

Abstract
Neurodegenerative diseases (NDD) are disorders characterized by the progressive loss of neurons affecting motor, sensory, and/or cognitive functions. The incidence of these diseases is increasing and has a great impact due to their high morbidity and mortality. Unfortunately, current therapeutic strategies only temporarily improve the patients’ quality of life but are insufficient for completely alleviating the symptoms. An interaction between the immune system and the central nervous system (CNS) is widely associated with neuronal damage in NDD. Usually, immune cell infiltration has been identified with inflammation and is considered harmful to the injured CNS. However, the immune system has a crucial role in the protection and regeneration of the injured CNS. Nowadays, there is a consensus that deregulation of immune homeostasis may represent one of the key initial steps in NDD. Dr. Michal Schwartz originally conceived the concept of “protective autoimmunity” (PA) as a well‐controlled peripheral inflammatory reaction after injury, essential for neuroprotection and regeneration. Several studies suggested that immunizing with a weaker version of the neural self‐antigen would generate PA without degenerative autoimmunity. The development of CNS‐related peptides with immunomodulatory neuroprotective effect led to important research to evaluate their use in chronic and acute NDD. In this review, we refer to the role of PA and the potential applications of active immunization as a therapeutic option for NDD treatment. In particular, we focus on the experimental and clinical promissory findings for CNS‐related peptides with beneficial immunomodulatory effects.
Keywords: CNS‐related peptides, neurodegeneration, protective autoimmunity
Considerable evidence indicated that the loss of immune homeostasis may represent one of the key initial steps in neurodegenerative disease (NDD). Several studies suggested that immunizing with a weaker version of the neural self‐antigen induces mechanisms that bring to protection and regeneration of the injured central nervous system (CNS). The development of CNS‐related peptides with immunomodulatory neuroprotective effects led to important research to evaluate their use in chronic and acute NDD. In this review, we refer to the potential applications of active immunization as a therapeutic strategy for NDD treatment. In particular, we focus on the experimental and clinical findings for CNS‐related peptides with beneficial immunomodulatory effects.
Abbreviations
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- Aβ
beta‐amyloid plaques
- CNS
central nervous system
- GA
glatiramer acetate
- MBP
myelin basic protein
- NDD
neurodegenerative diseases
- PA
protective autoimmunity
- PD
Parkinson's disease
- SCI
spinal cord injury
- SOD
superoxide dismutase
- TBI
traumatic brain injury
- Tregs
regulatory T cells
1. INTRODUCTION
Neurodegenerative diseases (NDD) are a heterogeneous group of diseases characterized by the progressive degeneration of the structure and function of the central nervous system (CNS) or the peripheral nervous system affecting motor, sensory, and/or cognitive functions. NDD can further be divided into acute and chronic classification. Chronic diseases such as amyotrophic lateral sclerosis (ALS), Alzheimer's disease (AD), and Parkinson's disease (PD) are the common conception of NDD in which the shared pathological hallmark is the accumulation of misfolded proteins. Moreover, acute traumatic lesions of the CNS, such as global or focal cerebral ischemia (stroke), spinal cord injury (SCI), and traumatic brain injury (TBI), cause widespread inflammation and other phenomena that lead to neurodegeneration.
Traditionally, most of the research on NDD is aimed towards the diseases that have the most significant impact on the population, such as ALS, AD, and PD. In brief, ALS is a motor neuron disease caused by gradual deterioration and death of motor neurons. It mainly involves the nerve cells (neurons) responsible for controlling voluntary muscle movement. In this disease, both the upper and lower motor neurons degenerate (or die) and stop sending messages to the muscles, which gradually weaken, start to twitch (fasciculations), and atrophy. Eventually, the brain loses its ability to initiate and control voluntary movements.1
AD is a progressive disorder with a gradual decline in memory, executive function, and ability to perform daily activities. The specific biomarkers associated with an AD diagnosis are the presence of beta‐amyloid plaques (Aβ) and neurofibrillary tangles composed of the aggregated microtubules‐associated protein tau.2
PD is a chronic progressive disease characterized by motor symptoms such as bradykinesia, tremor, and rigidity. The pathophysiological hallmark of PD is the specific loss of midbrain dopamine neurons in the substantia nigra, which leads to the described symptoms. PD is characterized by the accumulation of Lewy bodies, α‐synuclein, and related multimers in dopaminergic neurons.3
Even though NDD have high mortality, their greatest impact is on morbidity: the incidence of these diseases is rapidly increasing in an aging population like ours, creating an urgent need for treatment development. Thus, it is estimated that AD alone affected 40 million people worldwide in 2015 and that this number will increase to 135 million by 2050.4 For chronic NDD, several aetiologies have been studied, such as trauma, genetic mutation, and exposure to toxic and infectious agents, diet, and behavioral occupational factors. Moreover, a significant interaction between the immune system and CNS is widely associated with neuronal damage in NDD.
In acute NDD, it has been shown that the inflammatory response is strongly modulated by an autoimmune reaction directed against neural constituents, specifically against myelin basic protein (MBP), one of the most abundant immunogenic proteins in the CNS.5, 6
TBI and SCI are increasingly recognized as global health priorities given the preventability of most injuries and the complex and expensive medical care they require. The term SCI refers to damage to the spinal cord resulting from trauma or disease, such as cancer. Up to 90% of SCI have been traumatic in origin. These induce acute and chronic repercussions depending on the vertebral level and the severity of the injury.7 TBI is a disruption in the normal function of the brain that can be caused by a bump or jolt to the head or a penetrating head injury. TBI and SCI constitute a considerable portion of the global injury burden and are caused primarily by falls and road injuries.7 Stroke is a broad term that encompasses neurological injury resulting from any vascular cause. The most common type is ischemic stroke (about 87%), being one of the most prevalent vascular diseases globally and a major cause of disability and death worldwide.8
Commonly, immune cell infiltration has been identified with inflammation and is considered harmful to the injured CNS. However, the immune system induces cellular and humoral responses and boosts tissue repair, cellular healing, and clearance of cellular detritus. These findings indicate that immune cells have a crucial role in protecting and regenerating the injured CNS. Dr. Michal Schwartz from the Weizmann Institute of Science in Israel originally conceived the concept of “protective autoimmunity” (PA).9 This group started to modulate the action of myelin‐specific autoreactive lymphocytes by immunizing with MBP. This strategy improved tissue preservation, neuronal survival, and motor recovery after acute SCI.10, 11 However, immunization with self‐antigens induced an autoimmune disease known as experimental autoimmune encephalomyelitis. Therefore, research for eliciting PA without provoking an autoimmune disease led to the use of a weaker version of the self‐antigen for immunization. These types of antigens are known as CNS‐related peptides. Vaccinating with these peptides would generate PA without degenerative autoimmunity. The development of CNS‐related peptides with immunomodulatory neuroprotective effect led to significant research into their potential therapeutic use in chronic and acute NDD.
In this review, we described the concept and role of PA, the beneficial effects of active immunotherapy, and the potential applications of different CNS‐related peptides to the treatment of NDD.
2. THE ROLE OF THE IMMUNE SYSTEM IN BOTH THE DEGENERATION AND REGENERATION OF THE CNS: THE CONCEPT OF NEUROPROTECTIVE IMMUNITY
Classically, the CNS was considered an “immune‐privileged” site, in which the blood–brain barrier (BBB) kept the peripheral cells of both the innate and adaptive immune system out. Under this notion, the microglia, the resident CNS macrophages, were the only source of the innate immune cells in the CNS. Thus, autoimmunity and neurodegeneration were assumed to be automatic consequences of the immune cell infiltration and activation during BBB damage. However, several findings revealed that the CNS has an active interaction with the peripheral immune system.12 Evidence points out that CNS–immune system interactions are a normal ongoing mechanism for the maintenance of CNS integrity, promoting neurogenesis and improving cognitive function.13 In healthy individuals, this immune–CNS interaction is tightly controlled to keep a positive relationship. Both NDD and the normal aging process occur an immune dysregulation and abnormal immune responses. Aging and immunity are closely associated with the pathogenesis of NDD. Several studies demonstrated that immunosenescence can induce an overactivation of CNS immune cells, promoting neuroinflammation.14
It is accepted that regardless of the different triggering events, the common feature in NDD is neuroinflammation. Microglia can orchestrate a potent inflammatory response under pathological conditions. Traditionally, macrophage and microglial activation has been classified in two different and opposite states: pro‐inflammatory (M1) and anti‐inflammatory (M2). However, extensive activation profiles have been recently described, especially concerning NDD.15 Anyway, activated microglia play a potentially harmful role in releasing reactive oxygen species (ROS) and pro‐inflammatory cytokines (IL‐1ß, IL‐6, and TNF‐α).16 Once activated, microglia can be a potent immune effector that initiates innate and adaptive immune responses and produce several cytokines, chemokines, and growth factors.16
Nowadays, it is accepted that the well‐controlled peripheral inflammatory reaction after an injury is essential for neuroprotection and regeneration. In NDD, an uncontrolled and prolonged response occurs leading to a vicious cycle of glial priming promoting neuronal damage. In 1999, Dr. Michal Schwartz's laboratory reported that autoimmunity in the CNS, under certain circumstances, could be protecting injured neurons from disseminating damage17, 18 and facilitate the regeneration processes in the severed spinal cord.18 She proposed the new concept of PA to refer to “autoimmunity response that is evoked by CNS insult when non‐immunological local protective mechanisms cannot adequately buffer the injury‐induced toxicity.”19 This physiological mechanism could be boosted by immunizing with CNS‐related peptides as a therapeutic strategy for NDD.
