

Abstract
As a morphogen, Sonic Hedgehog (Shh) mediates signaling at a distance from its sites of synthesis. After secretion, Shh must traverse a distance through the extracellular matrix (ECM) to reach the target cells and activate the Hh response. ECM proteins, in particular, the heparan sulfate proteoglycans (HSPGs) of the glypican family, have both negative and positive effects on Shh signaling, all attributed to their ability to bind Shh. Using mouse embryonic stem cell-derived mosaic tissues with compartments that lack the glycosyltransferases Exostosin1 and Exostosin2, or the HSPG core protein Glypican5, we show that Shh accumulates around its source cells when they are surrounded by cells that have a mutated ECM. This accumulation of Shh is correlated with an increased noncell autonomous Shh response. Our results support a model in which Shh presented on the cell surface accumulates at or near ECM that lacks HSPGs, possibly due to the absence of these Shh sequestering molecules.
Keywords: Cell signalling, Developmental biology, Embryoid bodies, Embryonic stem cells
Introduction
Sonic Hedgehog (Shh) and its paralog Desert Hh (Dhh) and Indian Hh (Ihh) can function as morphogens, signaling molecules that are produced locally and form a concentration gradient as they spread through surrounding tissue. The graded signal is interpreted by cells in a dosage-dependent manner to control gene expression and cell fate specification. Hhs are essential for patterning and differentiation in most animals [1] and aberrant regulation of Hh pathway is associated with congenital anomalies, such as holoprosencephaly, and cancer [2, 3]. The importance of a graded Hh distribution in tissue patterning during embryogenesis has long been recognized, although it is poorly understood how these gradients are established.
Hh is synthesized as a precursor protein that undergoes autoproteolysis giving rise to C-terminal and N-terminal fragments [4]. As a consequence of this event, the active N-terminal domain (HhNp) is covalently modified by cholesterol at its C terminus and in addition, is palmitoylated at the N-terminus [5, 6]. These lipid modifications promote membrane association of HhNp [7, 8]. Subsequent release of HhNp into the extracellular space requires the Resistance, Nodulation, and Division antiporter Dispatched1 (Disp1) [9], the Cubulin domain protein Scube2 [10, 11], and members of a Disintegrin and Metalloprotease family of sheddases [12, 13].
In the extracellular space, Shh associates with extracellular matrix (ECM) components that shape Hh gradients including heparan sulfate proteoglycans (HSPGs). HSPGs consist of a protein core (such as Glypican [Gpc] and Syndecan) to which heparan sulfate (HS) glycosaminoglycan chains are attached [14]. HS glycosaminoglycan chains are added to a core protein by the sequential action of individual glycosyltransferases and modification enzymes in a three-step process involving chain initiation, polymerization, and modification [15]. Exostosin1 (Ext1) and Exostosin2 (Ext2) form a heteromeric complex of the glycosyltransferases that catalyze HS chain polymerization [16]. Previous studies showed that a gene trap mutation of Ext1 resulted in substantial reduction of HS chain length [17].
Genetic screens in Drosophila have shown that mutation of the Ext1/2 orthologs (tout velu, and brother of tout velu) or the Gpcs dally and dally-like impede Hh spread and reduce signaling range, indicating a role for HS-modified Gpcs facilitating Hh distribution [18–20]. However, reduced HS side-chain elongation in mice carrying a hypomorphic Ext1 allele results in an increased range of Ihh signaling during embryonic chondrocyte differentiation [21], consistent with a negative activity of HSPG on Hh distribution away from its source. HS-modified Gpcs either can act to sequester Hh ligands thus inhibiting signaling or can stabilize the association of the ligand with the Hh receptor Patched (Ptch) to promote signaling [22, 23]. For instance, Glypican3 (Gpc3) acts as a negative regulator of Shh activity by competing with Ptch1 for Shh binding [24], whereas Gpc5 has been identified as a Shh coreceptor and promotes downstream Shh signaling [25, 26].
As a morphogen, Shh directs neural patterning by conferring positional information to ventral neural progenitors through well-studied transcriptional responses [27]. In the developing spinal cord, Shh released from the notochord and floor plate distributes in a gradient along the dorsoventral (DV) axis of the neural tube that is required for normal DV patterning. These Shh-mediated signaling events can be modeled in vitro using neuralized embryoid bodies (nEBs) that are derived from mouse embryonic stem cells (mESCs) [28–30]. Here, we assess the role of Ext1/2 and Gpcs in Shh distribution and response. Using mosaic nEBs with defined contributions of Shh-expressing, Shh-transporting, and Shh-responding cells, we show that absence of Ext1/2 or Gpc5 in cells surrounding the Shh source results in accumulation of extracellular Shh, enhancing long-range signaling. Our results demonstrate that HS-modified Gpc5 is an inhibitor of Shh distribution and its loss phenocopies the loss of Ext1 or Ext2.
Materials and Methods
Cell Lines
HB9:GFP mESCs were a gift from Dr. Thomas Jessell (Columbia University). Their identity was confirmed by the presence of the Hb9:gfp transgene. Sim1:Cre/tdTomato mESCs were a gift from Dr. Samuel Pfaff (University of California, San Diego). Shh−/−; Ptch1+/LacZ and wild-type mESCs overexpressing Shh were previously described [29, 31]. mESC lines were maintained using standard conditions without feeder cells.
nEB Differentiation
mESCs were differentiated into nEBs using established procedures [28]. nEBs were aggregated for 24 hours in DFNB medium in Petri dishes rotated at 0.8 Hz. One micromolar retinoic acid (RA; Sigma, St. Louis, MO) was added at 24 hours. nEBs were fixed 72 hours after the addition of RA for antibody staining of neural progenitors. nEBs were fixed 72 hours after the addition of RA for imaging and quantifying HB9:GFP fluorescence. Sim1:Cre/tdTomato fluorescence was imaged 96 hours after the addition of RA.
Immunostaining
nEBs were fixed with 4% PFA, washed, permeabilized, and blocked. The nEBs were then incubated with primary antibodies. For extracellular Shh staining, mouse anti-Shh (5E1) was added to the culture medium 3 hours before fixation and secondary antibody treatment. Rabbit anti-Isl1/2 was a gift from Dr. Thomas Jessell (Columbia University). Rabbit anti-Olig2 (AB9610) was purchased from MilliporeSigma (St. Louis). The samples were then washed and incubated with the appropriate fluorescently labeled secondary antibody (Invitrogen, Carlsbad, CA, USA). nEBs were mounted in Fluormount-G and positive nuclei were quantified. Native HB9:GFP and Tomato+ fluorescence was imaged directly, after fixation and mounting, without antibody detection. Mounted nEBs were imaged with a Zeiss Observer fluorescence microscope with a ×20 objective. Within each experiment, stacks were deconvolved and resulting image files were scrambled for unbiased counting. Images were processed using the Fiji ImageJ and Photoshop software (Adobe, San Jose, CA, USA).
Genome Editing
sgRNAs were designed using the online CRISPR Design tool (http://tools.genome-engineering.org) and cloned into pX459 [32]. Target sequences of Ext1 are 5′-TCTTGCCCCACTAAATGGGA-3′ and 5′-GCTTGGGTCCTTCAGATTCC-3′. Target sequences of Ext2 are 5′-GTTCTATGTAGCAGACAAGC-3′ and 5′-ACTAGATTCCTAGATGGGTA-3′. Target sequences of Gpc5 are 5′-CGCAAGCCGAACACGAGCCG-3′ and 5′-TGGTGAGACGACGACCTTTC-3′. Shh−/−;Ptch1+/LacZ mESCs were transfected and transiently selected with puromycin. Individual clones were isolated and expanded for further analysis by DNA sequencing of the targeted region. Deletions of sequences were confirmed and sequenced after polymerase chain reaction using primers bracketing the deleted region. Sequence data for the edited cell lines are provided in Supporting Information S1.
