

Abstract
For the first time, derivatives of 3,7-diazabicyclo[3.3.1]nonane (bispidine) were proposed as potential inhibitors of the SARS-CoV-2 main viral protease (3-chymotrypsin-like, 3CLpro). Based on the created pharmacophore model of the active site of the protease, a group of compounds were modeled and tested for activity against 3CLpro. The 3CLpro activity was measured using the fluorogenic substrate Dabcyl-VNSTLQSGLRK(FAM)MA; the efficiency of the proposed approach was confirmed by comparison with literature data for ebselen and disulfiram. The results of the experiments performed with bispidine compounds showed that 14 compounds exhibited activity in the concentration range 1–10 μM, and 3 samples exhibited submicromolar activity. The structure–activity relationship studies showed that the molecules containing a carbonyl group in the ninth position of the bicycle exhibited the maximum activity. Based on the experimental and theoretical results obtained, further directions for the development of this topic were proposed.
Keywords: SARS-CoV-2 main viral protease; inhibition; 3,7-diazabicyclo[3.3.1]nonane; bispidinone; molecular docking
The global pandemic of a new 2019 coronavirus infection (COVID-19) caused by the SARS-CoV-2 virus began in the Chinese city of Wuhan in December 2019 and has since spread worldwide. To control human viral diseases, there are several directions. The first is disease prevention, which includes preventing the spread of the virus or prevaccination (successfully used in the case of some viral infections, such as smallpox). Today, vaccines are being successfully developed at several leading virological and research centers, and mass vaccination has already begun worldwide.
The other main direction in the fight against viral infections is the use of specific chemotherapeutic agents aimed at suppressing the replication of the virus in the host cell.1 The use of antiviral drugs at an early stage of disease development can significantly reduce the viral load on the body and the development of complications caused by the infectious disease. At the moment, the American pharmaceutical company Pfizer announced the start of Phase I clinical trials of its oral COVID-19 drug PF-07321332, developed on the basis of a protease inhibitor (https://cen.acs.org/acs-news/acs-meeting-news/Pfizer-unveils-oral-SARS-CoV/99/i13).
Each stage of the virus life cycle can be a potential target for drug therapy.2 Papain-like (PLpro) and 3-chymotrypsin-like (3CLpro), also called the major proteases, are vital for viral replication, and their function is to cleave the two translatable viral polyproteins (1A and 1AB) into functional components. Because of its small size, as well as its high homology to similar proteins of the coronaviruses that cause SARS and MERS atypical pneumonias, the SARS-CoV-2 major protease is the most characterized target for potential antiviral drugs. The main protease cleaves polyprotein 1AB at 11 specific sites. The site recognition sequence in most cases consists of the (Leu-Gln)-(Ser-Ala-Gly) chain site where the bond between glutamine and serine is cleaved. No proteases with the same cleavage specificity are known among human enzymes, which may indicate the probable lack of toxicity of potential inhibitors of the main protease SARS-CoV-2. Thus, the search for compounds inhibiting the work of this protease is one of the main directions in the field of SARS-CoV-2 chemotherapy.3
At the moment, a series of works devoted to the search for effective inhibitors of the basic protease have been published, among which the class of compounds belonging to peptidomimetics deserves special attention.4 The largest number of studies on possible inhibitors is devoted to molecules that can covalently bind the SH group of the catalytic amino acid residue Cys145 of the protease.4−13 The mechanisms of such covalent binding are represented in most cases by conjugated Michael addition to conjugated C=C double bond or nucleophilic addition to the carbonyl C=O double bond. Furthermore, the possibility of noncovalent inhibition of the catalytic function of viral proteases by small molecules is also considered.14−16 In this case, the active site of the enzyme is blocked, which prevents its interaction with the substrate.17 A significant number of publications are devoted exclusively to molecular modeling of known compounds of natural and synthetic origin against the main protease in order to find effective agents.18,19,28−33,20−27
The 3,7-diazabicyclo[3.3.1]nonan (bispidine) framework belongs to the so-called “privileged structures” in medicinal chemistry.34 This can be explained by a wide and diverse set of biological activities exhibited by compounds containing the bispidine framework. The key features of bispidines that allow their derivatives to be so widely used in medicinal chemistry and pharmacology are (1) their ability to selectively and independently functionalize at both nitrogen atoms;35 (2) the ability to control the conformational behavior of the bicyclic core;36 (3) and their pronounced complexing properties.37,38 Bispidines have been studied as potential antagonists of serine proteases, including thrombin and factor Xa.39,40 There are also known studies of symmetrical bispidine derivatives working as inhibitors of Japanese encephalitis virus.41 However, in terms of application to the problems of combating coronavirus infections, in particular to the creation of inhibitors of the main viral protease, bispidines and their analogues have not yet been applied.
