

Abstract
Reactive oxygen species (ROS) increase during adipogenesis and in obesity. Oxidants react with cysteine residues of proteins to form glutathione (GSH) adducts, S-glutathionylation, that are selectively removed by glutaredoxin-1 (Glrx). We have previously reported that Glrx knockout mice had increased protein S-glutathionylation and developed obesity by an unknown mechanism. In this study, we demonstrated that 3T3L1 adipocytes differentiation increased ROS and protein S-glutathionylation. Glrx ablation elevated protein S-glutathionylation and lipid content in 3T3L1 cells. Glrx replenishment decreased the lipid content of Glrx KO 3T3L1 cells. Glrx KO also increased protein expression and protein S-glutathionylation of the adipogenic transcription factor CCAAT enhancer-binding protein (C/EBP) β. Protein S-glutathionylation decreased the interaction of C/EBPβ and protein inhibitor of activated STAT (PIAS) 1, a small ubiquitin-related modifier (SUMO) E3 ligase that facilitates C/EBPβ degradation. Experiments with truncated mutant C/EBPβ demonstrated that PIAS1 interacted with the liver-enriched inhibitory protein (LIP) region of C/EBPβ. Furthermore, mass spectrometry analysis identified protein S-glutathionylation of Cys201 and Cys296 in the LIP region of C/EBPβ. The C201S, C296S double mutant C/EBPβ prevented protein S-glutathionylation and preserved the interaction with PIAS1. In summary, Glrx ablation stimulated 3T3L1 cell differentiation and adipogenesis via increased protein S-glutathionylation of C/EBPβ, stabilizing and increasing C/EBPβ protein levels.
Keywords: S-glutathionylation, glutaredoxin-1, CCAAT enhancer-binding protein, reactive oxygen species
Introduction
The global obesity epidemic poses a critical public health problem and is associated with an enhanced risk of diabetes, chronic kidney disease, cardiovascular disease, and cancer (1). Excessive fat accumulation causes adipose tissue inflammation and subsequent dysfunction (2). Adipocyte hypertrophy and differentiation from precursor cells significantly contribute to the expansion of adipose tissue in obesity (3). Reactive oxygen species (ROS) stimulate adipocyte differentiation in vitro (4, 5) and in vivo (6, 7), and are believed to accelerate adipose tissue dysfunction leading to metabolic syndrome (8). Thus, ROS may promote fat accumulation and adipogenesis in obesity in a vicious feed-forward cycle.
Antioxidants such as glutathione (GSH) scavenge ROS and should interrupt this feed-forward mechanism. We have previously studied glutamate-cysteine ligase modifier subunit (GCLM) knockout (KO) mice that causes chronic GSH depletion in all tissues. Interestingly, these mice had lower body weights compared to wild type (9) and were resistant to high-fat diet-induced obesity despite increased levels of ROS (10). However, GSH is not only an antioxidant but also participates in cell signaling. In the presence of ROS, GSH modifies proteins post-translationally, referred to as protein S-glutathionylation or GSH adducts (11). Thus, our GCLM KO also decreased protein S-glutathionylation levels in mice (12) and likely disrupted important redox signaling.
Glutaredoxin-1 (Glrx) removes GHS adducts of proteins selectively (13). In contrast to GCLM KO mice, Glrx KO mice had higher levels of protein S-glutathionylation and developed marked obesity (14). These studies indicate that ROS may stimulate adipogenesis and obesity by increasing protein S-glutathionylation. However, the impact of protein S-glutathionylation on adipogenesis remains elusive. In our current study, we demonstrated that protein S-glutathionylation stabilized a key adipogenic transcriptional factor C/EBPβ and stimulated adipogenesis in 3T3L1 cells.
Materials and Methods
Reagents.
Dexamethasone (D4902), 3-Isobutyl-1-methylxanthine (IBMX)(5879), insulin (I5500), 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (Tempol)(176141), reduced glutathione (G4251), oxidized glutathione (G4376) and doxycycline hyclate (D9891) were from Sigma Aldrich (St. Louis, MO, USA). Anti-β actin antibody (4970), anti- peroxisome proliferator-activated receptor (PPAR) γ antibody (2443) anti-C/EPBα antibody (8178), anti-C/EBPβ antibody (3087), anti-HA tag antibody (3724), anti-DYKDDDK (FLAG-tag) antibody (2368), anti-GFP antibody (2555) were from Cell Signaling Technology (Danvers, MA, USA), anti-GSH antibody (101-A100) was from Virogen (Watertown, MA, USA), and anti-Glrx antibody (ab45953) was from Abcam (Cambridge, UK). Myc-FLAG-tagged mouse C/EBPβ plasmid (MR227563), Myc -FLAG-tagged mouse PIAS1 plasmid (MR209770), and C-terminal HA-tag vector (PS100004) were from Origene (Rockville, MD, USA). OxiSelect™ Intracellular ROS Assay Kit was from Cell Biolabs Inc (San Diego, CA, USA). Dihydrofluorescein diacetate (H2FDA, 292648) was from Merck (Kenilworth, NJ, USA). MitoSOX Red (M36008) was from Thermo Fisher Scientific (Waltham, MA, USA). The adenovirus expressing tetracycline-responsive element–driven Glrx-conjugated GFP protein was a kind gift of Dr. R. Asmis University of Texas Health Science Center, San Antonio. The pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmid (#62988) was a kind gift of Feng Zhang, Broad Institute, Massachusetts Institute of Technology, Cambridge, MA, and obtained through Addgene (Watertown, MA, USA). Oil red O (154-02072) and formalin (061-00416) were obtained from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Anti-Myc tag monoclonal antibody magnetic beads (M047-11) were purchased from Medical and Biological Laboratories (Nagoya, Japan). DMEM 1.5 g/L glucose (10567-014), 4.5 g/L glucose (10313-021), FBS(10270), TrypLE™ Express (12605036), Halt™ protease/phosphatase inhibitor cocktail (78440), Zeba spin columns (89889), sample loading buffer (NP0007), Imperial protein stain (24617), Lipofectamine 2000 (11668019) and 3000 (L3000015), protein G magnetic Dynabeads™ (10004D), TaqMan primers and TaqMan™ Gene Expression Master Mix (4369016) were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Cell culture.
3T3L1 preadipocyte were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and were grown in DMEM containing 1.5 g/L glucose and 10% FBS. The medium was changed to DMEM containing 4.5 g/L glucose and 10% FBS to prepare cells for experiments. Adipogenesis of 3T3L1 cells was induced by switching the medium to DMEM containing 4.5 g/L glucose, 10% FBS, 0.5 mmol/L IBMX, 1 μmol/L dexamethasone, and 1 μmol/L insulin two days after cells reached confluency. The medium was replaced every other day with DMEM containing 10% FBS and 1 μmol/L insulin. In some experiments, 2 mmol/L Tempol was added to 3T3L1 cells at the time of adipogenesis induction. For transfection of 3T3L1 cells with Ad tet-Glrx, AdenoMag (AM70200; OZ BIOSCIENCES, San Diego CA, USA) was used one day after passaging cells according to the manufacturer’s protocol. Glrx expression was induced by adding 1 μg/mL doxycycline combined with the induction of adipocyte differentiation.
