

Abstract
Signaling between the C-C chemokine receptor 2 (CCR2) with its ligand, monocyte chemoattractant protein-1 (MCP-1) promotes cancer progression by directly stimulating tumor cell proliferation and downregulating the expression of apoptotic proteins. Additionally, the MCP-1/CCR2 signaling axis drives the migration of circulating monocytes into the tumor microenvironment, where they mature into tumor-associated macrophages (TAMs) that promote disease progression through induction of angiogenesis, tissue remodeling, and suppression of the cytotoxic T lymphocyte (CTL) response. In order to simultaneously disrupt MCP-1/CCR2 signaling and target CCR2-expressing cancer cells for drug delivery, KLAK-MCP-1 micelles consisting of a CCR2-targeting peptide sequence (MCP-1 peptide) and the apoptotic KLAKLAK peptide were synthesized. In vitro, KLAK-MCP-1 micelles were observed to bind and induce cytotoxicity to cancer cells through interaction with CCR2. In vivo, KLAK-MCP-1 micelles inhibited tumor growth (34 ± 11%) in a subcutaneous B16F10 murine melanoma model despite minimal tumor accumulation upon intravenous injection. Tumors treated with KLAK-MCP1 demonstrated reduced intratumor CCR2 expression and altered infiltration of TAMs and CTLs as evidenced by immunohistochemical and flow cytometric analysis. These studies highlight the potential application of CCR2-targeted nanotherapeutic micelles in cancer treatment.
Keywords: micelle, CCR2, peptide, cancer, tumor-associated macrophage
Graphical Abstract
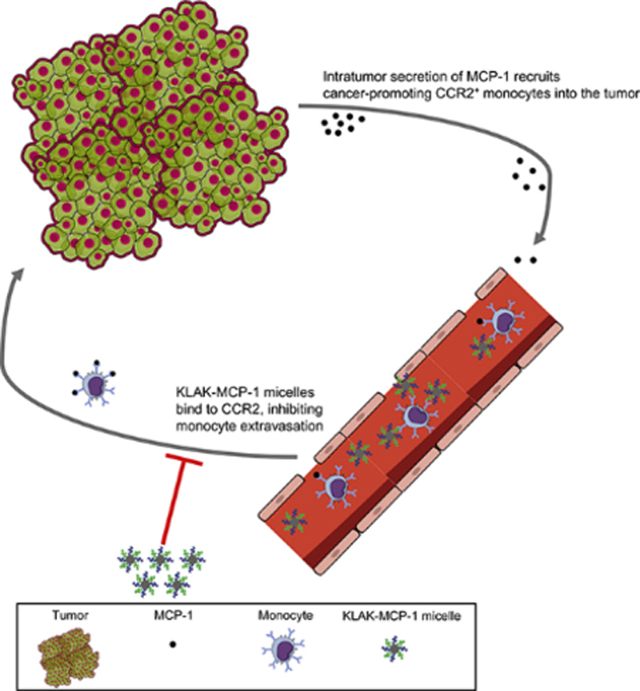
Introduction
Signaling between the C-C chemokine receptor 2 (CCR2) with its ligand, monocyte chemoattractant protein-1 (MCP-1) has been correlated with disease progression and metastasis in a variety of cancers including prostate cancer and melanoma [1–5]. Clinically, elevated serum levels of MCP-1 and intratumor CCR2 expression are associated with an unfavorable disease prognosis [6]. Tumor cells exploit MCP-1-CCR2 signaling to stimulate tumor cell proliferation and survival via downregulation of apoptotic pathways [7–12]. In addition to sustaining pro-survival signaling cascades in tumor cells, intratumor MCP-1 secretion also drives the recruitment of circulating CCR2+ monocytes that differentiate into tumor-associated macrophages (TAMs) [13, 14]. Clinical studies have linked high TAM infiltration to poor prognosis and cancer progression, as TAMs produce angiogenic cytokines, release enzymes that facilitate tissue remodeling and tumor cell migration, and establish an immunosuppressive tumor microenvironment through the production of anti-inflammatory cytokines and inhibition of cytotoxic T lymphocyte (CTL) activity [15–20].
Given the multiple roles of MCP-1/CCR2 signaling in cancer, disruption of this signaling axis has been proposed as an anti-cancer therapy. However, clinical trials utilizing monoclonal antibodies against MCP-1 have not been successful [21–23]. For example, in a phase II clinical trial using the anti-MCP-1 monoclonal antibody Carlumab in metastatic, castration-resistant prostate cancer patients, none of the 46 enrolled patients had an objective response to treatment. Although Carlumab was able to reduce free serum MCP-1 levels 2 hours post-administration, this reduction was only transient. Free serum MCP-1 levels quickly increased to 3- to 5-fold of baseline levels 48 hours post-administration and the absence of a durable effect of Carlumab on MCP-1 levels has been suggested to explain the lack of clinical effectiveness.
In addition to MCP-1 blockade, another approach to disrupt the MCP-1/CCR2 axis is to target MCP-1’s binding partner, CCR2. Noel et al. administered the CCR2 antagonist PF-04136309 in conjunction with nab-paclitaxel/gemcitabine to pancreatic ductal adenocarcinoma patients [24]. In 21 treated patients, 23.8% responded to treatment with a reduction in tumor size compared to the baseline. These clinical studies indicate that CCR2-blockade may be an alternative strategy for disrupting MCP-1/CCR2 signaling. Notably, CCR2-targeting therapy also offers the ability to directly target multiple aberrant cells including tumor cells and monocytes, which can be further leveraged to deliver cytotoxic drugs for therapeutic effect in addition to mitigating MCP-1/CCR2 signaling.
In an effort to target CCR2 for potential applications in cancer therapy, our group has previously developed a micelle comprised of a 23-residue peptide sequence derived from the binding motif (residues 13–35) of the MCP-1 chemokine (MCP-1 micelle) and reported its ability to bind to monocytes and induce toxicity against prostate cancer cells in vitro [25–27]. Incorporation of the CCR2-binding peptide (MCP-1 peptide) into a micelle can improve its secondary structure and in vivo stability by providing protection from serum peptidases [25, 26]. To test CCR2-targeting and evaluate its anticancer properties and potential to modulate the tumor microenvironment in vivo, herein, we incorporate the apoptotic peptide, KLAKLAK (KLAK peptide) into the MCP-1 micelle to augment its anticancer efficacy (KLAK-MCP-1 micelle). Specifically, we demonstrate that the addition of KLAKLAK improves micelle toxicity to cancer cells both in vitro and in vivo. Additionally, KLAK-MCP-1 micelles bind and are cytotoxic to multiple cancer cell lines, as well as monocytes, in a CCR2-dependent manner in vitro and were observed to inhibit tumor growth in subcutaneous B16F10 melanoma models by decreasing TAM and increasing CTL infiltration into the tumor.
2. Materials and Methods
2.1. Materials
MCP-1 [CYNFTNRKISVQRLASYRRITSSK] and scrambled MCP-1 [CYNSLVFRIRNSTQRKYRASIST] peptides were purchased from Ontores Biotechnologies (Zhejiang, China). 1,2-distearoyl-sn-glycero-3phosphoethanolamine-N-[maleimide-(polyethylene glycol)-2000] (DSPE-PEG2000-maleimide) and 1,2-distearoyl-sn-glycero-3phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (DSPE-PEG2000-amine) were purchased from Avanti Lipids (Alabaster, AL, USA). Cy7 mono-N-hydroxysuccinimide (NHS) ester was purchased from Lumiprobe (Hunt Valley, MD, USA). Culture media and reagents, including DMEM, Ham’s F-12K, RPMI-1640, Keratinocyte-SFM, fetal bovine serum (FBS), penicillin-streptomycin, and phosphate buffered saline (PBS) were purchased from Gibco, Waltham, MA, USA.
2.2. Amphiphile synthesis
KLAKLAK peptides were synthesized on an automated peptide synthesizer (PS3, Protein Technologies, Tucson, AZ, USA) with Fmoc-mediated solid phase peptide synthesis on a Wang-Lys resin and cleaved using a 94:2.5:2.5:1 vol% trifluoroacetic acid:1,2-ethanedithiol:water:triisopropylsilane solution. All peptides were synthesized with an N-terminal cysteine residue to facilitate covalent conjugation to maleimide-terminated lipid tails. Following cleavage, peptides were precipitated and washed twice with ice-cold diethyl ether and dissolved in water and lyophilized. Crude peptides were resuspended in water and purified using reverse-phase, high-pressure liquid chromatography (RP-HPLC, Prominence, Shimadzu, Columbia, MD, USA) on a Luna C18 column (Phenomenex, Torrance, CA, USA) at 55°C with 0.1% formic acid in water/acetonitrile mobile phases. The purity of the eluted peptides was characterized using matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF-MS, Bruker, MA, USA). The expected mass peak for the KLAKLAK peptide is 874 g/mol, while the expected mass peaks for the MCP-1 and scr-MCP-1 peptides are 2892 g/mol (Fig. S1).
Pure KLAKLAK, MCP-1, and scr-MCP-1 peptides were conjugated to a 10% molar excess of DSPE-PEG2000-maleimide through thioether linkage in water as previously described [25–31]. The reaction was performed under a pH of 7.2 at room temperature for 72 hours with constant agitation. Afterwards, peptide amphiphiles were purified and characterized through RP-HPLC and MALDI-TOF-MS. The expected mass peak for the KLAKLAK peptide amphiphile is 3716 g/mol, and the expected mass peaks for the MCP-1 and scr-MCP-1 peptide amphiphiles are 5834 g/mol (Fig. S2). Cy7 amphiphiles were synthesized by reacting DSPE-PEG2000-amine with a 3-fold molar excess of cy7 NHS ester in a 0.1M sodium bicarbonate buffer (pH 8.2) overnight. Cy7 amphiphiles were purified and characterized through RP-HPLC and MALDI-TOF-MS, with an expected mass peak of 3409 g/mol (Fig. S3).