In this work, we performed a comprehensive study of the inhibition of the main viral protease (3CLpro) by 1,5-dimethylbispidine and bispidine-9-one derivatives (Figure S2). The investigation included the creation of a pharmacophore model of the enzyme’s active site, the design and synthesis of potential inhibitors, the development of a test system, testing for biological activity, computer simulation of the results obtained, and studying the relationship between the structure of the substances described and the biological activity exhibited.
Initially, we created the pharmacophore model of potential inhibitors of the SARS-CoV-2 main protease based on the analysis of Protein Data Bank (Figure 1; see Supporting Information (SI) for details). XRD models of the main protease SARS-CoV-2 cocrystallized with covalent and noncovalent inhibitors were used for calculations (Figure S3).
Figure 1.

(A) Example of inhibitors: GC3736 (6WTK); GTPL112507 (6XHM); and GTPL1072010 (6Y2G). Pharmacophore hypothesis of covalent interaction with Cys145: (B) GC3737 - gray structure; GTPL112508 - green structure; and GTPL1072011 - orange structure. Acceptors are shown by red arrowed spheres, donors are shown by blue arrowed spheres, and hydrophobic centers are shown by green spheres. Gray spheres are tolerance radii of the hypothesis (2 Å). (C) Two-dimensional hypothesis using GC373 as an example.
An analysis of the chemical structure of the covalent inhibitors of the main protease published in the literature4−13 shows its high conservativity. All inhibitors have a typical scaffold in their structure in which there is an activated carbonyl group subjected to nucleophilic attack by the SH group of the catalytic amino acid residue Cys145. During C–S bond formation, the oxygen atom of the carbonyl group is converted into a hydroxyl group, which forms an additional hydrogen bond with Cys145. The resulting pharmacophore consists of six pharmacophore centers: three electron density acceptors (A1, A2, A3), one electron density donor (D1), and two hydrophobic centers (H1, H2) (Figure 1). We assume that the main scaffold required for the interaction of the covalent inhibitor with enzyme active site is a set of electron density acceptors, among which A1 is the S–H attachment center, and the others provide positioning of the molecule in the binding site by formation of hydrogen bonds. Two hydrophobic centers and an electron density donor provide additional stabilization.
The analysis of potentially active compounds among the family of N,N′-disubstituted bispidines 1–4 (Figure 2) was performed using the model obtained. Schemes of the target agent synthesis, experimental details, and data of physicochemical studies of the synthesized compounds are given in the SI. Compounds 1,38,422b,384c,434m,384r,434s,44 and 4t(44) have already been described. We investigated unsubstituted bispidinone (1), several bispidin-9-ones having different amide substituents at the nitrogen atoms (2), compounds lacking the keto group at the ninth position of bispidinone backbone amides (3), and a number of amines based on bispidin-9-one (4) (see Figures S2, S10). The usage of the carbonyl-containing compounds was based on the idea to explore the electrophilic nature of this group to enter the interaction with the SH-group of the Cys145 of the enzyme active site. The choice of structure type—amine or amide—and substituents at the nitrogen atoms was complicated due to the absence of any data on the use of bispidines as inhibitors of the major viral protease. Therefore, we were guided by data from the literature on the inhibition of serine proteases,39,40 on the results of the pharmacophore model we created, and on the synthetic possibilities of our laboratories. An attempt was made to vary the substituents at the nitrogen atoms of bispidine within a wide enough range in order to be able to study the “structure–activity” relationship for a series of compounds with the same framework, as well as to be able to compare the influence of the nature of the central framework on activity (see Figure S2). Symmetrically N,N′-disubstituted bispidines were chosen according to literature data for antiviral activity of such symmetrical analogues.41 Concerning the unsymmetrical compounds, this would be a matter of our future efforts. It is looks meaningful to say here that not all the designed structures were actually available synthetically, thus we were limited, to some extent, to those examples that would be isolable and pure enough to be studies in biological tests. That is why in the group of compounds 4 the 5-membered heterocycles predominated.
Figure 2.

Structures of bispidinone 1, bispidinone-based amides 2, bispidine-based amides 3, and bispidinone-based amines 4.