Constructs
cDNA of mouse Gpc5 was obtained from Dharmacon (cat No MMM1013–211693346, Lafayette, CO, USA), and then was inserted into pUB-IREShyg3. This plasmid was derived from pIREShyg3 that was modified by replacing the CMV promoter with the Ubiquitin C promoter derived from pUb/Bsd.
Quantifications
nEBs stained for extracellular Shh were imaged using a ×20 objective. The conditions resulting in the highest levels of Shh staining were used to define an exposure time that allowed the use of the full dynamic range of the camera. Within a single experiment, this exposure time was used to document all conditions and individual nEBs. Using ImageJ, domains of Shh staining were circled and quantified for average level over background and surface area. As the surface areas did not vary significantly between the various conditions, we present only the mean staining within the circled domain. Results are presented in a box-and-whiskers plots. Statistical analyses were performed using Dunnett’s post hoc following a significant one-way analysis of variance (ANOVA) result. All experiments were performed at least three times in independent biological replicates performed at different times.
Cells positive for Isl-1/2, Oig2, and cell positive for GFP or TdTomato were counted in each nEB. These counts are presented in box-and-whiskers plots. Statistical analyses were performed using Dunnett’s post hoc following a significant one-way ANOVA result, and significance is indicated in the graphs. All experiments were performed at least three times in independent biological replicates performed at different times. For each independent experiment, 3–30 randomly chosen nEBs were quantified. Combined data are presented as “box-and-whiskers” plots.
Results
Shh Synthesized in Wild-Type Cells Accumulates Around Cells Lacking Ext1/2 Function
The use of mosaic nEBs allowed us to precisely delineate roles of (mutant) cells in the production of, the transport of, and the response to Shh [29]. To assess the role of HSPGs in Shh distribution, we made genome edited mESC lines carrying homozygous null mutations in Ext1 and Ext2. The Ext1 and Ext2 nulls were identified by analysis of the genomic loci. Ext1−/− cells were further confirmed by the absence of Ext1 mRNA. In these experiments, we removed Ext1 and Ext2 from Shh−/−;Ptch1+/LacZ mES cells. As they lack a functional Shh locus, we guarantee that the observed Shh staining is not endogenous, while the Ptch1:LacZ allele is used later for quantification of the response. Mosaic nEBs comprised of a majority of Ext1−/− or Ext2−/− cells and 2% wild-type cells stably expressing a Shh transgene [29] provide an in vitro model in which we can assess the function of Ext1/2 in the distribution of, and response to Shh derived from a sparse source.
We first assayed the consequences of loss of Ext1/2 on Shh distribution by generating mosaic nEBs comprised of a large majority of Ext1−/− or Ext2−/− cells and 2% Shh-expressing (otherwise wild-type) cells as Shh source. Live staining with the anti-Shh monoclonal antibody 5E1 favors staining of Shh present in the extracellular space [31]. No Shh was detected in nEBs without Shh-expressing cells (Fig. 1A–1C). The areas in which we detected extracellular Shh in Ext1−/− and Ext2−/− nEBs (Fig. 1E, 1F) were much larger than those in nEBs where the bulk of the cells are wild-type for Ext1/2 (Fig. 1D, quantified in Fig. 1G). This demonstrates that HSPGs affect Shh distribution. It also indicates that Shh synthesized in cells with a HSPG competent matrix accumulates when adjacent cells lack HSPGs. Loss of Ext1 function in the Shh-expressing cells has a minor effect on the amount of Shh observed extracellularly (Fig. 1H, 1I, quantified in Fig. 1J).
Figure 1.
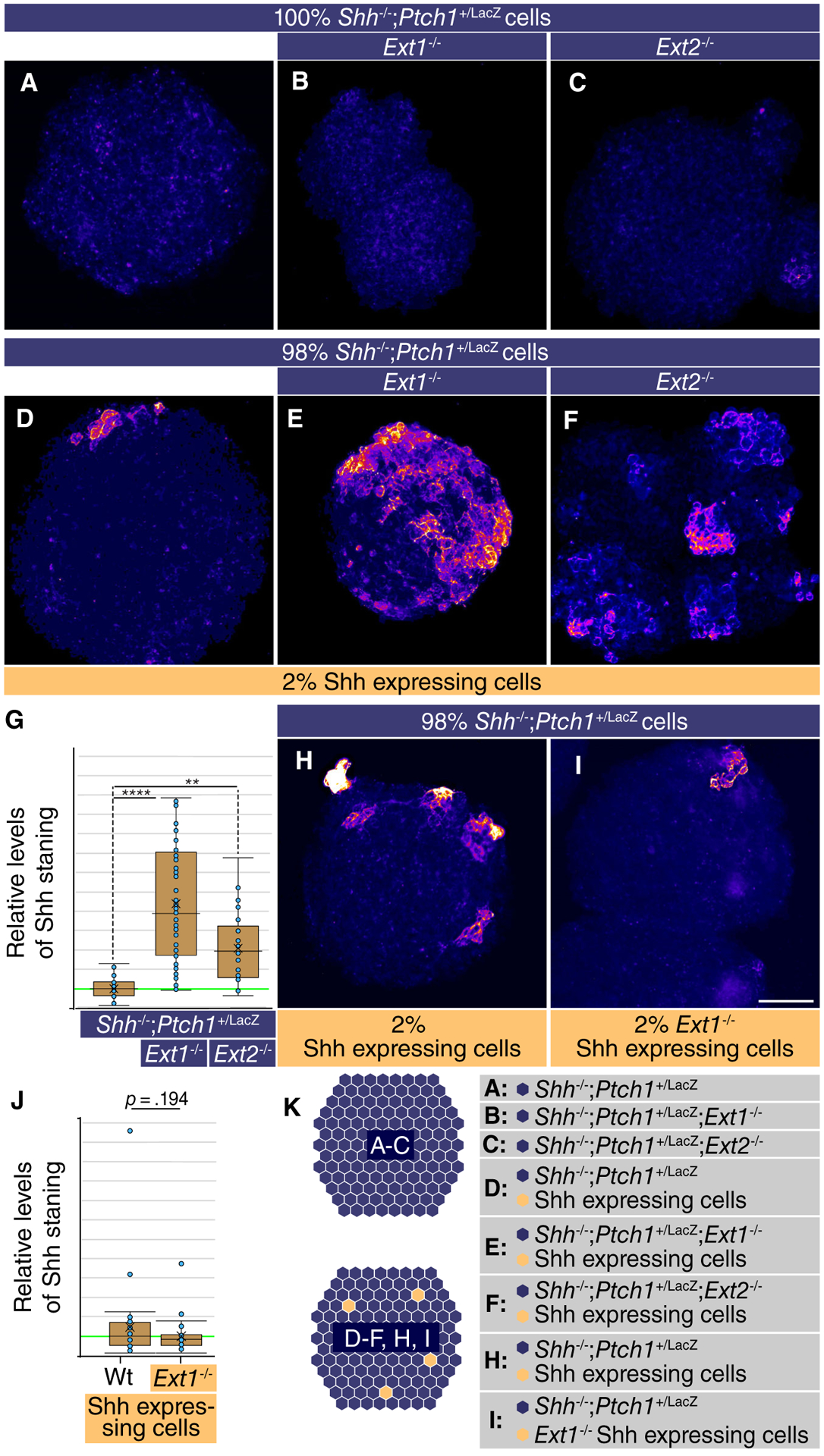
Loss of Ext1/2 function in surrounding cells increases Sonic Hedgehog (Shh) accumulation around the source cells. (A–E, H, I): Neuralized embryoid bodies (nEBs) derived from mouse embryonic stem cell lines with the indicated composition and genotype. Extracellular Shh distribution was assayed by live staining after 4 days. nEBs derived from Shh−/−;Ptch1+/LacZ cells (A), Shh−/−;Ptch1+/LacZ;Ext1−/− cells (B), and Shh−/−;Ptch1+/LacZ;Ext2−/− cells (C), without embedded Shh-expressing cells. (D–F): Mosaic nEBs consisting of 98% either Shh−/−;Ptch1+/LacZ cells (D), Shh−/−;Ptch1+/LacZ;Ext1−/− cells (E), or Shh−/−;Ptch1+/LacZ;Ext2−/− cells (F), incorporating 2% Shh-expressing cells were live stained for Shh. (G): Quantification of (E)–(F). The level of Shh staining in positive areas was measured. One-way analysis of variance with post hoc Dunnett’s test; **, p < .01; ****, p < .0001. (H, I): Mosaic nEBs consisting either 2% wild-type Shh-expressing cells (H), or Ext1−/− Shh-expressing cells (I) and 98% Shh−/−;Ptch1+/LacZ cells were generated and assayed by live staining for extracellular Shh distribution. (J): Quantification of (H) and (I). (K): Diagram showing the experimental approach. Scale bar is 100 μm.
