

Abstract
Liver cancer is the third leading cause of cancer‐related deaths throughout the world, and more than 0.6 million people die from liver cancer annually. Therefore, novel therapeutic strategies to eliminate malignant cells from liver cancer patients are urgently needed. Recent advances in high‐throughput genomic technologies have identified de novo candidates for oncogenes and pharmacological targets. However, testing and understanding the mechanism of oncogenic transformation as well as probing the kinetics and therapeutic responses of spontaneous tumors in an intact microenvironment require in vivo examination using genetically modified animal models. The zebrafish (Danio rerio) has attracted increasing attention as a new model for studying cancer biology since the organs in the model are strikingly similar to human organs and the model can be genetically modified in a short time and at a low cost. This review summarizes the current knowledge of epidemiological data and genetic alterations in hepatocellular carcinoma (HCC), zebrafish models of HCC, and potential therapeutic strategies for targeting HCC based on knowledge from the models.
Keywords: epidemiology of HCC, genetic alterations in HCC, hepatocellular carcinoma (HCC), therapeutic strategies for HCC, zebrafish
Liver tumors that developed in zebrafish are highly analogous to liver tumors in humans, and several transgenic zebrafish lines for modeling hepatocellular carcinoma (HCC) have been established. This review summarizes the current knowledge of epidemiological data and genetic alterations in HCC, zebrafish models of HCC, and potential therapeutic strategies for targeting HCC based on knowledge from the models.
1. INTRODUCTION
Liver cancer is the fifth most frequent cancer in the world. Despite aggressive therapy, the 5‐year survival rate of liver cancer patients remains low (below 12% in the United States). 1 Cancer is the third leading cause of death throughout the world and more than 0.6 million people die from liver cancer annually. 2 Liver cancer mortality is also due to its resistance to existing anticancer regents; therefore, new therapeutic strategies to eliminate malignant cells from liver cancer patients are urgently needed.
Recent advances in high‐throughput genomic technologies have made it possible to identify de novo oncogene candidates and pharmacological targets through analyzing large numbers of samples from cancer patients. Whole‐genome sequencing analysis of clinical HCC samples has revealed that etiological background influences somatic mutation patterns and the subsequent carcinogenesis of HCC. 3 Another genomic high‐resolution, copy‐number, and whole‐exon sequencing analysis has identified 135 homozygous deletions and 994 somatic mutations and predicted functional consequences from HCC samples. 4 However, determining whether identified genetic alterations truly confer malignant transformation of normal cells requires in vivo examinations of genetically modified animals with designed gene mutations that mimic human data. Since the mid‐1980s, a tremendous knowledge of cancer‐related genes has been accumulated by investigating genetically modified mice that either harbored an oncogene or lacked a tumor suppressor gene. These in vivo models have demonstrated transformations of normal cells into malignant cells by both gain‐ or loss‐of‐function cancer‐related genes, thus providing fundamental knowledge about the molecular malignant transformation mechanisms induced by oncogenes, as well as the kinetics and therapeutic responses of spontaneous tumors in intact tumor microenvironments. 5 The mouse model has several disadvantages despite these findings, including the high cost of animal husbandry, the difficulty of obtaining a large number of lab animals, and the labor intensity and time required to establish and perform in vivo assays, which prevent rapid and high‐throughput studies.
Zebrafish (Danio rerio) as a cancer model offers unique advantages. 6 , 7 , 8 For example, (a) zebrafish have orthologues for 86% of 1318 human drug targets, 71% of human proteins, and 82% of disease‐causing human proteins 9 , 10 ; (b) drugs are administered to zebrafish by just dissolving the drugs in water where zebrafish are maintained; (c) the transparency of zebrafish embryos enables to observe directly some processes during tumor development and regression 11 ; (d) zebrafish generate large numbers of progeny (100‐200 eggs per one female zebrafish), that is effective in statistical analyses; and (e) cost of maintaining zebrafish is low.
In the past few years, we have been studying liver cancers in zebrafish both via chemical carcinogens and transgenic approaches. 12 , 13 , 14 Comparative transcriptomic analyses have revealed high conservation of molecular signatures and progression between zebrafish and human liver tumors. 15 In this review, we summarized the epidemiological data and genetic alternations of human liver cancer, the current knowledge from zebrafish HCC analyses, and the potential of developing therapeutic strategies for the treatment of HCC.
2. EPIDEMIOLOGICAL STUDIES OF LIVER CANCER
Hepatocellular carcinoma (HCC) is the most common type of adult liver cancer and represents over 90% of all cases of primary liver cancer. 16 The majority of HCC cases result from infection with chronic hepatitis B virus (HBV) (54%) and hepatitis C virus (HCV) (31%). However, substantial geographic variation in these data exists. In Africa and Asia, more than 60% of HCC patients are HBV carriers, 20% are HCV carriers, and the remaining 20% cases are resultant from alcohol abuse or dietary exposure to aflatoxin B1. In contrast, in the United States, Europe, Egypt, and Japan, the rate of HCV and HBV carriers in HCC patients is more than 60% and 20%, respectively, and the remaining cases are caused by excessive alcohol consumption or metabolic liver diseases. 17
Clinical studies have revealed that HCC harbors a large number of clonally integrated HBV genomes and that the incidence of HCC development is proportional to serum HBV DNA level. 18 , 19 , 20 Experimental studies in transgenic mice expressing the HBV component protein demonstrated that HCC developed spontaneously. 21 , 22 , 23 Clinical studies indicate a strong association between HCV infection and HCC development. 24 Experimentally, transgenic mice expressing HCV core proteins also develop HCC. 25
Alcohol consumption is an avoidable risk factor for HCC development. Clinical studies have revealed that the risk leading to HCC development increases when daily alcohol consumption exceeds 80 grams per day for more than 10 years. 26 Ethanol is absorbed by the small intestine and metabolized mainly in the liver. Alcohol dehydrogenase oxidizes ethanol to acetaldehyde, which is converted into acetate by aldehyde dehydrogenase. When alcohol consumption is high, cytochrome P450 2E1 (CYP2E1) also catalyzes ethanol into acetaldehyde but produces reactive oxygen species (ROS). Acetaldehyde and ROS function as carcinogens and interfere with DNA synthesis and repair. 27
Dietary exposure to aflatoxin B1 is another major risk factor for HCC development. Aflatoxin B1 is a genotoxic hepatocarcinogen that causes cellular transformation by inducing DNA adducts, leading to genetic alternations in target cells. Aflatoxin B1 is metabolized by cytochrome‐P450 enzymes into aflatoxin B1‐8, 9‐epoxide, a reactive intermediate, which then binds to genomic DNA in liver cells and results in DNA adducts. 28 In geographic areas with high exposure to dietary aflatoxin B1, such as China and Africa, a transversion of guanine to thymine at the third position of codon 249 serine in TP53 is often observed. 29 , 30 , 31 Experimental studies by exposing human HCC cell lines to aflatoxin B1 have also induced similar transversion in codon 249 of TP53. 32 , 33
3. MOLECULAR PATHOGENESIS OF HCC
Studies over the past four decades have demonstrated that cancer cells develop by accumulating numerous genetic mutations, which leads to the overexpression of oncogenes and/or loss of tumor suppressor genes; these mutations confer proliferation, survival, and drug resistance to cancer cells. 34 , 35 These mutations are also observed in the development and progression of HCC, 36 , 37 as summarized in Table 1. This section describes the genetic alternations that are most frequently observed in HCC.
TABLE 1.
A list of genetic mutations which are observed in HCC
| Gene | Function of gene product | Cause of genetic alteration | Frequency | Reference |
|---|---|---|---|---|
| APC | Signaling suppressor | Loss of function mutations of APC gene | 3% | 15 |
| AXIN1 | Signaling suppressor | Loss of function mutations of AXIN gene | 5% | 60 |
| AXIN2 | Signaling suppressor | Somatic mutation at codon 688 of AXIN2 gene | 2% | 43 |
| BRAF | Kinase | Somatic mutation at codon 600 of B‐raf gene | 15% | 130 |
| BRCA2 | DNA repair regulator | Chromosomal deletion in 13q | 43% | 45 |
| C‐MET | Tyrosine kinase receptor | Overexpression | 69% | 131 |
| C‐MYC | Transcriptional factor | Chromosomal amplification in 8q | 48% | 95 |
| CCND1 | Cyclin‐dependent kinase | Chromosomal amplification in 11q13 | 13% | 132 |
| CDKN2A | Cyclin‐dependent kinase inhibitor | Decreased expression through promoter methylation | 62% | 133 |
| CTNNB1 | Transcriptional factor | Somatic mutation | 20‐40% | 61 |
| EGFR | G‐protein coupled receptor | Overexpression | 53% | 75 |
| ERBB3 | G‐protein coupled receptor | Overexpression | 63% | 75 |
| FLT1 | Tyrosine kinase receptor | Overexpression | 50% | 134 |
| HBV (DNA virus) | Virus proteins | HBV integration | 54‐60% | 17 |
| HCV (RNA virus) | Virus proteins | HCV integration | 20‐31% | 17 |
| IGF2 | Growth Factor | Elevated expression | 14% | 135 |
| JAK1 | Kinase | Somatic mutations | 9% | 136 |
| KDR2 | Tyrosine kinase receptor | Overexpression | 86% | 137 |
| KRAS | GTPase | Somatic mutation at codon 12 of k‐ras | 3% | 90 |
| PDGFRA | Tyrosine kinase receptor | Overexpression | 63% | 138 |
| PDGFRB | Tyrosine kinase receptor | Overexpression | 19% | 137 |
| PTEN | Tyrosine phosphatase | Chromosomal deletion in 10q23 | 44% | 139 |
| RB1 | Cell cycle regulator | Chromosomal deletion in 13q | 43% | 45 |
| SATB1 | Genomic organizer | Ecotopic expression | 57% | 140 |
| SOCS1 | Signaling suppressor | Decreased expression through promoter methylation | 65% | 141 |
| SOCS3 | Signaling suppressor | Decreased expression through promoter methylation | 33% | 142 |
| TGFA | Growth Factor | Elevated level in urine | 65% | 143 |
| TP53 | Transcriptional factor | Somatic mutation at codon 249 of p53 gene | 23% | 144 |
| VEGFA | Growth Factor | Chromosomal amplification in 6p21 | 4‐6% | 145 |
3.1. HBV and HCC
As mentioned above, the leading cause of HCC is HBV. This DNA virus is a noncytopathic, partially double‐stranded, and hepatotropic. The HBV DNA encodes reverse transcriptase/DNA polymerase (pol), the capsid protein known as hepatitis B core antigen, envelope proteins (L, M, and S), and protein X (HBx) (whose functions are not fully understood). 38
Comparative genomic hybridization and loss of heterozygosity analyses of HCC samples have revealed chromosomal instability in more than 80% of HBV‐associated tumors, including gains of chromosomes 1q, 5, 6q, 7q, 8q, 17q, and 20 and loss of chromosomes 1p, 4q, 6q, 8p, 13q, 16, 17p, and 21. 4 , 39 , 40 , 41 Representative oncogenes such as CCNA1 and Myc lie on 4q and 8p, respectively, whereas representative tumor suppressor genes such as RB1, TP53, AXIN1, and CDH1 lie on 13q, 17p, 16p, and 16q, respectively. 42 , 43 , 44 , 45
Experimental studies have demonstrated that transgenic mice expressing the HBx gene specifically succumb to progressive histopathological changes in the liver, initially with multifocal areas of altered hepatocytes, followed by the appearance of benign adenomas, and then the development of malignant carcinomas. 22 In contrast, transgenic mice producing only HBV L envelope protein uniformly develop HCC. 21 Other transgenic mice expressing HBsAg develop necroinflammatory liver disease, which progresses into HCC. 23 These observations suggest that HBV‐driven proteins themselves are sufficient to cause the malignant transformation of hepatic cells. In cooperation with their transformation ability, the integration of HBV DNA into the host genome in hepatic cells accelerates hepatic carcinogenesis by inducing genomic instability in hepatic cells. This results in the elevated expression of oncogenes and decreased expression of tumor suppressor genes.
