

Abstract
Melanotic neuroectodermal tumor of infancy (MNTI) is a rare, fast-growing, pigmented neoplasm of neural crest origin. Despite of its rapid and locally infiltrative growth, it is still considered benign with high recurrence rate and malignant potential, so early diagnosis is extremely important to limit its local expansion. Recurrences can be expected primarily because of incomplete excision, tumor dissemination, or due to its multicentric nature. We report a case of MNTI originating in the maxilla of a 7-month-old male infant, which was managed with complete surgical excision of the tumor, with no signs of recurrence after a 1-year follow-up period. In addition, a complete literature review has been reported, in an attempt to understand the origin, histopathologic and immunohistochemistry features with surgical modalities and advantages of radiotherapy and chemotherapy in cases where complete surgical extirpation is questionable.
Keywords: Infancy, melanotic neuroectodermal tumor, neural crest cells
INTRODUCTION
The melanotic neuroectodermal tumor of infancy (MNTI) is a rare neoplasm that occurs most often during the 1st year of life. The uncertainty of the histogenesis of this tumor is reflected in a multiplicity of names, which have included melanotic progonoma, pigmented admantinoma, congenital pigmented epulis, melanoblastoma and retinal anlage tumor.[1,2]
Locally aggressive, benign lesion known to be derived from cells of the neural crest cells, Borello and Gorlin[3] proposed the neural crest origin of this tumor in 1966. Neural crest cells are multipotent embryonic cells that ultimately differentiate into various structures, including the odontogenic ectomesenchyme, melanocytes and neural ganglia. These cells display mesodermal and ectodermal morphologic features at different stages of their ontogeny, explaining the difficulty in deciphering the embryologic origin of these tumors and possibly explaining the biphasic cellular phenotype.[4] The results of immunohistochemical investigations of histogenesis are inconsistent. MNTIs came with the detection of messenger RNA for melanotransferrin in the tumor cells of MNTI.[5]
We report a case of MNTI originating in the maxilla of a 7-month-old male infant, which was managed with complete excision of the pathology preserving the vital structures.
CASE REPORT
Clinical presentation and radiographic feature
A 7-month-old child presented to our unit with a painless swelling on the alveolar ridge of the anterior maxilla, that had been gradually increasing in size from past 2 months [Figure 1a and b]. The extraoral examination revealed a diffuse swelling noted in left infraorbital region, obliterating the left nasolabial folds, elevating alar base, with superior displacement of lateral nares. The intraoral examination showed well-defined submucosal mass, with a smooth intact overlying mucosa, buccal expansion with partial obliteration of vestibule with an intact mucosal surface in the entire left alveolus, hard palate [Figure 1c]. No biopsy or aspiration was performed. A clinical diagnosis of fibro-osseous lesion of left maxilla was made. Computed tomography showed a radiolucent, expansile osteolytic mass involving the left alveolus and maxilla [Figures 2 and 3], extending to the orbital rim and extending into and crossing the midline, unerupted tooth buds were displaced laterally. Sclerotic expansion of the bone was noted toward the nasal cavity. It was decided to do an excisional biopsy of the pathology.
Figure 1.

(a-c) Preoperative picture of the patient with the pathology
Figure 2.
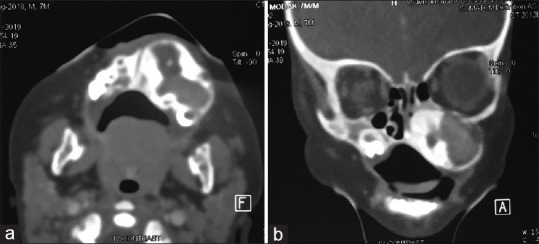
(a-b) CT images showing expansion of the maxilla
Figure 3.
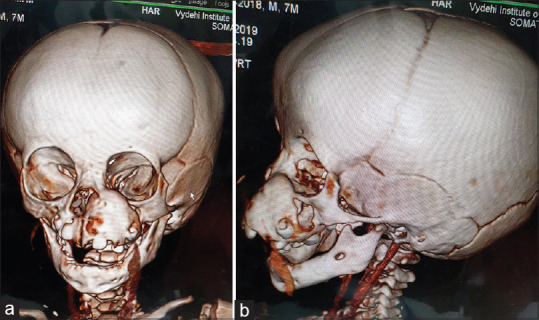
(a-b) 3D CT images showing the extent of involvement
Treatment and histopathologic features
The patient underwent surgical excision of the tumor with peripheral ostectomy. A vestibular incision was placed to expose the pathology extending till left nasal cavity and infraorbital area, the lesion was removed in pieces and 4 deciduous teeth 51,61,62,63 were removed. During excision, pathology showed a black-bluish pigmentation [Figure 4a-c].