Recently, the brain's choroid plexus was identified as a selective gate for leukocyte input to the CNS, which led to the recruitment of macrophages and T cells in both neural tissue injury20 and neurodegeneration.21 Systemic regulatory T cells (Tregs) are crucial for maintaining autoimmune homeostasis and protection from autoimmune diseases.22, 23 However, the research analyzing the role of Tregs in CNS repair has shown contradictory results, with both protective and destructive effects.24, 25, 26 It has been described, in an experimental model of AD,27 that a peripheral reduction in Foxp3+ Tregs is followed by their accumulation in the CNS, suggesting that peripheral and tissue‐infiltrating Tregs play distinct roles in the brain pathology. Besides, it has been demonstrated that transient depletion of Tregs results in elevated levels of leukocyte trafficking molecules by the brain's choroid plexus through the increased interferon (IFN)‐γ availability in this compartment and high levels of systemic IFN‐γ‐expressing cells in the spleen.27 It has been proposed that IFN‐γ induces microglia into a phenotype that promotes glutamate clearance, contributing to the restoration of homeostasis.28 In agreement with these findings, our group found a correlation between chronic stress‐induced cognitive deficits and decreased peripheral and central IFN‐γ production.29, 30 Interestingly, it has been reported elevated Tregs levels with enhanced suppressive activities in AD patients.31, 32
3. IMMUNOMODULATORY THERAPIES
As described above, an appropriate local immune response mediated by autoimmune T cells is necessary to reduce neuronal cell death after CNS injury. Although effective immunotherapeutic options for acute CNS injury and chronic neuroinflammation remain limited, interest in this field is rapidly increasing. Figure 1 shows active and passive immunization and the possible cellular mechanisms involved. In particular, active immunization has attracted attention. Thus, stimulating the immune system to produce antigen‐specific antibodies to aid in removing extracellular protein aggregates or through the boosting of PA seems to be promissory strategies. Tables 1 and 2 summarized the clinical and experimental findings for immunomodulatory therapies for NDD treatment.
FIGURE 1.
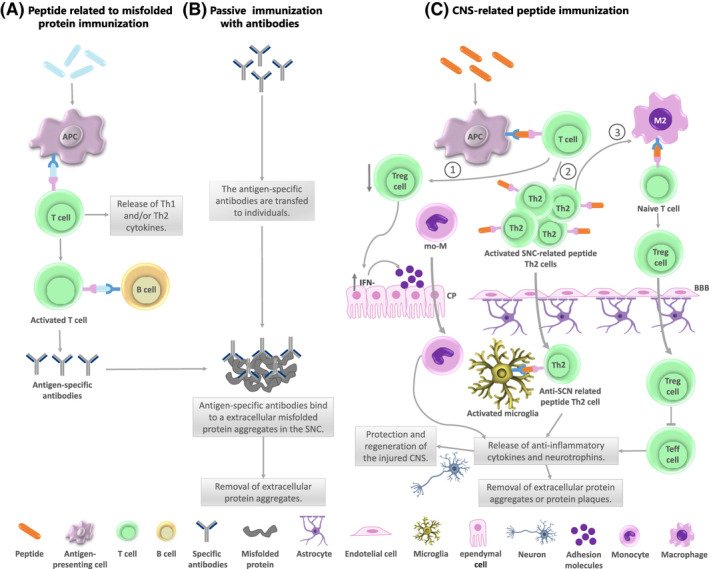
Immunomodulatory therapies. (A) Immunization to target epitopes from misfolded protein activates the immune system producing antigen‐specific antibodies. The antigen‐presenting cells (APCs) internalize and process the antigen and present it to T lymphocytes inducing T cell and B cell activation. The antigen‐specific antibodies produced bind to extracellular misfolded protein aggregates in the CNS to remove these protein aggregates. (B) In passive immunization the antigen‐specific antibodies are administered to individuals. The specific antibodies bind to extracellular misfolded protein aggregates in the CNS for clearance, without the need for immune response to be triggered. (C) The immunization with CNS‐related peptides produces T cell activation triggering three possible mechanisms: (1) a decrease of systemic T regulatory (Treg) cells and an increase in IFN‐γ producing cells at the choroid plexus (CP) increasing adhesion molecule levels leading to monocyte‐derived macrophages (mo‐ MΦ) trafficking through the CP; (2) the CNS‐related peptides reactive T cells infiltrate the CNS crossing the blood–brain barrier (BBB). In the CNS, these T cells come in contact with glial cells and activate microglia into a neuroprotective phenotype. Activated microglia present CNS‐related peptide to Th2 cells producing anti‐inflammatory cytokines and neurotrophins; (3) T cell activation, mediated by APCs, induces a prevalence of Th2 phenotype increasing the number and suppressor capacity of regulatory T cells and an anti‐inflammatory microglia profile controlling the ongoing inflammation. All these mechanisms bring to protection and regeneration of the injured CNS. The PA limits neuroinflammation, enhances expression of neurotrophic factors (BDNF, NT3) and insulin‐like growth factor‐1 (IGF‐1), releases anti‐inflammatory cytokines (IL‐4, IL‐10), improves cognitive performance and removal of extracellular protein aggregates or protein plaques, among others in a neurodegenerative context
TABLE 1.