To evaluate the inhibitory activity of the synthesized substances against the basic protease, we used its recombinant analogue obtained in the E. coli system (see the SI for details). The sequence encoding the basic protease SARS-CoV-2 (3CLpro) of the Wuhan-Hu-1 isolate (GenBank:MN908947) was used to obtain the enzyme. The fluorogenic substrate Dabcyl-VNSTLQSGLRK(FAM)MA, which includes the 3CLpro proteolysis site of SARS-CoV-2, was used for quantitative analysis of the degree of inhibition. Using the developed system, compounds 1–4 that we synthesized were examined (see the SI for details of the experimental data). The results of biological testing with respect to the main protease and the toxicity of the described agents on HEK293T cells are shown in Table 1. Validation of the system developed by us was performed using the drugs disulfiram and ebselen for comparison. The activity of these substances correlates with previously published data in the literature.4,45 In addition, based on previously published theoretical calculations,46 we used ibuprofen as a reference drug. This compound showed no activity at all against the main protease.
Table 1. Enzymatic Assay Results of Protease Inhibitors Investigated in This Study against the SARS CoV-2 Protease and Toxicity on the HEK293T Cell Line.
| Compound | IC50 (3CLpro) (μM) | IC50 (HEK293T) (μM) | Compound | IC50 (3CLpro) (μM) | IC50 (HEK293T) (μM) |
|---|---|---|---|---|---|
| Disulfiram | 6.1 ± 0.6a | NT | 3a | 17.9 ± 3.1 | 302 ± 16 |
| Ebselen | 1.7 ± 0.4a | NT | 3e | 46.6 ± 3.1 | 253 ± 11 |
| Ibuprofen | >500 | NT | 3k | 99.6 ± 10 | >1000 |
| 1 | 2.6 ± 0.39 | >1000 | 4c | 8.8 ± 2 | >1000 |
| 2a | 5.9 ± 2.2 | >1000 | 4d | 0.9 ± 0.2 | 359 ± 18 |
| 2b | 18.5 ± 3.1 | >1000 | 4l | 5.5 ± 1.2 | >700 |
| 2c | 5.5 ± 0.7 | 57.3 ± 4.2 | 4m | 7.1 ± 1.4 | >1000 |
| 2d | 3.3 ± 0.5 | 692 ± 25 | 4n | 33.7 ± 5.2 | 257 ± 18 |
| 2e | 4.8 ± 0.6 | >1000 | 4o | 5.2 ± 1.1 | >700 |
| 2f | 1.45 ± 0.2 | >900 | 4p | 2.02 ± 0.41 | 465 ± 21 |
| 2g | 4.99 ± 0.8 | 456 ± 14 | 4q | 0.83 ± 0.17 | 410 ± 11 |
| 2h | 1.4 ± 0.2 | >700 | 4r | 2.2 ± 0.5 | 560 ± 13 |
| 2i | 13.0 ± 2.1 | >800 | 4s | 16.3 ± 3.1 | >700 |
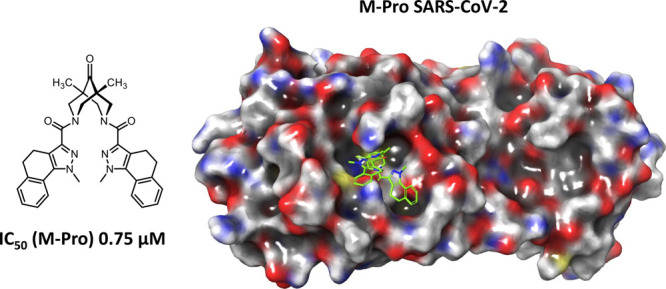
| 2j | 0.75 ± 0.2 | >800 | 4t | 13.6 ± 1.4 | NT |
According to the literature,4 the IC50 of disulfiram is 9.35 ± 0.18 μM, and the IC50 of ebselen is 0.67 ± 0.09 μM. NT, not tested. IC50, 50% inhibiting concentration as M ± SD, where M is the mean, and SD is the standard deviation; n ≥ 3.
The results of the biological tests show that bispidine derivatives are effective inhibitors of the main protease SARS-CoV-2. Most of the compounds we studied exhibited activity as basic protease inhibitors in the lower micromolar range. It is important to note that among these agents the bispidine-based amides of type 3 without the keto group at the ninth position showed the least activity. Among the bispidinone-based amides, compound 2j, a bis-amide containing dihydrobenzoindazole fragments, showed the highest activity in the submicromolar range. High agent activity, comparable with the references, was found in all amides except substances 2b and 2i. Amines with the bispidinone backbone of general formula 4 can also be considered as an extremely promising class of protease inhibitors. Thus, substances 4d and 4q containing phenyl and substituted triazole fragments, respectively, at the methylene linker showed submicromolar activity. Substances 4c, 4l, 4m, 4o, 4p, and 4r inhibited the protease in the lower micromolar range. In addition, we studied the cytotoxic properties of the above compounds on the HEK293T cell line. Except for amide 2c, all the agents we have described are nontoxic to the specified cell line.
