

Summary.
Background:
There are two basic carboxypeptidases in plasma. Carboxypeptidase B2 (CPB2) is activated from a circulating zymogen, proCPB2, and carboxypeptidase N (CPN) is constitutively active with both inactivating complement C3a and C5a.
Aims:
To test the roles of CPB2 and CPN in complement-driven mouse models of cobra venom factor (CVF) challenge and hemolytic-uremic syndrome (HUS).
Methods:
Cpb2−/−, Cpn−/− and wild-type (WT) mice were compared in an HUS model induced by Shiga toxin and lipopolysaccharide administration and following CVF administration.
Results:
HUS was exacerbated in Cpb2−/− mice more than in Cpn−/− mice, compared with WT mice. Cpb2−/− mice developed the HUS clinical triad of microangiopathic hemolytic anemia, uremia and thrombocytopenia. Treatment with anti-C5 antibody improved survival of both Cpb2−/− and Cpn−/− mice. In contrast, when challenged acutely with CVF, the reverse phenotype was observed. Cpn−/− mice had markedly worse disease than Cpb2−/− mice, whereas the WT mice were resistant.
Conclusions:
CPN and CPB2 play overlapping but non-redundant roles in regulating complement activation in vivo. The constitutively active CPN is key for inactivation of systemic C5a, whereas CPB2 functions as an on-demand supplementary anaphylatoxin inhibitor in inactivating excessive C5a formed locally.
Keywords: carboxypeptidase B2, carboxypeptidase N, cobra venom factor, complement activation, hemolytic-uremic syndrome
Introduction
Two basic carboxypeptidases are present in plasma, carboxypeptidase N (CPN), which is constitutively active, and procarboxypeptidase B2 (proCPB2, also known as thrombin-activatable fibrinolysis inhibitor, TAFI), a plasma proenzyme activated by the thrombin/thrombomodulin (TM) complex or plasmin to carboxypeptidase B2 (CPB2). CPB2 removes C-terminal lysine residues from fibrin, leading to reduced incorporation of plasminogen and tissue plasminogen activator into the partially digested clot, resulting in inhibition of fibrinolysis as well as reduction of prourokinase-mediated activation of plasminogen [1,2]. Both enzymes remove C-terminal arginine residues from bradykinin, complement C3a and C5a, thereby inactivating them [1,3–5]. Although CPN is a constitutively active inhibitor of anaphylatoxins [6], activation of proCPB2 by the thrombin/TM complex serves as a homeostatic negative feedback mechanism to dampen adverse effects of excessively generated pro-inflammatory mediators at sites of tissue injury [7].
Mice deficient in CPN (Cpn−/−) are killed by intravenous administration of C5a as well as cobra venom factor (CVF) that activates complement, generating C5a, whereas wild-type (WT) mice survive [8]. In studies on Cpb2−/− mice, a similar dose of CVF had no effect on survival [9], suggesting that CPN is responsible for systemic inactivation of the anaphylatoxins.
Studies using the Cpb2-deficient (Cpb2−/−) mouse confirm that the observed in vitro inactivation of complement C3a and C5a by CPB2 also occurs in vivo [10,11]. Cpb2−/− mice developed more severe alveolitis than WT mice upon tracheal instillation of C5a [12]. In a polymicrobial sepsis model, lack of inactivation of C3a by CPB2 in Cpb2−/− mice leads to protection of the mice from the disease [7]. In a C5a-mediated autoimmune arthritis model, Cpb2−/− mice developed much more severe arthritis than WT mice [10]. Furthermore, patients with rheumatoid arthritis (RA) who carry the Cpb2 allele variant encoding isoleucine instead of threonine at position 325, which results in an increased plasma half-life (~16 min vs. ~8 min) of CPB2, have a lower risk of developing severe RA [10]. Thus, CPB2 functions as a major regulator of C3a and C5a activity in vivo.
Hemolytic uremic syndrome (HUS) is primarily caused by enteric infection with Shiga toxin (Stx)-producing enterohemorrhagic Escherichia coli (EHEC) and has the clinical triad of microangiopathic hemolytic anemia, thrombocytopenia and acute renal failure. Retrograde trafficking of Stx from the intestine to glomerular endothelial and epithelial cells in the kidney, where the Stx receptor is highly expressed, leads to extensive cell injury within the microvasculature and post-glomerular filtration system in the kidney. Laboratory tests from HUS patients showed a substantial consumption of complement C3, indicating that complement activation plays a prominent role in the pathogenesis of HUS [13,14]. Eculizumab, a monoclonal anti-C5 antibody, is the first-line therapy for non-Stx-mediated atypical HUS [15,16] and is also beneficial in Stx-mediated HUS when administered early in the course of disease, although a randomized controlled trial has not yet been performed [17,18].