3.2. HCV and HCC
The second leading cause of HCC, HCV, is a completely cytoplasmic replicating RNA virus that maintains its genome as an episome associated with the endoplasmic reticulum. The HCV‐positive stranded RNA genome encodes the core protein, envelope proteins (E1 and E2), and nonstructural proteins (NS2, NS34, NS4A, NS5A, and NS5B) for viral replication, assembly, and maturation. Among these viral components, the HCV core proteins, E1, and E2 induce HCC in transgenic mice by overexpression. 25 , 38 , 46 , 47
The HCV core protein is reported to suppress the host immune responses. It inhibits Fas/TNF‐α‐mediated apoptosis in HCV‐infected hepatocytes by binding to the cytoplasmic domains of TNFR1 and lymphotoxin b receptor; it suppresses antiviral cytotoxic T‐lymphocyte responses through its interactions with the gC1q receptor. 48 , 49 , 50 , 51 Interference with the host immune responses results in infected hepatocyte survival, which promotes persistent HCV infection. Although transgenic mice expressing the HCV core protein developed HCC, the transgenic mice did not show impaired immune responses to adenovirus infections. 25 , 52 It has also been reported that transgenic mice expressing both E1 and E2 show accelerated diethylnitrosamine (DEN)‐induced tumor formation as a result of suppressed apoptosis. 46
3.3. Wnt/β‐catenin signaling pathway
The Wnt/β‐catenin signaling pathway regulates stem cell pluripotency and cell fate decisions during development. 53 , 54 Wnt ligands are secreted glycoproteins that bind to Frizzled receptors and confer β‐catenin transcriptional activity. The dual biological functions of β‐catenin are to mediate Wnt signaling and promote cell‐to‐cell adhesion by binding with E‐cadherin at adherens junction sites. In the absence of Wnt ligands, the cytoplasmic pool of β‐catenin is tightly regulated by proteasomal degradation, a process where β‐catenin is phosphorylated by a complex of casein kinase 1α (CK1α), the scaffold protein AXIN, and the tumor suppressor adenomatous polyposis coli (APC). The phosphorylated β‐catenin is then subjected to ubiquitylation and proteasomal degradation. Binding Wnt to its receptors prevents AXIN and APC from interacting with β‐catenin. This results in β‐catenin stabilization and translocation into the nucleus, where β‐catenin associates with T‐cell factors (TCF) to activate target genes. 53
Aberrant regulation of Wnt/β‐catenin signaling has been frequently observed in many types of cancers. 55 Clinical studies conducted by different groups have revealed that somatic mutations of the β‐catenin gene leading to β‐catenin stabilization occurs in 20‐40% of HCC cases. 43 , 56 , 57 , 58 Genetic alternations that cause constitutive stabilization of β‐catenin, such as through loss of function mutations of APC and AXIN genes, have also been observed in HCC. 59 , 60 , 61 Furthermore, experimental studies have demonstrated that 67% of APC knockout mice develop HCC via activated β‐catenin‐mediated transcription. 62 Transgenic mice expressing an oncogenic mutant form of β‐catenin (a truncated NH2 terminus) also rapidly develop hepatomegaly via inhibiting apoptosis. 63 Disruption of β‐catenin/Tcf4 complex with β‐catenin/Tcf inhibitors such as PKF118‐310, PKF115‐584, or CGP049090 also interferes with the proliferation of cultured human HCC cells (HepG2 and Huh7) in vitro; it also inhibits tumor growth in vivo in the HepG2 xenograft model by inducing apoptosis as well as decreasing the expression of Myc and CCND1. 64 These observations suggest that aberrant regulation of Wnt/β‐catenin signaling undoubtedly contributes to the development of HCC.
3.4. TP53
TP53 functions largely as a transcription factor and can trigger a variety of antiproliferative programs by activating or repressing key effector genes. As a cellular gatekeeper, TP53 regulates cell division‐related mechanisms to ensure the fidelity of cell division, cellular senescence (a permanent form of cell cycle arrest), apoptosis or programmed cell death, and autophagy (a catabolic process involving the degradation of the cell's components primarily through the lysosomal machinery). In cell cycle checkpoints, TP53 contributes to both G1 and G2/M phase arrests through inducing the transactivation of p21waf1/cip1 (a cyclin‐dependent kinase inhibitor) and interfering with the function of cyclin B1/cdc2 complex, respectively. 65 , 66 In cellular senescence, TP53 regulates senescence by transactivating p21waf1/cip1 and E2F7. 67 In apoptosis, the function of TP53 is both transcription dependent and transcription independent. In the former, which takes place in the nucleus, TP53 contributes to apoptosis by transactivating numerous genes such as Bax, PIG3, Killer/DR5, CD95 (Fas), p53AIP1, Prep, Noxa, and PUMA. 68 In the mitochondria, TP53 induces outer membrane permeabilization, thus leading to the release of proapoptotic factors from the mitochondrial intermembrane space. In controlling autophagy, TP53 has a dual function: nuclear TP53 initiates autophagy through transactivating AMPK, PTEN, Sestrins, and the damage‐regulated autophagy modulator (DRAM) gene that encodes lysosomal protein, 69 whereas cytoplasmic TP53 inhibits autophagic flux by an unknown mechanism.
Regulation of TP53 is caused by the disruption of its interaction with MDM2, a negative regulator that mediates the ubiquitin‐mediated degradation of TP53. In response to cellular stress, the amino terminus of TP53 is phosphorylated by various kinases, including ATM, ATR, DNA‐PK, CHK1, and CHK2, 70 which prevents MDM2 binding and leads to the stabilization of TP53.
It has been well documented that the loss of TP53 function is a common feature of many human cancers. 71 , 72 It has been shown from clinical studies that HCC samples derived from cancers attributable to HBV or HCV and/or exposure to aflatoxin B1 frequently exhibit a point mutation at the third position of codon 249 serine in TP53. 29 , 31 Experimental studies have demonstrated that liver‐specific deletions of TP53 lead to the formation of HCC. 73 Moreover, a Cre‐loxP‐based strategy to temporally recover Tp53 expression in Tp53 knockout mice has demonstrated that, without affecting normal tissue, restoration of endogenous Tp53 regressed radiation‐induced lymphomas and sarcomas by inducing apoptosis and cellular senescence, respectively. 36
These observations suggest that TP53 not only functions as an initial barrier against HCC development but also suppresses tumor growth through inducing a variety of antiproliferative programs.
3.5. ErbB receptor tyrosine kinase family
The ErbB receptor family consists of four cell surface receptors: ErbB1/EGFR/HER1, ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4. These are cell membrane receptor tyrosine kinases and are activated by ligand binding (ie, EGF, TGFa, AR, and Epigen) and receptor dimerization. They regulate cell proliferation, migration, differentiation, apoptosis, and cell motility by activating PI3K/Akt, MAPK, and many other signaling pathways. The coding genes are often overexpressed, amplified, or mutated in cancers of the lung, breast, ovarian, bladder, and others. 74
Immunohistochemistry analysis of clinical samples from 100 HCC patients has shown that EGFR expression is elevated in HCC lesions but not in adjacent nontransformed hepatic cells in 53 cases and ErbB3 overexpression in HCC lesions is observed in 64 cases. Furthermore, the elevated expression of EGFR and ErbB3 correlates with more malignant phenotypes such as high proliferating ability, advanced stage, intrahepatic metastasis, and poor disease‐free survival. 75 Experimentally, constitutive activation of EGFR‐mediated signaling in transgenic mice and zebrafish has demonstrated that EGFR activation indeed results in HCC and the suppression of the EGFR signaling causes regressed HCC. 14 , 76 , 77 Moreover, the blockade of EGFR signaling with gefitinib EGFR inhibitor also prevents DEN‐induced liver carcinogenesis in rats and inhibits the proliferation of human HCC cell lines Huh‐7 and HepG2 in vitro. 78 , 79 Thus, EGFR‐mediated signaling plays a critical role in HCC development and maintenance.
4. TRANSGENIC ZEBRAFISH MODELS FOR HCC
According to the current knowledge of molecular pathogenesis of HCC, there is no single dominant molecular pathogenesis underlying all HCCs. Therefore, different therapeutic strategies would be required for treating different HCC subclasses due to different genetic alternations. To devise the strategies, different HCC models will be required to mimic different HCC subclasses. Our early comparative transcriptomic analysis demonstrated that liver tumors that developed in zebrafish as a result of exposure to chemical carcinogens are highly analogous to liver tumors in humans. 15 Based on the molecular pathogenesis of human HCC, several transgenic zebrafish lines with different oncogenes have been established. These lines are summarized in Table 2. This section summarizes the knowledge obtained from studies of these transgenic zebrafish.