Figure 4.
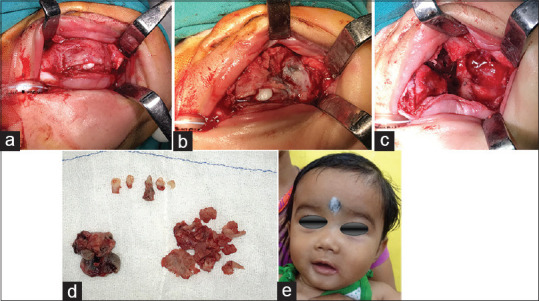
(a-b) Excision of pathology and immediate post op appearance
The surgical specimens consisted of a larger specimen, measuring 2.3 cm × 1.8 cm × 1 cm and a small specimen measuring 0.4 cm × 0.5 cm × 0.1 cm [Figure 4d].
Histopathologic sections of the decalcified hematoxylin and eosin section specimen showed cortical and cancellous bone showing trabeculae containing numerous osteocytes with osteoblastic rimming separated by fibrovascular connective tissue stroma. Cancellous bone showed resorption. Focal areas of connective tissue area showed resorption. Focal areas show infilteration with epithelioid cells. The section even showed biphasic population of cells, hyperchromatic nuclei resembling neuroblast-like cells centrally, cuboidal to flattened epithelioid cells containing melanin pigment peripherally. Few areas of hemorrhage also seen. A diagnosis of MNTI of maxilla was given. The postoperative period was uneventful [Figure 4e]. The patient has been followed up for 2 years and there have been no signs of recurrence [Figure 5].
Figure 5.
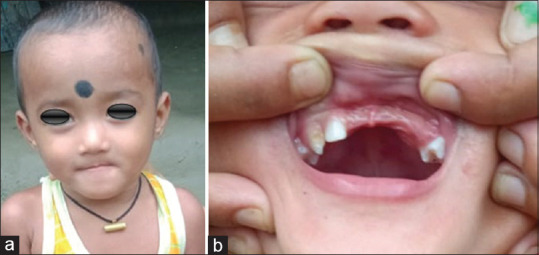
(a-b) Post op appearance after 2 years
DISCUSSION
MNTI is an uncommon, rapidly growing neoplasm of neural crest origin primarily developing in the jaws of infants. The lesion was originally described by Krompecher in 1918, as a congenital melanocarcinoma.[6]
Krompecher in 1918, called it a congenital melanocarcinoma, several theories for the origin of this tumor have been proposed, believing it arising from the retinal anlage, whereas few authors believed it to be of odontogenic origin.[7] Still others think it could represent a melanocarcinoma derived from the enclosed epithelial rests or simply neuroectodermal in origin. Few consider these tumors to be originating from the Jacobson vomeronasal organ. However, the common consensus favors the neuroectodermal origin of these tumors.[8]
In the past, a multiplicity of names has been used for this lesion owing to the confusing and conflicting histogenetic theories. These names included congenital melanocarcinoma, melanotic epithelial odontome, melanotic progonoma, pigmented adamantinoma, melanotic ameloblastoma, congenital pigmented epulis, melanoameloblastoma, retinal anlage tumor, melanocytoma and pigmented neuroectodermal tumor of infancy.[7]
Lamping et al.,[9] the tissue resembles closely to retinal structures and because of involvement of orbital structures, considering in the differential diagnosis of pigmented, orbital tumors, especially those occurring in infants. The alveolar structures lined by cells closely resembling retinal pigmented epithelium and containing either small neuroblastic cells reminiscent of those seen in retinoblastomas or more highly differentiated neural tissue.[9]
Vanillylmandelic acid (VMA) levels indicate the possibility of neuroectodermal origin. Biochemical analyses have shown high levels of urinary VMA in MNTI.[10] In our case, VMA levels were not checked, as was directly taken up for excisional biopsy. In a recent study done by Chrcanovic and Gomez, measurements of the levels of urinary VMA were done for 69 cases and elevated levels were seen in 24. Measurements of the levels of serum Alpha-fetoprotein were reported for only five cases, with elevated levels reported for four of them. There was information about the percentage of Ki-67 in 36 cases. There was no statistical correlation between Ki67 and several factors (recurrence, cortical bone perforation, age, lesion size, duration of the lesion, or VMA) and no statistically significant difference in mean percentage Ki-67 values between lesions that recurred and lesions that did not recur. At present, no reliable histological or immunophenotypical markers predict clinical aggressiveness or the need for complete excision. Moreover, although some recurrent tumors are aneuploid, recurrent diploid tumors have also been reported.[10]