Adipogenesis assay.
The adipogenesis assay was performed as previously described with slight modifications (15). To prepare an Oil red O solution, 60 mg of Oil red O was dissolved in 20 mL isopropanol. Then, 60 mL of deionized water was added, and the mixture filtered through a 0.22 μm syringe filter (SLGV033RS)(Merck, Kenilworth, NJ, USA). 3T3L1 cells were seeded in 12 well dishes, washed with PBS, and fixed with 10% formalin in PBS for 20 minutes, six days after adipocyte differentiation. Cells were washed three times with water, dried, and stained with 300 μL Oil red O solution for 20 minutes. After the stained cells were washed three times with water and dried, Oil red O was extracted with 200 μL of 100% isopropanol for 10 minutes. The Oil red O concentration was measured at 490 nm with a SpectraMax 340 (Molecular Devices Corporation, San Jose, CA, USA).
Glrx knockout by CRISPR/Cas9.
Glrx KO in 3T3L1 cells was achieved by using CRISPR/Cas9 with the pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmid system, as previously described with slight modifications (16). The guiding RNA for Glrx knockout consisted of the following sequence: CACCGTTCTTGGGTCTTTCTGCAGT and AAACACTGCAGAAAGACCCAAGAAC. After annealing, this oligonucleotide was inserted into the BbsI cloning site of PX459. The PX459 plasmid with and without (control) cloned guiding RNA were transfected into 3T3L1 cells using Lipofectamine 3000 according to the manufacturer’s protocol. One day after transfection, puromycin selection was carried out for two days, and selected cells were expanded and used for experiments.
Analysis of intracellular ROS.
Intracellular ROS were detected with H2FDA, and mitochondrial O2.− was measured with MitoSOX Red using flow cytometry (17). After 4 days of adipocyte differentiation, 3T3L1 cells were incubated with 10 μmol/L H2FDA or 5 μmol/L MitoSOX red or DMSO at 37 °C for 20 min. Cells were then washed with PBS and detached with TrypLE ™ Expreess. Cells were again washed twice with PBS and then resuspended in PBS with 2% FBS and 0.5 μmol/L EDTA. Flow cytometric analysis was performed with a FACSCalibur (BD Biosciences, San Jose, CA, USA). Quantification of ROS levels was perfomed by OxiSelect™ Intracellular ROS Assay Kit, a 2’, 7’-dichlorodihydrofluorescin diacetate (DCFH-DA) based ROS detection system, following manufacturer’s instructions.
Detection of protein S-glutathionylation on C/EBPβ.
For the detection of protein S-glutathionylation, cells were washed twice with PBS and immediately frozen by placing the dish in liquid nitrogen. Cells were then lysed in 25 mmol/L Tris-HCl, 150 mmol/L NaCl, 10 mmol/L EDTA, 1% Triton X-100, and 1x protease/phosphatase inhibitor cocktail. After 5min on ice, cell lysate was centrifuged at 12,000 x g for 10 minutes (4°C). Then the supernatant was desalted twice over Zeba spin columns, spun at 1500 x g for 2 minutes (4°C) for removing GSH and oxidized GSH(GSSG). Pulldown was performed by using a pan anti-glutathionylation antibody and protein G magnetic Dynabeads™. Anti-GSH antibody was cross-linked to the protein G magnetic Dynabeads™ following the manufacturer’s protocol. Samples were incubated with the modified beads at room temperature for 1 hour. Samples were eluted using 0.1 mol/L Glycine-HCl (pH 2.5) for 5 minutes, and then neutralized using 1 mol/L Tris-HCl, pH 8.5, and analyzed for C/EBPβ by Western blot.
Co-immunoprecipitation.
To create HA-tagged mouse C/EBPβ, full-length C/EBPβ from Myc-FLAG-tagged mouse C/EBPβ plasmid was subcloned into a C-terminal HA-tag vector. Liver-enriched inhibitory protein (LIP), deleted LIP domain (ΔLIP), C201S, C296S, and double mutant (C201S, C296S) of C/EBPβ in C-terminal HA-tag vector were synthesized by Eurofins Genomics (Tokyo, Japan). HEK293T cells were seeded in 6 cm dishes and transfected with 2 μg HA-tagged mouse C/EBPβ, LIP, ΔLIP, C201S, C296S, double mutant C/EBPβ, and Myc-FLAG-tagged mouse PIAS1 by using Lipofectamine 2000. The lysates of cells overexpressing these constructs were adjusted to 500 μg protein in 200 μL buffer and incubated with 10 mmol/L GSH and 90 mmol/L GSSG for 10 hours at 4 °C. Then, samples were desalted twice on Zeba spin columns to remove GSH and GSSG. These samples were mixed with 500 μg of lysates from Myc-FLAG-tagged mouse PIAS1 overexpressing HEK293T cells, and co-immunoprecipitation (Co-IP) was performed by using anti-Myc tag monoclonal antibody magnetic beads. Detection of HA-tagged and Myc-FLAG-tagged proteins were performed by immunoblotting using antibodies against tags, respectively.
Mass spectrometry.
HEK293T cells were cultured in 10 cm dishes and transfected with Myc-FLAG-tagged mouse C/EBPβ. The overexpressed Myc-FLAG-tagged mouse C/EBPβ was pulled down by anti-Myc tag monoclonal antibody magnetic beads cell lysates and eluted. Purified Myc-FLAG-tagged mouse C/EBPβ was incubated with 10 mmol/L GSH and 90 mmol/L GSSG for 10 hours at 4°C. After electrophoresis under nonreducing conditions, the gel was stained with Imperial™ protein stain (24615; Thermo Fisher Scientific ,Waltham, MA, USA), and the Myc-FLAG-tagged mouse C/EBPβ protein band excised from the gel. MS analysis was performed by MS Bioworks (MSB-10 PTM profiling plus)(Ann Arbor, MI, USA). In-gel digestion with trypsin was performed on the submitted sample using a robot (ProGest)(DigiLab, Hopkinton, MA, USA). The gel slices were washed with 25 mM ammonium bicarbonate followed by acetonitrile. Then the gel slices were digested with trypsin at 37 °C for 4 hours, quenched with formic acid, and the supernatant analyzed directly without further processing. Half of each digest was analyzed by nano LC/MS/MS with a Waters Nano Acquity HPLC system interfaced to a Thermo Fisher Q Exactive. Peptides were loaded on a trapping column and eluted from a 75 µm analytical column at 350 nL/min; both columns were packed with Luna C18 resin (Phenomenex, Torrance, CA, USA). The mass spectrometer was operated in data-dependent mode, with the Orbitrap operating at 70,000 FWHM and 17,500 FWHM for MS and MS/MS respectively. The fifteen most abundant ions were selected for MS/MS.
