

Abstract
The prolyl-alanyl-specific endoprotease (EndoPro) is an industrial enzyme produced in Aspergillus niger. EndoPro is mainly used for food applications but also as a protease in proteomics. In-depth characterization of this enzyme is essential to understand its structural features and functionality. However, there is a lack of analytical methods capable of maintaining both the structural and functional integrity of separated proteoforms. In this study, we developed an anion exchange (AEX) method coupled to native mass spectrometry (MS) for profiling EndoPro proteoforms. Moreover, we investigated purified EndoPro proteoforms with complementary MS-based approaches, including released N-glycan and glycopeptide analysis, to obtain a comprehensive overview of the structural heterogeneity. We showed that EndoPro has at least three sequence variants and seven N-glycosylation sites occupied by high-mannose glycans that can be phosphorylated. Each glycosylation site showed high microheterogeneity with ∼20 glycans per site. The functional characterization of fractionated proteoforms revealed that EndoPro proteoforms remained active after AEX-separation and the specificity of these proteoforms did not depend on N-glycan phosphorylation. Nevertheless, our data confirmed a strong pH dependence of EndoPro cleavage activity. Altogether, our study demonstrates that AEX-MS is an excellent tool to characterize complex industrial enzymes under native conditions.
Keywords: anion exchange chromatography, mass spectrometry, structure−function relationship, prolyl-alanyl-specific endoprotease, native separation
Introduction
Industrially produced enzymes are widely used in everyday processes, from cleaning clothes to making food such as bread and beer.1,2 Usually, these enzymes are produced through fermentation processes by microbes, including bacteria or fungi.3 Many of these microorganisms exhibit a remarkably versatile metabolism, and through secretory pathways, they produce proteins with various post-translational modifications (PTMs), including glycosylation.4 Such modifications may have either positive, negative, or no impact on functionality. For instance, the glycosylation of industrially produced lipases may affect thermal stability and catalytic activity.5 Therefore, both structural and functional characterization is essential for a deep understanding of structure–function relationships of protein products. In this regard, N-glycosylation is one of the most critical PTMs as it has a significant effect on many protein properties (e.g., secretion, activity, stability, and substrate specificity). However, due to the sheer molecular complexity of N-glycans and N-glycosylation meta-, macro-, and microheterogeneity,6 hundreds of proteoforms can be found on a single protein, making characterization challenging.7,8
An interesting industrial enzyme with excellent application potential is the highly glycosylated prolyl-alanyl-specific endoprotease (EndoPro). This enzyme is secreted by the filamentous fungus such as Aspergillus niger. EndoPro is mainly used in food applications, e.g., preventing chill-haze formation in beer by degrading proline-rich peptides and proteins.9 Moreover, EndoPro has proven to be useful for proteomics sample preparation (ProAlanase10) due to its rather unique specificity toward proline/alanine residues and the activity at low temperature and pH.11−13 Hence, the use of EndoPro for these applications emphasizes the need for its comprehensive characterization and better understanding. Šebela et al.13 already examined the glycosylation of this enzyme using MALDI-TOF-MS. The authors detected a large heterogeneity of high-mannose N-glycans and suggested the presence of phosphorylated glycans. These findings encouraged us to develop an analytical pipeline for in-depth structural characterization of different EndoPro glycoforms and investigate their activity.
It is challenging to determine the structure–function relationship of a highly heterogeneous enzyme such as EndoPro. A critical step is separating proteoforms using an approach compatible with both structural characterization and functional evaluation. Conventional liquid separation mechanisms (e.g., reversed-phase liquid chromatography; RPLC) are not suitable for this purpose due to denaturing conditions during the separation. In reaction to that, we have been witnessing rapid development of separation techniques maintaining native-like conformations of the protein analytes, including ion-exchange chromatography (IEX),14,15 size exclusion chromatography (SEC),16,17 and hydrophobic interaction chromatography (HIC).18,19 However, one of the primary bottlenecks for broader implementation of these approaches is the mobile phase incompatibility with mass spectrometry (MS) due to the high concentrations of nonvolatile salts required for the separation. Recently, IEX methods using pH gradients (i.e., chromatofocusing) have become popular for intact protein analysis as this technique uses low ionic strength and volatile mobile phases, enabling online hyphenation with MS. Elution is accomplished by changing the mobile phase pH until it equals the isoelectric point (pI) of each protein variant.20 Although such methods have already been utilized for the structural characterization of biopharmaceuticals,21−24 their compatibility with further assessment of functionality and activity of the separated proteoforms is less investigated.
Here, we developed an AEX method coupled to native MS to separate differentially charged proteoforms of EndoPro. We complemented the AEX-MS measurements by a comprehensive MS-based analysis of N-glycosylation (i.e., released N-glycan and glycopeptide analysis), which was previously shown to be a suitable approach for complex glycoproteins.25−27 The data interpretation revealed that EndoPro is an extremely complex protein exhibiting different sequence variants and containing seven N-glycosylation sites occupied by a variety of high-mannose glycans, including phosphorylated glycans at specific sites. Moreover, we evaluated the functional properties of separated EndoPro proteoforms by investigating their activity and cleavage specificity. Overall, the AEX-MS method shows great potential to characterize complex industrial enzymes under native conditions allowing further functional characterization.
Experimental Section
Materials
Ammonium formate was purchased from Fluka (Steinheim, Germany). Acetic acid, 2-aminobenzoic acid (2-AA), 2-picoline borane (2-PB), ammonium acetate, ammonium bicarbonate (ABC), bovine serum albumin (BSA), cesium iodide, cytochrome C (cyt C), dimethyl sulfoxide (DMSO), disodium hydrogen phosphate dihydrate, Tris (2-carboxyethyl)phosphine hydrochloride (TCEP), chloroacetamide (CAA), dithiothreitol (DTT), iodoacetamide (IAA), nonidet P-40 substitute (NP-40), potassium dihydrogen phosphate, sodium chloride, super-DHB, and urea were obtained from Sigma-Aldrich (Steinheim, Germany). Formic acid (FA) and 2-propanol (IPA) were supplied by Riedel-De Haen (Seelze, Germany). The HPLC-grade acetonitrile (ACN) was purchased from Biosolve (Valkenswaard, The Netherlands). Trifluoroacetic acid (TFA) was obtained from Merck (Darmstadt, Germany). Recombinant Peptide-N-Glycosidase F (PNGase F) was purchased from Roche Diagnostics or Promega. Milli-Q water was provided by Purelab ultra (ELGA Labwater, Ede, The Netherlands). EndoPro purified fromAspergillus nigerwas provided by DSM formulated in 50% glycerol.
Gel Electrophoresis
For the SDS-PAGE and IEF gel of the EndoPro reference sample, the EndoPro stock solution was diluted 80× with 50 mM ABC buffer and divided into 3 aliquots (A, B, and C). To sample A, 20 μL 50 mM ABC buffer was added and the sample was stored in the freezer immediately. For sample B, 20 μL 50 mM ABC buffer was added and the sample was incubated overnight at 37 °C and 1000 rpm. Sample C was deglycosylated by addition of 20 μL PNGaseF an incubation overnight at 37 °C and 1000 rpm. For the SDS-PAGE gel, 65 μL of each sample was mixed with 25 μL NuPAGE LDS sample buffer (4×) (Invitrogen) and 10 μL NuPAGE Sample Reducing Agent (10×) (Invitrogen). Samples were heated for 10 min at 70 °C prior to loading on the gel. For the IEF gel, 25 μL of each sample was mixed with 25 μL IEF pH 3–7 Sample buffer (2×) and 10 μL was loaded on gel.
For the SDS-PAGE gel of the collected fractions, samples were prepared by dissolving around 1 μg protein in 18 μL Milli-Q water and 6 μL Laemmli buffer (4×) containing mercaptoethanol. Subsequently, each sample was heated for 10 min at 100 °C. From the samples, 20 μL was applied on NuPage 4–12% Bis-Tris 10-well gel (Invitrogen). The gel was run at 200 V for 37 min using 20× diluted MES SDS running buffer (Novex). After that, the gel was rinsed three times with Milli-Q water and stained with SimplyBlue SafeStain (Invitrogen).