TABLE 2.
A list of transgenic zebrafish for modeling HCC
| Transgene | Promoter | Phenotype | Onset period | Frequency | Ref. |
|---|---|---|---|---|---|
| HBx‐mCherry | fabp10 | HBx‐mCherry transgenic zebrafish develops HCC in TP53−/− background, | 11 mpf | 44% | 80 |
| HCP | fabp10 | HCP transgenic zebrafish develops HCC only in combination with thioacetamide (TAA) | 6 weeks after TAA treatment | 83 | |
| Loss of APC (APC+/−) | Null | APC+/ zebrafish develops spontaneous liver tumor. DMBA treatment increases the frequency to 70.8%. | More than 15 mpf | 17.6% | 85 |
| ctnnb1/β‐catenin | fabp10 | The level of HCC in the fish was 78% and 80% at 6 and 12 mpf, respectively. | 6 mpf | 78% | 86 |
| CTNNB1mt and tcf7l2 | fabp10 | The double transgenic larvae shows significant hepatomegaly within 3 days from the transgene expression. | Unknown | Unknown | 87 |
| Xmrk | fabp10 | All of Xmrk transgenic zebrafish develops HCC within 3 weeks from the transgene expression. | 3 weeks from the transgene induction | 100% | 14 |
| EGFP‐KrasG12V | fabp10 | All of EGFP‐KRASG12V transgenic zebrafish develops robust and homogeneous tumors in the liver after 1 month of the transgene induction | 1 month from the transgene induction | 100% | 93 |
| EGFP‐myca | fabp10 | 5% of EGFP‐myca transgenic zebrafish develops multinodular HCC with cirrhosis at 8‐9 months from the transgene induction. | 6‐7 months from the transgene induction | 51% | 96 |
| EDN1 | fabp10 | 83% of EDN1 transgenic zebrafish shows steatosis by 5 months, 17‐18% of the fish developed hyperplasia by 7‐9 months, and 17‐20% of the fish exhibited HCC by 11mpf. | 7‐9 mpf | 17–20% | 101 |
| Tgfb1a | fabp10 | Tgfb1a transgenic zebrafish develops hyperplasia and HCC. | 6 weeks from the transgene induction | 70% | 104 |
| UHRF1‐GFP | fabp10 | 80% of UHRF1‐GFP transgenic zebrafish die by 20 dpf. Among the fish survived for more than 15 dpf, 76% of them develop HCC by 20 dpf | 20 dpf | 76% | 111 |
| Twist1‐ERT2 | fabp10 | 80% of Twist1‐ERT2/xmrk double‐transgenic zebrafish shows spontaneous dissemination of mCherry‐labeled hepatocytes from the liver to the entire abdomen region and the tail region within 5 days from Twist1‐ERT2 activation | 5 days from Twist1‐ERT2 activation | 80% | 115 |
4.1. HBx‐driven HCC models
Large numbers of clonally integrated HBV genomes, which were harbored by HCC and transgenic mice expressing HBx, developed HCC spontaneously. 19 , 20 , 22 In an HBx transgenic zebrafish model, a fusion protein of HBx and mCherry (HBx‐mCherry) was expressed under the control of the liver‐specific fabp10 promoter. Hematoxylin and eosin (H&E) staining analysis revealed that, in wild‐type (WT) background specimens, the zebrafish resulted in hyperplasia (10%), dysplasia (5%), or steatosis (40%) at 11 months postfertilization (mpf); however, in TP53 null mutation background (TP53−/− ), the zebrafish developed HCC (44%), dysplasia (17%), or steatosis (11%) at 11 mpf. Immunohistochemistry and quantitative RT‐PCR analyses showed that 29% of the zebrafish in TP53−/− had potent levels of nuclear PCNA accumulation in the liver and cell cycle‐related genes such as ccna1, ccnb1, ccng1, cdk1, and cdk2 were dramatically upregulated; conversely, none of the zebrafish in WT exhibited these levels nor the elevated expression of these genes. In hyperplasia and HCC derived from the zebrafish in TP53−/− , high Src expression and elevated phosphorylation levels of ERK and Akt were observed. In contrast, in the livers derived from the zebrafish in WT and TP53−/− , Src was not detected. By crossing HBx‐mCherry with Src transgenic zebrafish line that expresses Src under control of the liver‐specific fabp10 promoter, HBx‐mCherry and Src double‐transgenic zebrafish were generated and 50% of the double‐line developed HCC in presence of TP53 by 14 mpf. 80
These experimental data suggest that HBx‐mCherry itself could not confer malignant transformation on hepatic cells, but it may induce HCC development in cooperation with either TP53−/− background or Src‐mediated signalings. As HBx could compromise DNA damage checkpoints through binding and inhibiting TP53, HBx‐mCherry fusion protein might fail to interact with TP53. 81 Therefore, the transgenic zebrafish require a p53−/‐ condition to develop HCC.
4.2. HCV core protein‐driven HCC and ICC models
Clinical studies have indicated a strong association between HCV infection and HCC development. 24 Moreover, HCV‐ and HCV‐related cirrhoses are also known to be risk factors for ICC. 82
To date, two types of HCV core protein (HCP) transgenic zebrafish lines have been established. One is to constitutively express HCP under the hepatocyte‐specific promoter, fabp10. This transgenic line develops HCC only in combination with thioacetamide (TAA) treatment, which is known to induce fibrosis and cirrhosis in animal models. Histological analysis has revealed that livers from the HCP transgenic zebrafish alone do not show any pathologic changes; normal cellular structures with well‐preserved cytoplasm, nuclei, and nucleoli are well maintained. However, in the presence of TAA, the transgenic zebrafish develop HCC by 6 weeks. At 1 week of TAA treatment, the transgenic zebrafish show higher steatosis than control WT zebrafish. By 4 weeks of TAA treatment, the transgenic zebrafish show bile duct damage and, by 6 weeks of TAA treatment, the transgenic zebrafish showed nodules comprising highly differentiated HCC with trabecular structures. In contrast, similarly treated WT zebrafish show nodules only at more than 12 weeks of TAA treatment. 83
The other transgenic line is to co‐express HCP and HBx under the same fabp10a promoter by using a doxycycline repressible system. Interestingly, HCP‐HBx double transgenic lines develop ICC instead of HCC. At 1 month of induction of HCP‐HBx expression, several morphological abnormalities in the liver were observed, including cytoplasmic vacuolation, bile duct dilation, and formation of fibrosis in 25% of the fish. The percentage of fibrosis fish increased to 45% at 3 months and 35% of the zebrafish showed early and severe ICC. In contrast, HCP and HBx single transgenic zebrafish did not exhibit any abnormalities or fibrosis in the liver. Transcriptomic analysis revealed that a set of genes related to cytoskeletal remodeling, cell adhesion, development, and cell cycle regulation were highly expressed in ICC from HCP‐HBx zebrafish. In particular, intense signals of TGF‐β1, phosphorylated‐p38, phosphorylated‐pERK1/2, and Smad3L were observed in neoplastic bile duct epithelial cells from HCP‐HBx zebrafish, whereas they were not detectable in the livers from HCP or HBx single transgenic zebrafish. Remarkably, knockdown of TGF‐β1 via injecting tgfb1 morpholinos into abdomens of 2‐month‐old HCP‐HBx zebrafish reduced bile duct proliferation from 53% to 32%, fibrosis formation from 53% to 23%, and ICC development from 29% to 11%. 84
These experimental data suggest that HCP itself could not confer malignant transformation on hepatic and biliary epithelial cells, but it may accelerate HCC and ICC development in cooperated with other carcinogens (eg, TAA) and oncogenes (eg, HBx).
4.3. β‐Catenin‐driven HCC model
Aberrant regulation of Wnt/β‐catenin signaling has been frequently observed in HCC. 43 , 56 , 57 , 58 To date, three types of transgenic zebrafish lines that show aberrant regulation of Wnt/β‐catenin signaling have been reported. One line had a heterozygous mutation in the APC gene (APC+/− ) and the mutant allele expressed truncated APC. Spontaneous liver tumors developed in 17.6% (six out of 34) of aged APC+/− fish (>15 months). Hepatic adenomas showed an accumulation of nuclear β‐catenin, which is the hallmark of activated Wnt signaling, and a high degree of proliferation. An experiment using chemical carcinogen 7,12‐ dimethylbenz[a]anthracene (DMBA) showed that 70.8% of DMBA‐treated APC+/− fish developed neoplasms in the liver compared with 20.5% of DMBA‐treated WT fish. 85
Another line expressed a mutated form of Xenopus laevis ctnnb1/β‐catenin under control of the hepatocyte‐specific promoter, fabp10. This transgene contained four point mutations that affected putative phosphorylation sites (S33A, S37A, T41A, and S45A); these mutations are frequently mutated in human HCC and generate constitutive active form of β‐catenin by interfering with its phosphorylation and subsequent degradation. 56 The level of HCC in fish was 78% and 80% at 6 and 12 mpf, respectively. Cross‐species comparison of HCC derived from the fish and human HCC revealed an upregulation of several Wnt target genes (myca, lef1, pparda, and sp5), and striking transcriptional similarities were observed between liver tumors in the fish and human HCC. In vivo drug screenings of SP600125 and EMD 420123 fish identifications, both of which target c‐Jun N‐terminal kinase (JNK), have a suppressor effect on fish liver enlargement. Activated β‐catenin was related with hyperactivation of JNK signaling pathway in both zebrafish and human HCC. Inhibition of JNK decreased liver size, specifically in HCC cells expressing constitutive active form of β‐catenin. The β‐catenin‐specific growth inhibitory effect of targeting JNK was conserved in human liver cancer cells. The suppressor effects of SP600125 were confirmed in a mouse model of HCC. 86
The other line expressed human CTNNB1mt and zebrafish tcf7l2 in hepatocytes. By crossing Tg(TRE:CTNNB1mt‐P2A‐tcf7l2) with Tg(fabp10a:TetON;TRE:eGFP‐krasv12), the double transgenic larvae showed significant hepatomegaly within 3 days from Dox treatment. Although KRAS activation resulted in lipid droplet accumulation and steatosis in the liver of the fish, Wnt and Myc activities significantly attenuated the accumulation and cell senescence triggered by KRASv12 expression. In vivo drug screenings using the fish identified that MK8245 (SCD inhibitor) suppresses hepatomegaly in the fish; MK8245 also suppresses the proliferation of human HCC cells. 87
Zebrafish models of β‐catenin‐driven HCC demonstrate that activation of β‐catenin promotes liver overgrowth. That is consistent with mouse models where its activation leads to increased hepatocyte proliferation and liver. Furthermore, MK8245 and SP600125, which are identified through in vivo drug screening using the models, suppress the proliferation of human HCC. These findings suggest that new insights from the models would lead to potential therapeutic strategies for targeting HCC.