Tran Kiem et al., showed proliferation of a dual population of cells: Polygonal pigmented cells that were positive for HMB – Human melanoma black, and EMA – Epithelial membrane antigen; and small round cells groups with neurofilament background, that were positive for Synaptophysin and CD56, these findings were consistent with the diagnosis of MNTI.[11]
Melissari et al.,[12] among other authors, showed ultrastructural features of nerve cells in addition to the immunohistochemical findings of enolase and S-100 protein in some of the tumor cells.[13]
Microscopically, MNTIs are biphasic tumors composed of collections of larger, polygonal, epithelioid cells resembling melanocytes admixed with smaller, neuroblast-like round cells, with variable deposits of melanin. Good evidence that MNTIs are of neural crest origin came with the detection of messenger RNA for melanotransferrin in the tumor cells of MNTI. Other researchers have aimed to identify more dangerous MNTIs, and some cell cycle proteins and DNA ploidy, which detected 2 aneuploid MNTIs that recurred, have been analyzed. A histologic means of discriminating between aggressive and more typical, benign MNTIs would be advantageous.[12]
The optimal surgical management for MNTI is not entirely clear. Total excision provides the best assurance of complete control with low recurrence. The most investigators now favor a surgical approach that is conservative via a bucco-gingival sulcus incision or lateral rhinotomy approach. If technically possible, complete resection with a 5-mm margin is recommended, but when radical surgery would cause mutilation, local excision with curettage of the underlying bone is usually sufficient.[14]
Some authors have demonstrated a significant presurgical reduction of the “neuroblastic-like” component with chemotherapy. When metastasis develops (up to 7% of cases), it is the “neuroblastic-like” component that is regarded as the aggressive part of the neoplasm. These malignant MNTI develop widespread metastasis and cause death within a few months. Consequently, these tumors are histologic mimics of neuroblastoma rather than MNTI. In this setting, surgery with adjuvant radiation and/or chemotherapy is the usual treatment. The usefulness of DNA ploidy by flow cytometry in predicting tumor behavior is controversial, although aneuploidy is associated with tumor recurrence.[15]
The primary aim in MNTI treatment is to cure with the benefit of minimizing the number of surgical interventions and their sequelae an additional consideration. Successful surgery should remove the need for adjuvant therapy. Recurrence rates have previously been reported varying between 10% and 15%. There is no established surgical margin but a 5-mm macroscopic margin has been proposed. Complete resection seems to provide the most reliable cure, but a successful outcome can also be achieved with preservation of key anatomical landmarks and microscopically incomplete resection. Any significant surgical intervention to the facial skeleton in a child is likely to cause altered growth and development. This could burden the patient with long-term rehabilitative care and reconstructive needs. In certain anatomical sub-sites, if an extensive resection can deliver a macroscopically clear margin then this may be appropriate. However, in more anatomically sensitive sites, then the balance in favor of a more conservative approach may be more acceptable. This alternative may be coupled with a delayed approach to reconstruction and intensive short-term follow-up to aid clinical surveillance and allow early detection of potential recurrence.[16]
Maroun et al. administered neoadjuvant chemotherapy before surgery, thereby obtaining adequate shrinkage of the tumor to allow better resectability and easier surgical access, which minimized the need for wider margins.[17]
CONCLUSION
MNTI, a biphasic neoplastic lesion that occurs in infancy and may be treated by surgical excision without an incisional biopsy, thus avoiding further manipulation of the lesion. The distinguishing features of this biphasic tumor cell population with melanin pigment allow its separation from other pediatric neoplasms.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient (s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initial s will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Hupp JR, Topazian RG, Krutchkoff DJ. The melanotic neuroectodermal tumor of infancy. Report of two cases and review of the literature. Int J Oral Surg. 1981;10:432–46. doi: 10.1016/s0300-9785(81)80080-4. [DOI] [PubMed] [Google Scholar]