Intact Reversed Phase Chromatography–Mass Spectrometry
The RPLC analysis of intact deglycosylated EndoPro was performed on an Acquity I-class instrument (Waters), equipped with a binary pump, autosampler, and column thermostatted compartment. A Waters Acquity UPLC Protein BEH300 C4 column (1.7 μm, 300 Å, 2.1 × 50 mm) was used. The separation was performed at 75 °C using mobile phases composed of 0.1% FA in milli-Q (A) and 90% ACN + 0.1% FA (B). The gradient was programmed as follows: 3% B for 2 min, 17% B at 2.2 min, 60% B at 8 min and 95% B at 8.2 min, 10% B at 9 min, 95% B at 10 min, 10% B at 11 min, 95% B at 12 min, 3% B at 12.2 min, followed by 3% B for 2.8 min. The separation was performed at a flow rate of 0.2 mL/min and an injection volume of 2 μL. For the RPLC-MS measurement, the Synapt G2-S (Waters, Wilmslow, UK) was used, operated in positive mode. The source temperature, desolvation temperature, desolvation gas flow, and nebulizer gas flow were set to 120 °C, 400 °C, 700 L/h, and 6 bar, respectively. The scan time was 1 s, the capillary voltage was 1 kV, the sampling cone was 40, the source offset was 80, and the collision energy was 4 eV. The m/z range was 500–3500.
Intact Anion Exchange Chromatography Coupled to Mass Spectrometry
The AEX-MS measurements were performed with a biocompatible Ultimate 3000 instrument (Thermo Fisher Scientific) coupled to Bruker 15T solariX Fourier-transform ion cyclotron resonance (FTICR) MS (Bruker Daltonics, Bremen, Germany). A ProPac SAX-10 column (2.0 × 250 mm, 10 μm) was purchased from Thermo Scientific (Landsmeer, The Netherlands). The optimal mobile phases contained 50 mM ammonium formate pH 5.5 (A) and 50 mM FA pH 2.5 (B). The optimized gradient for the separation of EndoPro started at 30% B. After 0.1 min, the percentage of mobile phase B was increased to 75%, followed by a linear gradient from 75% to 90% B in 15 min. Finally, the column was cleaned (100% B for 5 min) and equilibrated (30% B for 20 min). The flow rate, column temperature, and injected amount were 0.25 mL/min, 25 °C, and 55 μg, respectively. For fraction collection, 100 μg of EndoPro was injected and peaks were collected at 10.1 min (Fraction 1), 12.3 min (Fraction 2), 14.3 min (Fraction 3), 19.4 min (Fraction 4), and 25.9 min (Fraction 5). The fractions were collected in tubes containing 1 M ammonium formate solution to increase the pH immediately after collection. Finally, the fractions were concentrated to 1 mg/mL using Vivaspin 500 spin filters with a 10 kDa cutoff (GE Healthcare, Munich, Germany). Prior to further analysis, the fractions were diluted in the appropriate solution.
MS detection was performed in positive-ion mode, and a flow splitter was used prior to ionization, reducing the flow five times. Calibration was performed by direct infusion of cesium iodide at the start of the measurement (2.0 mg/mL in 50% ACN with 0.1% FA). Dopant enriched nitrogen (DEN) gas was used to increase sensitivity with IPA as dopant. The ESI capillary voltage was 4000 V and end plate offset −500 V. The nebulizer gas flow rate was 0.8 bar, the dry gas flow rate was 3 L/min, and the dry gas temperature of the nitrogen was 200 °C. The ion funnel 1 was set at 180 V, radiofrequency amplitude at 300 Vpp, and skimmer 1 at 135 V. The In-source collision energy was 40 V and the collision voltage in the collision cell was set to −15 V. The acquisition m/z-range was between 1287.5–8000. The accumulation time was set to 1s and the data acquisition size to 32 000 data points (with transient time of 0.0918 s). The final mass spectrum was obtained by the summation of 10 spectra.
Released N-Glycan Analysis with MALDI-FTICR-MS
The following steps were performed: N-glycan release, the 2-AA labeling, cotton hydrophilic interaction liquid chromatography (HILIC) purification, and spotting on the MALDI target plate. The complete procedure was performed in triplicate for the nonseparated EndoPro and AEX fractions. For the N-glycans release, EndoPro and AEX fractions were diluted to 2 μg/μL in 1× PBS solution. The 1× PBS solution was prepared by diluting 5× PBS (28.5 mg/mL Na2HPO4·2H2O, 2.4 mg/mL KH2PO4 and 42.5 mg/mL NaCl dissolved in Milli-Q). The sample (5 μL) was added to 10 μL 1× PBS followed by addition of a 10 μL freshly prepared release mix (2.5 U PNGase F in 50:50 5× PBS:4% NP-40). The sample was incubated overnight at 37 °C. The 2-AA labeling was performed by addition of 10 μL released glycan sample to 10 μL of labeling solution (50:50 2-AA (48 mg/mL) in DMSO and acetic acid: 2-PB (107 mg/mL) in DMSO) after which the samples were incubated for 2 h at 60 °C. After that, 113 μL ACN was added, and the samples were vortexed. The cotton HILIC purification was performed as described previously.28,29 The HILIC tips were prepared by placing a 3 mm cotton thread (Pipoos, Utrecht, The Netherlands) in a 20 μL pipet tip. The cotton was flushed with Milli-Q water and followed by conditioning with 85% ACN. Subsequently, the sample was loaded on the cotton by pipetting the sample 20 times up and down. After that, the cotton was washed with 85% ACN containing 1% TFA and 85% ACN. The glycans were eluted from the cotton in 10 μL Milli-Q. Finally, 1 μL of the purified sample was spotted on the MALDI plate (800/384 MTP AnchorChip, Bruker Daltonics, Bremen, Germany) together with 1 μL of super-DHB matrix (5 mg/mL in 50% ACN), and the spots were dried to air, followed by MALDI FTICR-MS measurement.
The MALDI-FTICR measurements were performed in negative-ion mode using a 15T SolariX FTICR-MS with a ParaCell and a SmartBeam-II laser system (Bruker Daltonics), which was operated at a frequency of 500 Hz with 200 laser shots per spot. The laser power was set to 20%. The mass spectra of the spotted samples were an average of 20 acquired scans in the m/z-range from 1011.86 to 5000.00 with 1 M data points.
Glycopeptide Profiling of EndoPro AEX Fractions
For the in-solution digestion of the AEX fractions, each sample (5 μg in 100 mM ammonium formate, pH 8) was reduced with 5 mM TCEP at 56 °C for 30 min and subsequently alkylated with 10 mM CAA at room temperature for 30 min in the dark. The samples were digested overnight at 37 °C with chymotrypsin at an enzyme-to-protein ratio of 1:50 (w/w). The complete procedure of each fraction was performed in duplicate. All proteolytic digests containing glycopeptides were desalted using an Oasis PriME HLB plate, then dried and stored at −80 °C until MS analysis.
The peptide mixtures (typically 300 fmol of EndoPro peptides) were separated and analyzed using a Thermo Scientific Ultimate 3000 RSLCnano system (Thermo Fisher Scientific, Bremen, Germany) coupled online to an Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific). Peptides were first trapped on the trapping cartridge (PepMap100 C18, 5 μm, 5 mm × 300 μm; Thermo Scientific) before RP separation on an in-house packed analytical column (Poroshell EC-C18, 2.7 μm, 50 cm × 75 μm) at 40 °C. Trapping was performed for 1 min in mobile-phase solvent A consisted of 0.1% FA in water. Mobile-phase solvent B consisted of 0.1% FA in ACN. A 60 min gradient was used as follows: 0–45 min, 9–40% B; 45–46 min, 40% B; 46–53 min, 95% B; 53–59 min, 9% B; 59–60 min, 0% B. The flow rate was set to 300 nL/min. Nanospray was achieved using a coated fused silica emitter (New Objective, Cambridge, MA) (outer diameter, 360 μm; inner diameter, 20 μm; tip inner diameter, 10 μm) biased to 2 kV. The mass spectrometer was operated in positive ion mode, and the spectra were acquired in the data-dependent acquisition mode. For the MS scans, the mass range was set from 350 to 2000 m/z at a resolution of 60 000 and the AGC target was set to 4 × 105. For the MS/MS measurements, HCD and electron-transfer and higher-energy collision dissociation (EThcD) were used. HCD was performed with a normalized collision energy of 30%. The supplementary activation energy of 27% was used for EThcD. For the MS/MS scans, the lowest mass was fixed at 120 m/z and the resolution was set to 30 000; the AGC target was set to 5 × 105. The precursor isolation width was 1.6 Da and the maximum injection time was set to 200 ms.