4.4. Xmrk (EGFR)‐driven HCC model
Overexpression of EGFR is observed in 53% of the HCC patients. 75 A hyperactive form of EGFR homolog in fish of the genus Xiphophorus, Xmrk, induces receptor dimerization and constitutive activation of downstream signaling in a ligand‐independent manner. 88 Transgenic expression of Xmrk in the liver of zebrafish using a doxycycline (Dox) inducible system leads to HCC in essentially all juvenile fish within 3 weeks. All of the induced Xmrk transgenic zebrafish show HCC features, 13 including disrupted two‐cell sheet organization, variation in nuclear and cellular sizes, syncytial cells, and vesicular nuclei with prominent and multiple nucleoli (Figure 1). 14
FIGURE 1.
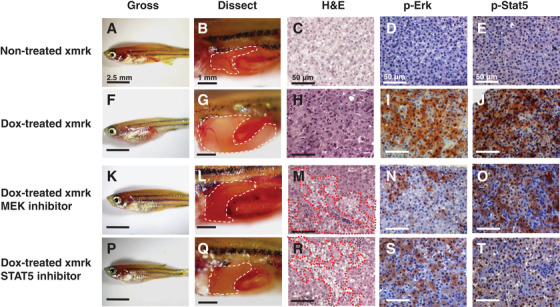
Effects of inhibition of p‐Erk and p‐Stat5 on HCC formation. (A‐E) Nontreated xmrk transgenic zebrafish as control. (F‐J) Doxycycline (Dox)‐treated xmrk transgenic zebrafish. (K‐O) Xmrk transgenic zebrafish treated with Dox and PD98059. (P‐T) Xmrk transgenic zebrafish treated with Dox and Stat5 inhibitor. Gross images of zebrafish, dissection to expose the liver, H&E staining, and IHC staining of phosphorylated Erk and Stat5 are indicated. Images are reprinted from ref. 14
An essential role in maintaining HCC in the zebrafish is Xmrk‐mediated signaling. Loss of Xmrk‐mediated signaling through the withdrawal of Dox regressed HCC in the induced transgenic zebrafish developing HCC. Histological examination revealed that neoplastic and normal hepatocytes coexist in the liver at 1 week from the Dox withdrawal, and the HCC features disappear by 2 weeks of Dox withdrawal. By 4 weeks of Dox withdrawal, all of the induced Xmrk transgenic zebrafish show normal liver morphology and histology. Moreover, resuming Dox treatment in HCC‐regressed transgenic zebrafish re‐induces the formation of HCC. Thus, the inducible Xmrk transgenic zebrafish model displays a clear oncogene‐addicted tumor formation. 14
Xmrk‐mediated signaling contributes to the proliferation of hepatocytes in the transgenic zebrafish as immunohistochemical staining has revealed a dramatic increase in PCNA‐positive cells in the liver after Dox induction. In the process of HCC regression, the number of PCNA‐positive hepatic cells rapidly drops, whereas the number of apoptotic cells increases dramatically. In HCC developed in the induced Xmrk transgenic zebrafish, activation of ERK and STAT5 is also observed. Pharmacological blocking with either MEK/ERK or STAT5 inhibitors for 3 weeks suppresses HCC progression; these treatments lead to smaller abdomen and liver size. Histological analysis reveals that in the liver of either inhibitor‐treated zebrafish, normal and malignant hepatocytes coexist (Figure 1). 14
These observations suggest that EGFR‐mediated signaling plays an essential role in not only HCC development but also in HCC maintenance. As clinical studies have indicated constitutive activation of Ras/ERK and Jak/STAT signaling in all cases of the clinical HCC samples analyzed (n > 80), 89 the inhibitors tested in the Xmrk transgenic zebrafish may also be capable of regressing human HCC.
4.5. KRAS‐driven HCC model
In clinical studies, approximately 3% (1/30 cases) of HCC patients possessed constitutively active mutation of KRAS in which codon 12 of KRAS is mutated from glycine to valine and constitutive activation of Ras/ERK signaling is observed in all cases of over 80 clinical samples. 89 , 90 In experimental studies, it has been demonstrated that the HCV core protein activates MAPK/ERK signaling and blocking Raf/ERK signaling with sorafenib prevents PLC∖PRF∖5 and human HepG2 cells from proliferating in vitro and in vivo. 91 , 92 Therefore, KRAS‐mediated signaling plays a critical role in developing and maintaining HCC. Using a mifepristone‐inducible transgenic system, an EGFP fusion protein with KRASG12V (EGFP‐KrasG12V) has been expressed in a liver‐specific manner. All of EGFP‐KRASG12V transgenic zebrafish developed robust and homogeneous tumors in the liver after 1 month of mifepristone induction. 93
EGFP‐KRASG12V expression in the liver induces malignant transform of hepatocytes through activating both Ras/ERK and PI3K/Akt pathways. Western blot and immunohistochemistry analyses have revealed that HCC derived from mifepristone‐treated KRAS transgenic zebrafish show activation of Ras/ERK and PI3K/Akt signaling pathways, and these active states are dramatically decreased after the withdrawal of mifepristone. The pharmacological block of Ras/ERK signaling with PD98059 (MEK inhibitor) suppresses hyperplastic liver growth in 49% of the zebrafish at the larval stage. Similarly, blocking PI3K/Akt/mTOR signaling with either LY294002 (PI3K inhibitor) or Rapamycin (mTOR inhibitor) inhibits the growth in over half of the larvae. Moreover, the combined treatment of PD98059 with either LY294002 or Rapamycin dramatically decreases the frequency of tumorigenesis. 13 These observations suggest that Ras/ERK and PI3K/Akt/mTOR signaling pathways might play essential roles in not only developing but also maintaining HCC. Thus, pharmacological block with multiple signaling pathways could be promising and effective for eliminating human HCC.
4.6. Myc‐driven HCC model
Chromosomal amplification at 8q, where the Myc gene is located, is observed in 48% (24/50 cases) of HCC patients and elevated expression of Myc‐regulated genes is strongly associated with a malignant conversion of HCC. 94 , 95 Therefore, Myc is thought to be an oncogenic driver in hepatocarcinogenesis. To date, two types of Myc transgenic zebrafish have been established: one expresses mouse Myc in a liver‐specific manner using a doxycycline‐inducible system and the other expresses EGFP‐fused gene with either zebrafish myca or mycb in the liver by using a mifepristone‐inducible system. 96
The induced liver expression of the mouse Myc in transgenic zebrafish causes liver hyperplasia, which is progressed to hepatocellular adenoma and carcinoma with prolonged transgene expression. Transcriptomic analysis reveals that upregulated genes in Myc‐driven zebrafish liver tumors are similar to those in human liver tumors. In particular, the ribosome biogenesis constitutes the key features of Myc‐driven carcinogenesis in the liver. 96
Transgenic zebrafish with myca expression show stronger phenotype than mycb transgenic zebrafish and develop different types of liver tumors. At 1 month from the transgene induction, 87% of the myca transgenic zebrafish show ascites like phenotype with yellow fluid in the abdomen cavity as well as fluid‐filled cysts in the liver. At 6‐7 months from the transgene induction, 51% of myca fish develop large tumors, 14% show large tumors with overt angiogenesis, and 7% retain ascites‐like phenotype with a pseudoglandular organization of tumor cells. At 8‐9 months from the transgene induction, about 5% of the induced fish develop multinodular HCC with cirrhosis. In this multinodular HCC, loss of E‐cadherin and β‐catenin accumulation in the nuclear compartment is observed. Furthermore, loss of the transgene through mifepristone withdrawal resulted in tumor regression through inducing apoptosis. 52
These observations suggest that myca plays an essential role in not only HCC development but also in HCC maintenance. Clinical studies show that elevated expression of Myc‐regulated genes is strongly associated with a malignant conversion of HCC. 94 , 95 Therefore, identifying the Myc‐regulated genes that play an essential role in maintaining HCC growth of the transgenic fish would lead to potential therapeutic strategies for targeting HCC.
4.7. Endothelin 1‐driven HCC model
Endothelin 1 (EDN1) is a potent vasoconstrictor peptide, which is composed of 21 amino acids. The biological effects of endothelins are mediated by plasma membrane‐bound receptors (ETA and ETB). Apart from being a recognized vasoconstrictor peptide, clinical studies reported that plasma concentration of EDN1 was markedly increased in both HCC patients and patients with metastasized tumors in the liver and that 70% (n = 14/20) of HCC samples showed EDN1 expression. 97 , 98 In a rat hepatoma model, it has been demonstrated that exogenous addition of EDN1 enhances hepatoma cell growth in a dose‐dependent manner, whereas an endothelin receptor antagonist inhibits tumor growth. 99 Moreover, in a HBV X antigen‐induced HCC mouse model, EDN1 is one of the genes that are induced by X antigen. 40 , 100 Therefore, EDN1 is thought to be an oncogenic driver in hepatocarcinogenesis. Transgenic EDN1 zebrafish that express human EDN1 under control of the liver‐specific fabp10a promoter are established. H&E staining analysis revealed that 83% of the zebrafish showed steatosis by 5 months, 17‐18% of the zebrafish developed hyperplasia by 7‐9 months, and 17‐20% of the zebrafish exhibited HCC by 11 mpf. Quantitative RT‐PCR analysis showed that elevated expression of lipogenic factor and enzymes (srebf1, cebpa, fasn, and adapt) and significant upregulation of cell cycle‐related genes (ccna1, ccnb1, ccne1, ccng1, cdk1, and cdk2) were observed in the liver of the zebrafish at 5 and 11 mpf, respectively. Immunohistochemistry analyses showed that potent levels of nuclear PCNA accumulation and elevated phosphorylation level of AKT were detected in the liver of the zebrafish at 9 and 11 mpf; conversely, only low levels of PCNA and phosphorylation level of AKT were detected in the liver of control zebrafish at the same age. 101
These experimental data suggest that EDN1 might promote cell proliferation through activating PI3K/AKT signaling pathway, and that might lead to HCC development.