Loss of Ext1/2 Increases the Noncell Autonomous Response to Shh
As HSPGs have been implicated as coreceptors for Shh, we assessed if the loss of Ext1 affected the response to Shh synthesized at a distal site. We assessed the Shh response by staining for Olig2 and Isl1/2, markers of ventral cell populations in the developing spinal cord. Consistent with elevated extracellular Shh levels in Ext1 and Ext2 null nEBs, but not supporting a role for HSPGs as Shh coreceptors, we observed higher levels of Shh-induced Olig2 and Isl1/2 in nEBs lacking Ext1 or Ext2 (Fig. 2A–2N), demonstrating that inactivation of Ext1/2 in the Shh responding cells resulted in enhanced response.
Figure 2.
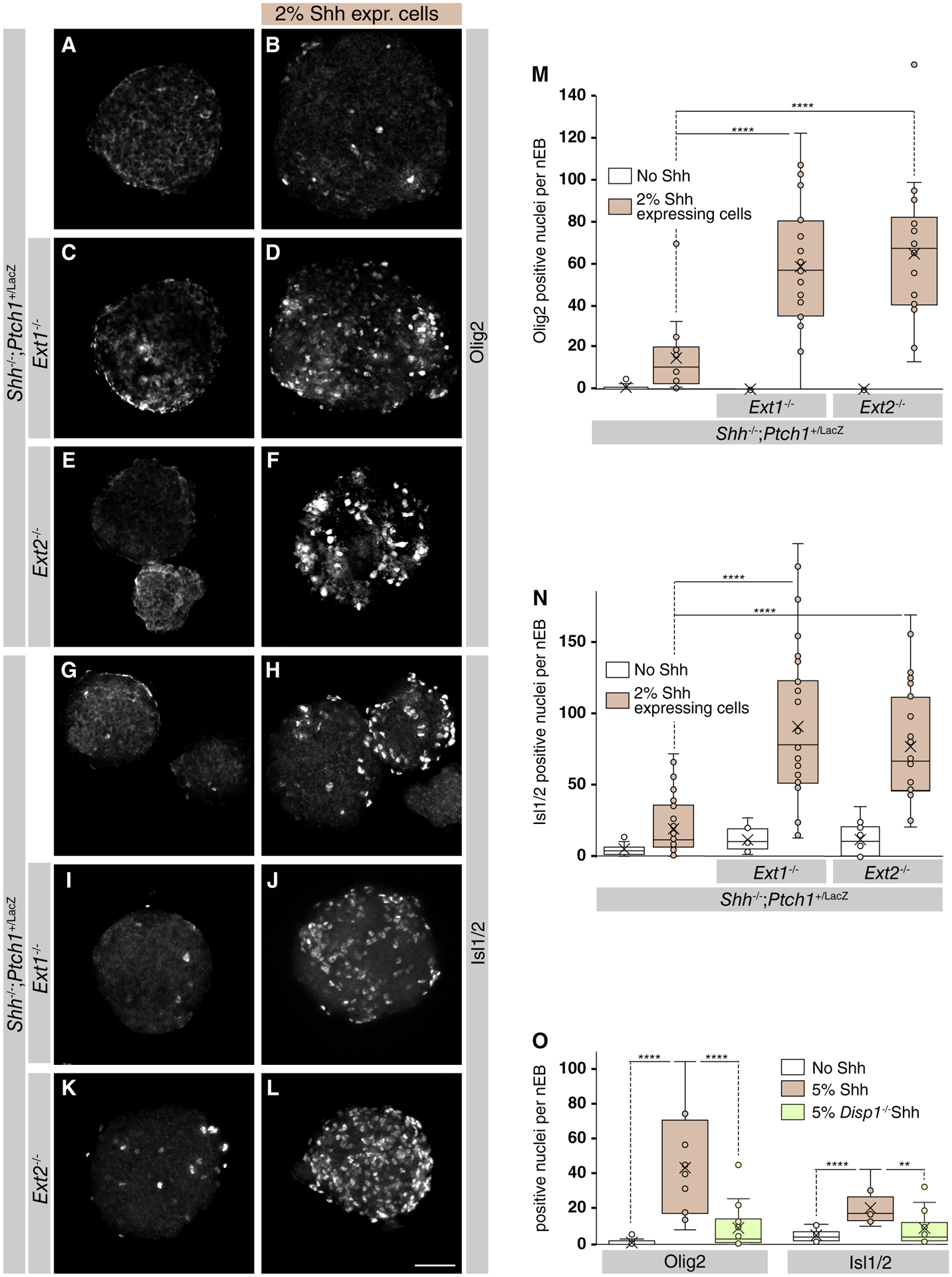
Loss of Ext1/2 in cells other than the Sonic Hedgehog (Shh) source results in an enhanced Shh response as measured by Isl1/2 and Olig2 induction. (A–F): Mosaic neuralized embryoid bodies (nEBs) composed of the indicated genotypes and stained for Olig2. (G–L): Mosaic nEBs composed of the indicated genotypes and stained for Isl1/2. (M): Quantification of the number of Olig2 positive nuclei per nEB. One-way analysis of variance (ANOVA) with post hoc Dunnett’s test; ****, p < .0001. (N): Quantification of the number of Isl1/2 positive nuclei per nEB. One-way ANOVA with post hoc Dunnett’s test; ****, p < .0001. (O): Mosaic nEBs consisting of 2% either wild-type or Disp1−/− Shh-expressing cells and 98% Shh−/−;Ptch1+/LacZ;Ext1−/− were cultured and assayed for Olig2 and Isl1/2 expression. One-way ANOVA with post hoc Dunnett’s test; **, p < .01; ****, p < .0001. Scale bar is 100 μm.
In order to confirm that the enhanced response to Shh we observed in Ext1/2 null cells was due to increased Shh release from its source, we inhibited Shh release by inactivating Disp1 in the Shh-expressing cells. Even though the exact molecular mechanism remains not fully understood, the involvement of Disp1 in mediating lipid-modified Shh from synthesizing cells is well established [31, 33, 34]. Disp1 inactivation in Shh-expressing cells led to a significant loss of the Shh response, revealed by Olig2 and Isl1/2 immunostaining (Fig. 2P), further supporting the notion that the observed changes in response are a direct consequence of Shh release. Together, our results indicate that Shh released from cells surrounded by normal HSPGs preferentially distributes into the extracellular space that borders the mutant ECM. Moreover, this accumulation near the sites of synthesis results in a significantly enhanced noncell autonomous Shh response. This shows that HSPGs affect Shh distribution and indicates that in mosaic nEBs, HSPGs are not required for the presentation of Shh to its cognate receptor.
Ext1/2-Dependent HS Chains Inhibit the Shh Response Noncell Autonomously
Inactivation of Ext1/2 in nEBs leads to an increased signaling indicating that Shh binding to HSPGs is inhibitory to Shh transport. To address this notion, we treated mosaic nEBs with ectopic HS and found that 20 μg/ml HS led to a significant reduction in Olig2 (Fig. 3A–3G) and Isl1/2 (Fig. 3H–3N) induction, indicating that the interaction of Shh with HS restricts its mobility, although alterations in receptor binding or solubilization in the medium might account for the reduced response.
Figure 3.