Digestion Speed and Specificity
The digestion specificity was determined for EndoPro and the AEX fractions. Cytochrome C (Cyt C) and bovine serum albumin (BSA) were selected as substrates for evaluating the digestion speed and specificity, respectively. For Cyt C, the digestion was directly performed in 50 mM ammonium formate buffer (pH 2 or 4) at 37 °C without denaturation, reduction or alkylation (this protein being devoid of disulfide bridges). The incubation time of the digestion at pH 2 was 0, 1, 2, 3, 4, 5, 7, 9, 11, or 30 min. For digestion at pH 4, the incubation time was 0, 15, 30, 90, 120, 200, 300, or 480 min. The obtained digests were prepared and loaded on a gel with the same procedure as for the SDS-PAGE gel of the AEX-fractions. These gels were stained with silverQuest staining (Invitrogen). The relative density of the band of the undigested Cyt C was extracted using ImageJ software.30
For BSA, the reduction and denaturation were performed by incubation of the protein in a 200 mM ammonium formate solution (pH 8) containing 4 M urea and 10 mM DTT for 30 min at 57 °C. After reduction, the samples were alkylated by adding 20 mM IAA and incubating for 30 min at room temperature in the dark. After that, the excess of IAA was quenched with the addition of 5 mM DTT. Subsequently, the sample was divided into two aliquots, and both were diluted five times. By titrating with FA, the pH of the first sample was lowered to 4 and the second sample to pH 2. The digestion was performed overnight at 37 °C after addition of EndoPro or one of the AEX fractions (enzyme-to-protein-ratio of 1:50 (w/w)). The obtained peptides were measured with RPLC-MS/MS analysis as described for the glycopeptide profiling. The complete procedure was performed 4 times for both pH values.
Data Analysis
The data analysis of the AEX-MS measurements was done using Compass data analysis from Bruker Daltonics. For the charge state deconvolution, the Maximum Entropy algorithm was used (mass range: 65 000–73 500 Da and resolving power: 3000). The base peak chromatogram (BPC) was smoothed with the Gauss smoothing algorithm (1 cycle). For calculation of the glycan compositions, average masses were used, including hexose (Hex, 162.14 Da), N-acetylhexosamine (HexNAc, 203.20 Da), and phosphorylation (Phos, 79.98 Da). For released N-glycans, the relative quantification of the mass spectra was performed by exporting the raw spectra as a text file (x,y) followed by processing with MassyTools (v1.0.2). The peptide-centric and digestion specificity data were interpreted using the Byonic software version 3.4.0 suite (Protein Metrics Inc., USA) and further validation was performed manually. The following parameters were used for data searches: precursor ion mass tolerance, 10 ppm; product ion mass tolerance, 20 ppm; fixed modification, Cys carbamidomethyl; variable modification: Met oxidation, and customized N-glycan database, which was based on the released glycan analysis (Table S1). Chymotrypsin (C-terminal, FYLWM) enzyme specificity search was chosen with a maximum of 3 missed cleavages. The database used contained the EndoPro protein amino acid sequence (Figure S1). Profiling and relative quantification of PTM modified EndoPro peptides were achieved by using the software Skyline.31 The information obtained from the released glycan analysis and MS/MS analysis was combined with manual interpretation of MS1 glycopeptide profiles extracted from corresponding retention time windows (Table S2). Quantification was performed by extracted ion chromatograms (EICs). For peak area calculations, the first three isotopes were taken from each manually validated peptide proteoform. Per each N-glycosylation site, all glycoforms’ integrated peak area was summed and set to 100%, so each glycoform was expressed as % area of the total area. The average peptide ratios from the two independently measured samples were taken as a final estimation of the abundance. The glycan structures of each glycoform were manually annotated. The reported glycan structures are depicted without the linkage type of glycan units since the acquired MS/MS patterns do not directly provide such information. For the digestion specificity of EndoPro, a nonspecific search was done by the Byonic software version 3.4.0 suite (Protein Metrics Inc., USA) with no maximum of missed cleavage. The following parameters were used for data searches: precursor ion mass tolerance, 10 ppm; product ion mass tolerance, 20 ppm; fixed modification, Cys carbamidomethyl; variable modification: Met oxidation. The fasta file used for the peptide searches contained BSA amino acid sequence (UniProt code: P02769, ALBU_BOVIN). Byonic Viewer (Protein Metrics Inc.) was used to filter the peptide spectral matches (PSMs) (|Log Prob| > 3). The cleavage specificity and peptide characteristics (i.e., peptide length and charge) were determined using in-house R scripts.
Combining Native MS, Glycopeptide-Centric Proteomic, and Glycomic Data
Annotation of the obtained proteoforms in Fraction 1 was assessed by an integrative approach combining the native MS and glycoproteomic data. This approach has been described previously by Yang et al.7 and further developed by implementing peak annotation in a site-specific manner by Lin et al.32 In short, in silico data construction of the native-like protein spectra was performed based on the masses and relative abundances of all site-specific PTMs derived from the glycopeptide analysis. First, a library of proteoforms with specific masses and probability ranks was generated using the data derived from the glycopeptide analysis. All proteoform mass combinations were ranked according to the intensity of glycopeptides from which they were constructed and the match between their theoretical and experimental mass. The proteoforms with the masses within 100 ppm, but different glycopeptide combinations, were sorted based on the better mass match and intensity of the glycopeptides (closer mass matches and higher intensities get higher ranks). The experimental masses of proteoforms were then matched to the generated library with the highest-ranked proteoforms. This procedure allows visualizing proteoforms in the native spectrum with their PTM compositions in a site-specific manner. Still, each proteoform peak is likely a mixture of many other glycoform combinations. Finally, the constructed spectra were compared to the experimental native spectrum of Fraction 1. Next to spectra simulations based on glycopeptide intensities, the simulations were also performed with intensities derived from the released N-glycan data. The scripts used for the spectra annotation are available at https://github.com/juer120/NativePTMannotation.
Results
The enzyme EndoPro is a 65–72 kDa glycoprotein. On the basis of the amino acid sequence, EndoPro has seven N-glycosylation sequons (NXT/S, where X is any amino acid except proline) (Figure S1). Šebela et al.13 showed that the glycan heterogeneity is mostly due to variation of the number of mannoses as for most glycoproteins fromAspergillus niger.33 Interestingly, it was suggested that these glycans might contain a phosphate group within their structure.33 Besides extensive glycosylation, the enzyme shows backbone mass heterogeneity (i.e., N-terminal truncation). At least three different variants are known, including the mature protein (M), a variant lacking an N-terminal alanine (M-A), and a variant lacking two N-terminal alanine residues (M-AA).13 Indeed, we performed an intact LC-MS analysis of deglycosylated EndoPro, confirming the presence of several sequence variants, including M, M-A, M-AA, and M+S. (Figure S2a). A structural model of EndoPro is not yet available; nevertheless, alignment analysis of the amino acid sequence of EndoPro showed high similarity with the S28 family of clan SC of serine proteases.34
Separating and Profiling of EndoPro Proteoforms by AEX-MS
EndoPro has a pI between 3.5 and 4.2. After deglycosylation, the pI increases to 5.2, indicating that glycosylation significantly influences the pI of this protein (Figure S2bc). To characterize the difference in charge of the glycosylation, we developed an AEX-MS method to separate proteoforms based on their pI using a decreasing pH gradient. The buffer composition and concentration were carefully optimized to find suitable conditions for both chromatographic separation and MS detection. Commonly used ammonium acetate buffers did not allow elution of EndoPro proteoforms (Figure S3a), while ammonium formate eluted even the most acidic species. Furthermore, our experiments showed that low salt concentrations (<50 mM) increased retention time and peak width resulting in lower overall separation quality (Figure S3b). We achieved the best results with 50 mM ammonium formate pH 5.5 as mobile phase A and 50 mM formic acid pH 2.5 as mobile phase B (Figure S4).