Summary of immunomodulatory therapies used to treat chronic neurodegenerative diseases in patients and in animal models
| Immunomodulatory therapies | ||||
|---|---|---|---|---|
| Model/condition | Intervention | Outcome | Study | |
| Chronic diseases | ||||
| ALS | Human with ALS | GA vaccination: 11 patients treated everyday. Ten patients treated every 2 weeks. | ‐Patients treated everyday shift to a Th2 cytokine production pattern. Meanwhile, patients treated every 2 weeks show a Th1 cytokine profile | Mosley (2007)33 |
| Human with ALS | GA vaccination: 184 patients were treated with 40‐mg GA daily for 52 weeks. |
‐GA was well tolerated. ‐No differences between treated and control groups were observed. |
Meininger (2009)34 | |
| AD | APP‐Tg mice (AD model) | Nasal vaccination: GA + IVX‐908 (25‐µg GA plus 1‐µg IVX‐908) was given on Days 1 and 5 the first week, and 25‐µg GA alone was given on Days 2 and 4. Then, mice received weekly boosts for 6 weeks or starting at age 5 months until age 14 months. |
‐Potently decreases Aβ plaques. ‐Activation of microglia. ‐Reduction in TGF‐β expression. |
Frenkel (2005)35 |
| AD double‐transgenic (APP/PS1) mice (Tg‐AD) and Tg‐AD chimeric mice | GA vaccination: five s.c. injection with a total of 100 µg of high‐molecular‐weight GA dissolved in 200 µl of PBS, twice during the first week and once per week thereafter. |
Neuroprotection and neurogenesis: ‐Recruitment of bone marrow‐derived dendritic cells. ‐Decrease in the number of Aβ‐immunoreactive plaques. ‐Induction of neurogenesis (increase). ‐Attenuated cognitive decline. |
Butovsky (2006)36; Butovsky (2007)37 |
|
| Double‐transgenic APPSWE/PS1∆E9 mice (AD‐tg) | GA immunization: s.c. injections twice a week for the first 2 weeks and then once a week for a total of 3 months (100 µg in 1 × PBS). |
EGR1 as a potential mediator for GA‐induced neuroprotection: ‐Nuclear EGR1 protein levels were elevated in the hippocampus. ‐Negative correlation between EGR1 nuclear protein and Aβ plaque burden in the hippocampus. |
Bakalash (2011)38 | |
| Double‐ transgenic APPSWE/PS1∆E9 mice (AD‐tg) |
GA immunization: weekly s.c. injections (100‐μg GA in PBS). Adoptive transfer: monthly i.v. injection CD115+‐MoBM (5–6 × 106 cells/mouse). |
Retention of cognitive function, synaptic preservation, plaque removal, restriction of astrogliosis, and modulation of the immune molecular milieu: ‐Enhancement of the recruitment of MΦBM, at least partially, due to the significant increase in brain levels of MCP1. ‐Improvement in spatial learning and memory. ‐Elevated levels of the IL‐10 and MMP‐9 (both concentrated around amyloid‐b plaques). ‐Increased ability of MΦBM to phagocytize amyloid‐β1–42 fibrils (high co‐expression of the scavenger receptors CD36 and SCARA1). Activated MΦBM can reduce oligomers and fibrils, and protect synaptic integrity and neuronal structure. |
Koronyo (2015)39; Li (2020)40 |
|
| APPSWE/PS1∆E9‐transgenic mice (AD‐tg) |
GA immunization: weekly s.c. injections (100 μg GA in PBS). Adoptive transfer: monthly i.v. injection CD115+‐MoBM (5–6 × 106 cells/mouse). |
GA induces the recruitment of peripheral phagocytic cells to amyloid plaques by OPN: ‐Upregulation of OPN expression in MΦBM ‐OPN promotes a highly phagocytic phenotype (increased uptake of Aβ fibrils and associated sequestering receptors), anti‐inflammatory (altered cell morphology, decreased iNOS, induction of IL‐10 and Aβ degrading enzyme MMP‐9), and pro‐scarring. |
Rentsendrj (2018)41 | |
| Patients with mild to moderate AD |
Aβ immunization: I.M. AN1792 225ug plus the adjuvant QS‐21.5 µg. As a single 0.5‐ml IM injection on day 0 and at Months 1, 3, 6, 9, and 12. |
‐Improved memory ‐CSF tau was decreased ‐Meningoencephalitis. |
Gilman (2005)42 | |
| Patients of mild‐to‐moderate AD |
Aβ immunization: UB‐311 (UBITh®). Three doses intramuscular route (300 mg/dose) at Weeks 0, 4, and 12. |
‐Generation of antibodies able to bind both Aβ1–42 monomers and oligomers. ‐It induced cognitive improvements in patients with early‐stage Alzheimer's dementia. |
Wang (2017)43 | |
| PD | SJK mice. MPTP PD model | Adoptive transfer of Cop‐1 immune cells |
‐Higher numbers of surviving SNpc dopaminergic neurons. ‐Neuroprotection mediated by Th2 cells. |
Benner (2004)44 |
| C57BL/6J mice. MPTP PD model | GA administration, 3.5 mg/kg (7 days/week) |
‐Motor recovery (grip strength and gait parameters). ‐Restoration of the nigrostriatal pathway |
Churchill (2019)45 | |
TABLE 2.
Summary of immunomodulatory therapies used to treat acute neurodegenerative diseases in patients and in animal models
| Immunomodulatory therapies | ||||
|---|---|---|---|---|
| Model/condition | Intervention | Outcome | Study | |
| Acute diseases | ||||
| Stroke |
Rat model of cerebral ischemia (tMCAo‐transient middle cerebral artery occlusion). |
Injection with Cop‐1: 200 μg after reperfusion. |
Neuroprotection and neurogenesis through the stimulation of PA: ‐Significant improvement in neurological deficit. ‐Lower percentage of infarct volume ‐Modulation of the CP microenvironment (upregulation of BDNF, IGF‐1, NT‐3 and IL‐10 in the SVZ, SGZ and CC, which correlated with an increase in neurogenesis; downregulation of IL‐17). |
Ibarra (2007)46 ; Cruz (2015)47 ; Cruz (2018)47 |
| Mouse model of tMCAo. | Injection with GA: 3.5 mg/kg of body weight before the induction of stroke. | No protective effect (stroke volume and functional parameters without significant differences) | Kraft (2014)48 | |
| Mouse model of permanent focal cerebral ischemia occlusion (pMCAo) and tMCAo | Injection with GA: 2 mg in 200 μl SS. after cerebral ischemia in the pMCAo group and immediately after reperfusion in the tMCAo group. |
No protective effect (does not reduce infarct volume or improve neurological deficit): ‐significant increase in neurogenesis in pMCAo ‐Reduction in microglial pro‐inflammatory cytokines (TNFα e IL‐1β) in tMCAo. Microglial cells were activated in both models. |
Poittevin 201349 | |
| SCI | Lewis rat. T7 or T9 spinal cord contusion | Passive (107 T cells specific to MBP) or Active (with MBP) immunization | ‐Enhanced motor and cellular recovery | Hauben (2000)10 |
| Sprague‐Dawley rats. T9‐T10 spinal cord contusion | GA immunization: High dose (2 mg/km, 28 days) or low dose (0.5 mg/ml, 28 days) |
‐GA in high doses has deleterious effects. ‐The response to GA in SCI depends on dose and time of administration |
Askarifirouzjaei (2019)50 | |
| Fischer rats and BALB/c mice. T9 spinal cord injury | GA immunization. Rats: 200 µg at the base of the tail. Mice: 150 µg subcutaneous injection. | ‐Nitric oxide (NO) and inducible nitric oxide synthase (iNOS) gene expression reduction at the site of injury | García (2012)51 | |
| TBI |
C57Bl/6J and BALB/c/OLA mice. Closed head injury model. |
Cop‐1 immunization: intramuscular injection of 100 μg. | Neuroprotective effect: reduction in the spread of damage. | Kinips (2003)52 |
4. IMMUNIZATION TO TARGET EPITOPES FROM MISFOLDED PROTEIN
Therapeutic vaccination for AD consists in producing specific antibodies targeting Aβ and tau proteins to increase the clearance by promoting phagocytosis and neutralize their toxic effects. The first vaccine tested in AD patients was the AN‐1792 with a full‐length Aβ1–42 peptide as immunogen associated with a Th1 adjuvant (QS‐21; saponin). This vaccine generated anti‐Aβ antibody responses in <25% of patients with improved memory and decreased tau protein levels in the cerebral spinal fluid, thus being a promissory result for the development of new Aβ vaccines. However, AN‐1792 was discontinued because it induced the onset of meningoencephalitis in 6% of treated patients, likely due to self‐reactive T‐cell activation and infiltration of Aβ‐reactive T‐cells into the CNS.42
Several second‐generation Aβ‐targeting vaccines were subsequently designed to minimize Aβ‐related T‐cell inflammation. However, these vaccines have not shown convincing clinical efficacy data53 or have been discontinued.54 CAD106, currently in phase III clinical trial, has completed two phases II clinical trials reporting acceptable safety and tolerability and evoking a robust serological response in 80% of patients, and brain PET imaging evidenced target engagement.55, 56 Aβ is the principal target of late‐stage development programs with relatively few agents in clinical trials for AD, suggesting a need to amplify the drug discovery ecosystem.43
Recently, a novel design of the UBITh® platform‐based, fully synthetic Aβ1–14 peptide vaccine (UB‐311) was described.43 This vaccine was designed as a chimeric peptide to maximize immunogenicity and formulated in a Th2‐biased delivery system to minimize T‐cell inflammatory reactivity. The administration of this peptide‐based vaccine showed to be safe and well‐tolerated, generating strong site‐specific (Aβ1–10) antibodies in all patients. Also, it was able to improve cognitive functions in patients with early‐stage Alzheimer's dementia.43
PD pathology in the brain is characterized by the presence of Lewy bodies, of which the principal constituent is the accumulated and aggregated misfolded synaptic protein—α‐synuclein. One of the most promising PD vaccines was recently developed using a short peptide (seven amino acids) to avoid an α‐synuclein‐specific T‐cell response but provides the T helper epitopes in carrier proteins to activate the B‐cell response.57, 58
ALS pathology is characterized by amyloid deposits from different proteins such as tar‐DNA‐binding protein 43 (TDP43), Chromosome 9 open reading frame 72 (C9ORF72) dipeptide repeats, phosphorylated high molecular weight neurofilament protein (pNFH), rho guanine nucleotide exchange factor (RGNEF), and fused in sarcoma (FUS).59 Recent reports have indicated that misfolded superóxide dismutase 1 (SOD1) can act like prions and spread the disease.60, 61 Therefore, it is a candidate protein for testing a possible vaccine. Two ALS vaccines against unfolded SOD1, tgG‐DSE2lim, and tgG‐DSE5b were investigated in a motor neuron disease mouse model expressing human SOD1‐G37R.62 Both vaccines showed a robust epitope‐specific antibody response with a desirable Th2‐biased immune response. However, the question is how this approach can be modified to address different mutants and conformations of SOD1 protein.63
In conclusion, active immunity by vaccination with a short peptide of misfolded protein takes advantage of eliciting an adequate immune response against a harmful self‐antigen, but there is a risk of causing adverse autoimmunity. There is an increasing interest in searching for safe and effective immunogens for vaccination to minimize autoimmune reactions. An alternative strategy is passive immunotherapy using a humanized monoclonal antibody, which is considered relatively safe.64, 65
5. HARNESSING “PA” FOR NEURO‐REGENERATION AND CNS INJURY RECOVERY WITH CNS‐RELATED PEPTIDES
As described above, PA is a physiological mechanism induced after CNS injury that could be boosted by peptides. The rationale behind this therapeutic strategy is that it is better to modulate the immune response than eliminate it. In chronic NDD, patients require a competent immune response to avoid complications due to infection. Boosting of PA is a strategy that has rendered encouraging results both in acute and chronic NDD, being the latter the most difficult stage of injury to carry out a therapeutic approach.