In animal models, HUS has been induced with either live E. coli expressing one of the Stx or by administration of purified Stx co-injected with lipopolysaccharide (LPS). Treatment with Stx induces expression of P-selectin on the luminal endothelial surface, which in turn binds and activates C3 via the alternative pathway generating C3a and C3b [19]. Additionally, Stx2 binds to complement factor H, delaying its binding to cell membranes, and down-regulates CD59 expression on the surface of glomerular endothelial and tubular epithelial cells, leading to enhanced formation of C5 convertase and the membrane attack complex (MAC), and resulting in uncontrolled complement activation [20–22]. Although the MAC plays a role in complement-mediated tissue injury, the anaphylatoxins, C3a and C5a, generated by the complement cascade, play an important role in leukocyte recruitment and activation, which also are significant contributors to the course of disease.
The relative roles of CPN and CPB2 in the control of complement activation in vivo have not been directly compared in a disease-relevant animal model. In this study, we examined the phenotypic response of the Cpb2−/− mice in comparison with the Cpn−/− mice in an Stx/LPS-induced HUS mouse model.
Methods
Mouse husbandry
Cpn−/− mice, generated by deleting the coding regions for CPN1 from the Cpn1 gene, and backcrossed >10 generations onto the C57BL/6 background, were obtained from Deltagen (San Mateo, CA, USA). Cpb2−/− mice, backcrossed > 11 generations onto the C57BL/6J background, have been described previously [23]. WT C57BL/6 mice were from Jackson Laboratory (Sacramento, CA, USA). Cpb2−/−/Cpn−/− double-deficient mice were generated by intercrossing the Cpb2−/− and Cpn−/− mice, and the resultant F1 Cpb2+/−/Cpn+/− mice were then crossed to generate lines of WT, Cpb2−/−, Cpn−/− and Cpb2−/−/Cpn−/− mice. The mice were housed at either Stanford University School of Medicine or at Palo Alto Veterans Affairs Health Care System (VAPAHCS) and experiments were performed under protocols approved by the Stanford University Committee of Animal Research or the VAPAHCS Institutional Animal Care and Use Committee in accordance with NIH guidelines. Animals were randomized to the different groups and analysis was blinded. All experiments were repeated at least twice independently and all mice that entered the experiments were accounted for. The health of animals during experiments was monitored using a seven-component scale ranging from 0 (healthy) to 28 [24].
Mouse model of HUS
We adapted a previously published protocol on an Stx/LPS-induced HUS model in Swiss/129 mice [25]. Twelve-week-old male mice received intraperitoneally (i.p.) 75 μg/kg LPS O111:B4 (Sigma-Aldrich, St. Louis, MO, USA) and 6 μg kg−1 Stx2 (Toxin Technology, Sarasota, FL, USA) dissolved in sterile saline to induce HUS. The endotoxin content of Stx2, determined by the ToxinSensor kit (Genscript, Piscataway, NJ, USA), was < 1700 endotoxin units (EU) μg−1, compared with LPS, which contained 190 000 EU μg−1. Untreated animals received the same dose of vehicle. All mice received twice-weekly subcutaneous (s.c.) 0.1 mg kg−1 buprenorphine SR from HUS induction. Survival was followed until death or mice were sacrificed at 48 h for blood collection. In some experiments, mice received i.p. 0.75 mg anti-C5 antibody BB5.1 produced in tissue culture, purified on protein A/G beads, with < 0.033 EU mg−1 endotoxin, 3 h prior to injection of Stx2 and LPS and subsequently every 24 h [26]. During this model, the health of the mice was monitored with a seven-component scoring system [24] every 6 h, and mice that had reached a score of > 23/28 were euthanized as their death would be imminent based on preliminary studies.
CVF challenge
Twelve-week-old male mice had 300 μg per mouse of CVF administered intraperitoneally (600 units, Comp-Tech, Tyler, TX, USA) and their health was monitored at 30 min, 1, 2 and 3 h after initiation before sacrifice at 3 h. In preliminary experiments all mice that were alive at 3 h were alive the next day. In some experiments, blood was collected by cardiac puncture 7 days before CVF administration and 20 min after.
Laboratory tests
Blood was collected by cardiac puncture into 3.2% sodium citrate in a 9 : 1 ratio in a heparinized Eppendorf tube at 48 h after disease induction and analyzed for levels of alanine transaminase (ALT), aspartate transaminase (AST), blood urea nitrogen (BUN), creatinine, lactate dehydrogenase (LDH), total bilirubin, and complete blood count (CBC). In some experiments, plasma was prepared and levels of C3, C5 and C5b-9 (LifeSpan Biosciences Inc., Seattle, WA, USA), fibrinogen (Innovative Research, Novi, MI, USA), D-dimer and thrombin-antithrombin complexes (TAT) determined by enzyme-linked immunosorbent assays (ELISAs; Kamiya Biomedical Company, Seattle, WA, USA). Levels of plasma CPB2 and CPN activity and protein were measured as previously described [23]. Blood smear slides were stained by the Wright-Giemsa method and examined under a microscope.