To understand which structural element of the N,N′-disubstituted bispidine molecule can be responsible for binding to certain sites of the enzyme active site, we performed molecular docking and attempted a structure–activity relationship analysis. All compounds carrying carbonyl groups in their structure were found to be capable of binding to Cys145.
Analysis of the matching of the structures of the new bispidine derivatives to our pharmacophoric hypothesis showed that there is a marked correlation between the biological activity of the compounds and the presence of the three main electron density acceptors in their structure, which can be located in the active site space of the main protease in a similar way as cocrystallized covalent inhibitors from XRD models of the main protease (GC3736 (6WTK); GTPL112507 (6XHM); and GTPL1072010 (6Y2G)). For all cocrystallized inhibitors and the most active new bispidine derivatives, the presence of the A1 acceptor in the structure is obligatory; in the case of bispidine-9-one molecules this corresponds to the hydroxy-group, formed because of the nucleophilic addition of Cys145 to the carbonyl at the ninth position. This fact is particularly evident when comparing pairs of compounds having similar substituents with significantly different biological activity (2a/3a and 2e/3e). Out of the 27 compounds, 16 form a covalent bond with Cys145 via a carbonyl group at the ninth position of the bispidine backbone. We consider this to be the most important modeling achievement, which allowed us to explain the experimental results and design/optimize the next generation of bispidine inhibitors.
The main structural difference in the group of bispidine-9-one 2 and bispidine 3 derivatives is the lack of the carbonyl group in the ninth position for the latter, which makes the formation of covalent bonds of derivatives 3 with Cys145 in this position impossible for compounds 3. For example, the study shows that 2e (−4.91 kcal/mol) forms a covalent bond at the carbonyl group of the bispidine backbone (Figure 3). Its structure has electron density acceptors A1 and A2, as well as a hydrophobic center H1 (one of the methyl groups of the bispidinone backbone). It is interesting that the estimated parameter for compound 3e (−5.39 kcal/mol) provides evidence for the seemingly better arrangement of the carbonyl group in the side substituent with respect to the amino acid residue Cys145 as compared to the carbonyl group of the bispidine backbone of compound 2e. However, the hydroxyl group formed because of nucleophilic addition shifts away from the important acceptor A1, bringing the proton closer to the donor D1. Comparison of the free energies of the covalent complexes shows a lower energy for the complex of compound 2e (−25.33 kcal/mol) as compared to the complex energy for the derivative 3e (−18.62 kcal/mol). An even more significant difference is observed for the covalent bonding energy components: 1.01 kcal/mol for the 2e derivative complex and 10.68 kcal/mol for the 3e derivative complex. Thus, it is possible to state that the carbonyl group at position 9 of the bispidine’s backbone has an advantage as a covalent bonding site for the compounds of this class.
Figure 3.

Two-dimensional image of the binding of 2e and 3e molecules to the SARS-CoV-2 main protease binding site.
It is well-known in bispidine’s bis-amide chemistry that at room temperature both anti- and syn-stereoisomers could be observed using NMR spectroscopy in some solvents (see footnote to Table S3).47−49 Analysis of the conformations of the side substituents in the series of bis-amides 2 and 3 shows that most molecules interact with the enzyme active site in the anti-conformation (Table S3).
Bispidinone derivative 2j (the most active agent among the compounds of general formula 2, Figure 4) possess all three electron acceptors (A1–A3) and an additional hydrophobic center H1 in its structure. Its homologue 2i in the conformation under consideration is not capable of offering the A2 acceptor but has two hydrophobic centers in its structure. One of its side substituents protrudes significantly beyond the enzyme’s active site and is exposed to solvent molecules (see Figure 4). Apparently, this specific conformation of the molecule 2i leads to a significant hindrance to the formation of the covalent bond. Conformations of compounds 2j (−5.93 kcal/mol) and 2i (−5.43 kcal/mol) are indeed quite different in terms of the free energy of the covalent complexes of these compounds and the main protease SARS-CoV-2. The value of dG for compound 2j was −46.68 kcal/mol, whereas for compound 2i this value was equal to −5.48 kcal/mol. The covalent bonding energy component for compound 2j was −0.99 kcal/mol, and for compound 2i it was 37.11 kcal/mol.
Figure 4.

