MUC1‐mediated metabolic alterations regulate response to radiotherapy in pancreatic cancer. Clin Cancer Res. 2017;23:5881‐5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shukla SK, Purohit V, Mehla K, et al. MUC1 and HIF‐1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32:71‐87.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gebregiworgis T, Purohit V, Shukla SK, et al. Glucose limitation alters glutamine metabolism in MUC1‐overexpressing pancreatic cancer cells. J Proteome Res. 2017;16:3536‐3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olou AA, King RJ, Yu F, Singh PK. MUC1 oncoprotein mitigates ER stress via CDA‐mediated reprogramming of pyrimidine metabolism. Oncogene. 2020;39:3381‐3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vernucci E, Abrego J, Gunda V, et al. Metabolic alterations in pancreatic cancer progression. Cancers. 2019;12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kaira K, Sunose Y, Arakawa K, et al. Prognostic significance of L‐type amino‐acid transporter 1 expression in surgically resected pancreatic cancer. Br J Cancer. 2012;107:632‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Coothankandaswamy V, Cao S, Xu Y, et al. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br J Pharmacol. 2016;173:3292‐3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678‐3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017;36:1302‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abrego J, Gunda V, Vernucci E, et al. GOT1‐mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. 2017;400:37‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. JPt Morris, Yashinskie JJ, Koche R, et al. alpha‐ketoglutarate links p53 to cell fate during tumour suppression. Nature. 2019;573:595‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Olivares O, Mayers JR, Gouirand V, et al. Collagen‐derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun. 2017;8:16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Daher B, Parks SK, Durivault J, et al. Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res. 2019;79:3877‐3890. [DOI] [PubMed] [Google Scholar]
- 42. Mayers JR, Wu C, Clish CB, et al. Elevation of circulating branched‐chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med. 2014;20:1193‐1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Katagiri R, Goto A, Nakagawa T, et al. Increased levels of branched‐chain amino acid associated with increased risk of pancreatic cancer in a prospective case‐control study of a large cohort. Gastroenterology. 2018;155:1474‐1482.e1. [DOI] [PubMed] [Google Scholar]
- 44. Sunami Y, Rebelo A, Kleeff J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancer. 2017;10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Swierczynski J, Hebanowska A, Sledzinski T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J Gastroenterol. 2014;20:2279‐2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guillaumond F, Bidaut G, Ouaissi M, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 2015;112:2473‐2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Oni TE, Biffi G, Baker LA, et al. SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. J Exp Med. 2020;217:e20192389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Commisso C, Davidson SM, Soydaner‐Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras‐transformed cells. Nature. 2013;497:633‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kamphorst JJ, Nofal M, Commisso C, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75:544‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107‐132. [DOI] [PubMed] [Google Scholar]
- 51. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461‐472. [DOI] [PubMed] [Google Scholar]
- 52. Towers CG, Thorburn A. Therapeutic targeting of autophagy. EBioMedicine. 2016;14:15‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25:1999‐2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rosenfeldt MT, O'Prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296‐300. [DOI] [PubMed] [Google Scholar]
- 56. Yang A, Rajeshkumar NV, Wang X, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4:905‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang A, Herter‐Sprie G, Zhang H, et al. Autophagy sustains pancreatic cancer growth through both cell‐autonomous and nonautonomous mechanisms. Cancer Discov. 2018;8:276‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Perera RM, Stoykova S, Nicolay BN, et al. Transcriptional control of autophagy‐lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wong P‐M, Feng Y, Wang J, Shi R, Jiang X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun. 2015;6:8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bar‐Sagi D, Feramisco JR. Induction of membrane ruffling and fluid‐phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 1986;233:1061‐1068. [DOI] [PubMed] [Google Scholar]