4.8. Transforming growth factor‐β‐driven HCC model
The pleiotropic cytokine transforming growth factor‐β (TGFβ) plays a bifunctional role in either inhibiting or promoting cell proliferation. During hepatocarcinogenesis, it acts as tumor suppressor and tumor promoter in the early and late stages, respectively. 102 Clinical studies reveal that HCC patients show a distinct signature of TGF‐β gene expression and the signature correlates with tumor invasiveness, the time before relapse, and long‐term survival of the patient. 103
An inducible tgfb1a transgenic zebrafish line that expresses zebrafish tgfb1a in the liver by using a mifepristone inducible system 96 has been established. Depending on the intensity of TGFβ1 induction, the fish developed different lesions in the liver; low and moderate levels of TGFβ1 caused hyperplasia and hepatocellular adenoma and high levels of TGFβ1 caused HCC to develop. In the HCC cells, TGFβ1‐mediated signaling switched from Smad to ERK‐mediated signaling. Although pharmacological inhibition of Smad‐mediated signaling with PD169316 (Smad2 inhibitor) does not affect HCC development in the fish, ERK‐mediated signaling with U0126 (MEK inhibitor) suppresses the proliferation of hepatic cells. 104
An inducible tgfb1a transgenic zebrafish demonstrated that overexpression of tgfb1a alone is sufficient to induce HCC. Clinical studies reveal that TGF‐β expression correlates with tumor invasiveness, the time before relapse, and long‐term survival of the patient with HCC 103 ; and constitutive activation of ERK signaling is observed in all cases of over 80 clinical samples. 89 , 90 Therefore, pharmacological inhibition of ERK‐mediated signaling with U0126 (MEK inhibitor) may be effective for eliminating TGF‐β expressing HCC.
4.9. Ubiquitin‐like with PHD and RING finger domains 1‐driven HCC model
Ubiquitin‐like with PHD and RING finger domains 1 (UHRF1) is a member of a subfamily of RING‐finger‐type E3 ubiquitin ligases, consisting of multiple domains: a N‐terminal ubiquitin‐like domain; tandem tudor and PHD domains for recognizing methylated histones; an SRA domain for interacting with hemimethylated DNA, DNA methyltransferase 1 (DNMT1), and histone deacetylase 1 (HDAC1); and a C‐terminal RING finger motif for E3‐ubiquitin ligase activity. 105 It contributes to not only cell cycle regulation and epigenetic modifications via recruiting DNMT1 and HDAC1, but also to TP53‐dependent DNA damage checkpoints. 106 In primary cultured human lung fibroblasts, UHRF1 expression peaked at late G1 and during the G2/M phase; conversely, in cancer cell lines such as HeLa, Jurkat, and A549, constant UHRF1 expression is observed throughout the entire cell cycle. 107 In clinical studies, elevated expression of UHRF1 is observed in several tumors from breast, lung, bladder, pancreas, colon, and prostate. 108 , 109 , 110 Therefore, UHRF1 is thought to be an oncogenic driver in tumorigenesis.
Recently, a transgenic zebrafish line that expresses the human UHRF1 gene fused to GFP (UHRF1‐GFP) in hepatocytes to induce HCC has been reported. In WT background, 74% of larval UHRF1‐GFP transgenic zebrafish have small livers and 80% of them die by 20 days postfertilization (dpf). In the small zebrafish livers at 5 dpf, intense senescence‐associated β‐galactosidase (SA‐β‐gal) staining is detected. Among the UHRF1 transgenic zebrafish survived for more than 15 dpf, 76% of them develop HCC by 20 dpf. Moreover, HCC incidence at 15 dpf in UHRF1 transgenic fish increased from 46% in the WT background to 87% in TP53 heterozygous mutation background. 111 These observations suggest that a bypass of TP53‐induced senescence would be required for UHRF1 to act as an oncogene.
Furthermore, clinical samples from 58 patients with dysplastic nodules (n = 18) or HCC (n = 40) have shown an average of 20‐ and 40‐fold overexpression of UHRF1 in advanced and very advanced HCC cases. Knockdown of UHRF1 by RNA interference in the human hepatic cancer cell line, HepG2, induces PARP cleavage and other markers of apoptosis. 111 Therefore, therapeutic strategies targeting UHRF1 might regress human HCC.
4.10. Twist1‐induced metastasis models
HCC is a rapidly growing tumor associated with a high propensity for vascular invasion and metastasis. Epithelial‐mesenchymal transition (EMT), which converts various types of epithelial cells into mesenchymal cells, plays a critical role in promoting metastasis. 112 Twist is an EMT‐inducible transcriptional factor; elevated expression of Twist is associated with poor survival rates in cancer patients. 113 Immunohistochemical microarray stainings of paired primary and metastatic HCC cells revealed that overexpression of Twist was correlated with HCC metastasis. 114
A tamoxifen‐controllable Twist1a‐ERT2 transgenic zebrafish is reported to model metastatic dissemination of hepatic cells. The activation of Twist1a‐ERT2 following 48 h of tamoxifen treatment induces EMT in the liver. By crossing this model with Xmrk transgenic zebrafish, approximately 80% of the double‐transgenic zebrafish show spontaneous dissemination of mCherry‐labeled hepatocytes from the liver within 5 days from the treatment initiation. This rapid and high‐frequency induction of the dissemination provides a novel way to screen chemicals/drugs in vivo for identification of antimetastasis drugs targeting metastatic dissemination of cancer cells (Figure 2). 115
FIGURE 2.
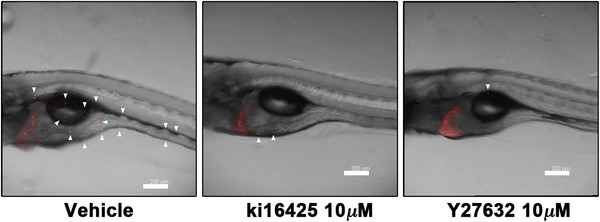
Reported antimetastasis drugs: ki16425 and Y27632 could suppress dissemination of mCherry‐labeled hepatic cells from the liver of Twist1a‐ERT2/xmrk double transgenic zebrafish. Representative images of the dissemination in the fish that were treated with doxycycline and 4‐OHT in presence of vehicle (left), 10 mmol/L of ki16425 (middle), or 10 mmol/L of Y27632 (right). Some disseminated mCherry‐positive cells are indicated by arrowheads. The images were shown as Z‐stack images using 100× magnification. Scale bar, 200 μm. Images are reprinted from ref. 115
In vivo drug screening using Twist1a‐ERT2/Xmrk double transgenic zebrafish identifies that adrenosterone, which inhibits hydroxysteroid (11‐beta) dehydrogenase 1 (HSD11β1), suppresses dissemination of mCherry‐labeled hepatic cells from the liver of Twist1a‐ERT2/xmrk double transgenic zebrafish. This suppressor effects is validated in a zebrafish xenotransplantation model where highly metastatic human HCC cell line HCCLM3 and breast cancer cell line MDA‐MB‐231 are inoculated into the duct of Cuvier of Tg (kdrl:EGFP) transgenic zebrafish. Pharmacological and genetic inhibition of HSD11β1 induces the re‐expression of E‐cadherin and other epithelial markers and the lost partial expression of mesenchymal markers via the downregulation of Snail and Slug. 115
These observations suggest, in this model, Twist1a‐ERT2‐driven EMT alone would not be sufficient to induce abdominal and distant cell dissemination and that cooperation of Xmrk‐driven cellular events would be required for dissemination.
5. INSIGHTS FROM ZEBRAFISH MODELS OF HCC FOR DEVISING NOVEL THERAPEUTIC STRATEGIES FOR HCC
The HCC tumor is complex and heterogeneous and generally has multiple genomic alterations. These alterations lead to the aberrant activation of cellular kinase‐signaling networks to confer malignant transformation and proliferation of cancer cells. Targeting a single kinase has proven successful in some cancer cases; for example, Imatinib (Gleevec, Novartis) eliminates Philadelphia chromosome‐positive B cell acute lymphoblastic leukemia by inhibiting BCR‐ABL and gefitinib (Iressa, AstraZeneca) inhibits the progression of nonsmall lung cancer by interrupting EGFR tyrosine kinase activity. 116 , 117 However, this approach has faced difficulties because cancer cells acquire resistance to these kinase inhibitors. 118 Recently, sorafenib (BAY43‐9006; Nexavar), a multikinase inhibitor, has been shown to have survival benefits in patients suffering from renal, melanoma, or HCC 119 cancers. To overcome the complexity of aberrant activation of the HCC signaling network, combination therapies that target multiple signaling pathways would be promising and effective.
5.1. Sorafenib and rapamycin combination therapy
Sorafenib is a kinase inhibitor targeting Raf kinase and several receptor tyrosine kinases, including platelet‐derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor 2 (VEGFR2), FLT3, c‐Kit, and Ret. Sorafenib has been considered the standard of care for patients with advanced HCC since 2007. 119 , 120 However, a phase II trial involving 137 patients with advanced HCC showed that sorafenib treatment resulted in a median overall survival of 9.2. 121 This suggests that a single sorafenib treatment resulted in modest efficiency. A combination therapy of sorafenib with other kinase inhibitors could be more effective.