Two-dimensional image of the binding of 2j and 2i molecules to the SARS-CoV-2 main protease binding site.
The pharmacophore feature of molecules that have shown pronounced antiprotease activity at micromolar concentrations (2j, 4d, 4q) is the presence in the structure of three acceptors of electron density (2j, 4q), or two acceptors in a more compact derivative 4d, allowing molecules to occupy the most favorable position near the catalytic amino acid residue Cys145 (Figures S8, S9).
ADMET properties analysis shows that most of the new bispidine derivatives (21 out of 26) have good drug-likeness (Table S4). The five molecules showing significant in vitro activity (2d, 4p, 2a, 4c, 4d) satisfy the strictest rule, showing ADMET values for good intestinal absorption and low toxicity. The combination of theoretically calculated parameters (docking score and drug-likeness) and the presence of pronounced in vitro 3CLpro inhibitory activity make compound 2d the most interesting potential inhibitor of SARS-CoV-2 main protease.
We understand our research is only the beginning of a long journey to find effective inhibitors of the virus SARS-CoV-2. Measuring the inhibition of the enzymatic activity of 3CLpro in vitro is the first step in the search for inhibitors capable of becoming an antiviral drug in the future. However, this approach is not without its drawbacks. The use of the recombinant 3CLpro protein in vitro does not allow taking into account the many biochemical processes occurring in the cell or even in the body that can affect the test substance inhibitor. Of the more obvious, the biochemical test does not take into account the toxicity of the compound, the ability to penetrate the cell wall, and possible transformations by the body’s enzymatic system. Our future work will be devoted to the study of bispidine derivatives using an infectious virus, and we have already begun this work. Further, the compounds we have identified may be imcompatible with in vivo studies; the limitations related to in vivo bioactivity, pharmokinetics, and potential toxicity will also be a focus of our future studies.
Besides the results obtained in this work, the following directions for the design of bispidine-based main viral protease inhibitors seem promising: (i) the introduction of chiral fragments to the nitrogen atoms, (ii) the introduction of amino acid substituents to the nitrogen atoms, (iii) the study of the effect of the substituents in the 9 position on the manifested activity, (iv) synthesis and study of N,N′-asymmetrically substituted bispidines. Further studies of the derivatives of this class of compounds may provide an opportunity to enter a fundamentally new class of inhibitors of the emergent virus that caused the 2019–2021 pandemic.
Acknowledgments
The reported study was funded by RFBR according to the Research Project No. 20-04-60215. The authors express sincere gratitude to Kudriashova Elena for help in creating graphical abstract.
Glossary
Abbreviations
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- 3CLpro
SARS-CoV-2 main viral protease
- COVID-19
Coronavirus disease 2019
- ADMET
Absorption, Distribution, Metabolism, Excretion, Toxicity
- IC50
50% inhibiting concentration
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00299.
Details for the synthetic procedures, compound characterization, computations, assays, and ADMET properties of new compounds (PDF)
Author Contributions
+ D.S. and D.B. contributed equally to this research.
The authors declare no competing financial interest.