The base peak chromatogram (BPC) of the optimized AEX-MS method showed six chromatographic peaks (Figure 1a). Comparison of MS and UV-data shows peak consistency concerning signal pattern and relative intensity except for the first peak eluting at 6 min (Figure S4a,b). After analyzing the peaks with SDS-PAGE, it was concluded that the first peak is most likely a nonproteinaceous species not related to EndoPro, while the other peaks are EndoPro proteoforms (Figure S4d). Next, we investigated high-resolution native mass spectra of the five EndoPro peaks, i.e., Fractions 1–5 (Figure 1b). A closer look at the spectra revealed that recorded protein-ion series have a native-like charge state distribution ranging from 11+ to 16+, indicating that the chromatographic conditions maintained the native protein conformation. Zero-charge deconvolution of the native spectra resolved in each fraction from ∼30 to more than ∼60 co-occurring analytes. The spectrum revealed two major distinct proteoform series differing by a mass corresponding to Alanine residue. Therefore, we used the sequences of the M and M-A variants containing three disulfide bonds in the mass calculations. This calculation resulted in average backbone masses of 54 289.11 Da (M) and 54 218.03 Da (M-A). We observed an array of EndoPro proteoforms differing in the total number of hexoses per protein in each peak. Although mass matching is typically a good approach to annotate native MS spectra of glycoproteins, the enormous complexity and ambiguous mass profiles of EndoPro proteoforms precluded us from assigning the peaks more precisely and reliably. The mechanism of EndoPro proteoform chromatographic separation remained unclear at this point. Nevertheless, we hypothesized that the proteoforms could be separated based on glycan phosphorylation levels. Therefore, we decided to profile the N-glycans and glycopeptides of the AEX fractions and thereby investigate the EndoPro proteoforms more in-depth.
Figure 1.
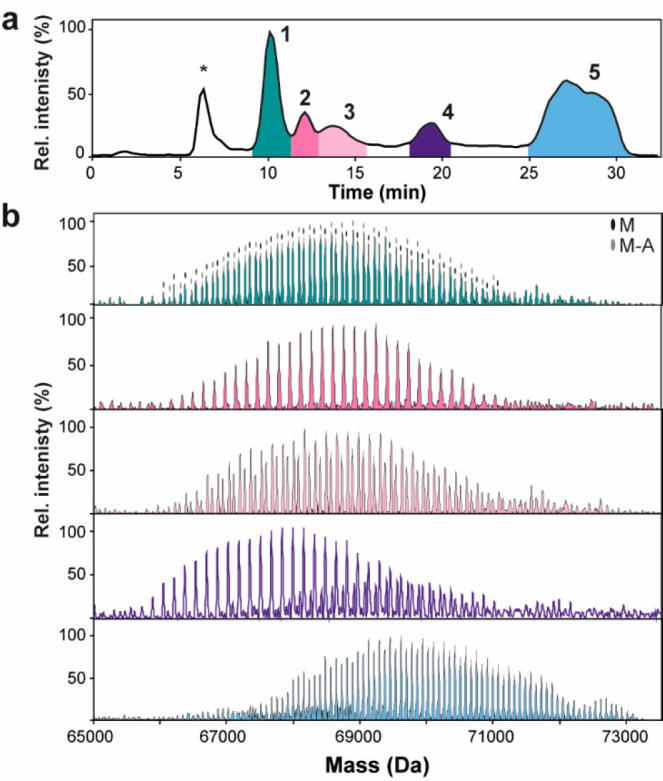
(a) BPC of the optimized AEX-MS method. The peak marked with * is an unknown species (see Figure S4). EndoPro proteoforms (numbered with 1–5) were separated based on a charge with a decreasing pH gradient. The pH was monitored using an inline pH meter, revealing a linear decrease in pH during elution of most EndoPro proteoforms (Figure S4c). (b) Zero charge deconvoluted native mass spectra of AEX-MS method: Fraction 1 (green), Fraction 2 (pink), Fraction 3 (pale pink), Fraction 4 (purple), and Fraction 5 (blue). In the spectrum of Fraction 1, the different sequence variants are indicated (i.e., M and M-A). Expanded spectra can be found in Figure S5.
Glycomics Analysis and Glycopeptide Profiling of EndoPro Proteoforms
The native MS analysis of AEX-separated EndoPro proteoforms provided a comprehensive overview of structural heterogeneity, revealing dozens of EndoPro proteoforms with extensive glycosylation. To further support our native MS data, we analyzed released N-glycans of nonseparated EndoPro and AEX-separated EndoPro proteoforms. We applied an established protocol for N-glycan release by PNGase F and subsequently labeling with 2-AA. The negative ion mode MALDI-MS spectra (Figure S6) provided N-glycan profiles with high mass accuracy, permitting high confidence assignment of glycan compositions. The obtained profiles revealed that a significant portion of N-glycans contains a phosphate group. Next, we quantified the glycan profiles of EndoPro and the AEX fractions (Figure 2, Table S3). As the native MS proteoform profiles of Fraction 1 indicate, glycans with 9 or 10 mannoses are the most abundant. Besides, glycans with 4 to 17 mannoses are partially phosphorylated (one phosphate group per glycan), from which glycans with 10 mannoses are most abundant. In Fraction 1, no phosphoglycans were detected. Moreover, phosphorylated glycans’ levels increased with retention time, indicating that the number of phosphoglycans per proteoform influences the AEX retention (Figure 2, inset).
Figure 2.

Relative quantitation of the released N-glycans of the AEX fractions measured in negative ion mode with MALDI-FTICR-MS. The N-glycans were released with PNGase F and labeled with 2-AA. All samples were measured in triplicate (n = 3) and the error bars show standard deviation. See the Experimental Section for details on the experimental procedure. H = hexose, N = N-acetylhexosamine, and P = phosphate. The inset shows phosphorylation level expressed as relative abundance of neutral and phosphoglycans for nonseparated EndoPro and Fraction 1–5. For the relative abundance of each glycoform, see Table S3.
Next, we examined the microheterogeneity and N-glycosylation site occupancy of the AEX-separated EndoPro proteoforms by glycopeptide analysis. EndoPro has seven known N-glycosylation sites (N60, N122, N186, N204, N288, N315, and N406). We performed several pilot experiments on nonseparated EndoPro to select a suitable protease (i.e., trypsin, chymotrypsin, pepsin, GluC, and AspN) that can cover as many EndoPro N-glycosylation sites as possible. These experiments were performed in different buffers and pH to satisfy various requirements for optimal protease performance. Chymotrypsin digestion was the only one that provided information on all seven N-glycosylation sites. For digesting AEX-separated EndoPro fractions, we decided to perform the chymotryptic digestion directly in the LC mobile phase (ammonium formate) due to the low concentration of the collected fractions and difficulties related to their concentrating. Before adding chymotrypsin, we titrated the samples to higher pH to satisfy optimal pH conditions for chymotrypsin, resulting in expected performance. The digestion was followed by analyzing the peptides with RPLC-MS/MS using HCD and EThcD fragmentation in parallel, yielding high confidence in glycan and peptide backbone sequencing.35 Data interpretation provided information about glycans composition and abundance located at all seven N-glycosylation sites for the AEX fractions. The MS/MS spectra were interpreted by the software Byonic and further validated by manual inspection. For example, the EICs of glycopeptides with amino acid sequence ANWFNSTILPDYCASY (site N288) reveals a clear shift in retention time between neutral and phosphorylated glycans (Figure S7a). Figures S7b,c display assigned HCD spectra of the neutral and corresponding phosphorylated glycopeptide. Both MS/MS spectra contain fragments confirming the glycan composition. Additionally, fragmentation of the phosphorylated glycopeptides generate diagnostic ions [HexPhos + H]+ (m/z 243.026), [Hex2Phos + H]+ (m/z 404.079), and [Hex3Phos + H]+ (m/z 567.132). These ions are not present in fragmentation spectra of glycopeptides with the neutral glycans, providing direct evidence about phosphate attachment. The list of assigned glycoforms used for relative quantification can be found in Table S4.