- 2.Steinberg B, Shuler C, Wilson S. Melanotic neuroectodermal tumor of infancy: Evidence for multicentricity. Oral Surg Oral Med Oral Pathol. 1988;66:666–9. doi: 10.1016/0030-4220(88)90314-3. [DOI] [PubMed] [Google Scholar]
- 3.Borello KD, Gorlin RJ. Melanotic neuroectodermal tumor of infancy: A neoplasm of neural crest origin. Cancer. 1966:19;96. doi: 10.1002/1097-0142(196602)19:2<196::aid-cncr2820190210>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Davis JM, DeBenedictis M, Frank DK, Lessin ME. Melanotic neuroectodermal tumor of infancy: A wolf in sheep's clothing. Ann Otol Rhinol Laryngol. 2015;124:97–101. doi: 10.1177/0003489414543070. [DOI] [PubMed] [Google Scholar]
- 5.Nitta T, Endo T, Tsunoda A, Kadota Y, Matsumoto T, Sato K. Melanotic neuroectodermal tumor of infancy: A molecular approach to diagnosis. Case report. J Neurosurg. 1995;83:145–8. doi: 10.3171/jns.1995.83.1.0145. [DOI] [PubMed] [Google Scholar]
- 6.Manojlović S, Virag M, Lukšić I, Müller D. Melanotic neuroectodermal tumour of infancy: Report of two cases and review of the literature. J Craniomaxillofac Surg. 2012;40:e103–7. doi: 10.1016/j.jcms.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 7.Batsakis JG. Melanotic neuroectodermal tumor of infancy. Ann Otol Rhinol Laryngol. 1987:45;743. doi: 10.1177/000348948709600132. [DOI] [PubMed] [Google Scholar]
- 8.Clarke BE, Parsons H. An embryological tumor of retinal anlage involving the skull. Cancer. 1951;4:78–85. doi: 10.1002/1097-0142(195101)4:1<78::aid-cncr2820040107>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 9.Lamping KA, Albert DM, Lack E, Dickersin GR, Chapman PH, Walton D. Melanotic neuroectodermal tumor of infancy. Ophthalmology. 1985:92:143–9. doi: 10.1016/s0161-6420(85)34080-0. [DOI] [PubMed] [Google Scholar]
- 10.Chrcanovic BR, Gomez RS. Melanotic neuroectodermal tumour of infancy of the jaws: An analysis of diagnostic features and treatment. Int J Oral Maxillofac Surg. 2019;48:1–8. doi: 10.1016/j.ijom.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Tran Kiem H, Nguyen T, Tran P, Nguyen L, Huu S, Canh D. Melanotic neuroectodermal tumor of infancy. J Pediatr Surg Case Rep. 2019;(46):101221. [Google Scholar]
- 12.Barrett AW, Morgan M, Ramsay AD, Farthing PM, Newman L, Speight PM. A clinicopathologic and immunohistochemical analysis of melanotic neuroectodermal tumor of infancy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;93:688–98. doi: 10.1067/moe.2002.124000. [DOI] [PubMed] [Google Scholar]
- 13.Melissari M, Tragni G. Gaetti L Melanotic neuroectodermal tumor of infancy. Immunohistochemical and ultrastructural study of a case. J Craniomaxillofac Surg. 1988;16:330. doi: 10.1016/s1010-5182(88)80073-8. [DOI] [PubMed] [Google Scholar]
- 14.Rustagi A, Roychoudhary A, Karak A. Melanotic neuroectodermal tumour of infancy of the maxilla: A case report with review of literature. J Oral Maxillofac Surg. 2011:69:1120–4. doi: 10.1016/j.joms.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 15.Pettinato G, Manivel JC, d'Amore ES, Jaszcz W, Gorlin RJ. Melanotic neuroectodermal tumor of infancy. A reexamination of a histogenetic problem based on immunohistochemical, flow cytometric, and ultrastructural study of 10 cases. Am J Surg Pathol. 1991;15:233–45. [PubMed] [Google Scholar]
- 16.Rickart AJ, Drummond-Hay V, Suchak A, Sadiq Z, Sebire NJ, Slater O, et al. Melanotic neuroectodermal tumour of infancy: Refining the surgical approach. Int J Oral Maxillofac Surg. 2019;48:1307–12. doi: 10.1016/j.ijom.2019.02.011. [DOI] [PubMed] [Google Scholar]
- 17.Maroun C, Khalifeh I, Alam E, Akl PA, Saab R, Moukarbel RV. Mandibular melanotic neuroectodermal tumor of infancy: A role for neoadjuvant chemotherapy. Eur Arch Otorhinolaryngol. 2016;273:4629–35. doi: 10.1007/s00405-016-4066-6. [DOI] [PubMed] [Google Scholar]