This article is made available via the ACS COVID-19 subset for unrestricted RESEARCH re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for the duration of the World Health Organization (WHO) declaration of COVID-19 as a global pandemic.
Supplementary Material
References
- Cho N. J.; Glenn J. S. Nat. Mater. 2020, 19, 813–816. 10.1038/s41563-020-0698-4. [DOI] [PubMed] [Google Scholar]
- Novikov F. N.; Stroylov V. S.; Svitanko I. V.; Nebolsin V. E. Russ. Chem. Rev. 2020, 89, 858–878. 10.1070/RCR4961. [DOI] [Google Scholar]
- Ullrich S.; Nitsche C. Bioorg. Med. Chem. Lett. 2020, 30, 127377. 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; Duan Y.; Yu J.; Wang L.; Yang K.; Liu F.; Jiang R.; Yang X.; You T.; Liu X.; Yang X.; Bai F.; Liu H.; Liu X.; Guddat L. W.; Xu W.; Xiao G.; Qin C.; Shi Z.; Jiang H.; Rao Z.; Yang H. Nature 2020, 1–9. [Google Scholar]
- Dai W.; Zhang B.; Su H.; Li J.; Zhao Y.; Xie X.; Jin Z.; Liu F.; Li C.; Li Y.; Bai F.; Wang H.; Cheng X.; Cen X.; Hu S.; Yang X.; Wang J.; Liu X.; Xiao G.; Jiang H.; Rao Z.; Zhang L.-K.; Xu Y.; Yang H.; Liu H. Science 2020, 368, 1331. 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong W.; Khan M. B.; Fischer C.; Arutyunova E.; Lamer T.; Shields J.; Saffran H. A.; McKay R. T.; van Belkum M. J.; Joyce M. A.; Young H. S.; Tyrrell D. L.; Vederas J. C.; Lemieux M. J. Nat. Commun. 2020, 11, 1–8. 10.1038/s41467-020-19339-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman R. L.; Kania R. S.; Brothers M. A.; Davies J. F.; Ferre R. A.; Gajiwala K. S.; He M.; Hogan R. J.; Kozminski K.; Li L. Y.; Lockner J. W.; Lou J.; Marra M. T.; Mitchell L. J.; Murray B. W.; Nieman J. A.; Noell S.; Planken S. P.; Rowe T.; Ryan K.; Smith G. J.; Solowiej J. E.; Steppan C. M.; Taggart B. J. Med. Chem. 2020, 63, 12725–12747. 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathnayake A. D.; Zheng J.; Kim Y.; Perera K. D.; Mackin S.; Meyerholz D. K.; Kashipathy M. M.; Battaile K. P.; Lovell S.; Perlman S.; Groutas W. C.; Chang K. O. Sci. Transl. Med. 2020, 12, eabc5332. 10.1126/scitranslmed.abc5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D. W.; Galanie S.; Phillips G.; O’Neill H. M.; Coates L.; Kovalevsky A. Structure 2020, 28, 1313–1320. 10.1016/j.str.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Science (Washington, DC, U. S.) 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Zhao Y.; Sun Y.; Zhang B.; Wang H.; Wu Y.; Zhu Y.; Zhu C.; Hu T.; Du X.; Duan Y.; Yu J.; Yang X.; Yang X.; Yang K.; Liu X.; Guddat L. W.; Xiao G.; Zhang L.; Yang H.; Rao Z. Nat. Struct. Mol. Biol. 2020, 27, 529–532. 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- Qiao J.; Li Y.-S.; Zeng R.; Liu F.-L.; Luo R.-H.; Huang C.; Wang Y.-F.; Zhang J.; Quan B.; Shen C.; Mao X.; Liu X.; Sun W.; Yang W.; Ni X.; Wang K.; Xu L.; Duan Z.-L.; Zou Q.-C.; Zhang H.-L.; Qu W.; Long Y.-H.-P.; Li M.-H.; Yang R.-C.; Liu X.; You J.; Zhou Y.; Yao R.; Li W.-P.; Liu J.-M.; Chen P.; Liu Y.; Lin G.-F.; Yang X.; Zou J.; Li L.; Hu Y.; Lu G.-W.; Li W.-M.; Wei Y.-Q.; Zheng Y.-T.; Lei J.; Yang S. Science 2021, 371, 1374–1378. 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. S.; Ma X. R.; Ma Y.; Alugubelli Y. R.; Scott D. A.; Vatansever E. C.; Drelich A. K.; Sankaran B.; Geng Z. Z.; Blankenship L. R.; Ward H. E.; Sheng Y. J.; Hsu J. C.; Kratch K. C.; Zhao B.; Hayatshahi H. S.; Liu J.; Li P.; Fierke C. A.; Tseng C. K.; Xu S.; Liu W. R. ChemMedChem 2021, 16, 942–948. 10.1002/cmdc.202000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther S.; Reinke P. Y. A.; Fernández-García Y.; Lieske J.; Lane T. J.; Ginn H. M.; Koua F. H. M.; Ehrt C.; Ewert W.; Oberthuer D.; Yefanov O.; Meier S.; Lorenzen K.; Krichel B.; Kopicki J.