2.3. Self-assembly of micelles
Micelles were self-assembled through thin-film hydration. Amphiphiles were dissolved and sonicated in methanol, before evaporation of the solution under a gentle nitrogen stream into a lipid film. The film was dried overnight under vacuum before hydration with water or PBS. The suspension was then sonicated, heated to 80°C for 30 minutes, and cooled to room temperature. KLAK-MCP-1 micelles were synthesized with KLAK and MCP-1 peptides at a 1:1 mole ratio. Fluorescent KLAK-MCP-1 micelles were synthesized by mixing KLAK, MCP-1, and cy7 amphiphiles at a 45:45:10 ratio [25].
2.4. Transmission electron microscopy (TEM)
7 μL of 50 μM KLAK-MCP-1 micelles suspended in MilliQ (MQ) water was placed on 400 mesh lacey carbon grids (Ted Pella, Redding, CA, USA) for 5 minutes, before excess liquid was wicked, and the samples were washed with MQ water. The sample was then stained with 2% uranyl acetate, washed once more with MQ water, and dried before imaging on a JEOL JEM 2100-F TEM (JEOL, Tokyo, Japan).
2.5. Dynamic light scattering (DLS) and zeta potential measurements
Micelle size and polydispersity was evaluated through DLS (Wyatt Technology Mobius system, Santa Barbara, CA, USA). 50 μM micelles in water or PBS (pH 7.4) was adjusted to pH 5, 5.5, 6.0, 6.5, or 7.0 through the addition of 1N hydrochloric acid (n = 3). Zeta potential measurements were made in MQ water using the same Mobius system with a platinum dip probe (n = 3).
2.6. Cell culture
All cell lines (B16F10, PC3, 22Rv1, WEHI-274.1, NCI-H460, and RWPE-1) were purchased from ATCC (Old Town Manassas, VA, USA) and screened for mycoplasma. B16F10 cells were cultured in DMEM supplemented with 10% v/v FBS and 1% penicillin-streptomycin. PC3 cells were cultured in Ham’s F-12K supplemented with 10% v/v FBS and 1% v/v penicillin-streptomycin. 22Rv1 and NCI-H460 cells were cultured in RPMI-1640 supplemented with 10% v/v FBS and 1% v/v penicillin-streptomycin. WEHI-274.1 cells were cultured in DMEM supplemented with 10% v/v FBS, 1% penicillin-streptomycin, and 0.05 mM 1,2-mercaptoethanol. RWPE-1 cells were cultured in keratinocyte-SFM basal media supplemented with 50 μg/mL bovine pituitary extract and 5 ng/mL human recombinant epidermal growth factor. All cells were cultured in a humidified chamber maintained at 37°C under 5% CO2 and media were renewed every 2–3 days.
2.7. In vitro micelle binding
To evaluate micelle binding to cancer cells and monocytes, 11.25 μM of cy7-labeled KLAK-MCP-1 or KLAK-scr-MCP-1 micelles were incubated for 4 hours with B16F10, PC3, 22Rv1, NCI-H460, or WEHI-274.1 cells that had been seeded onto glass cover slips (400,000 cells per slip), with or without a 1-hour pre-incubation with 250 μM MCP-1 peptide. After three PBS washes, cells were fixed with 4% PFA and blocked with 1% BSA, 22.52 mg/mL glycine, and 0.1% Tween-20 for 1 hour before incubation with anti-CCR2 primary antibodies for 1 hour (Abcam, Cambridge, UK, 1:100). Following three PBS washes, cells were incubated for 1 hour in the dark with a secondary antibody labelled with Alexa Fluor® 594 (Thermo Fisher Scientific, Waltham, MA, USA, 1:200) before counter-staining with DAPI (1 μg/mL). Cover slips were mounted onto microscope slides using the VectaMount aqueous mounting medium (Vector Laboratories, Burlingame, CA, USA) and imaged with a Leica DMi8 fluorescence microscope (Leica, Wetzlar, Germany).
2.8. MCP-1 and CCR2 mRNA expression
B16F10 cells were seeded into 12-well plates at a density of 100,000 cells/well. Cells were incubated with PBS, KLAK-MCP-1 micelles, MCP-1 micelles, KLAK-scr-MCP-1 micelles, KLAK micelles, or scr-MCP-1 micelles at a concentration of 5 μM for 4 hours before mRNA extraction using the RNeasy kit (Qiagen, Hilden, Germany) according to kit instructions. cDNA was generated using the RT2 First Strand kit from Qiagen. mRNA was generated using the RT2 SYBR Green qPCR Mastermix (Qiagen) before analysis using a CFX384 PCR detection system (Bio-Rad, Hercules, CA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene to normalize gene expression data, and the 2ΔΔCq method was used to quantify mRNA expression.
2.9. In vitro CCR2 protein expression of cell lines
CCR2 protein expression in the B16F10, PC3, 22Rv1, NCI-H460, and RWPE-1 cell lines was evaluated through an ELISA (Biomatik, Wilmington, DE, USA). Cells were plated at a density of 80,000 cells/well (n = 4) in a 24-well plate and allowed to adhere overnight. Cells were lysed using RIPA buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with the HALT protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA). Lysed cells were centrifuged at 12,000g for 10 minutes at 4°C and the supernatants were processed according to the manufacturer’s protocol. CCR2 protein expression was quantified by measuring the sample absorbance at 450 nm using a Varioskan Lux microplate reader (Thermo Fisher Scientific, Waltham, MA, USA) and converting absorbance measurements to protein expression (pg/mL) using a CCR2 standard curve provided by the ELISA kit.
2.10. In vitro cytotoxicity of micelles
The in vitro cytotoxicity of micelles was evaluated with the (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay (Biovision, Milpitas, CA, USA). B16F10, PC3, 22Rv1, NCI-H460, WEHI-274.1, and RWPE-1 cells were plated at a density of 2000 cells/well in 96-well plates. 0.5–50 μM of KLAK-MCP-1, KLAK-scr-MCP-1, KLAK, MCP-1, scr-MCP-1, and empty micelles were incubated with cells for 72 hours before addition of the MTS reagent and evaluation using a Varioskan Lux microplate reader at an absorbance wavelength of 490 nm. After subtracting the background absorbance of culture media + MTS reagent alone, cell viability data was normalized to PBS-treated cells, and IC50 values were calculated using GraphPad Prism 7 (GraphPad, San Diego, CA, USA). For each assay, n = 6 was used for each concentration.
2.11. In vivo efficacy
The in vivo efficacy of KLAK-MCP-1, KLAK-scr-MCP-1, and MCP-1 micelles was evaluated in a subcutaneous B16F10 murine melanoma model in 5-week-old, male C57BL/6J mice (n = 11 to 18, Jackson Laboratory, Bar Harbor, ME). On day 1, mice were inoculated with 1 × 105 B16F10 cells suspended in 100 μL of a 1:1 solution of culture medium:Matrigel (Corning, Corning, NY, USA) into the left flank. On days 2, 5, 8, and 11, mice were injected via the tail-vein with 100 μL of PBS or 500 μM MCP-1, KLAK-MCP-1, or KLAK-scr-MCP-1 micelles. On day 15, mice were sacrificed, and tumors and organs were harvested. Tumor volume was measured using digital calipers, according to: 0.5*(tumor length)*(tumor width)2.
2.12. Immunohistochemistry of excised tumors
Excised tumors were fixed in 10% formalin overnight at 4°C, embedded in paraffin, and sectioned (5 μm) for immunohistochemical (IHC) analyses. Sectioned tissues were incubated with primary antibodies for CCR2 (Abcam, Cambridge, UK, 1:100), cleaved caspase-3 (Cell Signaling Technology, Danvers, MA, USA, 1:500), F4/80 (Thermo Fisher Scientific, Waltham, MA, USA, 1:100), PDL1 (Cell Signaling Technology, Danvers, MA, USA, 1:100), or CD31 (Cell Signaling Technology, Danvers, MA, USA, 1:100) before the use of a horseradish peroxidase (HRP) staining kit (Cell Signaling Technology, Danvers, MA, USA). Sections were counterstained with hematoxylin and imaged using an EVOS M7000 fluorescent microscope (Invitrogen, Carlsbad, CA, USA).
2.13. Flow cytometric analysis of excised tumors
Excised tumors were passed through a cell strainer to obtain single cell suspensions. After incubation in a red blood cell lysis buffer, suspensions were counted, stained with CD4 (CST, Danvers, MA, USA, 1:160), CD8 (CST, Danvers, MA, USA, 1:160), and CD45 (Thermo Fisher Scientific, Waltham, MA, USA, 1:160), fixed, and immunophenotyped using a MACSQuant flow cytometer (n = 8, Miltenyi Biotec, Bergisch Gladbach, Germany). Intratumor cytotoxic T lymphocyte (CTL) populations were immunophenotyped as CD4−/CD8+/CD45+.