RT-PCR.
Total RNA was isolated from 3T3L1 cells using RNeasy Mini Kit (74104)(QIAGEN, Germantown, MD, USA). Reverse transcription was performed by using the ReverTra Ace® qPCR RT Master Mix (FSQ-201) (TOYOBO, Osaka, Japan). Samples were analyzed with quantitative real-time PCR using TaqMan primers: Rps18 (Mm02601777_g1), Pparg (Mm00440940_m1), Cebpa (Mm00514283_s1) and Cebpb (Mm00786711_s1), and the TaqMan™ Gene Expression Master Mix. Expression levels were analyzed by comparative Ct (ΔΔCT) with StepOne real-time PCR software (Thermo Fisher Scientific, Waltham, MA, USA).
Western blotting.
Samples were incubated with sample loading buffer for 5 minutes at 95°C. An equal amount of proteins was loaded and separated by SDS PAGE. For the detection of S-glutathionylation, cell lysates were desalted twice on Zeba spin columns to avoid artefactual protein modifications during sample preparation. Then, lysates were mixed with non-reducing sample buffer and analyzed by Western blot. After blocking with 5% (w/v) skim milk in PBS with Tween 20 (0.1%) for 1 hour, the membranes were incubated with specific primary antibodies overnight at 4°C, followed by incubation with respective horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature. Membranes were incubated with ECL prime (GE Healthcare UK Ltd, Little Chalfont, England) substrate and images obtained using a LAS-4000 (GE Healthcare UK Ltd, Little Chalfont, England). Densitometry analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical Analysis.
Results are expressed as means ± SEM. Statistical difference between the means of two groups was carried out using a Student’s unpaired t-test. Analyses among ≥3 groups were performed either by one-way ANOVA or two-way ANOVA, followed by the Tukey posthoc comparison test. A P-value of < 0.05 was considered significant.
Results
ROS and protein S-glutathionylation in adipocyte differentiation.
Four days after induction of adipogenesis, differentiated 3T3L1 adipocyte-like cells produced more ROS than undifferentiated cells. There was no difference in mitochondrial superoxide production, and thus ROS are likely derived from non-mitochondrial sources (Fig. 1A and B). To determine the impact of ROS on adipogenesis, we incubated 3T3L1 cells with the ROS scavenger Tempol during differentiation. Tempol significantly decreased the lipid content in differentiated 3T3L1 adipocyte-like cells (Fig. 1C). Protein S-glutathionylation increased during adipogenesis and were attenuated by Tempol, as detected by a pan antibody against GSH-protein adducts by Western blot (Fig. 1D) and relative quantification (Fig. 1E).
Figure 1:
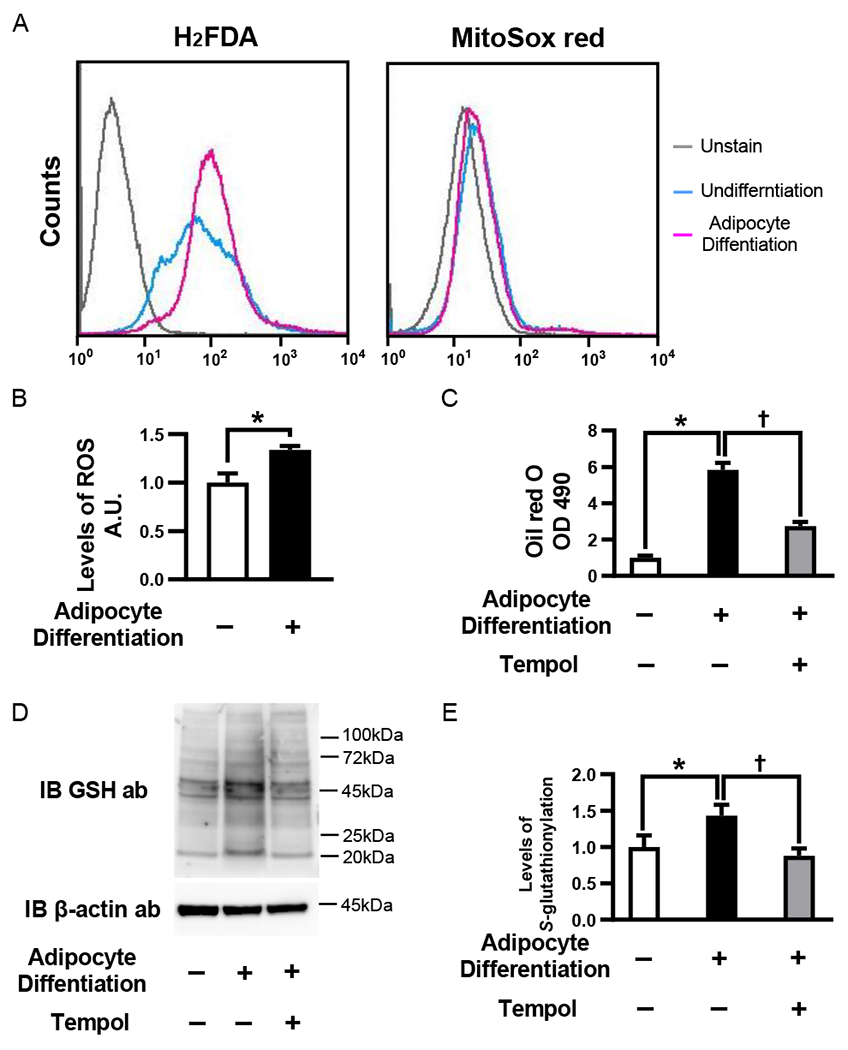
ROS and protein S-glutathionylation in adipocyte differentiation. (A) ROS detection by flow cytometry in 3T3L1 cells, 4days after adipocyte differentiation. Representative histograms of H2FDA (left) and MitoSOX red (right). Experiments were repeated three times with similar results. (B) The levels of ROS in 3T3L1 cells after adipocyte differentiation. The levels of ROS were measured by using OxiSelect™ Intracellular ROS Assay Kit, 4 days after adipocyte differentiation. (n=4 each group) (C) 3T3L1 cells were treated with 2 mmol/L Tempol or PBS at the induction of adipocyte differentiation. The lipid content of 3T3L1 cells was analyzed 6 days after adipocyte differentiation. (n=4 each group) (D and E) The levels of GSH-protein adducts after adipocyte differentiation with or without Tempol treatment. Protein S-glutathionylation were detected Western blotting analysis by using anti-GSH antibody 4 days after adipocytes differentiation with or without Tempol treatment. (D) Representative Western blot of protein S-glutathionylation and β-actin (E) Densitometry analysis of protein S-glutathionylation (n=4 each group). *P<0.05 compared with control cells, †P<0.05, compared with respective vehicle-treated cells.