- 61. Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell. 2015;162:259‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davidson SM, Jonas O, Keibler MA, et al. Direct evidence for cancer‐cell‐autonomous extracellular protein catabolism in pancreatic tumors. Nat Med. 2017;23:235‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yao W, Rose JL, Wang W, et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature. 2019;568:410‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ramirez C, Hauser AD, Vucic EA, Bar‐Sagi D. Plasma membrane V‐ATPase controls oncogenic RAS‐induced macropinocytosis. Nature. 2019;576:477‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bailey JM, Alsina J, Rasheed ZA, et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology. 2014;146:245‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Waddell N, Pajic M, Patch A‐M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47‐52. [DOI] [PubMed] [Google Scholar]
- 70. Birsoy K, Possemato R, Lorbeer FK, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Daemen A, Peterson D, Sahu N, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci USA. 2015;112:E4410‐4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim J, DeBerardinis RJ. Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metab. 2019;30:434‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sancho P, Burgos‐Ramos E, Tavera A, et al. MYC/PGC‐1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22:590‐605. [DOI] [PubMed] [Google Scholar]
- 74. Hui S, Ghergurovich JM, Morscher RJ, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kong SC, Nøhr‐Nielsen A, Zeeberg K, et al. Monocarboxylate transporters MCT1 and MCT4 regulate migration and invasion of pancreatic ductal adenocarcinoma cells. Pancreas. 2016;45:1036‐1047. [DOI] [PubMed] [Google Scholar]
- 76. Wu DH, Liang H, Lu SN, et al. miR‐124 suppresses pancreatic ductal adenocarcinoma growth by regulating monocarboxylate transporter 1‐mediated cancer lactate metabolism. Cell Physiol Biochem. 2018;50:924‐935. [DOI] [PubMed] [Google Scholar]
- 77. Bhagat TD, Von Ahrens D, Dawlaty M, et al. Lactate‐mediated epigenetic reprogramming regulates formation of human pancreatic cancer‐associated fibroblasts. eLife. 2019;8:e50663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sousa CM, Biancur DE, Wang X, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shibuya K, Okada M, Suzuki S, et al. Targeting the facilitative glucose transporter GLUT1 inhibits the self‐renewal and tumor‐initiating capacity of cancer stem cells. Oncotarget. 2015;6:651‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Le A, Cooper CR, Gouw AM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA. 2010;107:2037‐2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rajeshkumar NV, Dutta P, Yabuuchi S, et al. Therapeutic targeting of the Warburg effect in pancreatic cancer relies on an absence of p53 function. Cancer Res. 2015;75:3355‐3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Raez LE, Papadopoulos K, Ricart AD, et al. A phase I dose‐escalation trial of 2‐deoxy‐D‐glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71:523‐530. [DOI] [PubMed] [Google Scholar]
- 83. Rajeshkumar NV, Yabuuchi S, Pai SG, et al. Treatment of pancreatic cancer patient‐derived xenograft panel with metabolic inhibitors reveals efficacy of phenformin. Clin Cancer Res. 2017;23:5639‐5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chakrabarti G, Moore ZR, Luo X, et al. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP‐driven metabolic catastrophe induced by ß‐lapachone. Cancer Metab. 2015;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hatzivassiliou G, Zhao F, Bauer DE, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311‐321. [DOI] [PubMed] [Google Scholar]
- 86. Wolpin BM, Rubinson DA, Wang X, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist. 2014;19:637‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Boone BA, Bahary N, Zureikat AH, et al. Safety and biologic response of pre‐operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol. 2015;22:4402‐4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bryant KL, Stalnecker CA, Zeitouni D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25:628‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kinsey CG, Camolotto SA, Boespflug AM, et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS‐driven cancers. Nat Med. 2019;25:620‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS‐targeted therapies: is the undruggable drugged. Nat Rev Drug Discov. 2020;19:533‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Janes MR, Zhang J, Li LS, et al. Targeting KRAS mutant cancers with a covalent G12C‐specific inhibitor. Cell. 2018;172:578‐589.e17. [DOI] [PubMed] [Google Scholar]
- 92. Boudreau A, Purkey HE, Hitz A, et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol. 2016;12:779‐786. [DOI] [PubMed] [Google Scholar]
- 93. Davidson SM, Papagiannakopoulos T, Olenchock BA, et al. Environment impacts the metabolic dependencies of Ras‐driven non‐small cell lung cancer. Cell Metab. 2016;23:517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mehla K, Singh PK. Metabolic subtyping for novel personalized therapies against pancreatic cancer. Clin Cancer Res. 2020;26:6‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Karasinska JM, Topham JT, Kalloger SE, et al. Altered gene expression along the glycolysis‐cholesterol synthesis axis is associated with outcome in pancreatic cancer. Clin Cancer Res. 2020;26:135‐146. [DOI] [PubMed] [Google Scholar]