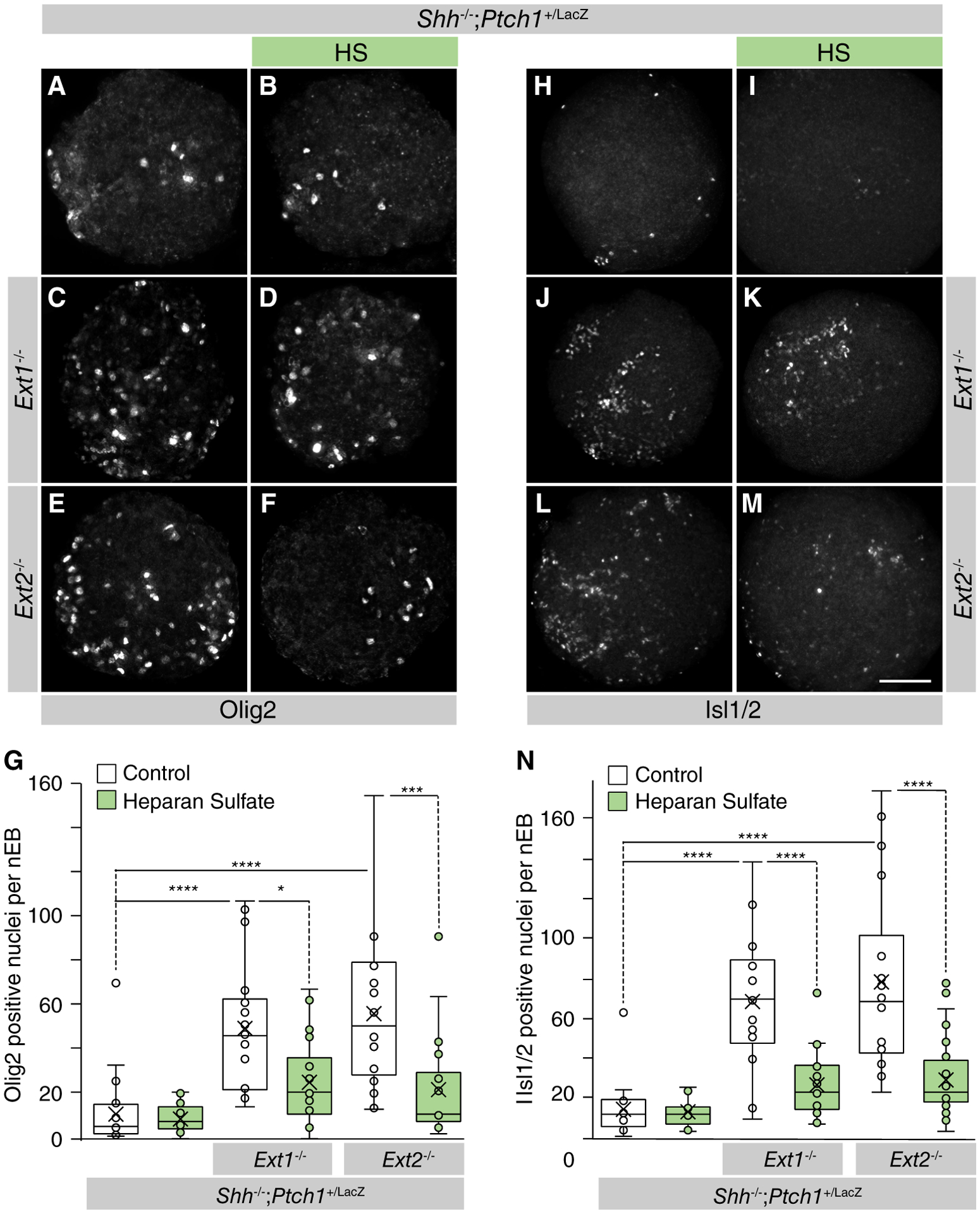
Heparan sulfate lowers Sonic Hedgehog (Shh) signaling in Ext1/2 null neuralized embryoid bodies (nEBs). (A–F, H–M): Mosaic nEBs of the indicated genotypes, including 2% Shh-expressing cells. Shh−/−;Ptch1+/LacZ cells (A, B, H, I) or Shh−/−;Ptch1+/LacZ;Ext1−/− cells (C, D, J, K) or Shh−/−;Ptch1+/LacZ;Ext2−/− cells (E, F, L, M) and 2% Shh-expressing cells were cultured in control medium (A, C, E, H, J, L) or in medium supplemented with heparan sulfate (B, D, F, I, K, M). (G, N): Quantification of the Shh-mediated induction of Olig2 (G) and Isl1/2 (N) per nEB. One-way analysis of variance with post hoc Dunnett’s test; *, p < .05;***, p < .001; ****, p < .0001. Scale bar is 100 μm.
In order to address the cell-autonomy of HSPGs for Shh signaling, mosaic nEBs comprised of equal numbers of Shh−/−; Ptch1+/LacZ and either Shh−/−;Ptch1+/LacZ;Ext1−/− or Shh−/−; Ptch1+/LacZ;Ext2−/− were generated (Fig. 4A). First, we examined extracellular Shh accumulation in chimeric nEBs and found that extracellular Shh accumulation in chimeric nEBs was indistinguishable from the Shh distribution in Shh−/−; Ptch1+/LacZ nEBs. This demonstrates that the negative function of HSPGs on Shh accumulation and signaling is dominant over the loss of function phenotype observed in nEBs lacking Ext1/2 (Fig. 4B). Second, we examined the Shh response in chimeric nEBs and found that the elevated Shh response observed in Ext1/2 null nEBs was suppressed to wild-type levels by HSPGs synthesized Shh−/−;Ptch1+/LacZ cells. Mosaic nEBs consisting of equal numbers of Shh−/−;Ptch1+/LacZ;Ext1−/− and Shh−/−;Ptch1+/LacZ; Ext2−/− cells have similar high levels of Olig2 and Isl1/2 positive cells as either cell-type grown alone (Fig. 4C, 4D), demonstrating that the mechanism by which the loss of Ext1 or Ext2 enhances Shh signaling is similar. Together, these results demonstrate HSPG function noncell autonomously inhibits Shh signaling, and furthermore show that the loss of HSPG does not affect the intrinsic Shh sensitivity of cells.
Figure 4.