The impact of Glrx and protein S-glutathionylation on adipocyte differentiation.
To determine the ROS-independent effects of protein S-glutathionylation on adipogenesis, we ablated Glrx in 3T3L1 cells using CRISPR/Cas9. Glutaredoxin-1 protein expression increased during adipocyte differentiation. After the knockdown, Glrx protein expression was effectively null (Fig. 2A). Glrx ablation further increased protein S-glutathionylation in differentiated 3T3L1 adipocyte-like cells compared with contorl cells (Fig. 2B and C), while ROS generation remained unaffected by Glrx ablation (Fig. 2D). However, differentiated Glrx KO cells contained significantly higher lipid content versus differentiated contorl cells (Fig. 2E). To confirm that Glrx caused the lipid-lowering effects, we replenished the KO cells with Glrx using adenoviral transduction of a tetracycline-inducible GFP-tagged Glrx (Ad tet-Glrx). After doxycycline treatment, Glrx replenished cells (Fig. 2F) contained significantly less lipid (Fig. 2G).
Figure 2.
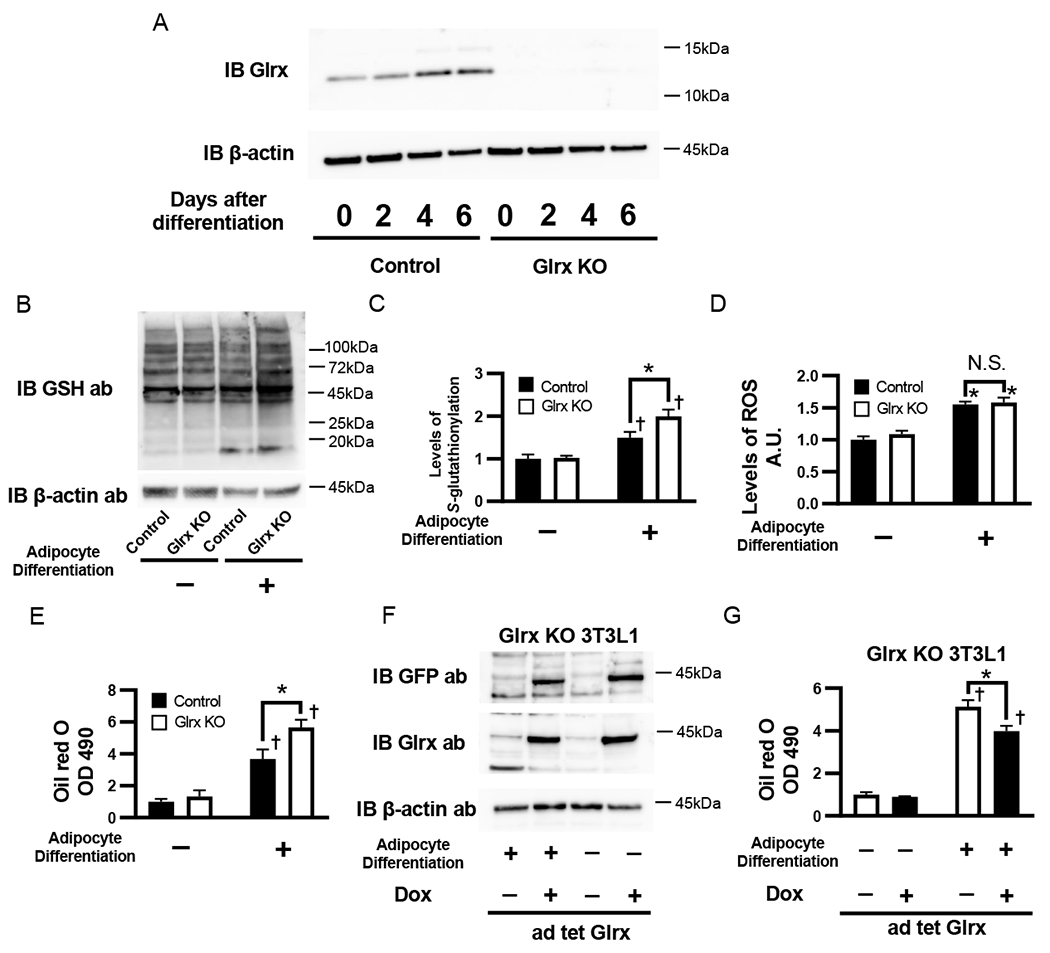
The impact of Glrx on protein S-glutathionylation and adipocyte differentiation. (A) Glrx expression during adipocyte differentiation and Glrx knockdown by CRISPR/Cas9. Representative Western blot of Glrx and β-actin in control and Glrx KO 3T3L1 cells, day 0, 2, 4, and 6 after adipocyte differentiation. (B and C) The levels of protein S-glutathionylation in control and Glrx KO 3T3L1 cells, 4 days after adipocyte differentiation. (B) Representative Western blot of protein S-glutathionylation and β-actin (C) Densitometry analysis of protein S-glutathionylation (n=4 each group). (D) Level of ROS in control and Glrx KO 3T3L1 cells measured by using OxiSelect™ Intracellular ROS Assay Kit , 4 days after adipocyte differentiation. (n=4 each group). (E) Lipid content in control and Glrx KO 3T3L1 cells 6 days after adipocyte differentiation. The lipid content of cell lysates from cells stained with Oil red O was measured at OD 490 (n=6 each group). (F and G) Effect of Glrx overexpression on lipid content in Glrx KO 3T3L1 cells. Glrx overexpression was induced by 1 µg/mL doxycycline in Ad tet-Glrx transduced Glrx KO 3T3L1 cells. Lipid content was analyzed 6 days after induction of adipocyte differentiation (n=4 each group). *P<0.05 compared with control cells, †P<0.05, compared with respective vehicle-treated cells.
Expression of adipogenic transcription factors in Glrx KO 3T3L1 cells.
Glrx KO increased RNA expression of the adipogenic transcription factors PPAR γ and C/EBPα, but C/EBPβ remained unchanged in 3T3L1 cells post-adipogenesis (Fig. 3A). Protein expression of PPARγ and C/EBPα also increased in differentiated Glrx KO cells and followed the expression pattern observed for mRNA. However, Glrx KO markedly upregulated C/EBPβ protein in differentiated cells (Fig. 3B-E), which likely depended on protein stability.