As mentioned above, experimental studies demonstrated that immunization with a weaker version of the self‐antigen could generate an appropriate PA without harmful autoimmunity. In the last years, much research was performed to develop these immunomodulatory peptides and their possible therapeutic applications. In particular, glatiramer acetate (GA) has been used to induce PA in several chronic and acute NDD with encouraging results.
5.1. GLATIRAMER ACETATE
GA also called copolymer 1 (Cop‐1), is a random copolymer of glutamic acid, lysine, alanine, and tyrosine approved by the FDA for the treatment of relapsing–remitting multiple sclerosis.66 It has a well long‐term safety profile and proved efficacy in a daily dose of 20 mg/ml.67 Its mechanism of action has not been fully elucidated, but it seems that GA has an immune‐modulatory effect and neuro‐protective properties.68 It has been shown that GA exerts an immunomodulatory effect on cells of the innate and adaptive immune systems by inhibiting the activation of MBP‐reactive T cells and inducing an anti‐inflammatory T‐cell environment. Also, it exerts an inhibitory effect on the pro‐inflammatory (M1) microglia phenotype and stimulates the anti‐inflammatory (M2) microglia phenotype. Moreover, it has been demonstrated that GA protects neurons and oligodendrocytes68 and affects three characteristic processes of neurogenesis: neuronal progenitor cell proliferation,69 migration,70 and differentiation.71 Moreover, it has been found that GA immunization enhanced the expression of hippocampal early growth response protein 1 (Egr1), which is necessary for synaptic plasticity and memory formation.38
Additionally, it was reported that GA can alleviate the deleterious effect induced by chronic stress exposure. Thus, weekly treatment with GA for 3 weeks was able to reverse the learning impairment induced by stress through a mechanism that likely involves regulating the cytokine balance and adult neurogenesis.72 Besides, it has been demonstrated that the treatment with CNS‐related peptides can ameliorate depressive behavior induced by chronic mild stress in rats. The behavioral outcome was accompanied by the restoration of hippocampal BDNF levels and neurogenesis.73
Considering the beneficial effects of GA in the CNS, this has been the most investigated CNS‐related peptide‐based therapy for the treatment of chronic and acute NDD. A single injection of GA is protective in acute models of CNS insults, whereas, in chronic models, several boosts are necessary for a long‐lasting protective effect. Below, we briefly described the experimental and clinical findings of the GA effect in NDD.
5.1.1. Amyotrophic lateral sclerosis
Neuroinflammation is a prominent pathological finding in ALS patients with microglial activation, astrogliosis, and infiltration of monocytes and T cells. It has been thought that regimens with anti‐inflammatory drugs could offer therapeutic benefits to ALS patients. Results in experimental models are scarce and contradictory. A phase II randomized controlled trial including 20 patients was performed to test the safety, tolerability, and immunogenicity of different doses of GA in human ALS.74 The authors found that the tolerability of GA was acceptable. In contrast to control, patients elicited a T‐cell proliferative response to GA, indicating that immunity was modulated similarly in patients with ALS and MS. In the same cohort,33 it was showed that GA immunization induces a strong humoral response. GA‐treated ALS patients exhibited significant levels of anti‐GA antibodies. Concerning cytokine levels, the authors found that patients treated daily with GA had a Th2 ‐type response, while in patients treated biweekly, the production of cytokines was towards Th1.33 Finally, in a double‐blind, randomized, multicenter, placebo‐controlled trial to study the efficacy of a 40 mg/day dose of GA in ALS patients, GA did not show any beneficial effect in this population.34 The authors found acceptable safety and tolerability of GA in this population.34 The most common events are reactions at the injection site for the daily 20‐ and 40‐mg GA doses.34 Currently, the efficacy with less frequent GA dosing regimens in patients with ALS has not been studied.
In addition, a wide range of compounds, representing at least 10 different defined mechanisms of action were tested, but, despite significant research efforts, most of the human clinical trials have failed to demonstrate clinical efficacy in ALS. These therapeutic failures are partly due to this disease's heterogeneous characteristics requiring stratified case monitoring. In this sense, identifying immunological biomarkers is necessary to provide precision medicine to patients.75, 76
5.1.2. Parkinson's disease
1‐Methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP)‐intoxicated mice constitute an appropriate experimental model of PD. In this model, it was reported that adoptive transfer of GA‐induced immune cells induces prompts a T‐cell accumulation in the inflamed brain regions with suppression of microglial response and increased local production of glial cell line‐derived neurotrophic factor (GDNF) by astrocytes.44 The process was T‐cell‐dependent and protected the nigrostriatal dopaminergic system. The authors also found that Tregs mediated neuroprotection through suppression of microglial responses to stimuli, including aggregated, nitrated alpha‐synuclein.24
It was recently demonstrated that the administration of GA could reverse motor dysfunction observed in MPTP‐intoxicated mice. GA treatment recovers tyrosine hydroxylase protein expression in the striatum, facilitates restoration of the mature form of BDNF, and decreases the microglial marker, IBA1, protein expression within the midbrain. These results indicated that GA treatment effectively reversed clinical and pathological features of PD in this murine model.45
5.1.3. Alzheimer's disease
As mentioned, AD is characterized by plaque formation, neuronal loss, and cognitive decline. Transgenic mouse models resembling pathological features of AD are used as tools for developing insights into the biological basis of this pathology.
The effect of GA vaccination in a double‐transgenic mouse was evaluated.36 Treated mice showed a decrease in the number of Aβ‐immunoreactive plaques, increased neurogenesis, and a reduced cognitive decline.36 The authors reported several mechanisms for the amelioration of pathological features. Among them was a phenotypic switch of the microglia to dendritic‐like cells CD11c+ producing insulin‐like growth factor 1 in the brain.36 The increase in local CD11c+ dendritic‐like cells is due to GA‐induced recruitment of bone marrow‐derived dendritic cells37 pointed to the choroid plexus of the brain as a selective leukocyte gateway to the CNS, enabling macrophages and T‐cell recruitment following neural tissue injury.27 Moreover, they present evidence that the suppression of the immune response by Tregs is a negative factor in AD pathology, reducing the gateway activity of the choroid plexus of the brain to orchestrate the recruitment of leukocytes into the CNS.27
At present, it is recognized that activated macrophages effectively remove Aβ42 oligomers and rescue VGluT1/PSD95 synapses, providing a rationale for harnessing macrophages to treat AD. In recent studies,39, 40, 41 it has been shown that either an enrichment bone marrow‐derived CD115+ monocyte subset or weekly administration of GA in a transgenic AD animal model enhances natural recruitment of blood‐borne monocytes to diseased brain parenchyma. This increased cerebral infiltration of monocytes resulted in a substantial attenuation of disease progression in murine AD models by mechanisms that involved enhanced cellular uptake and enzymatic degradation of toxic amyloid‐β, as well as regulation of brain inflammation. The combined treatment induced higher monocyte recruitment than either therapy alone. These observations encourage the investigation of autologous monocyte administration, alone or in combination with a safe immune‐modulation therapy, for therapeutic intervention in patients with AD.