The obtained glycopeptide profile showed differences in microheterogeneity for all AEX fractions (Figure 3 and S8). Consistent with the released N-glycan data, Fraction 1 contains only neutral high-mannose type glycans. All other fractions show phosphorylation of the glycans with an increase in the level of phosphoglycans at a higher retention time (Figure 3a). Noteworthy, the complexity of the glycoforms (i.e., the number of detected glycans) increases from Fraction 1 to 5. Regarding microheterogeneity, N186, N288, and N315 are less heterogeneous than other sites (Figure 3b). For example, N186 displays low heterogeneity in all fractions with 9 glycoforms in Fraction 1–3 and 11 glycoforms in Fraction 4 and 5. In contrast, the other sites (i.e., N60, N122, N204, and N406) are more heterogeneous, with 15–17 glycoforms per site. Notably, N204 contains glycans with a higher number of mannoses than the other sites (i.e., around 50% of the glycans has 13 or more mannoses, whereas the other sites have less than 12% of these glycans). Fractions containing phosphorylated glycans show, in general, a similar trend of microheterogeneity as Fraction 1, but with additional complexity added by phosphorylated glycans (except for N186, which contained almost no phosphorylated glycans across all fractions). The most abundant phosphorylated N-glycans are located at N60 and N122, especially in Fraction 5. In summary, we observed N-glycans on all seven EndoPro N-glycosylation sites. Partially occupied N-glycosylation sites were not detected, but their presence could not be entirely excluded. For example, Fraction 5, as the most heterogeneous sample, contains more than 160 glycans distributed among seven N-glycosylation sites, which is on average more than 20 glycans per site.
Figure 3.
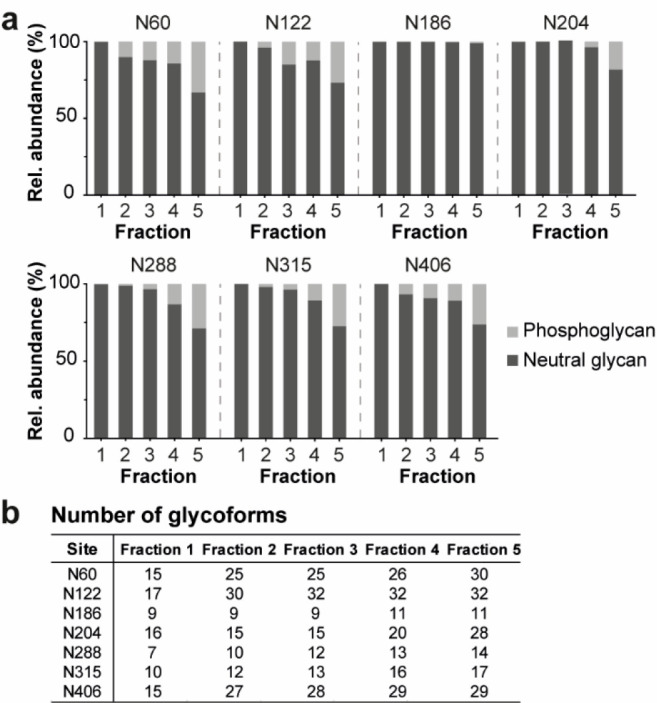
Glycopeptide profiling of the AEX fractions. (a) The phosphorylation level of the glycans expressed as relative abundance of neutral and phosphoglycans for all seven N-glycosylation sites (i.e., N60, N122, N186, N204, N288, N315, and N406). The bars represent the average of two replicates. See Table S2 for more details. (b) The microheterogeneity of each glycosylation site for all AEX fractions. The detailed glycopeptide profile per site of Fractions 1–5 is shown in Figure S8.
Integration of Native MS, Glycopeptide Profile, and Glycomics Data
Having the native MS, released N-glycan, and glycopeptide data of the AEX fractions, we attempted to cross-validate all data and untangle the enormous complexity of proteoform profiles. Our initial tentative annotation of native spectra was based on mass matching, but the complexity of proteoform profiles did not allow us to reveal the true nature of differences among the AEX-separated proteoforms. Similar to our previous studies,7,32 we generated a list of all possible combinations of proteoform masses based on site-specific glycopeptide-centric data resulting in a mass list for the annotation of the native spectra. However, due to the high number of PTM sites and their extensive microheterogeneity, the number of combinations exceeded our method’s computational capacity and precluded us from annotating proteoform profiles with phosphorylated glycans (Fraction 2–5). Still, we managed to successfully annotate the proteoform profile in Fraction 1 (Figure 4). We constructed native-like spectra of Fraction 1 in silico by using masses and relative abundances obtained from the glycopeptide data. Comparing the reconstructed mass profiles with the native spectra revealed a discrepancy between our glycopeptide-centric and native MS data (Figure 4a). For example, proteoforms generated in silico by combining N-glycans with fewer mannoses are not detected in the native proteoform profile. Also, simulated proteoforms containing large glycans with 11–15 mannoses have lower peak intensity than in the native profile. This observation indicates that glycopeptide analysis may be biased and underestimates the abundance of glycopeptides containing large N-glycans. Therefore, we also reconstructed native-like spectra by replacing glycopeptide relative intensities for the released glycan relative intensities. In this way, we assigned to each glycan at each site average relative abundance. Although this visualization does not respect site-specific glycan abundance, the reconstructed spectrum using average glycan abundances shows that the released glycan analysis, in this case, is less biased toward larger glycans compared to glycopeptide analysis. (Figure 4b). The list of all detected masses can be found in Table S5, where the proteoforms in Fraction 1 are also annotated. This data demonstrates the necessity for combining various MS approaches to characterize the complex proteoform profiles of EndoPro.
Figure 4.
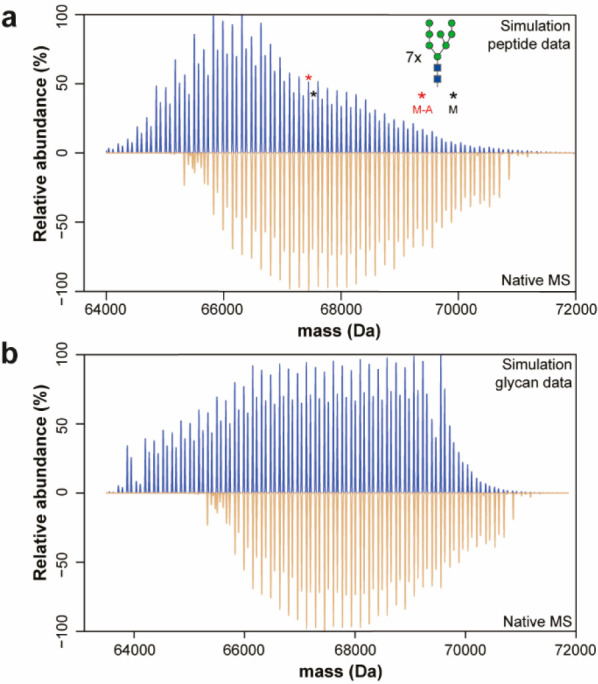
Comparison of Fraction 1 native MS spectrum with the in silico constructed spectrum. (a) The simulated spectrum is based on the peptide-centric proteomic data. (b) The glycomics data was used for the constructed spectrum by replacing peptide intensity for average glycan intensity obtained from released glycan analysis. The simulation algorithm generated theoretical proteoforms with smaller glycans that are not present in the native proteoform profile generated from both the glycopeptide and glycan data.
Activity and Specificity of Purified EndoPro Proteoforms
The enzymatic activity evaluation of the proteoforms is challenging due to maintaining their structures in the folded native state during and after chromatographic separation. Therefore, we investigated whether the AEX conditions are compatible with downstream enzyme activity experiments. First, the digesting activity of EndoPro was investigated at two commonly applied pH values (i.e., pH 2 and 4) as previous studies showed that EndoPro activity strongly depends on pH.10,11 We monitored the digestion of CytC by EndoPro with SDS-PAGE. CytC digestion’s time course revealed that nonseparated EndoPro digested CytC within 7 min at pH 2, while incubation for at least 90 min was required for full digestion at pH 4. For the following experiments with AEX fractions, we proceeded with pH 4 to evaluate their enzymatic activity. All five AEX fractions digested CytC at a similar rate as observed for nonseparated EndoPro (Figure 5a and S9). Overall, we successfully demonstrated that EndoPro proteoforms in the AEX fractions remained active after their chromatographic separation and showed similar digestion rates as nonseparated EndoPro.
Figure 5.