2.14. In vivo biodistribution of micelles
Subcutaneous B16F10 melanoma mouse models were established as described in Section 2.11. To evaluate biodistribution of micelles, 14 days after tumor inoculation, mice were intravenously injected with 100 μL of 500 μM of cy7-labelled MCP-1, KLAK-MCP-1, or KLAK-scr-MCP-1 micelles via tail-vein and sacrificed after 3 hours. Tissues (tumors, lymph nodes, brain, lungs, heart, intestines, spleen, liver, kidneys) were harvested and imaged ex vivo using an AMI HTX imaging system and analyzed using the Aura imaging software package (Spectral Instruments, Tucson, AZ, USA).
2.15. Histology
Harvested tissues were snap-frozen in blocks of optimum cutting temperature (OCT) compound (Sakura Finetek, Torrance, CA, USA), sectioned (CM3050 S Cryostat, Leica, Wetzlar, Germany), and 8 μm sections were stained using hematoxylin and eosin (H&E), and imaged using a Leica DMi8 fluorescence microscope (Leica, Wetzlar, Germany).
2.16. Serum analysis of liver and kidney function
Liver function was evaluated by using commercially available kits to assess the serum activity of alanine transaminase (ALT) and aspartate transaminase (AST, Sigma-Aldrich, St. Louis, MO, USA). Kidney function was evaluated by using commercially available kits to analyze serum samples for blood urea nitrogen (BUN, Bioo Scientific, Austin, TX, USA) and creatinine (Crystal Chem, Elk Grove Village, IL, USA).
2.17. Statistical analysis
Data are expressed as mean ± SD. Statistical analysis between two groups was performed using a Student’s t-test. Comparisons of three or means were performed with using analysis of variance (ANOVA) followed by post-hoc Tukey’s tests for multiple comparisons. A p-value ≤ 0.05 was considered to be statistically significant.
3. Results and Discussion
3.1. Synthesis and characterization of KLAK-MCP-1 micelles
KLAK-MCP-1 micelles were constructed using KLAK and MCP-1 peptide amphiphiles at a 50:50 ratio using methods previously reported by our group (Fig. S1, S2) [25–27]. Through TEM and DLS, KLAK-MCP-1 micelles were found to be spherical with a hydrodynamic diameter of 11.9 ± 2.3 nm (Fig. 1A, Table 1), which is within the reported range of 8–200 nm for favorable in vivo half-life [32–34]. To ensure KLAK-MCP-1 micelles are stable under acidic conditions that have been reported in tumor microenvironments, DLS measurements were taken in PBS buffers titrated from pH 5 to 7.4 [35–37]. As shown in Fig. 1B, KLAK-MCP-1 micelles maintained a size of 12–14 nm at 24 hours in various pH environments.
Figure 1:
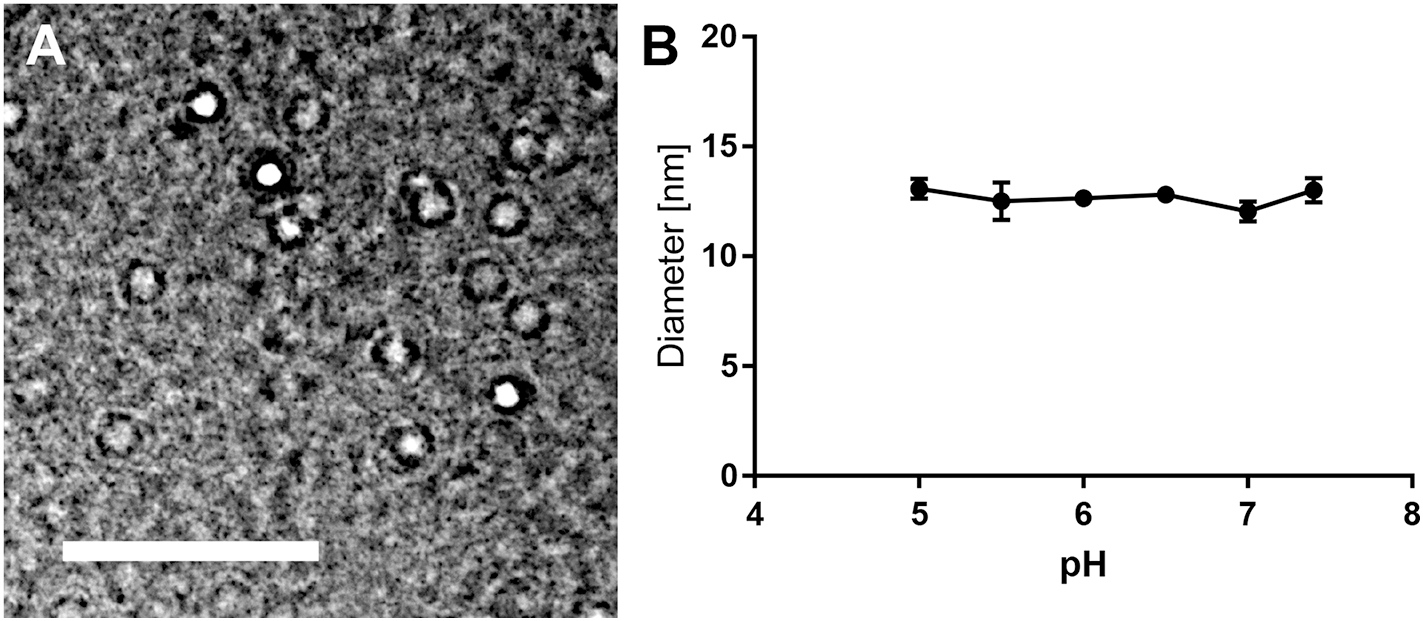
KLAK-MCP-1 micelle characterization. TEM of KLAK-MCP-1 micelles (A). DLS measurements demonstrate KLAK-MCP-1 micelle stability after 24 hours in various pH found in tumor tissue (n = 3) (B). Scale bar = 50 nm.
Table 1.
Characterization of micelles.
| Micelle (n = 3) | Diameter [nm] | PDI | Zeta Potential [mV] |
|---|---|---|---|
|
| |||
| KLAK-MCP-1 | 11.9 ± 2.3 | 0.14 ± 0.09 | 7.2 ± 1.1 |
| KLAK-scr-MCP-1 | 11.7 ± 1.5 | 0.14 ± 0.05 | 9.5 ± 2.2 |
| KLAK | 11.3 ± 0.7 | 0.19 ± 0.01 | 0.9 ± 0.4 |
| MCP-1 | 9.3 ± 2.1 | 0.10 ± 0.01 | 16.2 ± 0.8 |
| Scr-MCP-1 | 9.2 ± 0.5 | 0.18 ± 0.04 | 13.5 ± 2.0 |
| Empty | 8.6 ± 1.2 | 0.07 ± 0.04 | −9.6 ± 3.5 |
As shown in Table 1, KLAK-MCP-1 micelles were near neutral and had a zeta potential of 7.2 ± 1.1 mV. Non-targeting KLAK-scr-MCP-1 micelles consisting of KLAK and scr-MCP-1 peptides, KLAK micelles, MCP-1 micelles, scr-MCP-1 micelles, and empty DSPE-PEG2000-methoxy micelles were also synthesized and characterized in Table 1. All synthesized nanoparticles were similar in size, ranging from 8 to 12 nm with low polydispersity (< 0.2). While empty micelles had a slightly negative zeta potential of −9.6 ± 3.5 mV due to the negatively charged phosphate on the DSPE-PEG2000 tail, all other micelles had neutral to positive zeta potentials ranging from 0.9 mV to 16.2 mV, due to the basic lysine and arginine residues within the KLAK and MCP-1 peptides [38, 39].
3.2. KLAK-MCP-1 micelle binding to cancer cells in vitro
To evaluate micelle binding to cancer cells in vitro, cy7-labelled KLAK-MCP-1 or KLAK-scr-MCP-1 micelles were incubated with B16F10, PC3, 22Rv1, or NCI-H460 cancer cells at a non-cytotoxic concentration of 11.25 μM for 4 hours and examined via fluorescence microscopy. Immunostaining confirmed CCR2 expression in B16F10, PC3, and 22Rv1 cancer cells as shown in Fig. 2. As such, KLAK-MCP-1 micelles bound to B16F10, PC3, and 22Rv1 cells to a greater extent than KLAK-scr-MCP-1 micelles (2.35-, 8.20-, and 42.42-fold binding, respectively), indicating the MCP-1 peptide facilitated enhanced binding to CCR2-expressing cancer cells. To evaluate the specificity of KLAK-MCP-1 micelle binding to CCR2, a competitive binding assay was performed by incubating cells with KLAK-MCP-1 micelles following a 1 hour pre-treatment with 250 μM MCP-1 peptide. As shown in Fig. 2, preincubation of cells with MCP-1 peptide reduced KLAK-MCP-1 micelle binding to B16F10, PC3, and 22Rv1 cancer cells compared to cells incubated with only KLAK-MCP-1 micelles, demonstrating the specificity of KLAK-MCP-1 micelle binding to CCR2. Conversely, KLAK-MCP-1 micelles were unable to bind to NCI-H460 cancer cells, which do not express CCR2, further confirming the dependence of CCR2 expression for KLAK-MCP-1 micelle binding (Fig. 2D) [40].