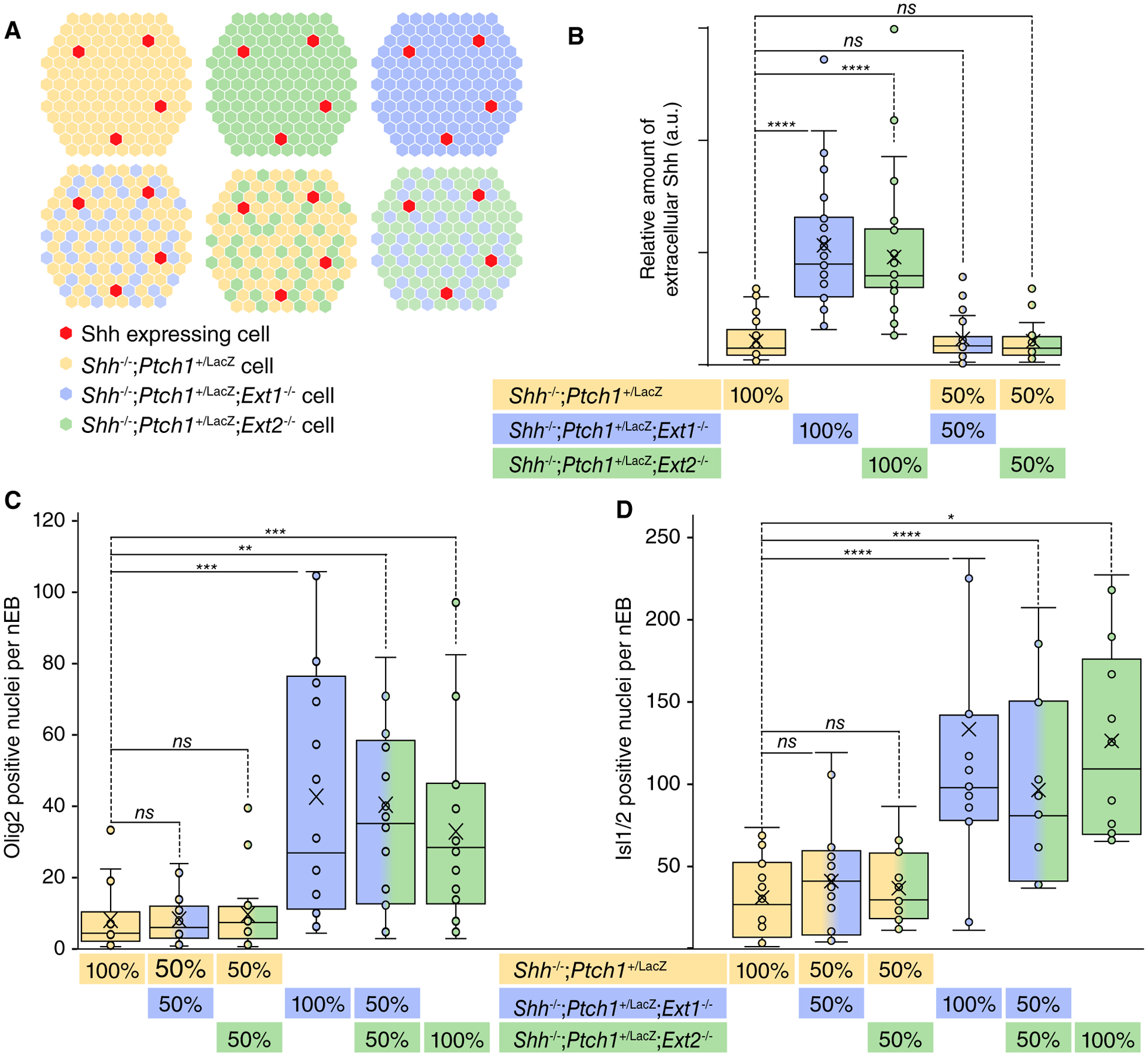
Confined loss of Ext function only in the Sonic Hedgehog (Shh)-transporting cells suffices to enhance the Shh response in Ext1/2 competent cells. (A): Schematic representations of the experiments. Neuralized embryoid bodies (nEBs) consisting of 1:1 of Shh−/−;Ptch1+/LacZ cells, Shh−/−;Ptch1+/LacZ;Ext1−/− cells, or Shh−/−;Ptch1+/LacZ;Ext2−/− cells were generated to assess Shh distribution and signaling response. (B): Quantification of Shh positive area per mosaic nEB composed of the indicated genotypes. One-way analysis of variance (ANOVA) with post hoc Dunnett’s test; ****, p < .0001. (C, D): The Shh-mediated induction of Olig2 (C) and Isl1/2 (D) per nEB was quantified. One-way ANOVA with post hoc Dunnett’s test; *, p < .05; **, p < .01; ***, p < .001; ****, p < .0001. Abbreviation: ns, not significant.
Glypican5 Is the Core Protein that Is Involved in HSPG-Mediated Shh Distribution and Response Regulation in nEBs
Ext1/2 catalyzes the glycanation of multiple distinct HSPG core proteins. To find the specific HSPG that primarily affects Shh distribution, we followed an informed approach to identify the required core protein. Three major families of proteoglycan (PG) core proteins have been characterized: the membrane-spanning Syndecans, the glycosylphosphatidylinositol-linked Gpcs, and the basement membrane-specific PGs Perlecan and Agrin [15, 35]. Gpcs are central for Hh distribution and signaling in Drosophila [18, 23]. We proceeded to mutate Glypican family members Gpc2, Gpc3, and Gpc5 that are expressed in nEBs and found that in particular, the loss of Gpc5 have similar phenotypes as Ext1/2 nulls. We observed that extracellular Shh was increased in Gpc5 null nEBs (Fig. 5A–5C) and consistent with the loss of Ext1/2; we observed that Olig2 and Isl1/2 induction by Shh-expressing cells was upregulated in Gpc5−/− nEBs as compared in nEBs that are wild-type for Gpc5 (Fig. 5D–5M). To address the sufficiency of Gpc5 to inhibit Shh distribution, a complementation experiment was conducted by creating Gcp5−/− mESCs stably expressing Gpc5. The results revealed that Gpc5 expression was able to restore the ability of Gcp5−/− mESCs to prevent Shh distribution away from the sites of synthesis in addition to suppressing Shh-mediated Olig2 and Isl1/2 induction (Fig. 5C, 5N). The similarity in phenotypes between the loss of Ext1/2 and Gpc5 indicates that Gpc5 is the core protein that is modified by Ext1/2 to limit Shh distribution and signaling.
Figure 5.
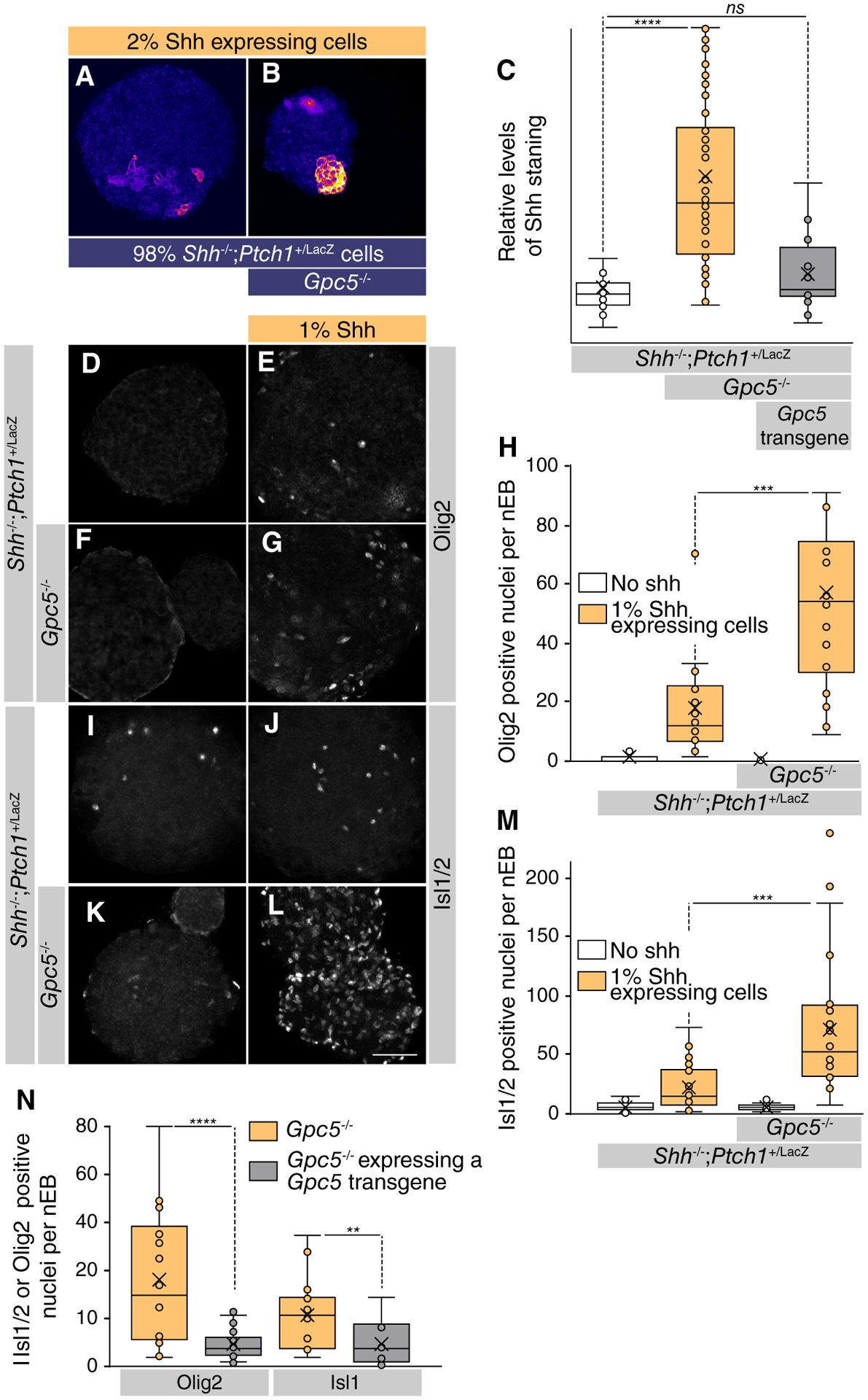
Gpc5 is the core protein that affects heparan sulfate proteoglycan-mediated Sonic Hedgehog (Shh) distribution and response in neuralized embryoid bodies (nEBs). (A, B): nEBs derived from mouse embryonic stem cell lines with the indicated composition and genotype. Extracellular Shh distribution was assayed by live staining after 4 days. Mosaic nEBs consisting either 98% Shh−/−;Ptch1+/LacZ (A) or Shh−/−;Ptch1+/LacZ;Gpc5−/− (B) and 2% Shh-expressing cells were stained for extracellular Shh. (C): Area of Shh positive per nEB was quantified. One-way analysis of variance with post hoc Dunnett’s test; ****, p < .0001. (D–L): Mosaic nEBs consisting either of only Shh−/−;Ptch1+/LacZ (D, I) or Shh−/−;Ptch1+/LacZ;Gpc5−/− (F, K) or incorporated 1% Shh-expressing cells (E, G, J, L) were stained for Olig2 (D–G) and Isl1/2 (I–L). (H): Quantification of (D)–(G). (M): Quantification of (I)–(K). Student’s t test; ***, p < .001. (N): A Gpc5 transgene was stably expressed in Shh−/−;Ptch1+/LacZ;Gpc5−/− cells. These cells were assessed for their ability to affect the Shh response in mosaic nEBs incorporating 2% Shh-expressing cells as compared with the maternal line. The expression of Olig2 and Isl1/2 positive cells per nEB was quantified. Student’s t test; **, p < .01; ****, p < .0001. Scale bar is 100 μm. Abbreviation: ns, not significant.