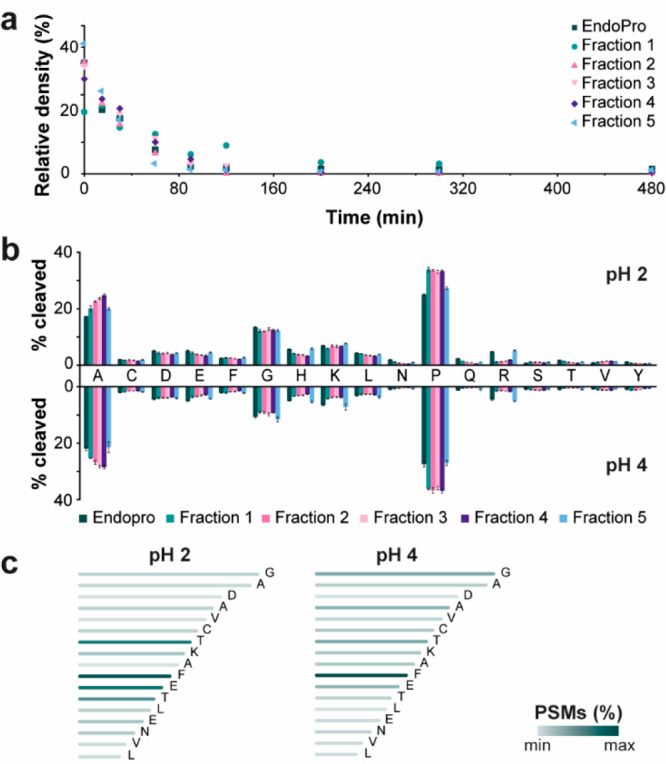
(a) Digestion of CytC by EndoPro or AEX fractions at pH 4 incubated for different times (i.e., 0, 15, 30, 60, 90, 120, 200, 300, and 480 min). The y-axis represents the relative density of the band of the undigested CytC and the x-axis the incubation time of the protease with CytC at 37 °C. The gel of digestion with EndoPro is shown in Figure S9. (b) The cleavage specificity of EndoPro and AEX fractions at pH 2 (upper part) and pH 4 (lower part). The x-axis shows the C-terminal amino acids of the BSA peptides generated by protease digestion. The data is a mean percentage (n = 4) of total cleavages of BSA per protease. The error bars show standard deviations. See the Experimental Section for details on the experimental procedure. (c) An example of C-terminal ragging for peptide “FDEHVKLVNELTEFAKTCVADESHAG”. The color indicates the relative number of PSMs detected for this peptide.
Subsequently, we investigated the cleavage specificity of EndoPro and AEX fractions by digesting BSA at both pH 2 and 4. After initial optimization of digestion conditions, we incubated BSA aliquots with EndoPro or AEX fractions in the protein to enzyme ratio 50:1 (w/w) overnight at 37 °C. After incubation, the digestion was quenched by loading and cleaning the peptides using Sep-Pak cartridges.10 The obtained BSA peptide mixtures were analyzed with RPLC-MS/MS using both EThcD and HCD for peptide fragmentation, similar to the work of van der Laarse et al.11Table S6 shows the search outcome, which was used for data interpretation. For both pH values, high sequence coverage was obtained for all samples (≥91% for pH 2 and ≥92% for pH 4). We used peptide spectral matches (PSMs) to quantitatively visualize the cleavage sites (Figure 5b) and peptide properties, such as length and charge (Figure S10). In general, EndoPro cleaved preferably after alanine (17% at pH 2 and 22% at pH 4) and proline (25% at pH 2 and 27% at pH 4), but also peptides with C-terminal glycine, lysine or leucine were quite abundant. Similarly to EndoPro, all AEX fractions showed increased specificity toward C-terminal cleavage of alanine (ranging from 20% to 25% for pH 2, and 21% to 28% for pH 4) and proline (ranging from 27% to 33% for pH 2, and 27% to 36% for pH 4). The relatively high abundance of nonspecific peptides encouraged us to investigate these peptides more in detail. We noticed that many BSA peptides were sequentially trimmed from their C-terminus, generating series of C-terminally truncated peptides (Figure 5c and S11). This “C-terminal ragging” was observed in all BSA digests at both pH values. Regarding the peptide properties, the digestion at both pH values did not reveal significant differences among samples and generated peptides with a broad peptide length distribution ranging from 5 to 75 amino acids (Figure S10a,b) and similar charge state distribution (Figure S10c,d). In conclusion, the performed experiments revealed consistent and comparable enzymatic activity of nonseparated EndoPro and AEX-separated EndoPro proteoforms. The enzymatic activity of EndoPro in AEX fractions did not show any significant dependence on their PTMs profile. We confirmed strong pH dependence on EndoPro cleavage activity rate and revealed a peculiar nonspecific activity generating C-terminally truncated peptides.
Discussion
EndoPro fromAspergillus nigeris a fascinating protein due to its complicated structure and broad application potential. Although EndoPro has been successfully utilized as a processing aid to improve food properties, e.g., to prevent chill-haze in beer,9 or as a digestion enzyme in proteomics,10−13 the detailed structure–function relationship of EndoPro proteoforms remain mostly unknown. Initial mass spectrometry-based studies demonstrated that EndoPro contains high-mannose type N-glycans.13 This glycosylation type is typical for proteins synthesized by fungi, includingAspergillus niger.33 Considering that the EndoPro sequence contains seven potential N-glycosylation sites, we hypothesized that EndoPro might be a highly heterogeneous protein. Indeed, our expectations were confirmed by recording high-resolution native spectra of AEX-separated EndoPro proteoforms. High complexity was observed, which encouraged us to investigate these proteoforms more in detail by performing released N-glycan and glycopeptide-centric analysis.
Integration of these three MS approaches revealed some discrepancies related to proteoform/glycoform/glycan abundances among native MS, glycopeptide and released N-glycan data. Our simulations showed that glycopeptide analysis was, in this case, suboptimal and revealed an underestimation of the abundance of larger glycans. Additionally, the data integration allowed us to annotate proteoforms in Fraction 1 in a site-specific manner. Although we could not annotate in detail the native spectra of Fractions 2–5 with phosphoglycans, our data exposed site-specific heterogeneity of all seven N-glycosylation sites on the level of separated EndoPro proteoforms. We observed that EndoPro contains dozens of high mannose N-glycans, which are partially phosphorylated on the specific sites. The phosphorylation level turned out to be one of the significant factors determining the AEX separation of EndoPro proteoforms. The negatively charged phosphate present on the glycans likely provides a stronger binding to the positively charged stationary phase. Therefore, proteoforms with more phosphoglycans have a longer retention time than species with less of these glycans. However, the difference in the number of phosphoglycans cannot wholly explain the observed result of AEX separation. For example, the level of phosphoglycans in Fraction 3 and 4 determined with released glycan analysis is very alike (i.e., 18.8% and 19.7%), but they differ around 5 min in retention time. We can speculate that site-specificity and certain glycoform combinations may play a role in the final apparent pI of the proteoforms. Interestingly, we noticed that the pI of EndoPro slightly increased after overnight incubation in ammonium bicarbonate buffer, while the molecular weight remained unaltered (Figure S2b). The increased pI after this incubation may be due to an altered conformation exposing fewer negative charges at the exterior of the protein, suggesting that the structural conformation of EndoPro plays a role in the net charge of the molecule. Previous studies dealing with the charge-based separation of acidic phospho/glycoproteins such as ovalbumin or erythropoietin showed that the chromatographic retention of proteoforms was affected by phosphorylation site occupancy or the number of sialic acids on the glycans.22,24 Notably, Füssl et al.22 observed that proteoforms with the same number of phosphates but located at different sites were separated, probably due to either the effect of phosphorylation on the local electrostatic amino acid environment or a change in protein conformation.
The presence of neutral high-mannose type N-glycans on EndoPro is in line with studies on glycoproteins in most filamentous fungi, whereas the detection of phosphorylated glycans is rarely reported. We attempted to determine the linkage type between phosphate and glycan, but our experiments, including treatment of EndoPro by phosphatase combined with mannosidase (as proposed by previous studies36−39), did not provide sufficient evidence to answer this question. Nevertheless, it is known that yeast’s glycosylation machinery can synthesize phosphorylated N-glycans, occurring in the form of mannose phosphodiester linkage.33,38,40 The gene Mnn6 encoding mannosyl-phosphate transferase in yeasts has orthologs across the filamentous fungal species, includingAspergillus niger. The filamentous fungusTrichoderma reeseihas been shown to attach diester-linked phosphates to glycans.37,39 Therefore, it is likely that alsoAspergillus nigercan synthesize phosphorylated high mannose glycans with this linkage type. Still, the direct evidence remains to be discovered as well as the function of phosphomannose residues.41
In addition to proteoform characterization, we also evaluated their enzymatic activity and specificity. We demonstrated that EndoPro proteoforms in the collected AEX fractions maintained their enzymatic activity and that the pH is a crucial parameter for digestion speed. Our results agree with previous studies showing that EndoPro acts primarily on the C-terminus of proline and alanine residues and minor cleavage can occur at glycine, leucine, arginine, serine, and tyrosine.10−13 Relatively broad digestion specificity may have several origins. However, our data suggest that the high number of nonspecific peptides results from the action of EndoPro. Moreover, Samadova et al.10 demonstrated that EndoPro is less specific with increasing digestion time. We evaluated more in detail a subset of BSA peptide mixtures generated by EndoPro, revealing many C-terminally truncated peptides detected as ladder-like peptide series. Interestingly, this C-terminal ragging was not observed on peptides generated by cleaving after proline. Additional structural studies need to be performed, including X-ray crystallography, to explain this peculiar enzymatic activity.