Figure 2:
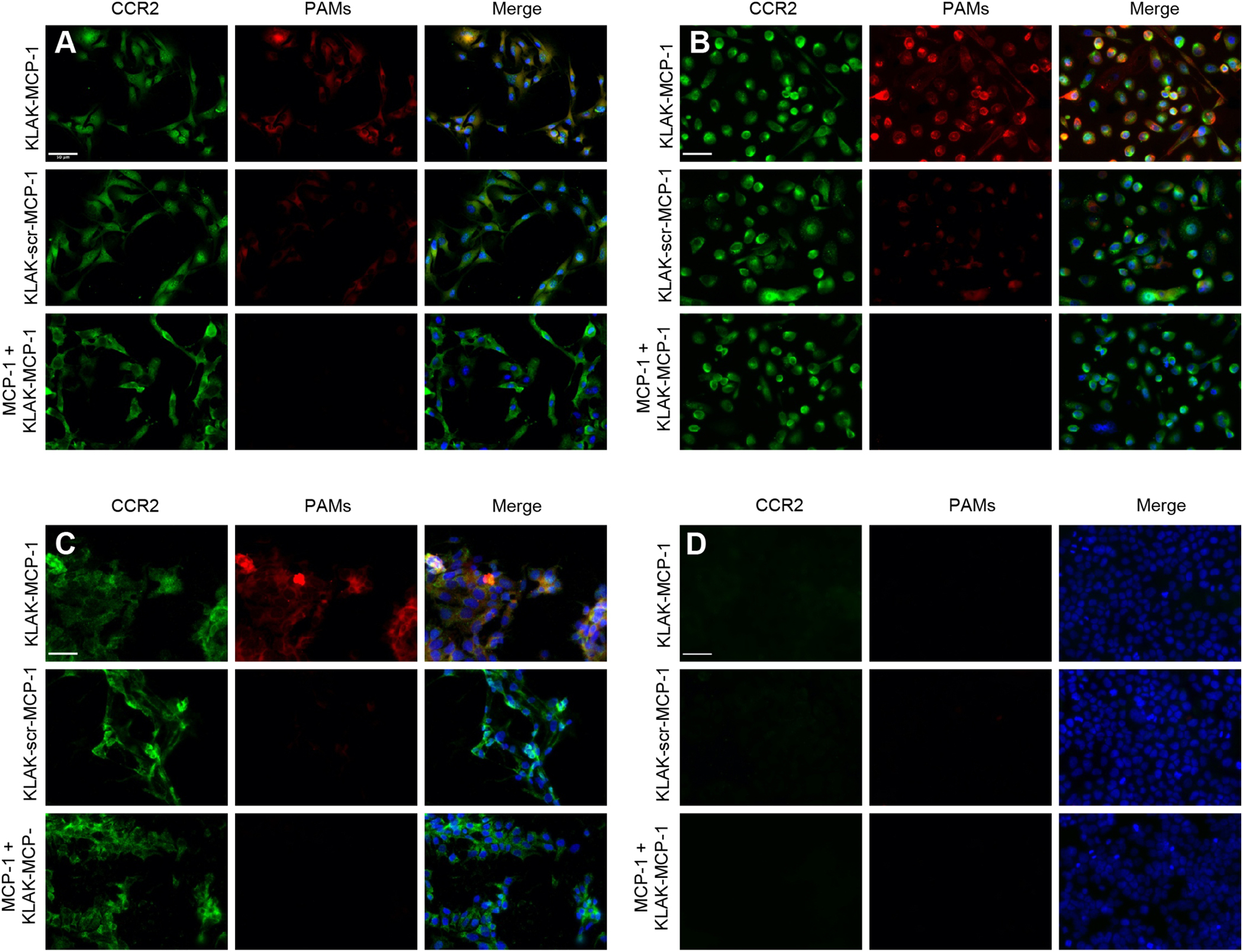
In vitro binding of micelles (red) to cancer cells. Fluorescence microscopy images show binding of B16F10 (A), PC3 (B), 22Rv1 (C), and NCI-H460 (D) cells after 4 hour treatment with KLAK-MCP-1 micelles (top), KLAK-scr-MCP-1 micelles (middle) or KLAK-MCP-1 micelles (11.25 μM) after pre-incubation with 250 μM MCP-1 peptides for 1 hour (bottom). Scale bar = 50 μm.
3.3. KLAK-MCP-1 micelles modulate CCR2 and MCP-1 mRNA expression
To assess the effects of micelle treatment on CCR2 and MCP-1 mRNA expression, qRT-PCR was performed using B16F10 cells treated with non-toxic concentrations of KLAK-MCP-1, KLAK-scr-MCP-1, KLAK, MCP-1, or scr-MCP-1 micelles. As shown in Fig. S6A, both KLAK-MCP-1 micelle and MCP-1 micelle treatment reduced CCR2 mRNA expression by 58 ± 19% and 41 ± 20% of the PBS control, respectively (p < 0.005 and p < 0.05), demonstrating that micelle treatment can downregulate CCR2 expression in addition to antagonizing its function. Additionally, KLAK-scr-MCP-1 micelle and KLAK micelle treatment also reduced CCR2 mRNA expression by 47 ± 19% (p < 0.01) and 42 ± 22% (NS), respectively, indicating that the KLAK peptide may also alter CCR2 expression. The downregulation of CCR2 expression resulting from micelle treatment at non-toxic concentrations demonstrates that KLAK-MCP-1 micelles may be capable of inducing therapeutic efficacy without directly exerting toxicity to cancer cells. Additional qRT-PCR analysis of MCP-1 mRNA expression was performed, although treatment with any micelles yielded no significant change compared to the PBS control (Fig. 6B).
Figure 6:
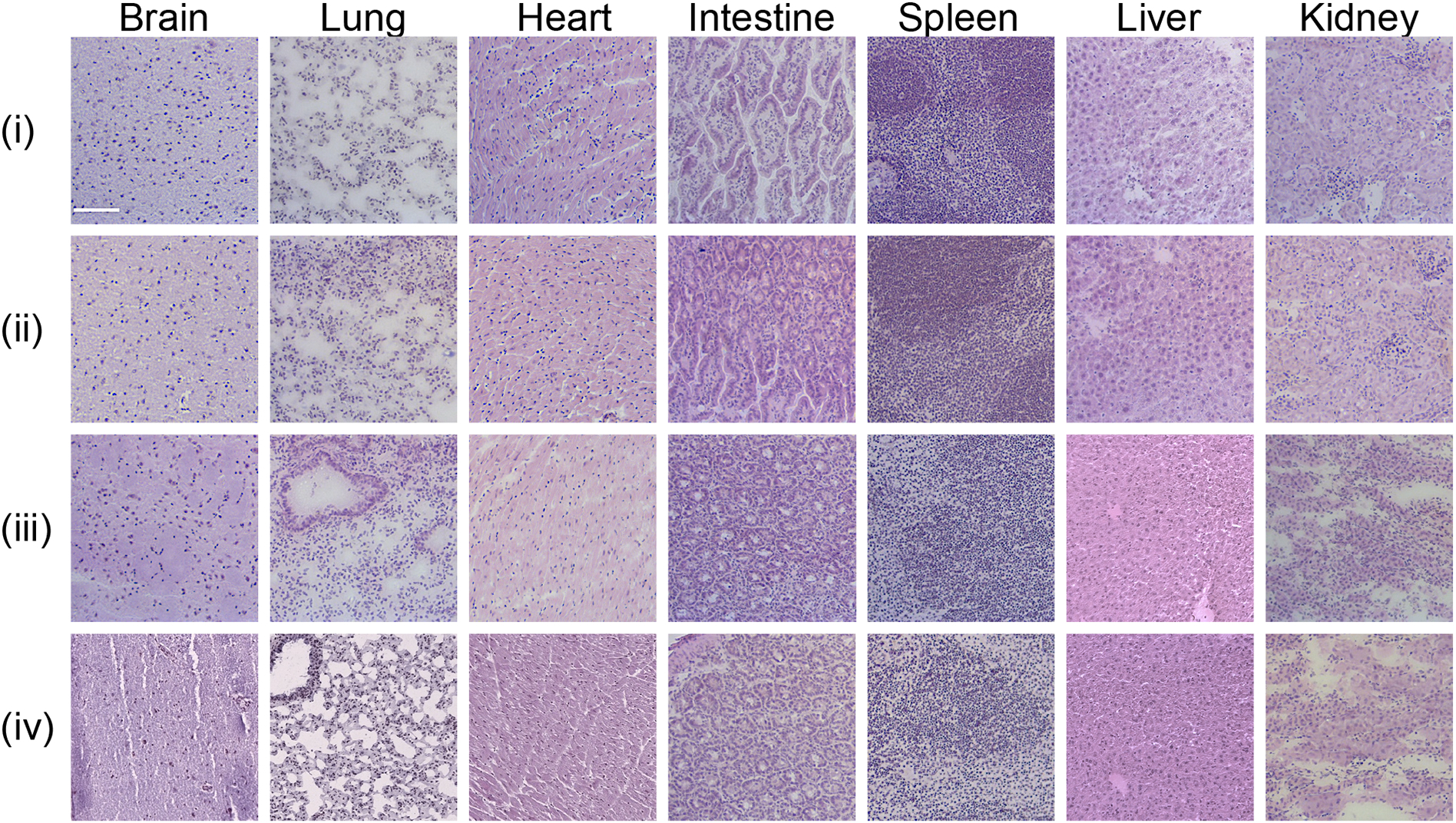
Evaluation of micelle biocompatibility through H&E staining of organs from mice treated with (i) PBS, (ii) MCP-1 micelles, (iii) KLAK-MCP-1 micelles, or (iv) KLAK-scr-MCP-1 micelles. Scale bar = 100 μm.