5.1.4. Spinal cord injury
GA effectiveness in SCI seems to depend on the dose used and the time of administration. It was studied the effect of 2‐week treatment of GA in low doses (0.5 mg/animal/day) in rats subjected to the avulsion of lumbar ventral roots.77 Results indicated that GA treatment induced a 40% increase in neuronal survival in the motor nucleus in the spinal cord along with a reduction of astroglial reactivity at the lesion site.
In a most recent study, GA treatment was carried out on female Sprague–Dawley rats using a high dose (2 mg/kg) for 28 consecutive days after the laminectomy. In contrast to other studies, the authors observed an impaired locomotor recovery, increased neuron loss in the acute phase after SCI, and a deleterious response against MBP.50
5.1.5. Stroke
It has been reported that the injection of low doses of GA 30 min after reperfusion generated a neuroprotective effect during the acute and chronic phases of stroke. Thus, immunization with GA significantly improved neurological function and decreased the percentage of infarct volume at 7 and 60 days after injection.46, 78 These effects are attributed to PA stimulation that alters the pro‐inflammatory cytokine profile observed after cerebral ischemia, inducing neuroprotection, and neurogenesis.47 The authors suggest that a possible GA‐induced mechanism to promote neurogenesis is through modulating the microenvironment of choroid plexus by the upregulation and release of BDNF, IGF‐1, NT‐3, and IL‐10 and downregulation of IL‐17.47 However, the immunization with high doses of GA immediately after cerebral ischemia did not show a protective effect. No reduction in infarct volume or improvement in the neural deficit was observed despite a significant increase in neurogenesis and a decrease in microglial activation.49 Consistent with these findings, prophylactic administration of GA 30 min before stroke induction resulted in similar stroke volumes between treated and controls, without differences in functional parameters on Day 1.48 Overall, these data demonstrate that GA did not have a protective effect on a very early stage after stroke in mice when administered in a prophylactic setting.
5.2. A91
A91 is a peptide derived from the immunogenic sequence (87–99) of the MBP, in which the lysine residue at position 91 has been replaced with alanine. The inoculation of A91 peptide induces the proliferation of CNS‐antigen‐specific T‐cells that exert protective actions through several mechanisms that promote neuroprotection.51, 79, 80, 81, 82 This therapeutic strategy has rendered encouraging results in both acute and chronic SCI, being the last one a challenging state to treat successfully.
It is necessary to consider the surrounding environment at chronic stages of injury. The glial scar formation, avoiding a correct reconnection of axons, is one of the main obstacles to allow the action of this therapeutic intervention. Another important feature of the chronic phase of injury is low activity at the damage site, followed by a progressive decline in the neurological function of injured individuals.
A beneficial effect of immunization with A91 after SCI has been reported. It was demonstrated that A91 acts by inhibiting lipid peroxidation,79 downregulating inducible nitric oxide synthase gene expression,51 and reducing apoptosis triggered by a traumatic injury.80 It also generates an improvement in the long‐term production of the neurotrophic factors BDNF and NT381 and GAP‐43.82 Besides, A91 induced significant production of anti‐inflammatory and regeneration‐associated proteins and a significant increase in neurogenesis in the chronic stages of injury.83 This favorable effect was associated with improved motor and sensitivity recovery in the chronic stage of SCI.82 Additionally, it is important to point out that this therapeutic strategy could be useful for other trauma‐related disorders such as TBI. Despite the promissory results described above for moderate injury, these beneficial effects were not observed in animals with severe SCI.83
Previous research has shown that scar removal84 or a matrix with bone marrow‐mesenchymal stem cells,85 used separately, promoted a significant tissue restoration and motor recovery after SCI. Considering the results obtained, Ibarra's laboratory investigated the effect of combining CNS‐related peptides with these alternative strategies to achieve a more efficient therapy. In a first approach, the authors explored both the immunization with A91 peptide alone or in combination with scar removal, on locomotor recovery, regeneration‐associated and cytokine gene expression, as well as the number of regenerating axons, in a model of chronic SCI.86 The results suggested that both treatments could substantially modify the nonpermissive microenvironment prevailing at the chronic phase of SCI, providing the opportunity to promote higher motor recovery. Treatment using scar removal combined with immunization with CNS‐related peptides therapy demonstrated greater beneficial effects.86
Afterward, the authors designed a combined therapy integrating surgical glial scar removal, a fibrin glue matrix with mesenchymal stem cells, and CNS‐derived peptides.87 The results showed that the combined therapy was able to modify the non‐permissive microenvironment post‐SCI but wasn't capable of inducing a proper axonal regeneration or neurogenesis as it was observed after treatment with CNS‐related peptides alone.87 However, they recently observed that combined therapy of monocyte locomotion inhibitor factor, A91 peptide, and glutathione monoethyl ester (GSH‐MEE) preserved spinal cord tissue integrity and promoted functional motor recovery in rats following chronicstage SCI.88
Also, it was reported that the vaccination with spinal cord homogenate‐pulsed dendritic cells89, 90 and in particular with A91‐pulsed dendritic cells91 enhanced the expression level of BDNF and NT‐3 and exerted neuroprotective effect in a SCI mice model.
It is important to note that it was proposing that the neuroprotective strategy using A91 peptide in combination with other immunomodulatory peptides could be useful for other neurodegenerative and neuroinflammatory acute or chronic pathologies.88 However, at present, there is a lack of evidence about the beneficial effect of A91 for others pathologies than SCI.
5.3. POLY‐YE
Poly‐YE is a copolymer formed by the dipeptide Glu‐Tyr that exerts regulatory effects on the immune system.92 Additionally, it has been observed that poly‐YE can decrease the activity of the regulatory T cells. Therefore, it has been used in several animal models of CNS injury to facilitate the spontaneous response of effector T cells by recognizing the antigens associated with cell damage. In an animal model of ischemic stroke, a single poly‐YE administration resulted in long‐lasting clinical and behavioral benefits, combined with neuroprotection and increased neurogenesis, from the subacute phase.93 In two animal models of glaucoma, the administration of Poly‐YE had a neuroprotective effect. Evidence shows that Poly‐YE acts through the regulation of the local microglia.94
6. CONCLUSIONS
Traditionally, it was thought that NDD of different etiology shared a common local neuroinflammatory component. However, several findings showed that the loss of immune homeostasis and/or early deregulation of immune responses may represent one of the key initial steps in disease pathogenesis leading to excessive inflammation or an inefficient reparative immune response. In the last years, it has become apparent that modulating the immune response was more important than eliminating it for the treatment of NDD. In this context, the concept of PA allowed the search of neural antigens that produce a weak immune response (CNS‐derived peptides) to fight with insult limiting the inflammatory response. The use of these peptides in experimental models of NDD has shown promissory results. In addition, clinical studies showed that GA has acceptable safety and tolerability.67 Nevertheless, the results have not shown the expected outcome. Combined therapy with other strategies that are capable of promoting significant tissue restoration is now under study. Considering the dual role of the immune system in NDD, a therapy that allows controlling excessive inflammation while maintaining the protective and regenerative immune functions will likely be crucial for the efficient treatment of NDD.
DISCLOSURES
The authors have indicated that they have no conflicts of interest regarding the content of this article.
ACKNOWLEDGMENTS
This work was supported by grants from the National Scientific and Technical Research Council of Argentina (CONICET, PIP 2015‐00163) and from the National Agency for the Promotion of Science and Technology (ANPCYT, PICT 2016‐2727) to AMG and from ANPCYT (PICT 2016‐0531) to MLP.