In conclusion, we developed a combined method for separating EndoPro proteoforms and their in-depth characterization. EndoPro consists of hundreds of proteoforms with several sequence variants and a long series of partially phosphorylated high-mannose N-glycans, making this enzyme one of the most heterogeneous proteins characterized. The observed enzymatic activity confirmed the preference of EndoPro cleaving after proline and alanine, but broader specificity and significant pH dependence should be considered for utilizing EndoPro in current and future applications.
Acknowledgments
The authors would like to thank Agnes L. Hipgrave Ederveen for the initial measurements of the released N-glycans of EndoPro. We acknowledge support from The Netherlands Organization for Scientific Research NWO (SATIN Project, Grant No. 731.017.202).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00663.
Figure S1: Sequence of EndoPro; Figure S2: Analysis of EndoPro using LC-MS and gel-based approaches; Figure S3: AEX-UV chromatograms of mobile phase optimization; Figure S4: Charge variant profile of EndoPro by AEX-UV and AEX-MS; Figure S5: Zero charge deconvoluted native mass spectra of AEX peaks; Figure S6: Assigned MALDI-FTICR mass spectra of released N-glycans; Figure S7: EICs and MS/MS spectra of phospho- and neutral glycans; Figure S8: Glycoproteomic analysis of AEX fractions; Figure S9: Gel of CytC digestion by EndoPro; Figure S10: BSA peptide characteristics after digestion with AEX fractions; Figure S11: C-terminal ragging of six BSA peptides (PDF)
Table S1: N-glycan database for skyline; Table S2: Skyline output for the glycosylation sites of all fractions; Table S3: Glycan compositions detected by MALDI-FTICR-MS for Fraction 1–5; Table S4: Glycopeptide profiling of Fraction 1–5; Table S5: The detected masses and intensity of intact AEX-MS analysis; Table S6: Search input and outcome characteristics of the EndoPro and fractions (XLSX)
Author Present Address
# ProQR Therapeutics, 2333 CK, Leiden, The Netherlands
Author Contributions
G.S., V.F., C.G., J.H.B.V., and O.S. conducted experiments. G.S., V.F., N.A., E.D.V., A.J.R.H., M.M.A.O., M.A., and M.W. conceived the ideas. G.S. and V.F. analyzed the data. G.S. and V.F. wrote the manuscript. All authors assisted with manuscript editing and have approved the final version of the manuscript.
The authors declare the following competing financial interest(s): Co-authors J.H.B.V., O.S., M.M.A.O., M.A., and N.A. are affiliated with DSM, a global company active in nutrition, health, and sustainable living.
Supplementary Material
References
- Vogel A.; May O.. Industrial Enzyme Applications; Wiley-VCH: Weinheim, 2019. [Google Scholar]
- Raveendran S.; Parameswaran B.; Ummalyma S. B.; Abraham A.; Mathew A. K.; Madhavan A.; Rebello S.; Pandey A. Applications of Microbial Enzymes in Food Industry. Food Technol. Biotechnol. 2018, 56 (1), 16–30. 10.17113/ftb.56.01.18.5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnau J.; Yaver D.; Hjort C. M.. Strategies and Challenges for the Development of Industrial Enzymes Using Fungal Cell Factories. In Grand Challenges in Fungal Biotechnology; Nevalainen H., Ed.; Springer: Cham, 2020; pp 179–210. [Google Scholar]
- Demain A. L.; Vaishnav Production of recombinant proteins by microbes and higher organisms. Biotechnol. Adv. 2009, 27, 297–306. 10.1016/j.biotechadv.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Yang M.; Yu X. W.; Zheng H.; Sha C.; Zhao C.; Qian M.; Xu Y. Role of N-linked glycosylation in the secretion and enzymatic properties of Rhizopus chinensis lipase expressed in Pichia pastoris. Microb. Cell Fact. 2015, 14, 40. 10.1186/s12934-015-0225-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čaval T.; Heck A. J. R.; Reiding K. R. Meta-heterogeneity: evaluating and describing the diversity in glycosylation between sites on the same glycoprotein. Mol. Cell. Proteomics 2021, 20, 100010. 10.1074/mcp.R120.002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Liu F.; Franc V.; Halim L. A.; Schellekens H.; Heck A. J. R. Hybrid mass spectrometry approaches in glycoprotein analysis and their usage in scoring biosimilarity. Nat. Commun. 2016, 7, 13397. 10.1038/ncomms13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlschlager T.; Scheffler K.; Forstenlehner I. C.; Skala W.; Senn S.; Damoc E.; Holzmann J.; Huber C. G. Native mass spectrometry combined with enzymatic dissection unravels glycoform heterogeneity of biopharmaceuticals. Nat. Commun. 2018, 9 (1), 1713. 10.1038/s41467-018-04061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M.; Edens L. Effective Prevention of Chill-Haze in Beer Using an Acid Proline-Specific Endoprotease from Aspergillus niger. J. Agric. Food Chem. 2005, 53, 7944–7949. 10.1021/jf0506535. [DOI] [PubMed] [Google Scholar]
- Samodova D.; Hosfield C. M.; Cramer C. N.; Giuli M. V.; Cappellini E.; Franciosa G.; Rosenblatt M. M.; Kelstrup C. D.; Olsen J. V. ProAlanase is an Effective Alternative to Trypsin for Proteomics Applications and Disulfide Bond Mapping. Mol. Cell Proteomics 2020, 19 (12), 2139–2157. 10.1074/mcp.TIR120.002129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laarse S. A. M.; van Gelder C. A. G. H.; Bern M.; Akeroyd M.; Olsthoorn M. M. A.; Heck A. J. R. Targeting Proline in (Phospho)Proteomics. FEBS J. 2020, 287 (14), 2979–2997. 10.1111/febs.15190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiatsiani L.; Akeroyd M.; Olsthoorn M.; Heck A. J. R. Aspergillus nigerProlyl Endoprotease for Hydrogen–Deuterium Exchange Mass Spectrometry and Protein Structural Studies. Anal. Chem. 2017, 89 (15), 7966–7973. 10.1021/acs.analchem.7b01161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šebela M.; Řehulka P.; Kábrt J.; Řehulková H.; Oždian T.; Raus M.; Franc V.; Chmelík J. Identification of N-glycosylation in prolyl endoprotease from Aspergillus niger and evaluation of the enzyme for its possible application in proteomics. J. Mass Spectrom. 2009, 44 (11), 1587–1595. 10.1002/jms.1667. [DOI] [PubMed] [Google Scholar]
- Muneeruddin K.; Nazzaro M.; Kaltashov I. A. Characterization of Intact Protein Conjugates and Biopharmaceuticals Using Ion-Exchange Chromatography with Online Detection by Native Electrospray Ionization Mass Spectrometry and Top-Down Tandem Mass Spectrometry. Anal. Chem. 2015, 87 (19), 10138–10145. 10.1021/acs.analchem.5b02982. [DOI] [PubMed] [Google Scholar]
- Fekete S.; Beck A.; Fekete J.; Guillarme D. Method development for the separation of monoclonal antibody charge variants in cation exchange chromatography, Part I: salt gradient approach. J. Pharm. Biomed. Anal. 2015, 102, 33–44. 10.1016/j.jpba.2014.08.035. [DOI] [PubMed] [Google Scholar]