3.4. In vitro cytotoxicity of KLAK-MCP-1 micelles to cancer cells is dependent on CCR2 expression
An ELISA was used to quantify CCR2 expression in B16F10, PC3, and 22Rv1 cells and correlate their reported metastatic potential and CCR2 levels (Table S1) [1–5]. B16F10 and PC3 are highly aggressive cell lines that readily metastasize in vivo, while 22Rv1 has been characterized as non-metastatic [41–43]. As shown in Table S1, B16F10 had the highest CCR2 expression (408.6 pg/mL), followed by PC3 (266.4 pg/mL), while 22Rv1 had the lowest (190.7 pg/mL), agreeing with clinical reports of a correlation between high CCR2 expression and disease progression [1–6]. The negative control, NCI-H460, which is a lung cancer cell line that is CCR2-, showed no detectable levels of CCR2 through ELISA (Table S1).
To determine if KLAK-MCP-1 micelle efficacy against cancer cells is dependent on the level of CCR2 expression, micelles were incubated with B16F10, PC3, 22Rv1, and NCI-H460 cells, and cytotoxicity was evaluated using an MTS cell proliferation assay. As shown in Fig. 3 and Table 2, KLAK-MCP-1 micelle treatment was most effective against B16F10 (IC50 = 1.4 ± 0.3 μM, p < 0.005 compared to PC3 or 22Rv1), which had the highest CCR2 expression, demonstrating that KLAK-MCP-1 efficacy increases with CCR2 expression. Additionally, PC3, which had higher CCR2 expression than 22Rv1 (266.4 ± 21.7 pg/mL vs. 190.7 ± 15.7 pg/mL, p<0.01), was observed to be more responsive to KLAK-MCP-1 micelle treatment, as shown in Table 2 (IC50 = 18.0 ± 1.4 μM vs. 31.3 ± 11.0 μM, p < 0.05). Since PC3 is often used as a model for late-stage, metastatic, androgen-independent prostate cancer, a type of cancer associated with a 5-year survival rate of just 29%, our results suggest that CCR2-targeted therapies may be an effective option for treating late-stage prostate cancers [42, 44, 45].
Figure 3:
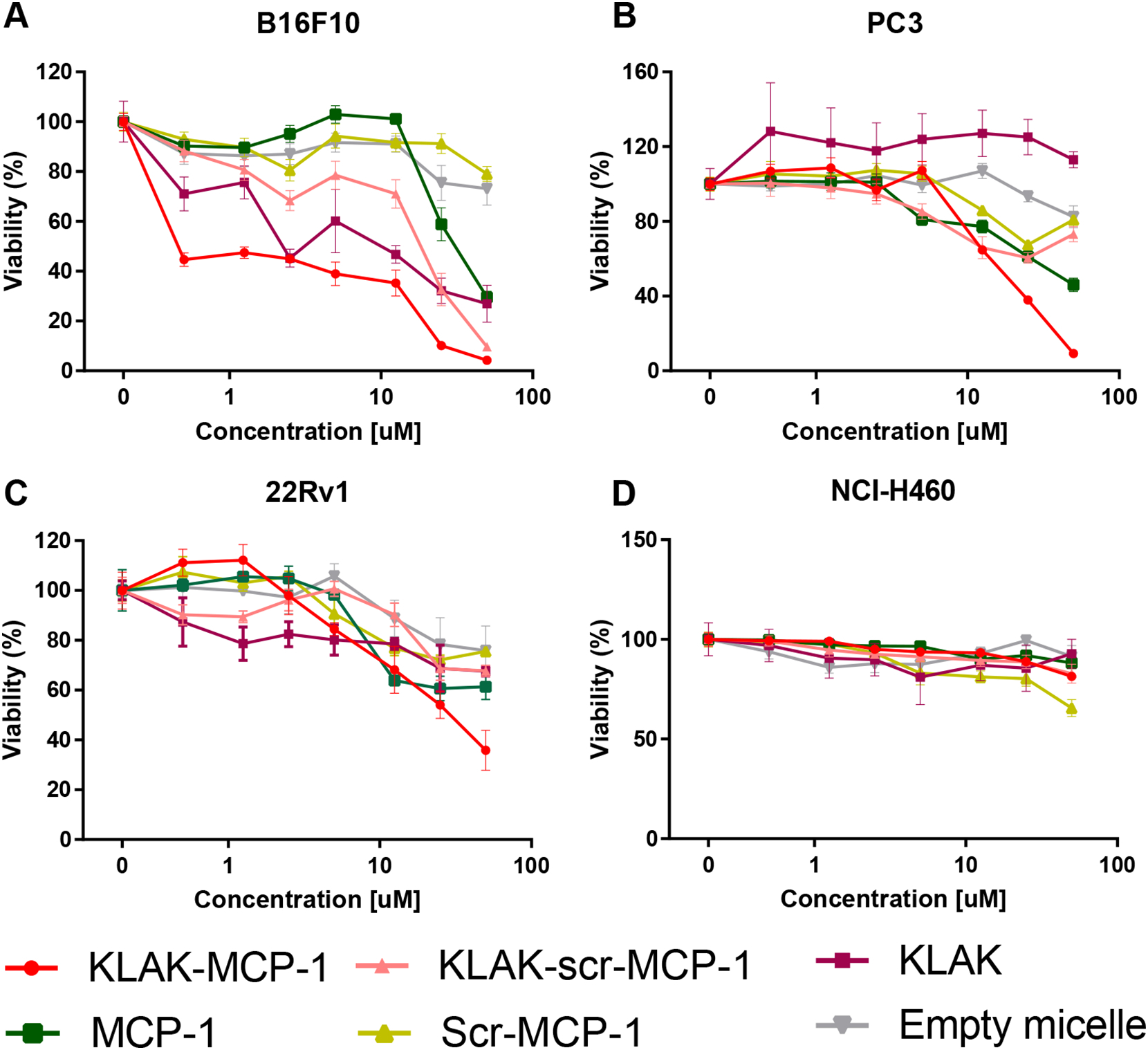
In vitro cytotoxicity of micelles (0.5 – 50 μM) against B16F10 (A), PC3 (B), 22Rv1 (C), and NCI-H460 (D) cells upon incubation for 72 hours (n = 6).
a p < 0.005 compared to KLAK-MCP-1 IC50 in PC3 and 22Rv1
b p < 0.0001 compared to KLAK-scr-MCP-1 and MCP-1 in B16F10
Table 2.
IC50 values of micelles (n = 6).
|
|
IC50 [μM] |
||||
|---|---|---|---|---|---|
| Micelle | Bl6F 10 | P1C3 | 22Rvl | NCI-H460 | RWPE-1 |
|
| |||||
| KLAK-MCP-1 | 1.4 ± 0.3a,b | 18.0 ± 1.4 | 31.3 ± 11.0 | >50 | >50 |
| KLAK-scr-MCP1 | 17.8 ± 2.2 | >50 | >50 | >50 | >50 |
| KLAK | 7.0 ± 2.7 | >50 | >50 | >50 | >50 |
| MCP-1 | 31.9 ± 7.0 | 47.5 ± 12.4 | >50 | >50 | >50 |
| Scr-MCP-1 | >50 | >50 | >50 | >50 | >50 |
| Empty | >50 | >50 | >50 | >50 | >50 |
p < 0.005 compared to KLAK-MCP-1 IC50 in PC3 and 22Rvl.
p < 0.0001 compared to KLAK-scr-MCP-1 and MCP-1 in Bl6Fl0.
Additionally, KLAK-MCP-1 micelles were observed to be more cytotoxic than KLAK-scr-MCP-1 micelle, KLAK micelle, MCP-1 micelle, scr-MCP-1 micelle, and empty micelle controls. For example, in B16F10, KLAK-MCP-1 micelles had significantly lower IC50 values compared to KLAK-scr-MCP-1 micelles (1.4 ± 0.3 μM vs. 17.8 ± 2.2 μM, p < 0.0001) and MCP-1 micelles (1.4 ± 0.3 μM vs. 31.9 ± 7.0 μM, p < 0.0001). The observed synergy between KLAK and MCP-1 may be explained through enhanced delivery of the KLAK peptide through interaction of the MCP-1 peptide with CCR2 present on the cancer cells, as the in vitro binding studies showed KLAK-MCP-1 micelles as having 2.35x more binding compared to KLAK-scr-MCP-1 micelles (Fig. 2).
CCR2 expression and micelle toxicity was also evaluated in the CCR2− NCI-H460 lung cancer cell line, as well as the RWPE-1 human prostate epithelial cell line. As shown in Table S1, neither NCI-H460 nor RWPE-1 cell lines had detectable levels of CCR2. No micelles showed any efficacy (IC50 > 50 μM) against either cell line at the maximum tested concentration (50 μM, Fig. 3D, Fig. S4). The lack of toxicity against these cell lines indicates CCR2 expression is necessary to achieve therapeutic efficacy.
3.5. In vivo efficacy of KLAK-MCP-1 micelles in a subcutaneous B16F10 melanoma model
After testing the in vitro efficacy of KLAK-MCP-1 micelles, we then evaluated their ability to inhibit tumor growth in a subcutaneous B16F10 murine melanoma model in C57BL/6J mice. The B16F10 model was chosen as it is a well-studied, metastatic melanoma model that expresses CCR2 to facilitate KLAK-MCP-1 efficacy (Tables 2, Fig. S1). After 24 hours following tumor inoculation, mice were treated with PBS or MCP-1, KLAK-MCP-1, or KLAK-scr-MCP-1 micelles via tail-vein injection. Mice were euthanized two weeks after tumor inoculation and tumors were excised. As shown in Fig. 4A, mice treated with KLAK-MCP-1 micelles were observed to have smaller tumor volumes compared to PBS-treated control mice (353 ± 181 mm3 vs. 538 ± 272 mm3, p < 0.05). Additionally, mice treated with MCP-1 micelles showed a modest decrease (17%) in tumor volume (447 ± 214 mm3 vs. 538 ± 272 mm3, NS) compared to the PBS control. Similar to the in vitro toxicity studies, the combination of KLAK and MCP-1 proved to be more efficacious than MCP-1 treatment alone.