Activation of Ras/ERK signaling has been observed in 100% of over 80 clinical HCC samples. 89 Aberrant mTOR signaling (p‐RPS6) is present in half of HCC specimens (n = 314). 122 In a KRASG12V transgenic zebrafish model, it has been shown that pharmacological blocking of Ras/MEK and PI3K/mTOR signalings with PD98059 (MEK inhibitor) and rapamycin (mTOR inhibitor) suppresses the enlargement of oncogenic livers. 13 Thus, we propose that a combination therapy of sorafenib with rapamycin may have a better effect in HCC treatment. An experimental study has demonstrated that the combination therapy strongly inhibits primary tumor growth and lung metastases in a mouse model xenografted with human HCC cells (LCI‐D20); the inhibition is resultant of therapy‐induced apoptosis and suppressed angiogenesis. 123
5.2. Combination therapy using gefitinib and ruxolitinib
Gefitinib is an EGFR inhibitor that interrupts EGFR‐mediated Ras/MEK, PI3K/AKT, and Jak/STAT signaling pathways by binding to the ATP‐binding site of EGFR tyrosine kinase. Blockading EGFR signaling with gefitinib prevents DEN‐induced liver carcinogenesis in rats and inhibits the proliferation of human HCC cell lines Huh‐7 and HepG2. 78 , 79 Despite these findings, a phase II study of erlotinib, which has a similar chemical compound as gefitinib, showed poor effects in regressing HCC (n = 38). 124
It has been reported that nonsmall lung cancer cells acquire resistance to gefitinib via bypassing downstream signaling of EGFR. 125 In our Xmrk transgenic zebrafish, we have demonstrated that the loss of EGFR‐mediated signaling regresses liver tumors and pharmacological blocking with MEK and STAT5 inhibitors also interrupts liver tumor formation (Figure 1). 14 These observations suggest that double blocking Ras/ERK and Jak/STAT signalings would be more effective in inhibiting the proliferation of HCC cells. We therefore propose a combination HCC therapy of gefitinib with an inhibitor that targets Jak/STAT signaling. Fortunately, the Jak1/2 inhibitor ruxolitinib (INC424; Novartis) has already been approved by the FDA for the treatment of intermediate or high‐risk myelofibrosis, and an experimental study has demonstrated that ruxolitinib has an antiproliferative effect on HCC. 126 Therefore, a drug repositioning of ruxolitinib would be desired for the combination therapy.
6. CONCLUSION
The zebrafish modeling system has been increasingly recognized as a platform for chemical screening because it provides the advantage of high‐throughput screening in an in vivo vertebrate setting with physiologic relevance to humans. 6 , 7 , 8 , 9 , 10 Our early comparative transcriptomic analysis demonstrated that liver tumors that developed in zebrafish as a result of exposure to chemical carcinogens are highly analogous to liver tumors in humans. 15 Based on the molecular pathogenesis of human HCC, several transgenic zebrafish lines with different oncogenes have been established. Some of them exist on the platform that allow the identification of chemicals that have suppressor effects on the proliferation of human HCC cells (Figure 3). A few chemicals identified in zebrafish screening have reached clinical trials. 127 , 128 , 129 Therefore, there is the possibility that the chemicals identified by zebrafish HCC modeling might reach clinical trials. Furthermore, studies using Xmrk and myca transgenic zebrafish demonstrate that EGFR‐mediated signaling and Myc‐regulated genes play an essential role not only in HCC development but also in HCC maintenance. Therefore, identifying the EGFR‐mediated signaling and the Myc‐regulated genes that play an essential role in maintaining HCC growth of these fish could lead to potential therapeutic strategies for targeting HCC.
FIGURE 3.
Liver tumors that developed in zebrafish as a result of exposure to chemical carcinogens are highly analogous to liver tumors in humans. Based on the molecular pathogenesis of human HCC, several transgenic zebrafish lines with different oncogenes have been established. Some of them exist on the platform that allow the identification of chemicals that have suppressor effects on the proliferation of human HCC cells. Image is reprinted from ref. 115
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We sincerely appreciate the European Association for the Study of the Liver (EASL) and the American Association for Cancer Research (AACR) for allowing us to reprint the images in Figures 1 and 2 in this review. The images are reprinted from a previous study of ours. The study, led by Z. Gong, was funded by the National Medical Research Council of Singapore (R‐154000547511) and the Ministry of Education of Singapore (R‐154000A23112).
Nakayama J, Gong Z. Transgenic zebrafish for modeling hepatocellular carcinoma. MedComm. 2020;1:140–156. 10.1002/mco2.29
REFERENCES
- 1. El‐Serag H. Hepatocellular carcinoma. N Engl J Med. 2011;365(11):1118‐1127. [DOI] [PubMed] [Google Scholar]
- 2. Gomaa A‐I. Hepatocellular carcinoma: epidemiology, risk factors and pathogenesis. World J Gastroenterol. 2008;14(27):4300‐4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fujimoto A, Totoki Y, Abe T, et al. Whole‐genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44(7):760‐764. [DOI] [PubMed] [Google Scholar]
- 4. Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy‐number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006;5(9):741‐754. [DOI] [PubMed] [Google Scholar]
- 6. White R, Rose K, Zon L. Zebrafish cancer: the state of the art and the path forward. Nat Rev Cancer. 2013;13(9):624‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nat Rev Drug Discov. 2005;4(1):35‐44. [DOI] [PubMed] [Google Scholar]
- 8. Letrado P, Miguel ID, Lamberto I, Díez‐Martínez R. Zebrafish: speeding up the cancer drug discovery process. Cancer Res. 2018;78(21):6048‐6058. [DOI] [PubMed] [Google Scholar]
- 9. Gunnarsson L, Jauhiainen A, Kristiansson E. Evolutionary conservation of human drug targets in organisms used for environmental risk assessments. Environ Sci Technol. 2008;42(15):5807‐5813. [DOI] [PubMed] [Google Scholar]
- 10. Howe K, Clark MD, Torroja CF, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496(7446):498‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakayama J, Makinoshima H. Zebrafish‐based screening models for the identification of anti‐metastatic drugs. Molecules. 2020;25(10):2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen AT, Emelyanov A, Koh CH, et al. A high level of liver‐specific expression of oncogenic Kras(V12) drives robust liver tumorigenesis in transgenic zebrafish. Dis Models Mech. 2011;4(6):801‐813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nguyen AT, Emelyanov A, Koh CH, Spitsbergen JM, Parinov S, Gong Z. An inducible kras(V12) transgenic zebrafish model for liver tumorigenesis and chemical drug screening. Dis Models Mech. 2012;5(1):63‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Z, Huang X, Zhan H, et al. Inducible and repressable oncogene‐addicted hepatocellular carcinoma in Tet‐on xmrk transgenic zebrafish. J Hepatol. 2012;56(2):419‐425. [DOI] [PubMed] [Google Scholar]
- 15. Lam SH, Wu YL, Vega VB, et al. Conservation of gene expression signatures between zebrafish and human liver tumors and tumor progression. Nat Biotechnol. 2006;24(1):73‐75. [DOI] [PubMed] [Google Scholar]
- 16. Goodman ZD. Neoplasms of the liver. Mod Pathol. 2007;20(Suppl 1):S49‐60. [DOI] [PubMed] [Google Scholar]
- 17. El‐Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142(6):1264‐1273.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen CJ, Yang HI, Su J, et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA. 2006;295(1):65‐73. [DOI] [PubMed] [Google Scholar]
- 19. Brechot C, Pourcel C, Louise A, Rain B, Tiollais P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature. 1980;286(5772):533‐535. [DOI] [PubMed] [Google Scholar]
- 20. Chakraborty PR, Ruiz‐Opazo N, Shouval D, Shafritz DA. Identification of integrated hepatitis B virus DNA and expression of viral RNA in an HBsAg‐producing human hepatocellular carcinoma cell line. Nature. 1980;286(5772):531‐533. [DOI] [PubMed] [Google Scholar]
- 21. Chisari FV, Filippi P, Buras J, et al. Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc Natl Acad Sci USA. 1987;84(19):6909‐6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351(6324):317‐320. [DOI] [PubMed] [Google Scholar]
- 23. Huang SN, Chisari FV. Strong, sustained hepatocellular proliferation precedes hepatocarcinogenesis in hepatitis B surface antigen transgenic mice. Hepatology. 1995;21(3):620‐626. [PubMed] [Google Scholar]
- 24. Saito I, Miyamura T, Ohbayashi A, et al. Hepatitis‐C virus‐infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci USA. 1990;87(17):6547‐6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moriya K, Fujie H, Shintani Y, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4(9):1065‐1067. [DOI] [PubMed] [Google Scholar]
- 26. Morgan TR, Mandayam S, Jamal MM. Alcohol and hepatocellular carcinoma. Gastroenterology. 2004;127(5 Suppl 1):S87‐96. [DOI] [PubMed] [Google Scholar]
- 27. Seitz HK, Stickel F. Molecular mechanisms of alcohol‐mediated carcinogenesis. Nat Rev Cancer. 2007;7(8):599‐612. [DOI] [PubMed] [Google Scholar]
- 28. Hamid AS, Tesfamariam IG, Zhang Y, Zhang ZG. Aflatoxin B1‐induced hepatocellular carcinoma in developing countries: geographical distribution, mechanism of action and prevention. Oncol Lett. 2013;5(4):1087‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350(6317):429‐431. [DOI] [PubMed] [Google Scholar]
- 30. Ming L, Thorgeirsson SS, Gail MH, et al. Dominant role of hepatitis B virus and cofactor role of aflatoxin in hepatocarcinogenesis in Qidong, China. Hepatology. 2002;36(5):1214‐1220. [DOI] [PubMed] [Google Scholar]
- 31. Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hot spot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350(4):427‐428. [DOI] [PubMed] [Google Scholar]
- 32. Aguilar F, Hussain SP, Cerutti P. Aflatoxin B1 induces the transversion of G→T in codon 249 of the p53 tumor suppressor gene in human hepatocytes. Proc Natl Acad Sci USA. 1993;90(18):8586‐8590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mace K, Aguilar F, Wang JS, et al. Aflatoxin B1‐induced DNA adduct formation and p53 mutations in CYP450‐expressing human liver cell lines. Carcinogenesis. 1997;18(7):1291‐1297. [DOI] [PubMed] [Google Scholar]
- 34. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789‐799. [DOI] [PubMed] [Google Scholar]