Disrupting Ext1/2 or Gpc5 in the Tissue Interposed Between the Shh Source and Responding Cells Is Sufficient to Enhance Signaling
The accumulation of Shh that we observed around Ext1/2 and Gpc5 null cells indicates that the ECM around cells that lack Ext1/2 or Gpc5 is more permeable to Shh. We used a culture system in which we can unambiguously assess the contribution of the cells that transport Shh in their ability to affect Shh signaling. We generated tripartite mosaic nEB consisting of (a) Shh-expressing wild-type cells (3% or 5%) as localized Shh sources, (b) 3% of reporter mESCs that have a genetically encoded Shh reporter and are HSPG competent, and (c) a pre-dominant compartment (92%–94%) of either Shh−/−;Ptch1+/LacZ or cells that lack Ext1/2 or Gpc5 and serve as the conduit for Shh. We used HB9:GFP cells [28] or V3 interneuron reporter Sim1:Cre/tdTomato [35] to confine the compartment in which we assess Hh pathway activation. The bulk of the cells in such nEBs is purely assessed for its role in Shh transport between the normal source and responding cells.
Tripartite mosaic nEBs largely consisting of cells that are wild-type for Ext1/2 (Shh−/−;Ptch1+/LacZ cells) showed no Tomato +V3 and low HB9:GFP expression in the absence of Shh-expressing cells and we observed minimal activation of the Hh response when a small number of Shh-expressing cells were included (Fig. 6A, 6B, 6J, 6K). In contrast, we observed robust Shh-dependent Tomato +V3 induction in mosaic nEBs comprised of 92% Shh−/−;Ptch1+/LacZ;Ext1−/− or Shh−/−;Ptch1+/LacZ;Ext2−/− or Shh−/−;Ptch1+/LacZ;Gpc5−/− cells (Fig. 6C–6I). Similar results were obtained using the HB9:GFP cells as reporters for Shh activity. Tripartite mosaic nEBs consisting of 94% Shh−/−;Ptch1+/LacZ; Ext1−/− or Shh−/−;Ptch1+/LacZ;Ext2−/− or Shh−/−;Ptch1+/LacZ; Gpc5−/− cells had many more GFP-expressing cells than mosaic nEBs that are principally comprised of Shh−/−;Ptch1+/LacZ cells (Fig. 6L–6R). These results demonstrate that HSPG deficiency in the ECM strongly facilitates long-range Shh signaling between the Shh source and responding cells surrounded by normal ECM indicating that HS-modified Gpc5 affects Shh transport, possibly by Shh sequestration.
Figure 6.
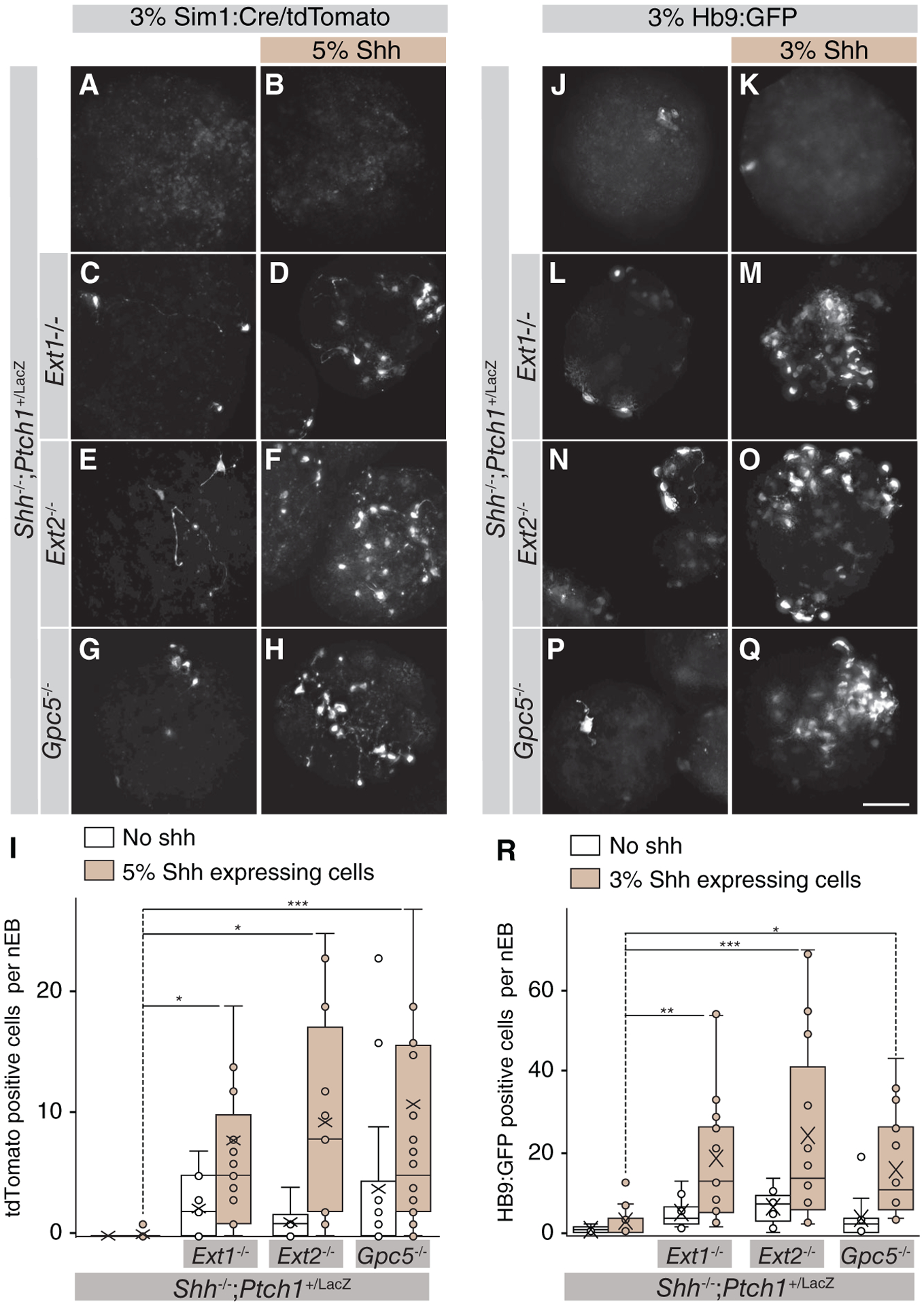
Los of Ext1/2 or Gpc5 only in the Sonic Hedgehog (Shh)-transporting cells suffices to enhance the Shh response. (A–H): Neuralized embryoid bodies, the bulk of which consists of the indicated genotype as well as 3% of Sim1:RFP cells, with (B, D, F, H) or without (A, C, E, G) 5% Shh-expressing cells. Images show tdTomato-expressing cells. (I): Quantification of (A)–(H). One-way analysis of variance (ANOVA) with post hoc Dunnett’s test; *, p < .05; ***, p < .001. (J–Q): Neuralized embryoid bodies, the bulk of which consists of the indicated genotype as well as 3% of Hb9:GFP cells, with (K, M, O, Q) or without (J, L, N, P) 3% Shh-expressing cells. Images show GFP-expressing cells. (R): Quantification of (J)–(Q). One-way ANOVA with post hoc Dunnett’s test; *, p < .05; **, p < .01; ***, p < .001. Scale bar is 100 μm.