-D.; Gelisio L.; Brehm W.; Dunkel I.; Seychell B.; Gieseler H.; Norton-Baker B.; Escudero-Pérez B.; Domaracky M.; Saouane S.; Tolstikova A.; White T. A.; Hänle A.; Groessler M.; Fleckenstein H.; Trost F.; Galchenkova M.; Gevorkov Y.; Li C.; Awel S.; Peck A.; Barthelmess M.; Schluenzen F.; Lourdu Xavier P.; Werner N.; Andaleeb H.; Ullah N.; Falke S.; Srinivasan V.; França B. A.; Schwinzer M.; Brognaro H.; Rogers C.; Melo D.; Zaitseva-Doyle J. J.; Knoska J.; Peña-Murillo G. E.; Mashhour A. R.; Hennicke V.; Fischer P.; Hakanpää J.; Meyer J.; Gribbon P.; Ellinger B.; Kuzikov M.; Wolf M.; Beccari A. R.; Bourenkov G.; von Stetten D.; Pompidor G.; Bento I.; Panneerselvam S.; Karpics I.; Schneider T. R.; Garcia-Alai M. M.; Niebling S.; Günther C.; Schmidt C.; Schubert R.; Han H.; Boger J.; Monteiro D. C. F.; Zhang L.; Sun X.; Pletzer-Zelgert J.; Wollenhaupt J.; Feiler C. G.; Weiss M. S.; Schulz E.-C.; Mehrabi P.; Karničar K.; Usenik A.; Loboda J.; Tidow H.; Chari A.; Hilgenfeld R.; Uetrecht C.; Cox R.; Zaliani A.; Beck T.; Rarey M.; Günther S.; Turk D.; Hinrichs W.; Chapman H. N.; Pearson A. R.; Betzel C.; Meents A. Science 2021, 372, 642 10.1126/science.abf7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. H.; Stone E. A.; Deshmukh M.; Ippolito J. A.; Ghahremanpour M. M.; Tirado-Rives J.; Spasov K. A.; Zhang S.; Takeo Y.; Kudalkar S. N.; Liang Z.; Isaacs F.; Lindenbach B.; Miller S. J.; Anderson K. S.; Jorgensen W. L. ACS Cent. Sci. 2021, 7, 467. 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockbaum G. J.; Reyes A. C.; Lee J. M.; Tilvawala R.; Nalivaika E. A.; Ali A.; Kurt Yilmaz N.; Thompson P. R.; Schiffer C. A. Viruses 2021, 13, 174. 10.3390/v13020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Karagiannis C.; Pitsillou E.; Darmawan K. K.; Ng K.; Hung A.; Karagiannis T. C. Comput. Biol. Chem. 2020, 89, 107372. 10.1016/j.compbiolchem.2020.107372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega J. T.; Serrano M. L.; Pujol F. H.; Rangel H. R. EXCLI Journal 2020, 19, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. W.; Yiu C.-P. B.; Wong K.-Y. F1000Research 2020, 9, 129. 10.12688/f1000research.22457.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandeel M.; Al-Nazawi M. Life Sci. 2020, 251, 117627. 10.1016/j.lfs.2020.117627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. J. Chem. Inf. Model. 2020, 60, 3277. 10.1021/acs.jcim.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner Ž.; Hatamipour M.; Banach M.; Pirro M.; Al-Rasadi K.; Jamialahmadi T.; Radenkovic D.; Montecucco F.; Sahebkar A. Arch. Med. Sci. 2020, 16, 490–496. 10.5114/aoms.2020.94655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Wang X.-J.. bioRxiv, 2020, 2020.01.29.924100. [Google Scholar]
- Li Y.; Zhang J.; Wang N.; Li H.; Shi Y.; Guo G.; Liu K.; Zeng H.; Zou Q.. bioRxiv, 2020, 2020.01.28.922922. [Google Scholar]
- Ton A. T.; Gentile F.; Hsing M.; Ban F.; Cherkasov A. Mol. Inf. 2020, 39, 2000028. 10.1002/minf.202000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Qin J. J.; Li X.; Tao G.; Wang Q.; Wu X.; Zhou J.; Zi X.; Zhang R. Carcinogenesis 2018, 39, 1026–1036. 10.1093/carcin/bgy063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesh; Selvaraj C.; Singh S. K.; Dubey V. K.; Kundu D. J. Biomol. Struct. Dyn. 2020, 1–9. 10.1080/07391102.2020.1763202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile D.; Patamia V.; Scala A.; Sciortino M. T.; Piperno A.; Rescifina A. Mar. Drugs 2020, 18, 225. 10.3390/md18040225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sk M. F.; Roy R.; Jonniya N. A.; Poddar S.; Kar P. J. Biomol. Struct. Dyn. 2021, 1–21. 10.1080/07391102.2021.1897680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D.; Gao K.; Chen J.; Wang R.; Wei G.. bioRxiv, 2020, 2020.02.05.936013. [Google Scholar]