- 35. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐674. [DOI] [PubMed] [Google Scholar]
- 36. Villanueva A. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27(1):55‐76. [DOI] [PubMed] [Google Scholar]
- 37. Minguez B, Tovar V, Chiang D, Villanueva A, Llovet JM. Pathogenesis of hepatocellular carcinoma and molecular therapies. Curr Opin Gastroenterol. 2009;25(3):186‐194. [DOI] [PubMed] [Google Scholar]
- 38. Block TM, Mehta AS, Fimmel CJ, Jordan R. Molecular viral oncology of hepatocellular carcinoma. Oncogene. 2003;22:5093‐5107. [DOI] [PubMed] [Google Scholar]
- 39. Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31(4):339‐346. [DOI] [PubMed] [Google Scholar]
- 40. Midorikawa Y, Yamamoto S, Tsuji S, et al. Allelic imbalances and homozygous deletion on 8p23.2 for stepwise progression of hepatocarcinogenesis. Hepatology. 2009;49(2):513‐522. [DOI] [PubMed] [Google Scholar]
- 41. Chochi Y, Kawauchi S, Nakao M, et al. A copy number gain of the 6p arm is linked with advanced hepatocellular carcinoma: an array‐based comparative genomic hybridization study. J Pathol. 2009;217(5):677‐684. [DOI] [PubMed] [Google Scholar]
- 42. Yumoto Y, Hanafusa T, Hada H, et al. Loss of heterozygosity and analysis of mutation of p53 in hepatocellular carcinoma. J Gastroenterol Hepatol. 1995;10(2):179‐185. [DOI] [PubMed] [Google Scholar]
- 43. Taniguchi K, Roberts LR, Aderca IN, et al. Mutational spectrum of beta‐catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene. 2002;21(31):4863‐4871. [DOI] [PubMed] [Google Scholar]
- 44. Matsumura T, Makino R, Mitamura K. Frequent down‐regulation of E‐cadherin by genetic and epigenetic changes in the malignant progression of hepatocellular carcinomas. Clin Cancer Res. 2001;7(3):594‐599. [PubMed] [Google Scholar]
- 45. Lin YW, Sheu JC, Liu LY, et al. Loss of heterozygosity at chromosome 13q in hepatocellular carcinoma: identification of three independent regions. Eur J Cancer. 1999;35(12):1730‐1734. [DOI] [PubMed] [Google Scholar]
- 46. Kamegaya Y, Hiasa Y, Zukerberg L, et al. Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology. 2005;41(3):660‐667. [DOI] [PubMed] [Google Scholar]
- 47. Lerat H, Honda M, Beard MR, et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122(2):352‐365. [DOI] [PubMed] [Google Scholar]
- 48. Matsumoto M, Hsieh TY, Zhu N, et al. Hepatitis C virus core protein interacts with the cytoplasmic tail of lymphotoxin‐beta receptor. J Virol. 1997;71(2):1301‐1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen CM, You LR, Hwang LH, Lee YH. Direct interaction of hepatitis C virus core protein with the cellular lymphotoxin‐beta receptor modulates the signal pathway of the lymphotoxin‐beta receptor. J Virol. 1997;71(12):9417‐9426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu N, Ware CF, Lai MM. Hepatitis C virus core protein enhances FADD‐mediated apoptosis and suppresses TRADD signaling of tumor necrosis factor receptor. Virology. 2001;283(2):178‐187. [DOI] [PubMed] [Google Scholar]
- 51. Kittlesen DJ, Chianese‐Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T‐lymphocyte proliferation. J Clin Invest. 2000;106(10):1239‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun L, Nguyen AT, Spitsbergen JM, Gong Z. Myc‐induced liver tumors in transgenic zebrafish can regress in tp53 null mutation. PLoS One. 2015;10(1):0117249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. MacDonald BT, Tamai K, He X. Wnt/beta‐catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13(7):513‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11‐26. [DOI] [PubMed] [Google Scholar]
- 56. Miyoshi Y, Iwao K, Nagasawa Y, et al. Activation of the beta‐catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res. 1998;58(12):2524‐2527. [PubMed] [Google Scholar]
- 57. Huang H, Fujii H, Sankila A, et al. β‐Catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am J Pathol. 1999;155(6):1795‐1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. de La Coste A, Romagnolo B, Billuart P, et al. Somatic mutations of the beta‐catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA. 1998;95(15):8847‐8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Laurent–Puig P, Legoix P, Bluteau O, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120(7):1763‐1773. [DOI] [PubMed] [Google Scholar]
- 60. Satoh S, Daigo Y, Furukawa Y, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus‐mediated transfer of AXIN1. Nat Genet. 2000;24(3):245‐250. [DOI] [PubMed] [Google Scholar]
- 61. Zucman‐Rossi J, Benhamouche S, Godard C, et al. Differential effects of inactivated Axin1 and activated beta‐catenin mutations in human hepatocellular carcinomas. Oncogene. 2007;26(5):774‐780. [DOI] [PubMed] [Google Scholar]
- 62. Colnot S, Decaens T, Niwa‐Kawakita M, et al. Liver‐targeted disruption of Apc in mice activates beta‐catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci USA. 2004;101(49):17216‐17221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cadoret A, Ovejero C, Saadi‐Kheddouci S, et al. Hepatomegaly in transgenic mice expressing an oncogenic form of beta‐catenin. Cancer Res. 2001;61(8):3245‐3249. [PubMed] [Google Scholar]
- 64. Wei W, Chua MS, Grepper S, So S. Small molecule antagonists of Tcf4/beta‐catenin complex inhibit the growth of HCC cells in vitro and in vivo. Int J Cancer. 2010;126(10):2426‐2436. [DOI] [PubMed] [Google Scholar]
- 65. St Clair S, Manfredi JJ. The dual specificity phosphatase Cdc25C is a direct target for transcriptional repression by the tumor suppressor p53. Cell Cycle. 2006;5(7):709‐713. [DOI] [PubMed] [Google Scholar]
- 66. Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation‐induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377(6549):552‐557. [DOI] [PubMed] [Google Scholar]
- 67. Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32(43):5129‐5143. [DOI] [PubMed] [Google Scholar]
- 68. Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53‐regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402‐412. [DOI] [PubMed] [Google Scholar]
- 69. Crighton D, Wilkinson S, O'Prey J, et al. DRAM, a p53‐induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126(1):121‐134. [DOI] [PubMed] [Google Scholar]
- 70. Appella E, Anderson CW. Post‐translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268(10):2764‐2772. [DOI] [PubMed] [Google Scholar]
- 71. Sherr CJ. Principles of tumor suppression. Cell. 2004;116(2):235‐246. [DOI] [PubMed] [Google Scholar]
- 72. Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild‐type p53. Science. 1990;249(4971):912‐915. [DOI] [PubMed] [Google Scholar]
- 73. Katz SF, Lechel A, Obenauf AC, et al. Disruption of Trp53 in livers of mice induces formation of carcinomas with bilineal differentiation. Gastroenterology. 2012;142(5):1229‐1239.e3. [DOI] [PubMed] [Google Scholar]
- 74. Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366(1):2‐16. [DOI] [PubMed] [Google Scholar]
- 75. Ito Y, Takeda T, Sakon M, et al. Expression and clinical significance of erb‐B receptor family in hepatocellular carcinoma. Br J Cancer. 2001;84(10):1377‐1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jhappan C, Stahle C, Harkins RN, Fausto N, Smith GH, Merlino GT. TGF alpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell. 1990;61(6):1137‐1146. [DOI] [PubMed] [Google Scholar]
- 77. Webber EM, Wu JC, Wang L, Merlino G, Fausto N. Overexpression of transforming growth factor‐alpha causes liver enlargement and increased hepatocyte proliferation in transgenic mice. Am J Pathol. 1994;145(2):398‐408. [PMC free article] [PubMed] [Google Scholar]
- 78. Hopfner M, Sutter AP, Huether A, Schuppan D, Zeitz M, Scherubl H. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J Hepatol. 2004;41(6):1008‐1016. [DOI] [PubMed] [Google Scholar]
- 79. Schiffer E, Housset C, Cacheux W, et al. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology. 2005;41(2):307‐314. [DOI] [PubMed] [Google Scholar]
- 80. Lu JW, Yang WY, Tsai SM, et al. Liver‐specific expressions of HBx and src in the p53 mutant trigger hepatocarcinogenesis in zebrafish. PLoS One. 2013;8(10):e76951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang XW, Gibson MK, Vermeulen W, et al. Abrogation of p53‐induced apoptosis by the hepatitis B virus X gene. Cancer Res. 1995;55(24):6012‐6016. [PubMed] [Google Scholar]
- 82. Yamamoto S, Kubo S, Hai S, et al. Hepatitis C virus infection as a likely etiology of intrahepatic cholangiocarcinoma. Cancer Sci. 2004;95(7):592‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rekha RD, Amali AA, Her GM, et al. Thioacetamide accelerates steatohepatitis, cirrhosis and HCC by expressing HCV core protein in transgenic zebrafish Danio rerio. Toxicology. 2008;243(1‐2):11‐22. [DOI] [PubMed] [Google Scholar]
- 84. Liu W, Chen JR, Hsu CH, et al. A zebrafish model of intrahepatic cholangiocarcinoma by dual expression of hepatitis B virus X and hepatitis C virus core protein in liver. Hepatology. 2012;56(6):2268‐2276. [DOI] [PubMed] [Google Scholar]
- 85. Haramis AP, Hurlstone A, van der Velden Y, et al. Adenomatous polyposis coli‐deficient zebrafish are susceptible to digestive tract neoplasia. EMBO Rep. 2006;7(4):444‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Evason KJ, Francisco MT, Juric V, et al. Identification of chemical inhibitors of beta‐catenin‐driven liver tumorigenesis in zebrafish. PLoS Genet. 2015;11(7):e1005305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yao Y, Sun S, Wang J, et al. Canonical Wnt signaling remodels lipid metabolism in zebrafish hepatocytes foLLOWING Ras oncogenic insult. Cancer Res. 2018;78(19):5548‐5560. [DOI] [PubMed] [Google Scholar]
- 88. Gomez A, Wellbrock C, Gutbrod H, Dimitrijevic N, Schartl M. Ligand‐independent dimerization and activation of the oncogenic Xmrk receptor by two mutations in the extracellular domain. J Biol Chem. 2001;276(5):3333‐3340. [DOI] [PubMed] [Google Scholar]
- 89. Calvisi DF, Ladu S, Gorden A, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130(4):1117‐1128. [DOI] [PubMed] [Google Scholar]
- 90. Tsuda H, Hirohashi S, Shimosato Y, Ino Y, Yoshida T, Terada M. Low incidence of point mutation of c‐Ki‐ras and N‐ras oncogenes in human hepatocellular carcinoma. Jpn J Cancer Res. 1989;80(3):196‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li L, Yichen C, Charles C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851‐11858. [DOI] [PubMed] [Google Scholar]
- 92. Hayashi J, Aoki H, Kajino K, Moriyama M, Arakawa Y, Hino O. Hepatitis C virus core protein activates the MAPK/ERK cascade synergistically with tumor promoter TPA, but not with epidermal growth factor or transforming growth factor alpha. Hepatology. 2000;32(5):958‐961. [DOI] [PubMed] [Google Scholar]