Figure 4:
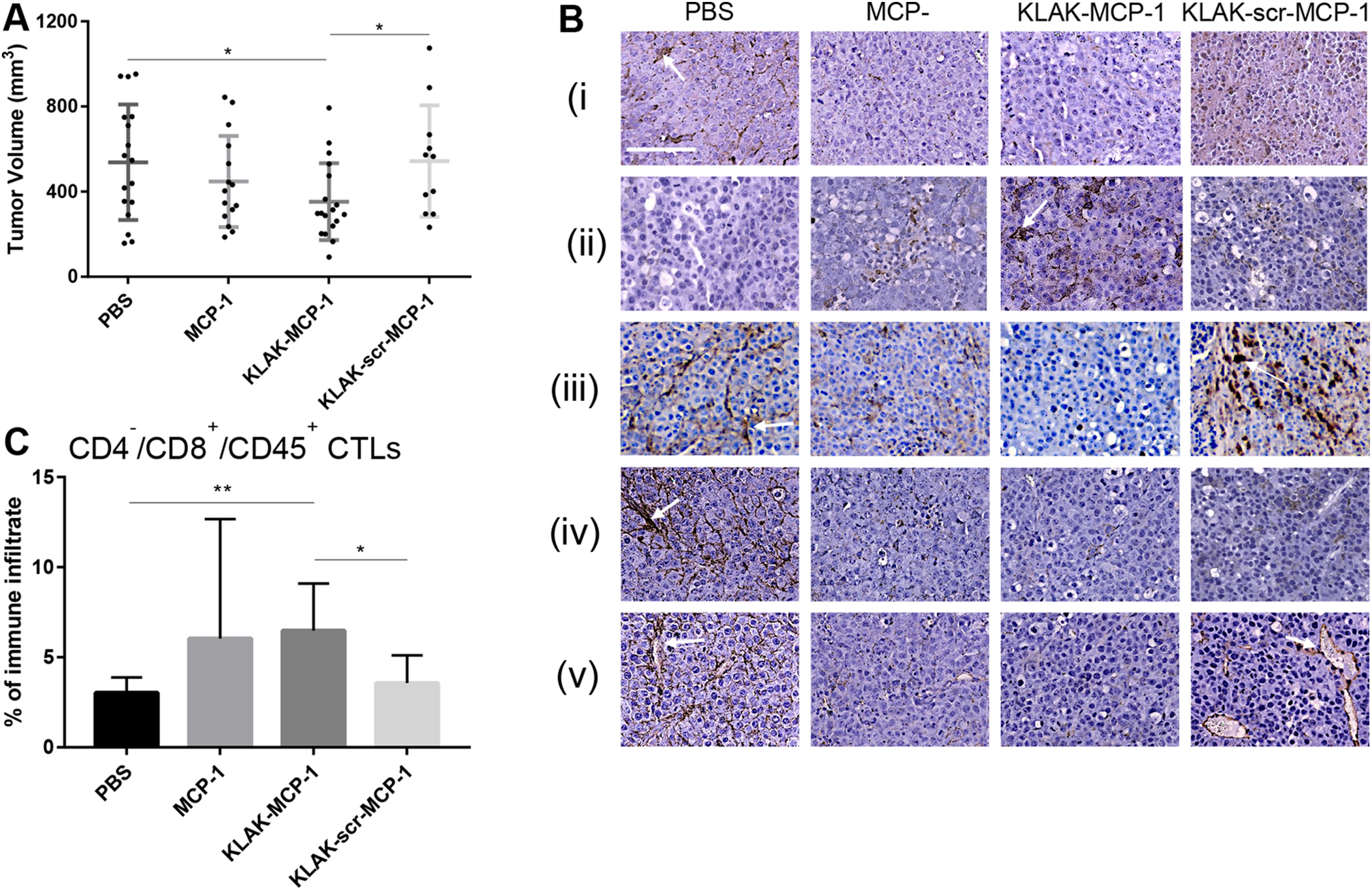
In vivo efficacy of micelles (n = 11 to 18). Tumor volume measurements of mice treated with PBS, MCP-1, KLAK-MCP-1, or KLAK-scr-MCP-1 micelles (A). IHC staining of tumors treated with PBS, MCP-1, KLAK-MCP-1, or KLAK-scr-MCP-1 for (i) CCR2, (ii) cleaved caspase-3, (iii) F4/80 (iv) PDL1, or (v) CD31 (B). Quantification of infiltrating cytotoxic T lymphocytes (CTLs) (C). *p < 0.05, **p < 0.01. Scale bar = 100 μm.
3.6. Immunohistochemical (IHC) analysis of tumor tissue sections
In order to elucidate the mechanisms behind the observed tumor inhibition, we performed IHC analysis of tumor tissue sections (Fig. 4B). As our in vitro mRNA expression studies showed that KLAK-MCP-1 micelle treatment was able to reduce CCR2 in B16F10 cells (Fig. S4A), we performed IHC staining of tumors for CCR2 to evaluate if KLAK-MCP-1 micelle treatment also decreased intratumor CCR2 expression in a similar manner. As shown in Fig. 4B(i), KLAK-MCP-1 and MCP-1 micelle groups had less CCR2 expression than PBS control tumors, demonstrating the ability of KLAK-MCP-1 and MCP-1 micelles to reduce intratumor CCR2 expression. Furthermore, we observed that smaller tumors (KLAK-MCP-1 and MCP-1 micelle groups) had less CCR2 expression, in agreement with clinical studies reporting the correlation between CCR2 expression and disease progression [1–6]. As intratumor CCR2 expression and signaling has been reported to downregulate apoptotic signaling, we performed IHC staining for cleaved caspase-3 to assess intratumor apoptosis [7–12]. In agreement with these reports, tumors from KLAK-MCP-1 and MCP-1 micelle groups, which had reduced CCR2 expression relative to the PBS control, were observed to have increased cleaved caspase-3 expression, indicating higher levels of tumor apoptosis (Fig. 4B(ii)).
Since our in vitro mRNA expression studies also indicated that KLAK-MCP-1 micelle treatment may reduce cancer cell production of MCP-1, we performed IHC staining for the pan-macrophage marker F4/80 to evaluate the effect of micelle treatment on MCP-1 driven TAM infiltration into the tumor. As shown in Fig. 4B(iii), we observed reduced staining in KLAK-MCP-1 and MCP-1 micelle groups compared to the PBS control. As TAM infiltration is known to mediate cancer progression through mechanisms such as immunosuppression and angiogenesis, we also evaluated expression of programmed death ligand 1 (PDL1), a major immune checkpoint molecule expressed by TAMs that contributes to an immunosuppressive microenvironment [20, 46], as well as for CD31, a protein expressed on the surface of endothelial cells that is indicative of angiogenesis [15, 47]. As shown in Fig. 4B(iv), tumors from the KLAK-MCP-1 and MCP-1 micelle groups were observed to have less PDL1 staining than the PBS control, indicating a less immunosuppressive tumor microenvironment, which correlates with the reduction in F4/80 staining in these tumors. The decreased PDL1 expression may lead to a stronger anti-tumor cytotoxic T lymphocyte (CTL) response and hence reduced tumor growth. As shown in Fig. 4B(v), IHC staining for CD31 showed a similar trend to PDL1, in which KLAK-MCP-1 and MCP-1 micelle groups exhibited reduced staining compared to the PBS control, indicating the presence of fewer blood vessels in these treatment groups to fuel tumor growth. IHC analysis of PDL1 and CD31 reveal an attenuation in tumor-promoting immunosuppression and angiogenesis in tumors treated with KLAK-MCP-1 or MCP-1 micelles.
3.7. Flow cytometric analysis of micelle-treated tumors
Since reduced IHC staining of PDL1 in KLAK-MCP-1 and MCP-1 micelle groups suggested an amelioration in immunosuppression, we hypothesized that micelle treatment may also induce a concomitant recovery in CTL activity, as numerous studies have reported greater CTL infiltration upon PDL1-depletion [48–50]. We analyzed tumor infiltration of CD4−/CD8+/CD45+ CTLs through flow cytometry. As shown in Fig. 4C, KLAK-MCP-1 micelle treatment was observed to increase CTL infiltration by approximately two-fold relative to the PBS control (6.5 ± 2.6% vs. 3.0 ± 0.9%, p < 0.05). Additionally, CTL infiltration in the KLAK-MCP-1 group was greater than that of the KLAK-scr-MCP-1 group (6.5 ± 2.6% vs. 3.6 ± 1.6%, p < 0.05), demonstrating the importance of the MCP-1 peptide in modulating the immune response. As CTLs are a major component of the anti-cancer immune response, the increased CTL activity observed in KLAK-MCP-1-treated tumors may have contributed to the tumor inhibition seen in this group.