Palumbo ML, Moroni AD, Quiroga S, Castro MM, Burgueño AL, Genaro AM. Immunomodulation induced by central nervous system‐related peptides as a therapeutic strategy for neurodegenerative disorders. Pharmacol Res Perspect. 2021;9:e00795. 10.1002/prp2.795
DATA AVAILABILITY STATEMENT
None to declare.
REFERENCES
- 1.Ragagnin AMG, Shadfar S, Vidal M, Jamali MS, Atkin JD. Motor neuron susceptibility in ALS/FTD. Frontiers Neuroscience. 2019;13:532. 10.3389/fnins.2019.00532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jack CR, Bennett DA, Blennow K, et al.; Contributors . NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535‐562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386(9996):896‐912. 10.1016/S0140-6736(14)61393-3 [DOI] [PubMed] [Google Scholar]
- 4.Alzheimer's disease facts and figures. Alzheimers Dement. 2021;17(3):327‐406. 10.1002/alz.12328 [DOI] [PubMed] [Google Scholar]
- 5.Butovsky O, Hauben E, Schwartz M. Morphological aspects of spinal cord autoimmune neuroprotection: colocalization of T cells with B7–2 (CD86) and prevention of cyst formation. FASEB J. 2001;15(6):1065‐1067. 10.1096/fj.00-0550fje [DOI] [PubMed] [Google Scholar]
- 6.Popovich PG, Stokes BT, Whitacre CC. Concept of autoimmunity following spinal cord injury: possible roles for T lymphocytes in the traumatized central nervous system. J Neurosci Res. 1996;45(4):349‐363. [DOI] [PubMed] [Google Scholar]
- 7.James SL, Theadom A, Ellenbogen RG, Bannick MS; GBD 2016 Traumatic Brain Injury and Spinal Cord Injury Collaborators . Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990‐2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(1):56–87. 10.1016/S1474-4422(18)30415-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benjamin EJ, Muntner P, Alonso A, et al. Heart disease and stroke statistics‐2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56‐e528. 10.1161/CIR.0000000000000659 [DOI] [PubMed] [Google Scholar]
- 9.Schwartz M. Physiological approaches to neuroprotection. boosting of protective autoimmunity. Surv Ophthalmol. 2001;45(Suppl 3):S256‐S260. 10.1016/S0039-6257(01)00208-9 [DOI] [PubMed] [Google Scholar]
- 10.Hauben E, Butovsky O, Nevo U, et al. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J Neurosci. 2000;20(17):6421‐6430. 10.1523/JNEUROSCI.20-17-06421.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauben E, Nevo U, Yoles E, et al. Autoimmune T cells as potential neuroprotective therapy for spinal cord injury. Lancet. 2000;355(9200):286‐287. 10.1016/s0140-6736(99)05140-5 [DOI] [PubMed] [Google Scholar]
- 12.Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48‐65. 10.1111/j.1600-065X.2006.00441.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziv Y, Ron N, Butovsky O, et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9(2):268‐275. 10.1038/nn1629 [DOI] [PubMed] [Google Scholar]
- 14.Costantini E, D'Angelo C, Reale M. The role of immunosenescence in neurodegenerative diseases. Mediators Inflamm. 2018;2018:6039171. 10.1155/2018/6039171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bachiller S, Jiménez‐Ferrer I, Paulus A, et al. Microglia in neurological diseases: a road map to brain‐disease dependent‐inflammatory response. Front Cell Neurosci. 2018;12:488. 10.3389/fncel.2018.00488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsukahara T, Haniu H, Uemura T, Matsuda Y. Therapeutic potential of porcine liver decomposition product: new insights and perspectives for microglia‐mediated neuroinflammation in neurodegenerative diseases. Biomedicines. 2020;8(11):446. 10.3390/biomedicines8110446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moalem G, Leibowitz‐Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999;5(1):49‐55. 10.1038/4734 [DOI] [PubMed] [Google Scholar]
- 18.Schwartz M, Moalem G, Leibowitz‐Amit R, Cohen IR. Innate and adaptive immune responses can be beneficial for CNS repair. Trends Neurosci. 1999;22(7):295‐299. 10.1016/s0166-2236(99)01405-8 [DOI] [PubMed] [Google Scholar]
- 19.Schwartz M, Kipnis J. Protective autoimmunity: regulation and prospects for vaccination after brain and spinal cord injuries. Trends Mol Med. 2001;7(6):252‐258. 10.1016/s1471-4914(01)01993-1 [DOI] [PubMed] [Google Scholar]
- 20.Shechter R, Miller O, Yovel G, et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013;38(3):555‐569. 10.1016/j.immuni.2013.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunis G, Baruch K, Miller O, Schwartz M. Immunization with a Myelin‐derived antigen activates the brain's choroid plexus for recruitment of immunoregulatory cells to the CNS and attenuates disease progression in a mouse model of ALS. J Neurosci. 2015;35(16):6381‐6393. 10.1523/JNEUROSCI.3644-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775‐787. 10.1016/j.cell.2008.05.009 [DOI] [PubMed] [Google Scholar]
- 23.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8(2):191‐197. 10.1038/ni1428 [DOI] [PubMed] [Google Scholar]
- 24.Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson's disease. J Leukoc Biol. 2007;82(5):1083‐1094. 10.1189/jlb.0507296 [DOI] [PubMed] [Google Scholar]
- 25.Walsh JT, Kipnis J. Regulatory T cells in CNS injury: the simple, the complex and the confused. Trends Mol Med. 2011;17(10):541‐547. 10.1016/j.molmed.2011.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walsh JT, Zheng J, Smirnov I, Lorenz U, Tung K, Kipnis J. Regulatory T cells in central nervous system injury: a double‐edged sword. J Immunol. 2014;193(10):5013‐5022. 10.4049/jimmunol.1302401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baruch K, Rosenzweig N, Kertser A, et al. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer's disease pathology. Nat Commun. 2015;6:7967. 10.1038/ncomms8967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaked I, Tchoresh D, Gersner R, et al. Protective autoimmunity: interferon‐gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem. 2005;92(5):997‐1009. 10.1111/j.1471-4159.2004.02954.x [DOI] [PubMed] [Google Scholar]
- 29.Palumbo ML, Di Rosso ME, Simon EH, Gonzalez Murano MR, Genaro AM. Altered interferon‐γ expression in lymphocytes as a potential peripheral marker of chronic stress‐induced cognitive deficit. Cytokine. 2018;107:26‐34. 10.1016/j.cyto.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 30.Palumbo ML, Canzobre MC, Pascuan CG, Ríos H, Wald M, Genaro AM. Stress induced cognitive deficit is differentially modulated in BALB/c and C57Bl/6 mice: correlation with Th1/Th2 balance after stress exposure. J Neuroimmunol. 2010;218(1–2):12‐20. 10.1016/j.jneuroim.2009.11.005 [DOI] [PubMed] [Google Scholar]
- 31.Rosenkranz D, Weyer S, Tolosa E, et al. Higher frequency of regulatory T cells in the elderly and increased suppressive activity in neurodegeneration. J Neuroimmunol. 2007;188(1–2):117‐127. 10.1016/j.jneuroim.2007.05.011 [DOI] [PubMed] [Google Scholar]
- 32.Torres KC, Araújo Pereira P, Lima GS, et al. Increased frequency of T cells expressing IL‐10 in Alzheimer disease but not in late‐onset depression patients. Prog Neuropsychopharmacol Biol Psychiatry. 2013;47:40‐45. 10.1016/j.pnpbp.2013.07.021 [DOI] [PubMed] [Google Scholar]
- 33.Mosley RL, Gordon PH, Hasiak CM, Van Wetering FJ, Mitsumoto H, Gendelman HE. Glatiramer acetate immunization induces specific antibody and cytokine responses in ALS patients. Amyotroph Lateral Scler. 2007;8(4):235‐242. 10.1080/17482960701374601 [DOI] [PubMed] [Google Scholar]
- 34.Meininger V, Drory VE, Leigh PN, Ludolph A, Robberecht W, Silani V. Glatiramer acetate has no impact on disease progression in ALS at 40 mg/day: a double‐ blind, randomized, multicentre, placebo‐controlled trial. Amyotroph Lateral Scler. 2009;10(5–6):378‐383. 10.3109/17482960902803432 [DOI] [PubMed] [Google Scholar]