- 93. Emelyanov A, Parinov S. Mifepristone‐inducible LexPR system to drive and control gene expression in transgenic zebrafish. Dev Biol. 2008;320:113‐121. [DOI] [PubMed] [Google Scholar]
- 94. Kaposi‐Novak P, Libbrecht L, Woo HG, et al. Central role of c‐Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009;69(7):2775‐2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Guan XY, Fang Y, Sham JS, et al. Recurrent chromosome alterations in hepatocellular carcinoma detected by comparative genomic hybridization. Genes Chromosom Cancer. 2010;29(2):110‐116. [PubMed] [Google Scholar]
- 96. Li Z, Zheng W, Wang Z, et al. A transgenic zebrafish liver tumor model with inducible Myc expression reveals conserved Myc signatures with mammalian liver tumors. Dis Models Mech. 2013;6(2):414‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kar S, Yousem SA, Carr BI. Endothelin‐1 expression by human hepatocellular carcinoma. Biochem Biophys Res Commun. 1995;216(2):514‐519. [DOI] [PubMed] [Google Scholar]
- 98. Ishibashi M, Fujita M, Nagai K, et al. Production and secretion of endothelin by hepatocellular carcinoma. J Clin Endocrinol Metab. 1993;76(2):378‐383. [DOI] [PubMed] [Google Scholar]
- 99. Pal SK, Figlin RA, Reckamp K. Targeted therapies for non‐small cell lung cancer: an evolving landscape. Mol Cancer Ther. 2010;9(7):1931‐1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lu JW, Hsia Y, Yang WY, et al. Identification of the common regulators for hepatocellular carcinoma induced by hepatitis B virus X antigen in a mouse model. Carcinogenesis. 2012;33(1):209‐219. [DOI] [PubMed] [Google Scholar]
- 101. Lu JW, Liao CY, Yang WY, et al. Overexpression of endothelin 1 triggers hepatocarcinogenesis in zebrafish and promotes cell proliferation and migration through the AKT pathway. PLoS One. 2014;9(1):e85318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mancarella S, Krol S, Crovace A, et al. Validation of hepatocellular carcinoma experimental models for TGF‐beta promoting tumor progression. Cancers. 2019;11(10):1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Coulouarn C, Factor VM, Thorgeirsson SS. Transforming growth factor‐β gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology. 2008;47(6):2059‐2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yan C, Yang Q, Shen H‐M, Spitsbergen JM, Gong Z. Chronically high level of tgfb1a induction causes both hepatocellular carcinoma and cholangiocarcinoma via a dominant Erk pathway in zebrash. Oncotarget. 2017;8:77096‐77109. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 105. Bronner C, Krifa M, Mousli M. Increasing role of UHRF1 in the reading and inheritance of the epigenetic code as well as in tumorogenesis. Biochem Pharmacol. 2013;86(12):1643‐1649. [DOI] [PubMed] [Google Scholar]
- 106. Tien AL, Senbanerjee S, Kulkarni A, et al. UHRF1 depletion causes a G2/M arrest, activation of DNA damage response and apoptosis. Biochem J. 2011;435(1):175‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mousli M, Hopfner R, Abbady AQ, et al. ICBP90 belongs to a new family of proteins with an expression that is deregulated in cancer cells. Br J Cancer. 2003;89(1):120‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wang F, Yang YZ, Shi CZ, et al. UHRF1 promotes cell growth and metastasis through repression of p16(ink(4)a) in colorectal cancer. Ann Surg Oncol. 2012;19(8):2753‐2762. [DOI] [PubMed] [Google Scholar]
- 109. Unoki M, Daigo Y, Koinuma J, Tsuchiya E, Hamamoto R, Nakamura Y. UHRF1 is a novel diagnostic marker of lung cancer. Br J Cancer. 2010;103(2):217‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jin W, Chen L, Chen Y, et al. UHRF1 is associated with epigenetic silencing of BRCA1 in sporadic breast cancer. Breast Cancer Res Treat. 2010;123(2):359‐373. [DOI] [PubMed] [Google Scholar]
- 111. Mudbhary R, Hoshida Y, Chernyavskaya Y, et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014;25(2):196‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tsai JF, Yang J. Epithelial–mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192‐2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Yang J, Mani SA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927‐939. [DOI] [PubMed] [Google Scholar]
- 114. Lee TK, Poon RTP, Yuen AP. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial‐mesenchymal transition. Clin Cancer Res. 2006;12(18):5369‐5376. [DOI] [PubMed] [Google Scholar]
- 115. Nakayama J, Lu JW, Makinoshima H, Gong Z. A novel zebrafish model of metastasis identifies the HSD11beta1 inhibitor adrenosterone as a suppressor of epithelial‐mesenchymal transition and metastatic dissemination. Mol Cancer Res. 2020;18(3):477‐487. [DOI] [PubMed] [Google Scholar]
- 116. Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR‐ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344(14):1038‐1042. [DOI] [PubMed] [Google Scholar]
- 117. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380‐2388. [DOI] [PubMed] [Google Scholar]
- 118. Weisberg E, Manley PW, Cowan‐Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR‐ABL for the treatment of imatinib‐resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007;7(5):345‐356. [DOI] [PubMed] [Google Scholar]
- 119. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378‐390. [DOI] [PubMed] [Google Scholar]
- 120. Marisi G, Cucchetti A, Ulivi P, et al. Ten years of sorafenib in hepatocellular carcinoma: are there any predictive and/or prognostic markers? World J Gastroenterol. 2018;24(36):4152‐4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Abou‐Alfa GK, Schwartz L, Ricci S, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24(26):4293‐4300. [DOI] [PubMed] [Google Scholar]
- 122. Villanueva A, Chiang DY, Newell P, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135(6):1972‐1983, 1983.e1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wang Z, Zhou J, Fan J, et al. Effect of rapamycin alone and in combination with sorafenib in an orthotopic model of human hepatocellular carcinoma. Clin Cancer Res. 2008;14(16):5124‐5130. [DOI] [PubMed] [Google Scholar]
- 124. Philip PA, Mahoney MR, Allmer C, et al. Phase II study of Erlotinib (OSI‐774) in patients with advanced hepatocellular cancer. J Clin Oncol. 2005;23(27):6657‐6663. [DOI] [PubMed] [Google Scholar]
- 125. Nguyen KS, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non‐small‐cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer. 2009;10(4):281‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wilson GS, Tian A, Hebbard L, et al. Tumoricidal effects of the JAK inhibitor Ruxolitinib (INC424) on hepatocellular carcinoma in vitro. Cancer Lett. 2013;341(2):224‐230. [DOI] [PubMed] [Google Scholar]
- 127. North T, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. White RM, Cech J, Ratanasirintrawoot S, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471(7339):518‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wang C, Tao W, Wang T, et al. Rosuvastatin, identified from a zebrafish chemical genetic screen for antiangiogenic compounds, suppresses the growth of prostate cancer. Eur Urol. 2010;58(3):418‐426. [DOI] [PubMed] [Google Scholar]
- 130. Colombino M, Sperlongano P, Izzo F, et al. BRAF and PIK3CA genes are somatically mutated in hepatocellular carcinoma among patients from South Italy. Cell Death Dis. 2012;3:e259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Suzuki K, Hayashi N, Yamada Y, et al. Expression of the c‐met protooncogene in human hepatocellular carcinoma. Hepatology. 1994;20(5):1231‐1236. [PubMed] [Google Scholar]
- 132. Zhang YJ, Jiang W, Chen CJ, et al. Amplification and overexpression of cyclin D1 in human hepatocellular carcinoma. Biochem Biophys Res Commun. 1993;196(2):1010‐1016. [DOI] [PubMed] [Google Scholar]
- 133. Liew CT, Li HM, Lo KW, et al. High frequency of p16INK4A gene alterations in hepatocellular carcinoma. Oncogene. 1999;18(3):789‐795. [DOI] [PubMed] [Google Scholar]
- 134. Li T, Zhu Y, Qin CY, et al. Expression and prognostic significance of vascular endothelial growth factor receptor 1 in hepatocellular carcinoma. J Clin Pathol. 2012;65(9):808‐814. [DOI] [PubMed] [Google Scholar]
- 135. Breuhahn K, Vreden S, Haddad R, et al. Molecular profiling of human hepatocellular carcinoma defines mutually exclusive interferon regulation and insulin‐like growth factor II overexpression. Cancer Res. 2004;64(17):6058‐6064. [DOI] [PubMed] [Google Scholar]
- 136. Kan Z, Zheng H, Liu X, et al. Whole‐genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23(9):1422‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Chu JS, Ge FJ, Zhang B, et al. Expression and prognostic value of VEGFR‐2, PDGFR‐beta, and c‐Met in advanced hepatocellular carcinoma. J Exp Clin Cancer Res. 2013;32(32):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Stock P, Monga D, Tan X, Micsenyi A, Loizos N, Monga SP. Platelet‐derived growth factor receptor‐alpha: a novel therapeutic target in human hepatocellular cancer. Mol Cancer Ther. 2007;6(7):1932‐1941. [DOI] [PubMed] [Google Scholar]
- 139. Bae JJ, Rho JW, Lee TJ, et al. Loss of heterozygosity on chromosome 10q23 and mutation of the phosphatase and tensin homolog deleted from chromosome 10 tumor suppressor gene in Korean hepatocellular carcinoma patients. Oncol Rep. 2007;18(4):1007‐1013. [PubMed] [Google Scholar]
- 140. Tu W, Luo M, Wang Z, et al. Upregulation of SATB1 promotes tumor growth and metastasis in liver cancer. Liver Int. 2012;32(7):1064‐1078. [DOI] [PubMed] [Google Scholar]
- 141. Yoshikawa H, Matsubara K, Qian G, et al. SOCS‐1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth‐suppression activity. Nat Genet. 2001;28(1):29‐35. [DOI] [PubMed] [Google Scholar]
- 142. Niwa Y, Kanda H, Shikauchi Y, et al. Methylation silencing of SOCS‐3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005;24(42):6406‐6417. [DOI] [PubMed] [Google Scholar]
- 143. Yeh YC, Tsai JF, Chuang LY, et al. Elevation of transforming growth factor alpha and its relationship to the epidermal growth factor and alpha‐fetoprotein levels in patients with hepatocellular carcinoma. Cancer Res. 1987;47(3):896‐901. [PubMed] [Google Scholar]
- 144. Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26(15):2166‐2176. [DOI] [PubMed] [Google Scholar]
- 145. Chiang DY, Villanueva A, Hoshida Y, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68(16):6779‐6788. [DOI] [PMC free article] [PubMed] [Google Scholar]