3.8. In vivo biodistribution of micelles
To assess the biodistribution of micelles, cy7-labeled MCP-1, KLAK-MCP-1, and KLAK-scr-MCP-1 micelles were intravenously injected in the subcutaneous B16F10 melanoma model two weeks after tumor inoculation and evaluated 3 hours post-injection (Fig. 5). Although we hypothesized that interaction with the CCR2-expressing tumors would facilitate tumor accumulation of KLAK-MCP-1 micelles, ex vivo imaging showed minimal tumor retention (2.5 ± 0.1%). As shown in Fig. 5, KLAK-MCP-1 micelles mostly accumulated in the liver (35.0 ± 1.0%) and kidneys (24.1 ± 0.7%), which is in agreement with previous micelle studies [27–29]. MCP-1, KLAK-MCP-1, and KLAK-scr-MCP-1 micelles had similar biodistribution profiles, with the exception of lymph node accumulation (Fig. 5A). MCP-1 micelles were found to have higher lymph node accumulation compared to the KLAK-scr-MCP-1 micelles (21.2 ± 4.6% vs. 10.6 ± 5.8, p < 0.05). KLAK-MCP-1 micelles also showed enhanced lymph node accumulation compared to KLAK-scr-MCP-1 (13.4 ± 2.3% vs. 10.6 ± 5.8%, NS). Increased lymph node accumulation and therapeutic efficacy despite low tumor retention may indicate that KLAK-MCP-1 micelles induced a therapeutic response by modulating the anticancer immune response, rather than directly exerting toxicity to the tumor, a phenomenon that has been observed in other cancer studies [51–53]. For example, Korangath et al. demonstrated nanoferrite particles inhibited breast cancer growth in immunocompetent mice with low tumor retention. Furthermore, their studies showed an increase in CTL infiltration into the tumor, demonstrating the ability of nanoparticles to induce therapeutic efficacy by eliciting a systemic immune response.
Figure 5:
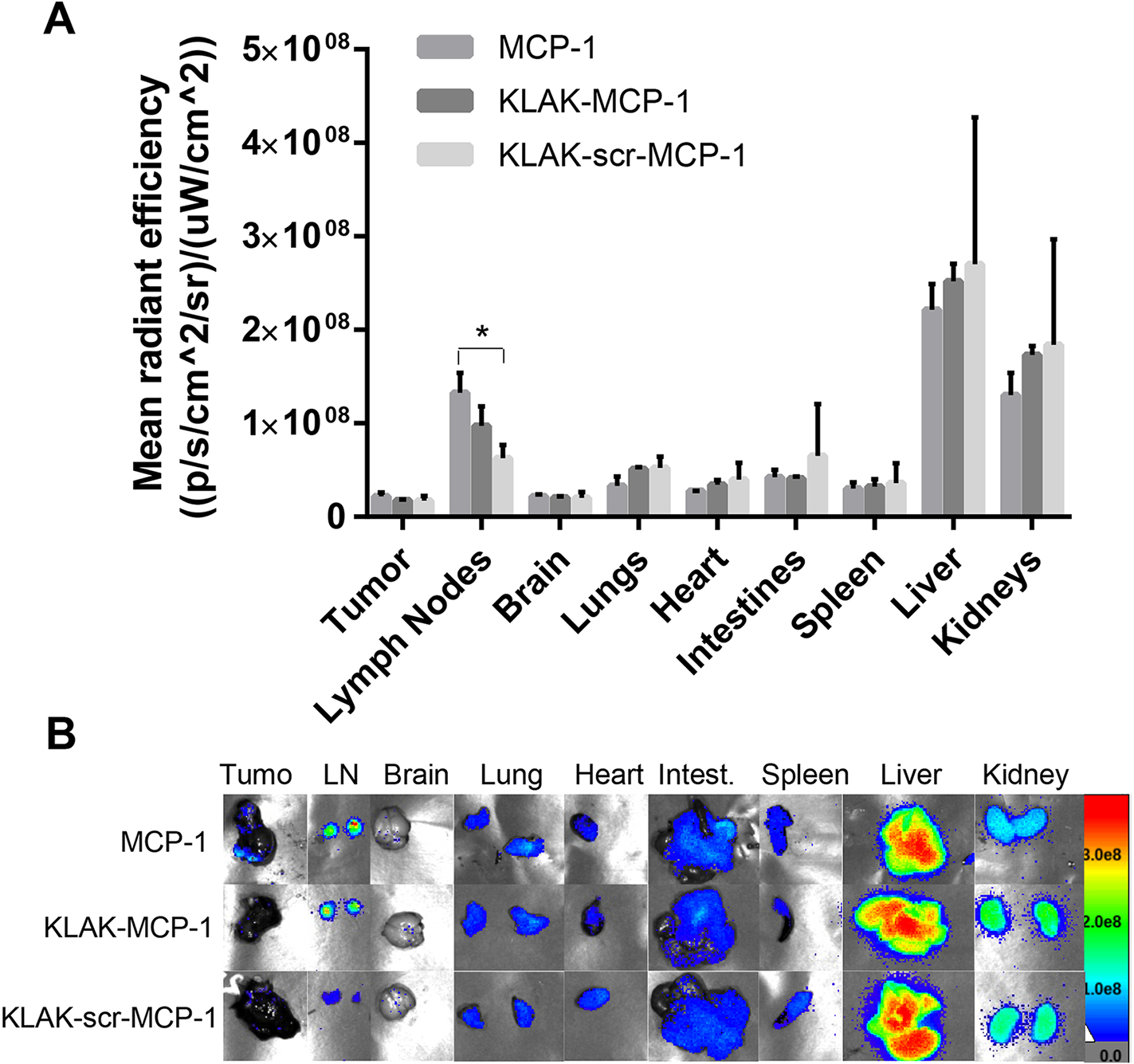
In vivo biodistribution of 500 μM cy7-labeled micelles 3 hours after intravenous injection. (n = 3, *p < 0.05).
Similarly, our flow cytometry and IHC studies demonstrated an ability of KLAK-MCP-1 micelles to induce tumor CTL infiltration (Fig.4C) and reduce tumor macrophage infiltration, supporting KLAK-MCP-1 micelles induce an immunomodulatory effect that can lead to therapeutic outcomes (Fig. 4B(iii)). In addition, KLAK-MCP-1 micelles may bind and induce cytotoxicity to CCR2+ circulating monocytes, inhibiting their recruitment to the tumor immune microenvironment. In vitro, KLAK-MCP-1 micelles were found to bind and exert cytotoxicity against murine monocytes (WEHI-274.1) and had an IC50 value of 5.5 ± 0.3 μM, which suggests monocytes are vulnerable to KLAK-MCP-1 micelle treatment (Fig. S4). These preliminary results show that KLAK-MCP-1 micelle treatment is capable of affecting monocyte populations in vitro, although additional studies will need to be performed in the future to probe its effects on circulating monocytes in vivo.
3.9. Biocompatibility of micelles in vivo
To assess any off-target toxicity upon KLAK-MCP-1 micelle treatment, biocompatibility was evaluated through histological analysis, as well as evaluation of serum chemistry markers pertinent to liver and kidney health. Upon H&E staining of the liver, kidneys, spleen, lungs, heart, intestines, and brain, no signs of morphological damage or differences in tissue morphology compared to the PBS control group were observed (Fig. 6).
As micelle accumulation was highest in the liver and kidneys, liver function was evaluated by assessing serum activity of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) [54, 55] and kidney health assessed via serum levels of blood urea nitrogen (BUN) and creatinine [56, 57]. As shown in Table S2, no statistical differences were observed between treatment groups in any of the biomarkers, indicating no adverse effects upon micelle treatment.
Conclusion
In this study, we designed and synthesized KLAK-MCP-1 micelles to evaluate the potential of CCR2-targeted nanotherapies for the treatment of CCR2+ cancers which are clinically associated with unfavorable outcomes. Upon incubation with cancer cells in vitro, the KLAK-MCP-1 micelle was found to bind and induce cytotoxicity to multiple cancer cell lines in a CCR2-dependent manner. Administration of KLAK-MCP-1 micelles led to reduced tumor growth in a B16F10 melanoma model, and immunohistochemical (IHC) staining showed reduced TAM infiltration into the tumor, as well as reduced expression of markers of immunosuppression and angiogenesis within the tumor. Additionally, flow cytometry analysis of tumors showed KLAK-MCP-1 micelle treatment was able to increase the number of tumor-infiltrating cytotoxic T lymphocytes. Interestingly, biodistribution analyses showed micelle accumulation in the lymph nodes and low tumor retention. This study also corroborated recent reports of achieving therapeutic efficacy through systemic immune responses without the observation of significant nanoparticle accumulation within the tumor. Given the ability of KLAK-MCP-1 micelles to induce a systemic anticancer response, future studies will evaluate their therapeutic application in metastatic models of cancer. Additionally, the ability of the micelles to accumulate in the lymph nodes is an interesting result that merits further study. It is possible that the micelles and their peptide components are being processed by antigen presenting cells and used to generate an adaptive immune response. In summary, our studies demonstrate the application of CCR2-targeted micelles in inhibiting tumor growth by modulating tumor infiltration of immune cell populations.
Supplementary Material
Highlights.
KLAK-MCP-1 micelles combine apoptosis-inducing peptides (KLAK) with CCR2-targeting ligands (MCP-1) for targeted drug delivery to CCR2+ cancer cells.
KLAK-MCP-1 micelles exhibited CCR2-dependent binding and toxicity to cancer cells and monocytes, and had little effect on CCR2− cells in vitro.
KLAK-MCP-1 micelles inhibited tumor growth and modulated the immune response by altering tumor-infiltrating macrophage and cytotoxic T lymphocyte populations.