Figure 3:
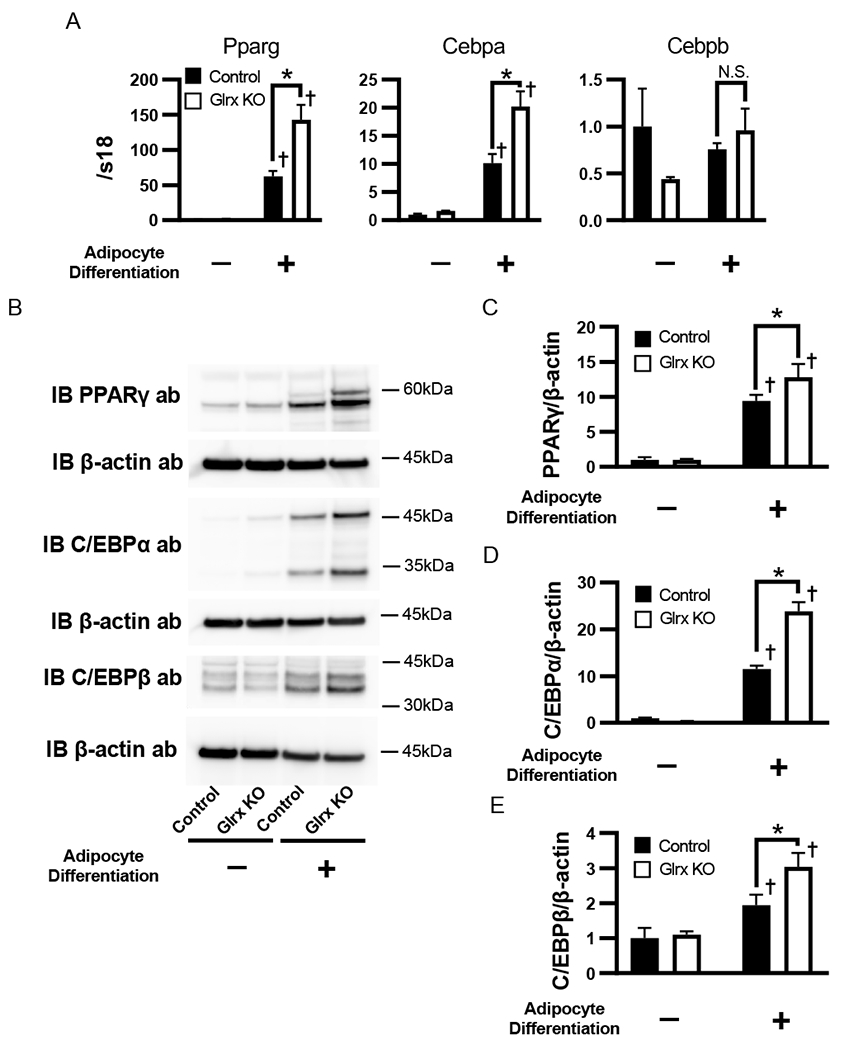
Expression of adipogenic transcription factors in Glrx KO 3T3L1 cells. (A) Expression levels of mRNA of PPARγ, C/EBPα and C/EBPβ. (B-E) Protein levels of PPARγ, C/EBPα and C/EBPβ. (B) Representative western blot of PPARγ, C/EBPα and C/EBPβ and β-actin in control and Glrx KO 3T3L1 cells 4 days after initiation of adipocyte differentiation. There were two bands in blot of C/EBPα, upper band was p42, lower band was p30 of C/EBPα, respectively. Densitometry analysis of PPARγ (C) C/EBPα (D) and C/EBPβ (E). n=4 each group *P<0.05 compared with control cells, †P<0.05 compared with respective vehicle-treated cells.
Regulation of C/EBPβ protein stability by S-glutathionylation.
To examine if C/EBPβ protein stability is affected by S-glutathionylation, we immunoprecipitated all GSH-modified proteins in either differentiated control or Glrx KO 3T3L1 cell lysates and probed for C/EBPβ. We detected S-glutathionylation of C/EBPβ only in Glrx KO cells (Fig. 4A). As previously reported, SUMOylated C/EBPβ interacts with SUMO E3 ligase PIAS1 (Fig. 4B), targeting C/EBPβ for ubiquitination and proteasomal degradation (18). To elucidate whether S-glutathionylation affects the interaction between C/EBPβ and PIAS1, we treated lysates of HA-tagged mouse C/EBPβ-expressing HEK293T cells with a mixture of reduced (GSH) and oxidized glutathione (GSSG) to promote protein S-glutathionylation (Fig. 4C). After removing the GSH/GSSG mixture, we combined the HA-tagged mouse C/EBPβ-containing lysate with a Myc-FLAG-tagged mouse PIAS1-containing lysate and performed a Co-IP. The Co-IP revealed that S-glutathionylation of C/EBPβ decreased the interaction with PIAS1 (Fig. 4D), likely preventing C/EBPβ downstream processing and degradation. C/EBPβ mRNA encodes the 38-kDa full-length protein but also produces three N-terminally truncated isoforms: 34-kDa liver-enriched activating protein (LAP), 21-kDa LIP and a 14-kDa isoform (19). To narrow down the PIAS1 interaction site in C/EBPβ, we synthesized HA-tagged LIP and HA-tagged C/EBPβ with deleted LIP domain (ΔLIP). Myc-FLAG-tagged mouse PIAS1, HA-tagged LIP and C/EBPβ with deleted LIP domain (ΔLIP) were overexpressed in HEK293T cells for Co-IP experiments (Fig. S1). Co-IP indicated that PIAS1 interacted with the LIP domain of C/EBPβ (Fig. 4E). To localize the position of S-glutathionylation in C/EBPβ, we treated the HA-tagged mouse C/EBPβ overexpressing lysates again with the same mixture of GSH/GSSG and performed mass spectrometry. LC/MS/MS analysis identified 332 matching peptides which covered 95.3% of the protein sequence (Fig. S2). We found five out of six cysteine residues modified with GSH, including Cys201 and Cys296 of the LIP domain (Fig. 4F). To confirm that the cysteines were essential for PIAS1 interaction, we generated single C201S and C296S mutants as well as the C201S, C296S double mutant. The Co-IP demonstrated that S-glutathionylation attenuated the interaction of PIAS1 with wild type, C201S, and C296S single mutant but not with the C201S, C296S double mutant C/EBPβ (Fig. 4G). Thus, S-glutathionylation on both Cys201 and Cys296 are essential for inhibiting the interaction between C/EBPβ and PIAS1.