Discussion
Taking advantage of the properties of mosaic nEBs, where we control the genetic makeup of Shh source cells, transporting cells, and responding cell independently, we demonstrate the HS-modified Gpc5 affects distribution of Shh, but not the presentation of this ligand to its receptor. Our results support a model in which Shh produced in cells with a normal ECM preferentially accumulates at the interface with ECM that lacks the HS modifications. This accumulation of extracellular Shh results in increased signaling, likely caused by altered Shh distribution, but not by changes in sensitivity of the responding cells.
The roles of HSPGs to regulate Hh signaling was first discovered in Drosophila, where loss of either the Ext1/2 orthologs tout-velu or sister of tout-velu was found to negatively affect the Hh response. The loss of the Glypican homologs dally and dally-like had a similar phenotype to the loss of tout-velu [20, 37, 38]. These observations have to be reconciled with the finding that both Ext1/2 and some Gpcs are tumor suppressors, and thus would inhibit rather than facilitate signaling. Shh has a Cardin–Weintraub Motif that mediates its binding to HS [39] and HSPGs. This binding could negatively affect Hh signaling by ligand sequestration and limiting distribution [24, 40], or it could positively affect Hh signaling by serving as a coreceptor for Hh binding to the target cells [26]. Noncell autonomous signaling involved three main events: (a) the synthesis and release of the ligand from the source cells, (b) the transport of the ligand through a tissue, and (c) the activation of a receptor in the responding cells. Our results demonstrate that in case of Shh signaling, the HSPG function positively affects the presentation of Shh by the source cells, negatively affects the transport of Shh, and has no discernable effect on the ability of cells to respond to Shh, thus providing an explanation for the positive and negative effects of HSPG/Gpcs on Hh signaling. The loss of Shh-sequestration by cells surrounded by an ECM lacking the proper HSPG complement would provide a simple explanation for why there is more Shh detected in the mutant ECM as well as the enhanced ability of Shh to distribute more efficiently. As the overall Hh response increases due to the lack of Ext1/2 or Gpc5, it appears that there is no major role for HSPG in the Shh presentation to its receptor, a notion further supported by our finding that in the tripartite mosaics the normal ECM surrounding the responding cells does not appear to affect the responsiveness.
The enhanced distribution of Shh we observe in tissues surrounded by an ECM that lacks the proper HSPGs would provide an elegant explanation why Ext1 and Gpc5 can function as tumor suppressors. Nonsmall-cell lung carcinoma often has upregulated Shh expression [41, 42]. As many Shh-induced tumors have distinct Shh-expressing and Shh-transporting/responding compartments [43], it is predicted that suppression of Gpc expression in the non-Shh-expressing cells enhances Shh accumulation and response, and thus the perniciousness of the tumor. It might be no surprise that in nonsmall-cell lung carcinomas, the loss of Gpc5 function is not uncommon [44]. Ext1/2 function as tumor suppressors for exostoses, cartilage capped bone tumors that appear next to growth plates [16]. The location is consistent with a role for Ihh that expressed in the growth plate and is required for growth plate maintenance [45] and bone growth. Loss of Ext1/2 would create a domain in which the transport of Ihh is facilitated, and the response unaffected, resulting in the typical bone tumors that characterize somatic loss of Ext1/2 function.
Although our findings are generally consistent with the observations of HSPG function in mammals, they are less so with the observations in Drosophila, where it appears that Ext1/2 and Gpcs facilitate Hh transport away from the source. Although the clonal experiments in the wing disk [37] most closely resemble or experiments using tripartite nEBs (with a normal Shh source, mutant Shh-transporting cells and normal responding cells), we get different results. One explanation for this difference is the reliance in Drosophila on cytonemes to distribute Hh [46]. Although Shh-carrying filopodia have been observed in the chicken wing bud [47], cytonemes are not immediately apparent in mammalian embryos. Nevertheless, our observation of a correlation between Shh accumulation associated with an ECM discontinuity caused by the absence of Ext1, Ext2, or Gpc5 and an increased Shh response at distal cells leaves open the possibility of long-range cell-to-cell contacts that mediate Shh signaling.
Conclusion
Our work provides evidence that extracellular HSPGs both affect Shh distribution and noncell autonomous signaling. The simplest explanation for the Shh accumulation at the interface of normal ECM and that lacking HSPGs is a direct effect on Shh transport. The correlation between local Shh accumulation and an increased long-range response is consistent with the role of GPCs as tumor suppressors. However, the mechanism by which accumulation of Shh near the sites of synthesis results in increased long-range signaling remains unclear.
Supplementary Material
Significance Statement.
Sonic Hedgehog (Shh) is a signaling molecule that despite its association with molecules in the extracellular matrix (ECM) signals over several cell diameters. The authors used mosaic neural organoids comprised of genetically distinct cells to assess the requirement of heparan sulfate proteoglycans exclusively in the Shh-producing cells, the Shh-transporting cells, and the cells responding to Shh. The results show that Shh transport, is inhibited by heparan sulfate modified Glypican5, a component of the ECM. The facilitated Shh transport observed by Glypican5 null cells provides an elegant explanation for why Glypican5 is a tumor suppressor.