Acknowledgements
The authors would like to acknowledge the financial support from Women in Science and Engineering (WiSE) Gabilan Assistant Professorship, Ming Hsieh Institute for Research on Engineering-Medicine for Cancer, L. K. Whittier Foundation, the National Heart, Lung, and Blood Institute (NHLBI, R00HL124279), NIH New Innovator Award (DP2-DK121328), and the University of Southern California startup funds awarded to E.J.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang H, et al. , Monocyte chemotactic protein-1 expression as a prognosic biomarker in patients with solid tumor: a meta analysis. Int J Clin Exp Pathol, 2014. 7(7): p. 3876–86. [PMC free article] [PubMed] [Google Scholar]
- 2.Lu Y, et al. , CCR2 expression correlates with prostate cancer progression. J Cell Biochem, 2007. 101(3): p. 676–85. [DOI] [PubMed] [Google Scholar]
- 3.Ueno T, et al. , Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin Cancer Res, 2000. 6(8): p. 3282–9. [PubMed] [Google Scholar]
- 4.Saji H, et al. , Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer, 2001. 92(5): p. 1085–91. [DOI] [PubMed] [Google Scholar]
- 5.Hefler L, et al. , Monocyte chemoattractant protein-1 serum levels in ovarian cancer patients. Br J Cancer, 1999. 81(5): p. 855–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ou B, et al. , A positive feedback loop of beta-catenin/CCR2 axis promotes regorafenib resistance in colorectal cancer. Cell Death Dis, 2019. 10(9): p. 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu Y, et al. , Monocyte chemotactic protein-1 (MCP-1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate, 2006. 66(12): p. 1311–8. [DOI] [PubMed] [Google Scholar]
- 8.Li MQ, et al. , Chemokine CCL2 enhances survival and invasiveness of endometrial stromal cells in an autocrine manner by activating Akt and MAPK/Erk1/2 signal pathway. Fertil Steril, 2012. 97(4): p. 919–29. [DOI] [PubMed] [Google Scholar]
- 9.Fang WB, et al. , CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J Biol Chem, 2012. 287(43): p. 36593–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuper C, Beck FX, and Neuhofer W, Autocrine MCP-1/CCR2 signaling stimulates proliferation and migration of renal carcinoma cells. Oncol Lett, 2016. 12(3): p. 2201–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brummer G, et al. , Chemokine Signaling Facilitates Early-Stage Breast Cancer Survival and Invasion through Fibroblast-Dependent Mechanisms. Mol Cancer Res, 2018. 16(2): p. 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Natsagdorj A, et al. , CCL2 induces resistance to the antiproliferative effect of cabazitaxel in prostate cancer cells. Cancer Sci, 2019. 110(1): p. 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franklin RA, et al. , The cellular and molecular origin of tumor-associated macrophages. Science, 2014. 344(6186): p. 921–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Movahedi K, et al. , Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res, 2010. 70(14): p. 5728–39. [DOI] [PubMed] [Google Scholar]
- 15.Riabov V, et al. , Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol, 2014. 5: p. 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim SY, et al. , Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget, 2016. 7(19): p. 28697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Q, et al. , New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy. J Immunol Res, 2016. 2016: p. 9720912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noy R and Pollard JW, Tumor-associated macrophages: from mechanisms to therapy. Immunity, 2014. 41(1): p. 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin Y, Xu J, and Lan H, Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol, 2019. 12(1): p. 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartley GP, et al. , Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol Res, 2018. 6(10): p. 1260–1273. [DOI] [PubMed] [Google Scholar]
- 21.Sandhu SK, et al. , A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol, 2013. 71(4): p. 1041–50. [DOI] [PubMed] [Google Scholar]
- 22.Vela M, et al. , Chemokine receptor-specific antibodies in cancer immunotherapy: achievements and challenges. Front Immunol, 2015. 6: p. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brana I, et al. , Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol, 2015. 10(1): p. 111–23. [DOI] [PubMed] [Google Scholar]
- 24.Noel M, et al. , Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs, 2020. 38(3): p. 800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung EJ, et al. , Monocyte-targeting supramolecular micellar assemblies: a molecular diagnostic tool for atherosclerosis. Adv Healthc Mater, 2015. 4(3): p. 367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poon C, et al. , Protein Mimetic and Anticancer Properties of Monocyte-Targeting Peptide Amphiphile Micelles. ACS Biomater Sci Eng, 2017. 3(12): p. 3273–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joo J, et al. , Shape Effects of Peptide Amphiphile Micelles for Targeting Monocytes. Molecules, 2018. 23(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung EJ, et al. , Fibrin-binding, peptide amphiphile micelles for targeting glioblastoma. Biomaterials, 2014. 35(4): p. 1249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, et al. , Design and in vivo characterization of kidney-targeting multimodal micelles for renal drug delivery. Nano Research, 2018. 11(10): p. 5584–5595. [Google Scholar]
- 30.Chin DD, et al. , Hydroxyapatite-binding micelles for the detection of vascular calcification in atherosclerosis. Journal of Materials Chemistry B, 2019. 7(41): p. 6449–6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chin DD, et al. , Collagenase-Cleavable Peptide Amphiphile Micelles as a Novel Theranostic Strategy in Atherosclerosis. Advanced Therapeutics, 2020. 3(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du B, et al. , Glomerular barrier behaves as an atomically precise bandpass filter in a sub-nanometre regime. Nat Nanotechnol, 2017. 12(11): p. 1096–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Longmire M, Choyke PL, and Kobayashi H, Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine (Lond), 2008. 3(5): p. 703–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoshyar N, et al. , The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine (Lond), 2016. 11(6): p. 673–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato Y, et al. , Acidic extracellular microenvironment and cancer. Cancer Cell Int, 2013. 13(1): p. 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen LQ and Pagel MD, Evaluating pH in the Extracellular Tumor Microenvironment Using CEST MRI and Other Imaging Methods. Adv Radiol, 2015. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huber V, et al. , Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol, 2017. 43: p. 74–89. [DOI] [PubMed] [Google Scholar]
- 38.Marsich L, et al. , Poly-L-lysine-Coated Silver Nanoparticles as Positively Charged Substrates for Surface-Enhanced Raman Scattering. Langmuir, 2012. 28(37): p. 13166–13171. [DOI] [PubMed] [Google Scholar]
- 39.Sun MS, et al. , Surface density of polyarginine influence the size, zeta potential, cellular uptake and tissue distribution of the nanostructured lipid carrier. Drug Delivery, 2017. 24(1): p. 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An J, et al. , Targeting CCR2 with its antagonist suppresses viability, motility and invasion by downregulating MMP-9 expression in non-small cell lung cancer cells. Oncotarget, 2017. 8(24): p. 39230–39240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burke AJ, et al. , Sensitivity Profiles of Human Prostate Cancer Cell Lines to an 80 Kinase Inhibitor Panel. Anticancer Res, 2016. 36(2): p. 633–41. [PubMed] [Google Scholar]
- 42.Laniado ME, et al. , Expression and functional analysis of voltage-activated Na+ channels in human prostate cancer cell lines and their contribution to invasion in vitro. Am J Pathol, 1997. 150(4): p. 1213–21. [PMC free article] [PubMed] [Google Scholar]
- 43.Overwijk WW and Restifo NP, B16 as a mouse model for human melanoma. Curr Protoc Immunol, 2001. Chapter 20: p. Unit 20 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moreira DM, et al. , Predicting Time From Metastasis to Overall Survival in Castration-Resistant Prostate Cancer: Results From SEARCH. Clin Genitourin Cancer, 2017. 15(1): p. 60–66 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Damodaran S, Kyriakopoulos CE, and Jarrard DF, Newly Diagnosed Metastatic Prostate Cancer: Has the Paradigm Changed? Urol Clin North Am, 2017. 44(4): p. 611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sumitomo R, et al. , PD-L1 expression on tumor-infiltrating immune cells is highly associated with M2 TAM and aggressive malignant potential in patients with resected non-small cell lung cancer. Lung Cancer, 2019. 136: p. 136–144. [DOI] [PubMed] [Google Scholar]
- 47.Lee C, et al. , Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J Immunother Cancer, 2019. 7(1): p. 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Juneja VR, et al. , PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med, 2017. 214(4): p. 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu-Monette ZY, et al. , PD-1/PD-L1 Blockade: Have We Found the Key to Unleash the Antitumor Immune Response? Front Immunol, 2017. 8: p. 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beyrend G, et al. , PD-L1 blockade engages tumor-infiltrating lymphocytes to co-express targetable activating and inhibitory receptors. J Immunother Cancer, 2019. 7(1): p. 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korangath P, et al. , Nanoparticle interactions with immune cells dominate tumor retention and induce T cell-mediated tumor suppression in models of breast cancer. Sci Adv, 2020. 6(13): p. eaay1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phung CD, et al. , Reprogramming the T cell response to cancer by simultaneous, nanoparticle-mediated PD-L1 inhibition and immunogenic cell death. J Control Release, 2019. 315: p. 126–138. [DOI] [PubMed] [Google Scholar]
- 53.Huang Y, et al. , Enzyme responsiveness enhances the specificity and effectiveness of nanoparticles for the treatment of B16F10 melanoma. J Control Release, 2019. 316: p. 208–222. [DOI] [PubMed] [Google Scholar]
- 54.Liu T, et al. , Molecular serum markers of liver fibrosis. Biomark Insights, 2012. 7: p. 105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McGill MR, The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J, 2016. 15: p. 817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keppler A, et al. , Plasma creatinine determination in mice and rats: an enzymatic method compares favorably with a high-performance liquid chromatography assay. Kidney Int, 2007. 71(1): p. 74–8. [DOI] [PubMed] [Google Scholar]
- 57.Rodrigues WF, et al. , Establishing standards for studying renal function in mice through measurements of body size-adjusted creatinine and urea levels. Biomed Res Int, 2014. 2014: p. 872827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.