- 35.Frenkel D, Maron R, Burt DS, Weiner HL. Nasal vaccination with a proteosome‐based adjuvant and glatiramer acetate clears beta‐amyloid in a mouse model of Alzheimer disease. J Clin Invest. 2005;115(9):2423‐2433. 10.1172/JCI23241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Butovsky O, Koronyo‐Hamaoui M, Kunis G, et al. Glatiramer acetate fights against Alzheimer's disease by inducing dendritic‐like microglia expressing insulin‐like growth factor 1. Proc Natl Acad Sci USA. 2006;103(31):11784‐11789. 10.1073/pnas.0604681103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Butovsky O, Kunis G, Koronyo‐Hamaoui M, Schwartz M. Selective ablation of bone marrow‐derived dendritic cells increases amyloid plaques in a mouse Alzheimer's disease model. Eur J Neurosci. 2007;26(2):413‐416. 10.1111/j.1460-9568.2007.05652.x [DOI] [PubMed] [Google Scholar]
- 38.Bakalash S, Pham M, Koronyo Y, et al. Egr1 expression is induced following glatiramer acetate immunotherapy in rodent models of glaucoma and Alzheimer's disease. Invest Ophthalmol Vis Sci. 2011;52(12):9033‐9046. 10.1167/iovs.11-7498 [DOI] [PubMed] [Google Scholar]
- 39.Koronyo Y, Salumbides BC, Sheyn J, et al. Therapeutic effects of glatiramer acetate and grafted CD115+ monocytes in a mouse model of Alzheimer's disease. Brain. 2015;138(Pt 8):2399‐2422. 10.1093/brain/awv150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S, Hayden EY, Garcia VJ, et al. Activated bone marrow‐derived macrophages eradicate Alzheimer's‐related Aβ42 oligomers and protect synapses. Front Immunol. 2020;11:49. 10.3389/fimmu.2020.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rentsendorj A, Sheyn J, Fuchs D‐T, et al. A novel role for osteopontin in macrophage‐mediated amyloid‐β clearance in Alzheimer's models. Brain Behav Immun. 2018;67:163‐180. 10.1016/j.bbi.2017.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553‐1562. 10.1212/01.WNL.0000159740.16984.3C [DOI] [PubMed] [Google Scholar]
- 43.Wang CY, Wang P‐N, Chiu M‐J, et al. UB‐311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer's disease. Alzheimers Dement. 2017;3(2):262‐272. 10.1016/j.trci.2017.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benner EJ, Mosley RL, Destache CJ, et al. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson's disease. Proc Natl Acad Sci USA. 2004;101(25):9435‐9440. 10.1073/pnas.0400569101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Churchill MJ, Cantu MA, Kasanga EA, Moore C, Salvatore MF, Meshul CK. Glatiramer acetate reverses motor dysfunction and the decrease in tyrosine hydroxylase levels in a mouse model of Parkinson's disease. Neuroscience. 2019;414:8‐27. 10.1016/j.neuroscience.2019.06.006 [DOI] [PubMed] [Google Scholar]
- 46.Ibarra A, Avendaño H, Cruz Y. Copolymer‐1 (Cop‐1) improves neurological recovery after middle cerebral artery occlusion in rats. Neurosci Lett. 2007;425(2):110‐113. 10.1016/j.neulet.2007.08.038 [DOI] [PubMed] [Google Scholar]
- 47.Cruz Y, García EE, Gálvez JV, et al. Release of interleukin‐10 and neurotrophic factors in the choroid plexus: possible inductors of neurogenesis following copolymer‐1 immunization after cerebral ischemia. Neural Regen Res. 2018;13(10):1743‐1752. 10.4103/1673-5374.238615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kraft P, Göbel K, Meuth SG, Kleinschnitz C. Glatiramer acetate does not protect from acute ischemic stroke in mice. Exp Transl Stroke Med. 2014;6(1):4. 10.1186/2040-7378-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poittevin M, Deroide N, Azibani F, et al. Glatiramer Acetate administration does not reduce damage after cerebral ischemia in mice. J Neuroimmunol. 2013;254(1–2):55‐62. 10.1016/j.jneuroim.2012.09.009 [DOI] [PubMed] [Google Scholar]
- 50.Askarifirouzjaei H, Khajoueinejad L, Salek Farrokhi A, et al. Implications of immunotherapy with high‐dose glatiramer acetate in acute phase of spinal cord injury in rats. Immunopharmacol Immunotoxicol. 2019;41(1):150‐162. 10.1080/08923973.2019.1566362 [DOI] [PubMed] [Google Scholar]
- 51.García E, Silva‐García R, Mestre H, et al. Immunization with A91 peptide or copolymer‐1 reduces the production of nitric oxide and inducible nitric oxide synthase gene expression after spinal cord injury. J Neurosci Res. 2012;90(3):656‐663. 10.1002/jnr.22771 [DOI] [PubMed] [Google Scholar]
- 52.Kipnis J, Nevo U, Panikashvili D, et al. Therapeutic vaccination for closed head injury. J Neurotrauma. 2003;20(6):559‐569. 10.1089/089771503767168483 [DOI] [PubMed] [Google Scholar]
- 53.Marciani DJ. A retrospective analysis of the Alzheimer's disease vaccine progress ‐ the critical need for new development strategies. J Neurochem. 2016;137(5):687‐700. 10.1111/jnc.13608 [DOI] [PubMed] [Google Scholar]
- 54.Agadjanyan MG, Petrovsky N, Ghochikyan A. A fresh perspective from immunologists and vaccine researchers: active vaccination strategies to prevent and reverse Alzheimer's disease. Alzheimers Dement. 2015;11(10):1246‐1259. 10.1016/j.jalz.2015.06.1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Farlow MR, Andreasen N, Riviere M‐E, et al. Long‐term treatment with active Aβ immunotherapy with CAD106 in mild Alzheimer's disease. Alzheimers Res Ther. 2015;7(1):23. 10.1186/s13195-015-0108-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vandenberghe R, Riviere M‐E, Caputo A, et al. Active Aβ immunotherapy CAD106 in Alzheimer's disease: a phase 2b study. Alzheimers Dement. 2017;3(1):10‐22. 10.1016/j.trci.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.von Euler CM, Vorup‐Jensen T. Targets and mechanisms in prevention of Parkinson's disease through immunomodulatory treatments. Scand J Immunol. 2017;85(5):321‐330. 10.1111/sji.12542 [DOI] [PubMed] [Google Scholar]
- 58.Boost Vaccination Data Encourage Continued Development of AFFiRiS Therapeutic Parkinson's Disease Vaccine against Alpha Synuclein. Vienna, Austria: AFiRiS; 2016. [Google Scholar]
- 59.Blokhuis AM, Groen EJN, Koppers M, van den Berg LH, Pasterkamp RJ. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125(6):777‐794. 10.1007/s00401-013-1125-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Healy EF. A prion‐like mechanism for the propagated misfolding of SOD1 from in silico modeling of solvated near‐native conformers. PLoS One. 2017;12(5):e0177284. 10.1371/journal.pone.0177284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sibilla C, Bertolotti A. Prion properties of SOD1 in amyotrophic lateral sclerosis and potential therapy. Cold Spring Harb Perspect Biol. 2017;9(10): 10.1101/cshperspect.a024141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao B, Marciniuk K, Gibbs E, Yousefi M, Napper S, Cashman NR. Therapeutic vaccines for amyotrophic lateral sclerosis directed against disease specific epitopes of superoxide dismutase 1. Vaccine. 2019;37(35):4920‐4927. 10.1016/j.vaccine.2019.07.044 [DOI] [PubMed] [Google Scholar]
- 63.Malik R, Wiedau M. Therapeutic approaches targeting protein aggregation in amyotrophic lateral sclerosis. Front Mol Neurosci. 2020;13:98. 10.3389/fnmol.2020.00098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bales KR, O'Neill SM, Pozdnyakov N, et al. Passive immunotherapy targeting amyloid‐β reduces cerebral amyloid angiopathy and improves vascular reactivity. Brain. 2016;139(Pt 2):563‐577. 10.1093/brain/awv313 [DOI] [PubMed] [Google Scholar]