Acknowledgments
This work was supported by National Institute of Health grant 1R01GM117090 to H.R. and a postdoctoral fellowship by the Siebel Stem Cell Institute to W.G. Rabbit anti-Isl1/2 and HB9:GFP mESCs were a gift from Dr. Thomas Jessell (Columbia University). Sim1:Cre/tdTomato mESCs were a gift from Dr. Sam Pfaff (Salk, San Diego). We thank Naveen Natesh for his help with generating Ext1/2 sgRNA constructs and Eric Chen for his help with generating Disp1 sgRNA constructs. We also thank Jessica Dong for generating Disp1−/− Shh-expressing ES cells and providing the data used in Figure 2O.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 2013;14:416–429. [DOI] [PubMed] [Google Scholar]
- 2.Hui C-C, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol 2011;27:513–537. [DOI] [PubMed] [Google Scholar]
- 3.Jiang J, Hui C-C. Hedgehog signaling in development and cancer. Dev Cell 2008;15: 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter JA, von Kessler DP, Ekker SC et al. The product of hedgehog autoproteolytic cleavage active in local and long-range signalling. Nature 1995;374:363–366. [DOI] [PubMed] [Google Scholar]
- 5.Bumcrot DA, Takada R, McMahon AP. Proteolytic processing yields two secreted forms of Sonic Hedgehog. Mol Cell Biol 1995;15: 2294–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pepinsky RB, Zeng C, Wen D et al. Identification of a palmitic acid-modified form of human Sonic Hedgehog. J Biol Chem 1998; 273:14037–14045. [DOI] [PubMed] [Google Scholar]
- 7.Rietveld A, Neutz S, Simons K et al. Association of sterol- and glycosylphosphatidylinositol-linked proteins with drosophila raft lipid microdomains. J Biol Chem 1999;274: 12049–12054. [DOI] [PubMed] [Google Scholar]
- 8.Chen M-H, Li Y-J, Kawakami T et al. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev 2004;18:641–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burke R, Nellen D, Bellotto M et al. Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified Hedgehog from signaling cells. Cell 1999;99: 803–815. [DOI] [PubMed] [Google Scholar]
- 10.Jakobs P, Exner S, Schurmann S et al. Scube2 enhances proteolytic Shh processing from the surface of Shh-producing cells. J Cell Sci 2014;127:1726–1737. [DOI] [PubMed] [Google Scholar]
- 11.Creanga A, Glenn TD, Mann RK et al. Scube/You activity mediates release of dually lipid-modified Hedgehog signal in soluble form. Genes Dev 2012;26:1312–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damhofer H, Veenstra VL, Tol JAMG et al. Blocking Hedgehog release from pancreatic cancer cells increases paracrine signaling potency. J Cell Sci 2015;128:129–139. [DOI] [PubMed] [Google Scholar]
- 13.Dierker T, Dreier R, Petersen A et al. Heparan sulfate-modulated, metalloprotease-mediated Sonic Hedgehog release from producing cells. J Biol Chem 2009;284:8013–8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernfield M, Götte M, Park PW et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 1999;68:729–777. [DOI] [PubMed] [Google Scholar]
- 15.Yan D, Lin X. Shaping morphogen gradients by proteoglycans. Cold Spring Harb Perspect Biol 2009;1:a002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senay C, Lind T, Muguruma K et al. The EXT1/EXT2 tumor suppressors: Catalytic activities and role in heparan sulfate biosynthesis. EMBO Rep 2000;1:282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Österholm C, Barczyk MM, Busse M et al. Mutation in the heparan sulfate biosynthesis enzyme EXT1 influences growth factor signaling and fibroblast interactions with the extracellular matrix. J Biol Chem 2009;284:34935–34943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han C, Belenkaya TY, Wang B et al. Drosophila glypicans control the cell-to-cell movement of Hedgehog by a dynamin-independent process. Development 2004;131:601–611. [DOI] [PubMed] [Google Scholar]
- 19.Lin X Functions of heparan sulfate proteoglycans in cell signaling during development. Development 2004;131:6009–6021. [DOI] [PubMed] [Google Scholar]
- 20.Bellaiche Y, The I, Perrimon N. Tout-velu is a Drosophila homologue of the putative tumour suppressor EXT-1 and is needed for Hh diffusion. Nature 1998;394:85–88. [DOI] [PubMed] [Google Scholar]
- 21.Koziel L, Kunath M, Kelly OG et al. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev Cell 2004;6:801–813. [DOI] [PubMed] [Google Scholar]
- 22.Ramsbottom SA, Pownall ME. Regulation of Hedgehog signalling inside and outside the cell. J Dev Biol 2016;4:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filmus J, Capurro M. The role of glypicans in Hedgehog signaling. Matrix Biol 2014;35: 248–252. [DOI] [PubMed] [Google Scholar]
- 24.Capurro MI, Xu P, Shi W et al. Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev Cell 2008;14:700–711. [DOI] [PubMed] [Google Scholar]
- 25.Witt RM, Hecht M-L, Pazyra-Murphy MF et al. Heparan sulfate proteoglycans containing a glypican 5 core and 2-O-sulfo-iduronic acid function as Sonic Hedgehog co-receptors to promote proliferation. J Biol Chem 2013;288: 26275–26288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li F, Shi W, Capurro M et al. Glypican-5 stimulates rhabdomyosarcoma cell proliferation by activating Hedgehog signaling. J Cell Biol 2011;192:691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dessaud E, McMahon AP, Briscoe J. Pattern formation in the vertebrate neural tube: A Sonic Hedgehog morphogen-regulated transcriptional network. Development 2008;135: 2489–2503. [DOI] [PubMed] [Google Scholar]
- 28.Wichterle H, Lieberam I, Porter JA et al. Directed differentiation of embryonic stem cells into motor neurons. Cell 2002;110:385–397. [DOI] [PubMed] [Google Scholar]
- 29.Roberts B, Casillas C, Alfaro AC et al. Patched1 and Patched2 inhibit smoothened noncell autonomously. Elife 2016;5:e17634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meinhardt A, Eberle D, Tazaki A et al. 3D reconstitution of the patterned neural tube from embryonic stem cells. Stem Cell Rep 2014;3:987–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Etheridge LA, Crawford TQ, Zhang S et al. Evidence for a role of vertebrate Disp1 in long-range Shh signaling. Development 2010;137: 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ran FA, Hsu PD, Wright J et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caspary T, García-García MaJ, Huangfu D et al. Mouse dispatched homolog1 is required for long-range, but not Juxtacrine Hh signaling. Curr Biol 2002;12:1628–1632. [DOI] [PubMed] [Google Scholar]
- 34.Ma Y, Erkner A, Gong R et al. Hedgehog-mediated patterning of the mammalian embryo requires transporter-like function of dispatched. Cell 2002;111:63–75. [DOI] [PubMed] [Google Scholar]
- 35.Esko JD, Selleck SB. Order out of chaos: Assembly of ligand binding sites in Heparan sulfate. Annu Rev Biochem 2002;71:435–471. [DOI] [PubMed] [Google Scholar]
- 36.Sternfeld MJ, Hinckley CA, Moore NJ et al. Speed and segmentation control mechanisms characterized in rhythmically-active circuits created from spinal neurons produced from genetically-tagged embryonic stem cells. Elife 2017;6: e21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The I, Bellaiche Y, Perrimon N. Hedgehog movement is regulated through tout velu-dependent synthesis of a heparan sulfate proteoglycan. Mol Cell 1999;4:633–639. [DOI] [PubMed] [Google Scholar]
- 38.Bornemann DJ, Duncan JE, Staatz W et al. Abrogation of heparan sulfate synthesis in Drosophila disrupts the Wingless, Hedgehog and Decapentaplegic signaling pathways. Development 2004;131:1927–1938. [DOI] [PubMed] [Google Scholar]
- 39.Farshi P, Ohlig S, Pickhinke U et al. Dual roles of the Cardin-Weintraub motif in multimeric Sonic Hedgehog. J Biol Chem 2011;286: 23608–23619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Capurro MI, Shi W, Filmus J. LRP1 mediates the Shh-induced endocytosis of the GPC3-Shh complex. J Cell Sci 2012;125:3380–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang WG, Ye L, Ruge F et al. Expression of Sonic Hedgehog (SHH) in human lung cancer and the impact of YangZheng XiaoJi on SHH-mediated biological function of lung cancer cells and tumor growth. Anticancer Res 2015;35:1321–1331. [PubMed] [Google Scholar]
- 42.Vestergaard J, Pedersen MW, Pedersen N et al. Hedgehog signaling in small-cell lung cancer: Frequent in vivo but a rare event in vitro. Lung Cancer 2006;52:281–290. [DOI] [PubMed] [Google Scholar]
- 43.Theunissen J-W, de Sauvage FJ. Paracrine Hedgehog signaling in cancer. Cancer Res 2009; 69:6007–6010. [DOI] [PubMed] [Google Scholar]
- 44.Guo L, Wang J, Zhang T et al. Glypican-5 is a tumor suppressor in non-small cell lung cancer cells. Biochem Biophys Rep 2016;6: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson GW, Kaste SC, Chemaitilly W et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017;8: 69295–69302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guerrero I, Kornberg TB. Hedgehog and its circuitous journey from producing to target cells. Semin Cell Dev Biol 2014;33:52–62. [DOI] [PubMed] [Google Scholar]
- 47.Sanders TA, Llagostera E, Barna M. Specialized filopodia direct long-range transport of SHH during vertebrate tissue patterning. Nature 2013;497:628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.