Figure 4:
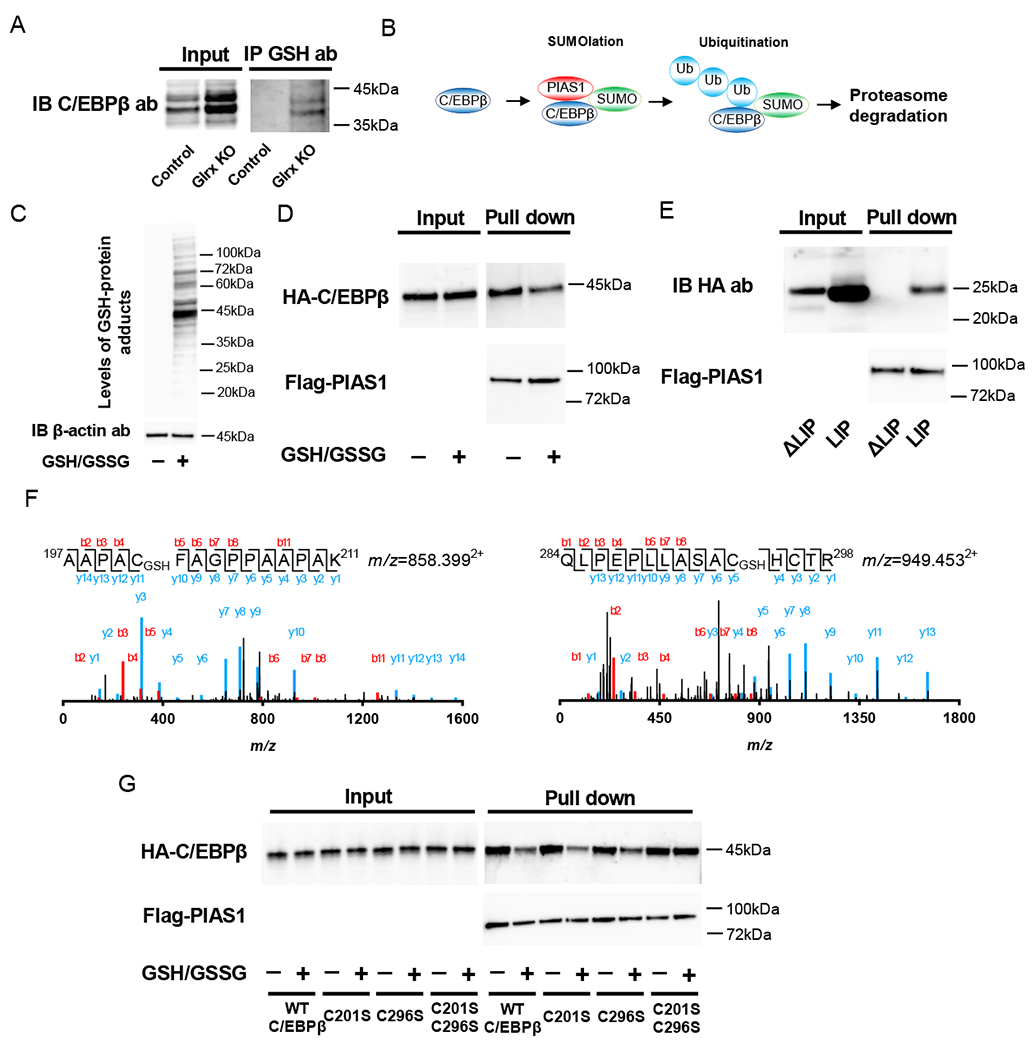
Regulation of C/EBPβ protein stability by S-glutathionylation. (A) Detection of S-glutathionylation on C/EBPβ. Cell lysate of control and Glrx KO 3T3L1 cells after adipocyte differentiation was immunoprecipitated with an anti-GSH antibody. Then Western blot analysis was performed by using an anti-C/EBPβ antibody. (B) Scheme of C/EBPβ regulation by SUMO E3 ligase PIAS1, SUMOlation, ubiquitination, and proteasome degradation. (C) Induction of S-glutathionylation in HEK293T cell lysate. HEK293T cell were treated with GSH/GSSG mixture to promote S-glutathionylation. Western blot was performed by using anti-GSH antibody. (D) Inhibition of PIAS1 attachment by S-glutathionylation on C/EBPβ. Western blotting analysis following Co-IP of Myc-FLAG-tagged mouse PIAS1 and GSH/GSSG mixture or vehicle treated HA-tagged mouse C/EBPβ by anti-Myc antibody-conjugated magnetic beads. Detection of HA-tagged mouse C/EBPβ and Myc-FLAG-tagged mouse PIAS1 was performed by anti-HA antibody and anti-Flag antibody, respectively. (E) Identification of a region of PIAS1 attachment in C/EBPβ. HA-tagged LIP isoform or LIP deleted full-length mutant (ΔLIP) overexpressed HEK293T cell lysate was mixed with Myc-FLAG-tagged mouse PIAS1 overexpressed HEK293T cell lysate followed by Co-IP using anti-Myc antibody-conjugated magnetic beads. Western blot analysis was performed using anti-HA antibody for LIP and ΔLIP, anti-FLAG antibody for PIAS1. (F) Detection of S-glutathionylation on Cys201 (Upper) and Cys296 (lower) by MS. S-glutathionylation of Cys201 and Cys296 were detected from trypsin fragments 197AAPACFAGPPAAPAK211 and 289QLPEPLLASAGCTR298. The actual mass of these fragments was 1,714.78 (m/z=858.3992+) and 1,896.89 (m/z=949.4532+), respectively; the MW increased by 305 Da for S-glutathionylation. (G) C201S and C296S double mutation of C/EBPβ diminished effect of S-glutathionylation on inhibition of PIAS1 attachment. HA-tagged mouse C/EBPβ, HA-tagged C201S, C296S single mutant C/EBPβ, and HA-tagged C201S and C209S double mutant C/EBPβ were transfected to HEK293T cells. These cell lysate incubated with vehicle or GSH/GSSG mixture for induction S-glutathionylation. Then, Co-IP experiment was performed using these cells lysate, and Myc and FLAG-tagged mouse PIAS1 overexpressed cell lysate. Experiments were repeated three times with similar results.
Discussion
Reactive oxygen species are recognized mainly for their damaging effect during inflammation or pathologies associated with oxidative stress. Antioxidant therapy often proved ineffective in attenuating advanced diseases (11), and in rare cases such as certain types of cancer, even promoted progression (20). Low levels of ROS, however, introduce reversible oxidative modifications to specific protein residues that participate in cell regulation, also referred to as redox signaling (11). Genetic mouse studies such as GCLM or Glrx KO mice further highlight the impact redox signaling plays in essential cellular processes, metabolism, and pathophysiology. Nevertheless, our understanding of redox signaling remains incomplete.
We focused on protein S-glutathionylation in adipogenesis because our previous studies suggested a critical role for this oxidative post-translational modification in adipogenesis and obesity (9, 14). Our significant findings are: i) protein S-glutathionylation increased during adipogenesis of 3T3L1 cells, ii) the level of protein S-glutathionylation related to the adipocyte differentiation status of 3T3L1 cells, and iii) S-glutathionylation stabilized the adipogenic transcription factor C/EBPβ by preventing the interaction with SUMO E3 ligase PIAS1.