- Haberger M.; Leiss M.; Heidenreich A. K.; Pester O.; Hafenmair G.; Hook M.; Bonnington L.; Wegele H.; Haindl M.; Reusch D.; Bulau P. Rapid characterization of biotherapeutic proteins by size-exclusion chromatography coupled to native mass spectrometry. mAbs 2016, 8 (2), 331–339. 10.1080/19420862.2015.1122150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J.; Pack L.; Hunter J. H.; Valliere-Douglass J. F. Native size-exclusion chromatography-mass spectrometry: suitability for antibody-drug conjugate drug-to-antibody ratio quantitation across a range of chemotypes and drug-loading levels. mAbs 2020, 12 (1), 1682895. 10.1080/19420862.2019.1682895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehkirch A.; D’Atri V.; Rouviere F.; Hernandez-Alba O.; Goyon A.; Colas O.; Sarrut M.; Beck A.; Guillarme D.; Heinisch S.; Cianferani S. An Online Four-Dimensional HICxSEC-IMxMS Methodology for Proof-of-Concept Characterization of Antibody Drug Conjugates. Anal. Chem. 2018, 90 (3), 1578–1586. 10.1021/acs.analchem.7b02110. [DOI] [PubMed] [Google Scholar]
- Wei B.; Han G.; Tang J.; Sandoval W.; Zhang Y. T. Native Hydrophobic Interaction Chromatography Hyphenated to Mass Spectrometry for Characterization of Monoclonal Antibody Minor Variants. Anal. Chem. 2019, 91 (24), 15360–15364. 10.1021/acs.analchem.9b04467. [DOI] [PubMed] [Google Scholar]
- Fekete S.; Beck A.; Veuthey J. L.; Guillarme D. Ion-exchange chromatography for the characterization of biopharmaceuticals. J. Pharm. Biomed. Anal. 2015, 113, 43–55. 10.1016/j.jpba.2015.02.037. [DOI] [PubMed] [Google Scholar]
- Füssl F.; Cook K.; Scheffler K.; Farrell A.; Mittermayr S.; Bones J. Charge Variant Analysis of Monoclonal Antibodies Using Direct Coupled pH Gradient Cation Exchange Chromatography to High-Resolution Native Mass Spectrometry. Anal. Chem. 2018, 90 (7), 4669–4676. 10.1021/acs.analchem.7b05241. [DOI] [PubMed] [Google Scholar]
- Füssl F.; Criscuolo A.; Cook K.; Scheffler K.; Bones J. Cracking Proteoform Complexity of Ovalbumin with Anion-Exchange Chromatography-High-Resolution Mass Spectrometry under Native Conditions. J. Proteome Res. 2019, 18 (10), 3689–3702. 10.1021/acs.jproteome.9b00375. [DOI] [PubMed] [Google Scholar]
- Leblanc Y.; Faid V.; Lauber M. A.; Wang Q.; Bihoreau N.; Chevreux G. A generic method for intact and subunit level characterization of mAb charge variants by native mass spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2019, 1133, 121814. 10.1016/j.jchromb.2019.121814. [DOI] [PubMed] [Google Scholar]
- van Schaick G.; Gstöttner C.; Büttner A.; Reusch D.; Wuhrer M.; Domínguez-Vega E. Anion Exchange Chromatography – Mass Spectrometry for Monitoring Multiple Quality Attributes of Erythropoietin Biopharmaceuticals. Anal. Chim. Acta 2021, 1143, 166–172. 10.1016/j.aca.2020.11.027. [DOI] [PubMed] [Google Scholar]
- Loke I.; Packer N. H.; Thaysen-Andersen M. Complementary LC-MS/MS-Based N-Glycan, N-Glycopeptide, and Intact N-Glycoprotein Profiling Reveals Unconventional Asn71-Glycosylation of Human Neutrophil Cathepsin G. Biomolecules 2015, 5 (3), 1832–1854. 10.3390/biom5031832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Franc V.; Heck A. J. R. Glycoproteomics: A Balance between High-Throughput and In-Depth Analysis. Trends Biotechnol. 2017, 35 (7), 598–609. 10.1016/j.tibtech.2017.04.010. [DOI] [PubMed] [Google Scholar]
- Tjondro H. C.; Ugonotti J.; Kawahara R.; Chatterjee S.; Loke I.; Chen S.; Soltermann F.; Hinneburg H.; Parker B. L.; Venkatakrishnan V.; Dieckmann R.; Grant O. C.; Bylund J.; Rodger A.; Woods R. J.; Karlsson-Bengtsson A.; Struwe W. B.; Thaysen-Andersen M. Hyper-truncated Asn355- and Asn391-glycans modulate the activity of neutrophil granule myeloperoxidase. J. Biol. Chem. 2021, 296, 100144. 10.1074/jbc.RA120.016342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman M. H.; Hemayatkar M.; Deelder A. M.; Wuhrer M. Cotton HILIC SPE microtips for microscale purification and enrichment of glycans and glycopeptides. Anal. Chem. 2011, 83 (7), 2492–2499. 10.1021/ac1027116. [DOI] [PubMed] [Google Scholar]
- Reiding K. R.; Blank D.; Kuijper D. M.; Deelder A. M.; Wuhrer M. High-throughput profiling of protein N-glycosylation by MALDI-TOF-MS employing linkage-specific sialic acid esterification. Anal. Chem. 2014, 86 (12), 5784–5793. 10.1021/ac500335t. [DOI] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9 (7), 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26 (7), 966–8. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. H.; Zhu J.; Meijer S.; Franc V.; Heck A. J. R. Glycoproteogenomics: A Frequent Gene Polymorphism Affects the Glycosylation Pattern of the Human Serum Fetuin/alpha-2-HS-Glycoprotein. Mol. Cell Proteomics 2019, 18 (8), 1479–1490. 10.1074/mcp.RA119.001411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering T. L.; Cummings R. D.; Aebi M.. Fungi. In Essentials of Glycobiology, 3rd ed.; Varki A. C. R., Esko J. D.,. et al. , Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2017. [Google Scholar]
- Edens L.; Dekker P.; van der Hoeven R.; Deen F.; de Roos A.; Floris R. Extracellular Prolyl Endoprotease from Aspergillus niger and Its Use in the Debittering of Protein Hydrolysates. J. Agric. Food Chem. 2005, 53, 7950–7957. 10.1021/jf050652c. [DOI] [PubMed] [Google Scholar]
- Reiding K. R.; Bondt A.; Franc V.; Heck A. J. R. The benefits of hybrid fragmentation methods for glycoproteomics. TrAC, Trends Anal. Chem. 2018, 108, 260–268. 10.1016/j.trac.2018.09.007. [DOI] [Google Scholar]
- Pazur J. H. The determination of the anomeric configuration of glycosyl-phosphoryl linkages of immunogenic phosphoglycans. Anal. Biochem. 1985, 145, 385–392. 10.1016/0003-2697(85)90378-1. [DOI] [PubMed] [Google Scholar]
- Stals I.; Sandra K.; Geysens S.; Contreras R.; Van Beeumen J.; Claeyssens M. Factors influencing glycosylation of Trichoderma reesei cellulases. I: Postsecretorial changes of the O- and N-glycosylation pattern of Cel7A. Glycobiology 2004, 14 (8), 713–724. 10.1093/glycob/cwh080. [DOI] [PubMed] [Google Scholar]
- Thieme T. R.; Ballou C. E. Nature of the Phosphodiester Linkage of the Phosphomannan from the Yeast Kloeckera brevis. Biochemistry 1971, 10 (22), 4121–4129. 10.1021/bi00798a017. [DOI] [PubMed] [Google Scholar]
- Maras M.; De Bruyn A.; Schraml J.; Herdewijn P.; Claeyssens M.; Fiers W.; Contreras R. Structural characterization of N-linked oligosaccharides from cellobiohydrolase I secreted by filamentous fungus Trichoderma reesei RUTC 30. Eur. J. Biochem. 1997, 245, 617–625. 10.1111/j.1432-1033.1997.00617.x. [DOI] [PubMed] [Google Scholar]
- Gemmill T. R.; Trimble R. B. Overview of N- and O-linked oligosaccharide structures found in various yeast species. Biochim. Biophys. Acta, Gen. Subj. 1999, 1426, 227–237. 10.1016/S0304-4165(98)00126-3. [DOI] [PubMed] [Google Scholar]
- Deshpande N.; Wilkins M. R.; Packer N.; Nevalainen H. Protein glycosylation pathways in filamentous fungi. Glycobiology 2008, 18 (8), 626–637. 10.1093/glycob/cwn044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.