- 65.Montoliu‐Gaya L, Villegas S. Aβ‐Immunotherapeutic strategies: a wide range of approaches for Alzheimer's disease treatment. Expert Rev Mol Med. 2016;18:e13. 10.1017/erm.2016.11 [DOI] [PubMed] [Google Scholar]
- 66.Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing‐remitting multiple sclerosis: results of a phase III multicenter, double‐blind placebo‐controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45(7):1268‐1276. 10.1212/wnl.45.7.1268 [DOI] [PubMed] [Google Scholar]
- 67.Ziemssen T, Ashtamker N, Rubinchick S, Knappertz V, Comi G. Long‐term safety and tolerability of glatiramer acetate 20 mg/ml in the treatment of relapsing forms of multiple sclerosis. Expert Opin Drug Saf. 2017;16(2):247‐255. 10.1080/14740338.2017.1274728 [DOI] [PubMed] [Google Scholar]
- 68.De Kleijn KMA, Martens GJM. Molecular effects of FDA‐approved multiple sclerosis drugs on glial cells and neurons of the central nervous system. Int J Mol Sci. 2020;21(12):4229. 10.3390/ijms21124229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liblau R. Glatiramer acetate for the treatment of multiple sclerosis: evidence for a dual anti‐inflammatory and neuroprotective role. J Neurol Sci. 2009;287:S17‐S23. 10.1016/S0022-510X(09)71296-1 [DOI] [PubMed] [Google Scholar]
- 70.Arnon R, Aharoni R. Neurogenesis and neuroprotection in the CNS–fundamental elements in the effect of Glatiramer acetate on treatment of autoimmune neurological disorders. Mol Neurobiol. 2007;36:245‐253. 10.1007/s12035-007-8002-z [DOI] [PubMed] [Google Scholar]
- 71.Dhib‐Jalbut S. Mechanisms of action of interferons and glatiramer acetate in multiple sclerosis. Neurology. 2002;58(8 Suppl 4):S3‐S9. 10.1212/wnl.58.8_suppl_4.s3 [DOI] [PubMed] [Google Scholar]
- 72.Palumbo ML, Trinchero MF, Zorrilla‐Zubilete MA, Schinder AF, Genaro AM. Glatiramer acetate reverts stress‐induced alterations on adult neurogenesis and behavior. Involvement of Th1/Th2 balance. Brain Behav Immun. 2012;26(3):429‐438. 10.1016/j.bbi.2011.12.006 [DOI] [PubMed] [Google Scholar]
- 73.Lewitus GM, Wilf‐Yarkoni A, Ziv Y, et al. Vaccination as a novel approach for treating depressive behavior. Biol Psychiatry. 2009;65(4):283‐288. 10.1016/j.biopsych.2008.07.014 [DOI] [PubMed] [Google Scholar]
- 74.Gordon PH, Doorish C, Montes J, et al. Randomized controlled phase II trial of glatiramer acetate in ALS. Neurology. 2006;66(7):1117‐1119. 10.1212/01.wnl.0000204235.81272.e2 [DOI] [PubMed] [Google Scholar]
- 75.Béland L‐C, Markovinovic A, Jakovac H, et al. Immunity in amyotrophic lateral sclerosis: blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020;2(2):fcaa124. 10.1093/braincomms/fcaa124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Petrov D, Mansfield C, Moussy A, Hermine O. ALS clinical trials review: 20 years of failure. are we any closer to registering a new treatment? Front Aging Neurosci. 2017;9(68):1‐11. 10.3389/fnagi.2017.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scorisa JM, Zanon RG, Freria CM, de Oliveira ALR. Glatiramer acetate positively influences spinal motoneuron survival and synaptic plasticity after ventral root avulsion. Neurosci Lett. 2009;451(1):34‐39. 10.1016/j.neulet.2008.12.017 [DOI] [PubMed] [Google Scholar]
- 78.Cruz Y, Lorea J, Mestre H, et al. Copolymer‐1 promotes neurogenesis and improves functional recovery after acute ischemic stroke in rats. PLoS One. 2015;10(3):e0121854. 10.1371/journal.pone.0121854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ibarra A, García E, Flores N, et al. Immunization with neural‐derived antigens inhibits lipid peroxidation after spinal cord injury. Neurosci Lett. 2010;476(2):62‐65. 10.1016/j.neulet.2010.04.003 [DOI] [PubMed] [Google Scholar]
- 80.Rodríguez‐Barrera R, Fernández‐Presas AM, García E, et al. Immunization with a neural‐derived peptide protects the spinal cord from apoptosis after traumatic injury. Biomed Res Int. 2013;2013:827517. 10.1155/2013/827517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martiñón S, García‐Vences E, Toscano‐Tejeida D, et al. Long‐term production of BDNF and NT‐3 induced by A91‐immunization after spinal cord injury. BMC Neurosci. 2016;17(1):42. 10.1186/s12868-016-0267-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rodríguez‐Barrera R, Flores‐Romero A, García E, et al. Immunization with neural‐derived peptides increases neurogenesis in rats with chronic spinal cord injury. CNS Neurosci Ther. 2020;26(6):650‐658. 10.1111/cns.13368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.García E, Silva‐García R, Flores‐Romero A, Blancas‐Espinoza L, Rodríguez‐Barrera R, Ibarra A. The severity of spinal cord injury determines the inflammatory gene expression pattern after immunization with neural‐derived peptides. J Mol Neurosci. 2018;65(2):190‐195. 10.1007/s12031-018-1077-3 [DOI] [PubMed] [Google Scholar]
- 84.Bradbury EJ, Burnside ER. Moving beyond the glial scar for spinal cord repair. Nat Commun. 2019;10(1):3879. 10.1038/s41467-019-11707-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morales I‐I, Toscano‐Tejeida D, Ibarra A. Non pharmacological strategies to promote spinal cord regeneration: a view on some individual or combined approaches. Curr Pharm Des. 2016;22(6):720‐727. 10.2174/1381612822666151204001103 [DOI] [PubMed] [Google Scholar]
- 86.Rodríguez‐Barrera R, Flores‐Romero A, Fernández‐Presas AM, et al. Immunization with neural derived peptides plus scar removal induces a permissive microenvironment, and improves locomotor recovery after chronic spinal cord injury. BMC Neurosci. 2017;18(1):7. 10.1186/s12868-016-0331-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rodríguez‐Barrera R, Flores‐Romero A, Buzoianu‐Anguiano V, et al. Use of a combination strategy to improve morphological and functional recovery in rats with chronic spinal cord injury. Front Neurol. 2020;11:189. 10.3389/fneur.2020.00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parra‐Villamar D, Blancas‐Espinoza L, Garcia‐Vences E, et al. Neuroprotective effect of immunomodulatory peptides in rats with traumatic spinal cord injury. Neural Regen Res. 2021;16(7):1273‐1280. 10.4103/1673-5374.301485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Y, Wang K, Chao R, et al. Neuroprotective effect of vaccination with autoantigen‐pulsed dendritic cells after spinal cord injury. J Surg Res. 2012;176(1):281‐292. 10.1016/j.jss.2011.06.066 [DOI] [PubMed] [Google Scholar]
- 90.Wang K, Chao R, Guo QN, et al. Expressions of some neurotrophins and neurotrophic cytokines at site of spinal cord injury in mice after vaccination with dendritic cells pulsed with homogenate proteins. NeuroImmunoModulation. 2013;20(2):87‐98. 10.1159/000345522 [DOI] [PubMed] [Google Scholar]
- 91.Wang Y, Li J, Kong P, et al. Enhanced expression of neurotrophic factors in the injured spinal cord through vaccination with myelin basic protein‐derived peptide pulsed dendritic cells. Spine. 2015;40(2):95–101. 10.1097/BRS.0000000000000694 [DOI] [PubMed] [Google Scholar]
- 92.Cady CT, Lahn M, Vollmer M, et al. Response of murine gamma delta T cells to the synthetic polypeptide poly‐Glu50Tyr50. J Immunol. 2000;165(4):1790‐1798. 10.4049/jimmunol.165.4.1790 [DOI] [PubMed] [Google Scholar]
- 93.Ziv Y, Finkelstein A, Geffen Y, et al. A novel immune‐based therapy for stroke induces neuroprotection and supports neurogenesis. Stroke. 2007;38(2 Suppl):774‐782. 10.1161/01.STR.0000255784.27298.23 [DOI] [PubMed] [Google Scholar]
- 94.Bakalash S, Yoles E, Geffen Y. Glu‐Tyr polypeptide (Poly–YE) vaccination for acute and chronic glaucoma. Invest Ophthalmol Vis Sci. 2004;45(13):910.14985310 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
None to declare.