Mitochondria (5) and NADPH oxidases (21) are primary sources of ROS during adipogenesis. ROS can change the GSH/GSSG ratio promoting protein S-glutathionylation formation (22). We observed increased levels of protein S-glutathionylation during adipogenesis. Antioxidants such as Tempol attenuated ROS, protein S-glutathionylation formation, and blunted adipogenesis. Interestingly, adipogenesis also upregulated the expression of Glrx, despite the increase in protein S-glutathionylation. Thus, Glrx might compensate for elevated protein S-glutathionylation and terminate S-glutathionylation-mediated redox signaling. Indeed, Glrx KO increased protein S-glutathionylation and promoted adipogenesis. Glrx KO did not attenuate the cellular antioxidant system because cellular ROS levels remained unchanged after the induction of adipogenesis. Conversely, adenoviral-mediated Glrx replenishment of Glrx KO cells decreased adipogenesis. In summary, the initiation of adipocyte differentiation increased ROS and protein S-glutathionylation to support adipogenesis, and Glrx moderated this process. Most studies manipulated intracellular protein S-glutathionylation by genetic or pharmacologic manipulations. In a recent publication, however, Janssen-Heininger and colleagues administered recombinant Glrx into the airways of mice, which decreased Fas S-glutathionylation and attenuated lung fibrosis (23). Therefore, recombinant Glrx could be a potential therapeutic for obesity, but a future clinical evaluation is required.
C/EBPβ plays an essential role in adipogenesis and obesity (24–27). C/EBPβ KO causes adipose tissue atrophy (26) and protects against diet-induced obesity in mice (27). Also, knockdown of C/EBPβ in 3T3L1 cells decreases adipogenesis (24). Regulation C/EBPβ occurs at multiple levels, including transcriptional regulation and post-transcriptional modifications (25), especially SUMOlation through the SUMO E3 ligase PIAS1 targets C/EBPβ for ubiquitination and proteasomal degradation (18). ROS activate C/EBPβ and stimulate adipogenesis (4) and obesity (6) by an unknown mechanism. We demonstrated that Glrx KO increased C/EBPβ expression with increased S-glutathionylation. PIAS1 interacts with C/EBPβ through its LIP domain, which contains Cys201 and Cys296. S-glutathionylation on these LIP domain cysteines decreased the C/EBPβ/PIAS1 interaction. The double cysteine-to-serine mutant C/EBPβ, however, interacted with PIAS1 undistrurbed under conditions of increased protein S-glutathionylation. Therefore, S-glutathionylation of both cysteines, Cys201 and Cys296, are critical for C/EBPβ to evade ubiquitination and proteasomal degradation.
The modification of proteins by ubiquitin and SUMO, which is one of the ubiquitin-like modifiers, is via a set of reactions that activate, transfer, and bind ubiquitin to cellular proteins, catalyzed by E1, E2, and E3 enzymes (28, 29). The substrate specificity depends on E3 ligases which recognize target proteins (30). Previously we showed that S-glutathionylation on hypoxia-inducible factor (HIF)-1α inhibited protein interaction with the ubiquitin E3 ligase VHL which resulted in HIF-1α stabilization (31). Other groups reported that S-glutathionylation on Kelch-like ECH-associated protein 1 (Keap1) also suppressed ubiquitin E3 ligase attachment to the target protein (32). In this study, we showed that S-glutathionylation on C/EBPβ inhibited the interaction with SUMO E3 ligase PIAS1. Hence, protein S-glutathionylation may generally modulate ubiquitin and SUMO signaling pathways by inhibition of E3 ligases interaction to target proteins .
Our study has limitations. First, we did not perform a comprehensive screening to identify S-glutathionylation on other potential proteins which may participate in adipogenesis. Nevertheless, C/EBPβ is a critical regulator and an excellent example of how redox regulation controls cell differentiation and adipogenesis. Secondly, our study only interrogated redox regulation of adipogenesis in vitro and the biochemical effect of S-glutathionylation on mouse C/EBPβ protein interactions, but we have previously demonstrated that Glrx KO mice develop dyslipidemia and obesity, providing further evidence that S-glutathionylation of C/EBPβ and regulation by Glrx may impact obesity in vivo.
In conclusion, increased protein S-glutathionylation promoted adipogenesis in Glrx KO 3T3L1 cells. S-glutathionylation on both Cys201 and Cys296 stabilized C/EBPβ by inhibiting the interaction with the SUMO E3 ligase PIAS1. Dysregulation of this interaction may increase lipid accumulation, which could potentially accelerate obesity in vivo.
Supplementary Material
Acknowledgments
We appreciate the help of Ms. Dominique Croteau for proofreading and revising our manuscript. We thank Dr. Feng Zhang for providing PX459 V2.0. This work was supported by the Uehara Memorial Foundation and the Yamanashi Prefectural Satoshi Omura Human Resources Fund to Y.W, NIH grants R01 DK103750 to M.M.B., R01 HL133013 to R.M., and American Heart Association “Grant in Aid” 16GRNT27660006 to M.M.B.
This work was supported by the Uehara Memorial Foundation and the Yamanashi Prefectural Satoshi Omura Human Resources Fund to Y.W, NIH grants R01 DK103750 to M.M.B, R01 HL133013 to R.M, and American Heart Association “Grant in Aid” 16GRNT27660006 to M.M.B.