- Gao K.; Nguyen D. D.; Wang R.; Wei G.-W.. bioRxiv, 2020, 2020.01.30.927889. [Google Scholar]
- Zhavoronkov A.; Aladinskiy V.; Zhebrak A.; Zagribelnyy B.; Terentiev V.; Bezrukov D. S.; Polykovskiy D.; Shayakhmetov R.; Filimonov A.; Orekhov P.; Yan Y.; Popova O.; Vanhaelen Q.; Aliper A.; Ivanenkov Y. Insilico Medicine Hong Kong Ltd A 2020, 307, E1. [Google Scholar]
- Joshi T.; Joshi T.; Sharma P.; Mathpal S.; Pundir H.; Bhatt V.; Chandra S. European Review for Medical and Pharmacological Sciences 2020, 24, 4529–4536. [DOI] [PubMed] [Google Scholar]
- Tomassoli I.; Gündisch D. Curr. Top. Med. Chem. 2016, 16, 1314–1342. 10.2174/1568026615666150915111434. [DOI] [PubMed] [Google Scholar]
- Medved’ko A.; Dalinger A.; Nuriev V.; Semashko V.; Filatov A.; Ezhov A.; Churakov A.; Howard J.; Shiryaev A.; Baranchikov A.; Ivanov V.; Vatsadze S. Nanomaterials 2019, 9, 89. 10.3390/nano9010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zefirov N. S.; Palyulin V. A. In Topics in Stereochemistry; Iliel E. L., Wilen S. H., Eds.; 1991; Vol. 20, pp 171–230. [Google Scholar]
- Comba P.; Kerscher M.; Schiek W. Prog. Inorg. Chem. 2008, 55, 613–704. 10.1002/9780470144428.ch9. [DOI] [Google Scholar]
- Medved’ko A.; Egorova B.; Komarova A.; Rakhimov R.; Krut’ko D.; Kalmykov S.; Vatsadze S. ACS Omega 2016, 1, 854–867. 10.1021/acsomega.6b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryavtsev K. V.; Shulga D. A.; Chupakhin V. I.; Sinauridze E. I.; Ataullakhanov F. I.; Vatsadze S. Z. Tetrahedron 2014, 70, 7854–7864. 10.1016/j.tet.2014.09.009. [DOI] [Google Scholar]
- Vatsadze S. Z.; Shulga D. A.; Loginova Y. D.; Vatsadze I. A.; Wang L.; Yu H.; Kudryavtsev K. V. Mendeleev Commun. 2016, 26, 212–213. 10.1016/j.mencom.2016.04.011. [DOI] [Google Scholar]
- Haridas V.; Rajgokul K. S.; Sadanandan S.; Agrawal T.; Sharvani V.; Gopalakrishna M. V. S.; Bijesh M. B.; Kumawat K. L.; Basu A.; Medigeshi G. R. PLoS Neglected Trop. Dis. 2013, 7, e2005 10.1371/journal.pntd.0002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatsadze S. Z.; Semashko V. S.; Manaenkova M. A.; Krut’Ko D. P.; Nuriev V. N.; Rakhimov R. D.; Davlyatshin D. I.; Churakov A. V.; Howard J. A. K.; Maksimov A. L.; Li W.; Yu H. Russ. Chem. Bull. 2014, 63, 895–911. 10.1007/s11172-014-0526-6. [DOI] [Google Scholar]
- Vatsadze S. Z.; Medved’ko A. V.; Bodunov A. A.; Lyssenko K. A. Mendeleev Commun. 2020, 30, 344–346. 10.1016/j.mencom.2020.05.028. [DOI] [Google Scholar]
- Dalinger A. I.; Medved’ko A. V.; Balalaeva A. I.; Vatsadze I. Z.; Dalinger I. L.; Vatsadze S. Z. Chem. Heterocycl. Compd. 2020, 56, 180–191. 10.1007/s10593-020-02643-2. [DOI] [Google Scholar]
- Ma C.; Hu Y.; Townsend J. A.; Lagarias P. I.; Marty M. T.; Kolocouris A.; Wang J. ACS Pharmacology and Translational Science 2020, 3, 1265–1277. 10.1021/acsptsci.0c00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente C. M.; Freiberger M. I.; Ravetti S.; Beltramo D. M.; Garro A. G. J. Biomol. Struct. Dyn. 2021, 1–12. 10.1080/07391102.2021.1872420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palyulin V. A.; Emets S. V.; Chertkov V. A.; Kasper C.; Schneider H. J. Eur. J. Org. Chem. 1999, 1999, 3479–3482. . [DOI] [Google Scholar]
- Wang Z.; Islam M. J.; Vukotic V. N.; Revington M. J. J. Org. Chem. 2016, 81, 2981–2986. 10.1021/acs.joc.6b00250. [DOI] [PubMed] [Google Scholar]
- Churakov A. V.; Medved’ko A. V.; Prikhodchenko P. V.; Krut’ko D. P.; Vatsadze S. Z. Mendeleev Commun. 2021, 31, 352–355. 10.1016/j.mencom.2021.04.023. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.