Glossary
- C/EBP
CCAAT enhancer-binding protein
- Co-IP
co-immunoprecipitation
- DOX
doxycycline
- Glrx
glutaredoxin-1
- GSH
glutathione
- GSSG
oxidized glutathione
- HIF
hypoxia-inducible factor
- Keap
kelch-like ECH-associated protein
- LAP
liver-enriched activating protein
- LC/MS/MS
liquid chromatography-tandem mass spectrometry
- LIP
liver-enriched inhibitory protein
- PIAS
protein inhibitor of activated STAT
- PPAR
peroxisome proliferator-activated receptor
- ROS
reactive oxygen species
- SUMO
small ubiquitin-related modifier
- Tempol
4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl
References
- 1.Fruh SM (2017) Obesity: Risk factors, complications, and strategies for sustainable long-term weight management. J. Am. Assoc. Nurse Pract. 29, S3–S14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Unamuno X, Gómez-Ambrosi J, Rodríguez A, Becerril S, Frühbeck G, and Catalán V (2018) Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Invest. 48, e12997. [DOI] [PubMed] [Google Scholar]
- 3.Hausman DB, DiGirolamo M, Bartness TJ, Hausman GJ, and Martin RJ (2001) The biology of white adipocyte proliferation. Obes. Rev. 2, 239–254 [DOI] [PubMed] [Google Scholar]
- 4.Lee H, Lee YJ, Choi H, Ko EH, and Kim J-W (2009) Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J. Biol. Chem. 284, 10601–10609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tormos KV, Anso E, Hamanaka RB, Eisenbart J, Joseph J, Kalyanaraman B, and Chandel NS (2011) Mitochondrial Complex III ROS Regulate Adipocyte Differentiation. Cell Metab. 14, 537–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang YC, Yu YH, Shew JY, Lee WJ, Hwang JJ, Chen YH, Chen YR, Wei PC, Chuang LM, and Lee WH (2013) Deficiency of NPGPx, an oxidative stress sensor, leads to obesity in mice and human. EMBO Mol. Med. 5, 1165–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Youn J, Siu KL, Lob HE, Itani H, Harrison DG, and Cai H (2014) Role of vascular oxidative stress in obesity and metabolic syndrome. Diabetes 63, 2344–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, and Shimomura I (2004) Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 114, 1752–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi T, Watanabe Y, Saito Y, Fujioka D, Nakamura T, Obata JE, Kitta Y, Yano T, Kawabata K, Watanabe K, Mishina H, Ito S, and Kugiyama K (2010) Mice lacking the glutamate-cysteine ligase modifier subunit are susceptible to myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 85, 785–795 [DOI] [PubMed] [Google Scholar]
- 10.Kendig EL, Chen Y, Krishan M, Johansson E, Schneider SN, Genter MB, Nebert DW, and Shertzer HG (2011) Lipid metabolism and body composition in Gclm(−/−) mice. Toxicol. Appl. Pharmacol. 257, 338–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe Y, Cohen RA, and Matsui R (2016) Redox Regulation of Ischemic Angiogenesis – Another Aspect of Reactive Oxygen Species –. Circ. J. 80, 1278–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe Y, Watanabe K, Kobayashi T, Saito Y, Fujioka D, Nakamura T, Obata JE, Kawabata K, Mishina H, and Kugiyama K (2013) Chronic depletion of glutathione exacerbates ventricular remodelling and dysfunction in the pressure-overloaded heart. Cardiovasc. Res. 97, 282–292 [DOI] [PubMed] [Google Scholar]
- 13.Chrestensen CA, Starke DW, and Mieyal JJ (2000) Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J. Biol. Chem. 275, 26556–26565 [DOI] [PubMed] [Google Scholar]
- 14.Shao D, Han J, Hou X, Fry J, Behring JB, Seta F, Long MT, Roy HK, Cohen RA, Matsui R, and Bachschmid MM (2017) Glutaredoxin-1 Deficiency Causes Fatty Liver and Dyslipidemia by Inhibiting Sirtuin-1. Antioxid. Redox Signal. 27, 313–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho H, Kim KM, Han S, Choe J, Park SG, Choi SS, and Kim YK (2012) Staufen1-Mediated mRNA Decay Functions in Adipogenesis. Mol. Cell 46, 495–506 [DOI] [PubMed] [Google Scholar]
- 16.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han YH, Moon HJ, You BR, Kim SZ, Kim SH, and Park WH (2010) The effects of N-acetyl cysteine on the MG132 proteasome inhibitor-treated lung cancer cells in relation to cell growth, reactive oxygen species and glutathione. Int. J. Mol. Med. 25, 657–662 [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Zhang Y-D, Guo L, Huang H-Y, Zhu H, Huang J-X, Liu Y, Zhou S-R, Dang Y-J, Li X, and Tang Q-Q (2013) Protein inhibitor of activated STAT 1 (PIAS1) is identified as the SUMO E3 ligase of CCAAT/enhancer-binding protein β (C/EBPβ) during adipogenesis. Mol. Cell. Biol. 33, 4606–4617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luedde T, Duderstadt M, Streetz KL, Tacke F, Kubicka S, Manns MP, and Trautwein C (2004) C/EBP β isoforms LIP and LAP modulate progression of the cell cycle in the regenerating mouse liver. Hepatology 40, 356–365 [DOI] [PubMed] [Google Scholar]
- 20.Klein EA, Thompson IM, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL, Gaziano JM, Karp DD, Lieber MM, Walther PJ, Klotz L, Parsons JK, Chin JL, Darke AK, Lippman SM, Goodman GE, Meyskens FL, and Baker LH (2011) Vitamin E and the Risk of Prostate Cancer. JAMA 306, 1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han CY, Umemoto T, Omer M, Den Hartigh LJ, Chiba T, LeBoeuf R, Buller CL, Sweet IR, Pennathur S, Abel ED, and Chait A (2012) NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. J. Biol. Chem. 287, 10379–10393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vigilanza P, Aquilano K, Baldelli S, Rotilio G, and Ciriolo MR (2011) Modulation of intracellular glutathione affects adipogenesis in 3T3-L1 cells. J. Cell. Physiol. 226, 2016–2024 [DOI] [PubMed] [Google Scholar]
- 23.Anathy V, Lahue KG, Chapman DG, Chia SB, Casey DT, Aboushousha R, van der Velden JLJ, Elko E, Hoffman SM, McMillan DH, Jones JT, Nolin JD, Abdalla S, Schneider R, Seward DJ, Roberson EC, Liptak MD, Cousins ME, Butnor KJ, Taatjes DJ, Budd RC, Irvin CG, Ho Y-S, Hakem R, Brown KK, Matsui R, Bachschmid MM, Gomez JL, Kaminski N, van der Vliet A, and Janssen-Heininger YMW (2018) Reducing protein oxidation reverses lung fibrosis. Nat. Med. 24, 1128–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y-Y, Li X, Qian S-W, Guo L, Huang H-Y, He Q, Liu Y, Ma C-G, and Tang Q-Q (2011) Transcriptional activation of histone H4 by C/EBPβ during the mitotic clonal expansion of 3T3-L1 adipocyte differentiation. Mol. Biol. Cell 22, 2165–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo L, Li X, and Tang Q-Q (2015) Transcriptional regulation of adipocyte differentiation: a central role for CCAAT/enhancer-binding protein (C/EBP) β. J. Biol. Chem. 290, 755–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka T, Yoshida N, Kishimoto T, and Akira S (1997) Defective adipocyte differentiation in mice lacking the C/EBPβ and/or C/EBPδ gene. EMBO J. 16, 7432–7443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Millward CA, Heaney JD, Sinasac DS, Chu EC, Bederman IR, Gilge DA, Previs SF, and Croniger CM (2007) Mice With a Deletion in the Gene for CCAAT/Enhancer-Binding Protein β Are Protected Against Diet-Induced Obesity. Diabetes 56, 161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hershko A and Ciechanover A (1998) THE UBIQUITIN SYSTEM. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 29.Herrmann J, Lerman LO, and Lerman A (2007) Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res. 100, 1276–1291 [DOI] [PubMed] [Google Scholar]
- 30.Berndsen CE and Wolberger C (2014) New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 21, 301–307 [DOI] [PubMed] [Google Scholar]
- 31.Watanabe Y, Murdoch CE, Sano S, Ido Y, Bachschmid MM, Cohen RA, and Matsui R (2016) Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1α and improve limb revascularization. Proc. Natl. Acad. Sci. U. S. A. 113, 6011–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carvalho AN, Marques C, Guedes RC, Castro-Caldas M, Rodrigues E, van Horssen J, and Gama MJ (2016) S -Glutathionylation of Keap1: a new role for glutathione S -transferase pi in neuronal protection. FEBS Lett. 590, 1455–1466 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.