Flow cytometry analysis of leukocyte–platelet aggregates
Citrated mouse whole blood was fixed with 1% paraformaldehyde and stained with anti-CD41, Ly-6G, CD11b (BD Bioscience, San Jose, CA, USA) [27]. The peripheral blood neutrophils were defined as CD11bhigh- Ly6Ghigh and monocytes as CD11b+Ly6G−. Events in neutrophil and monocyte gates that were also CD41-positive were leukocyte–platelets aggregates.
Histopathology
Kidney tissue was collected from mice in the HUS model that were either sacrificed at 48 h or immediately ante-mortem as determined by having a health score > 23. Tissues were fixed in 10% neutral buffered formalin and subsequently dehydrated and embedded in paraffin for routine histology. Sections were stained with hematoxylin and eosin (H&E), periodic acid-Schiff or Jones silver stain and evaluated by light microscopy.
Statistics
Data were analyzed with Prism v6 for Mac. Comparisons between two groups were analyzed with two-tailed Student’s t-test and two-way analysis of variance (anova), with post hoc Tukey’s correction used for comparison of three or more groups. Error bars show ± standard error of the mean (SEM). Kaplan–Meier survival curves were analyzed by the log-rank (Mantel-Cox) test and in experiments with more than two groups by multiple two-way comparisons by the log-rank (Mantel-Cox) test, followed by the Bonferroni correction for the number of comparisons. P < 0.05 was considered as statistically significant.
Results
Cpb2−/−, Cpn−/− and Cpb2−/−/Cpn−/− mice
To define the relative roles of the two basic carboxypeptidases in plasma, CPB2 and CPN, we used Cpb2−/−, Cpn−/− and Cpb2−/−/Cpn−/− mice [8,23]. When Cpb2+/−/Cpn+/− mice were crossed, all expected genotypes were recovered in the expected Mendelian ratios, including the double-deficient Cpb2−/−/Cpn−/− mice. Both male and female Cpb2−/−/Cpn−/− mice are fertile and the female mice carry to term with normal-sized litters and have no apparent phenotype before challenge. Thus, the presence of a plasma basic carboxypeptidase is not required for murine life.
Plasma levels of basic carboxypeptidases in Cpb2−/− and Cpn−/− mice
We measured basic carboxypeptidase activity by a chromogenic assay in plasma samples, both with and without activation by the thrombin/TM complex, and showed that Cpb2−/− mice did not have increased CPN activity and Cpn−/− mice did not have increased CPB2 activity, whereas there was no detectable carboxypeptidase activity in the plasma from Cpb2−/−/Cpn−/− double-deficient mice (Table 1).
Table 1.
Plasma basic carboxypeptidase activity
| CPB2 activity (mOD/min) | CPN activity (mOD/min) | |
|---|---|---|
| WT (n = 3) | 0.15 ± 0.06 | 1.60 ± 0.16 |
| Cpb2−/− (n = 3) | 0.02 ± 0.03 | 1.80 ± 0.22 |
| Cpn−/− (n = 3) | 0.17 ± 0.05 | 0.01 ± 0.01 |
| Cpb2−/− / Cpn−/− (n = 5) | 0.04 ± 0.05 | 0.03 ± 0.04 |
Citrated plasma, collected from the four genotypes, was assayed for carboxypeptidase N (CPN) and carboxypeptidase B2 (CPB2) activity. WT, wild type.
Cpb2−/− mice were more susceptible to Stx2 and LPS than WT or Cpn−/− mice
A survival study following the induction of HUS with Stx2 and LPS showed both Cpb2−/− and Cpn−/− mice had exacerbated disease, resulting in earlier death than WT mice (Fig. 1A, B). Cpb2−/− mice had a median survival of 60 h vs. 96 h in WT mice (n = 15 and 42, respectively; P < 0.0001), whereas Cpn−/− mice had a median survival of 81.5 h (n = 28; P = 0.0002). The hazard ratio for CPB2 deficiency is 11.1 compared with WT mice (95% confidence interval [CI], 4.2–29.5) and is 3.33 (95% CI, 1.79–6.2) for CPN deficiency compared with WT mice. Loss of CPB2 leads to worse disease than loss of CPN (P = 0.0083), with a hazard ratio of 3.07 (95% CI, 1.34–7.04).
Fig. 1.
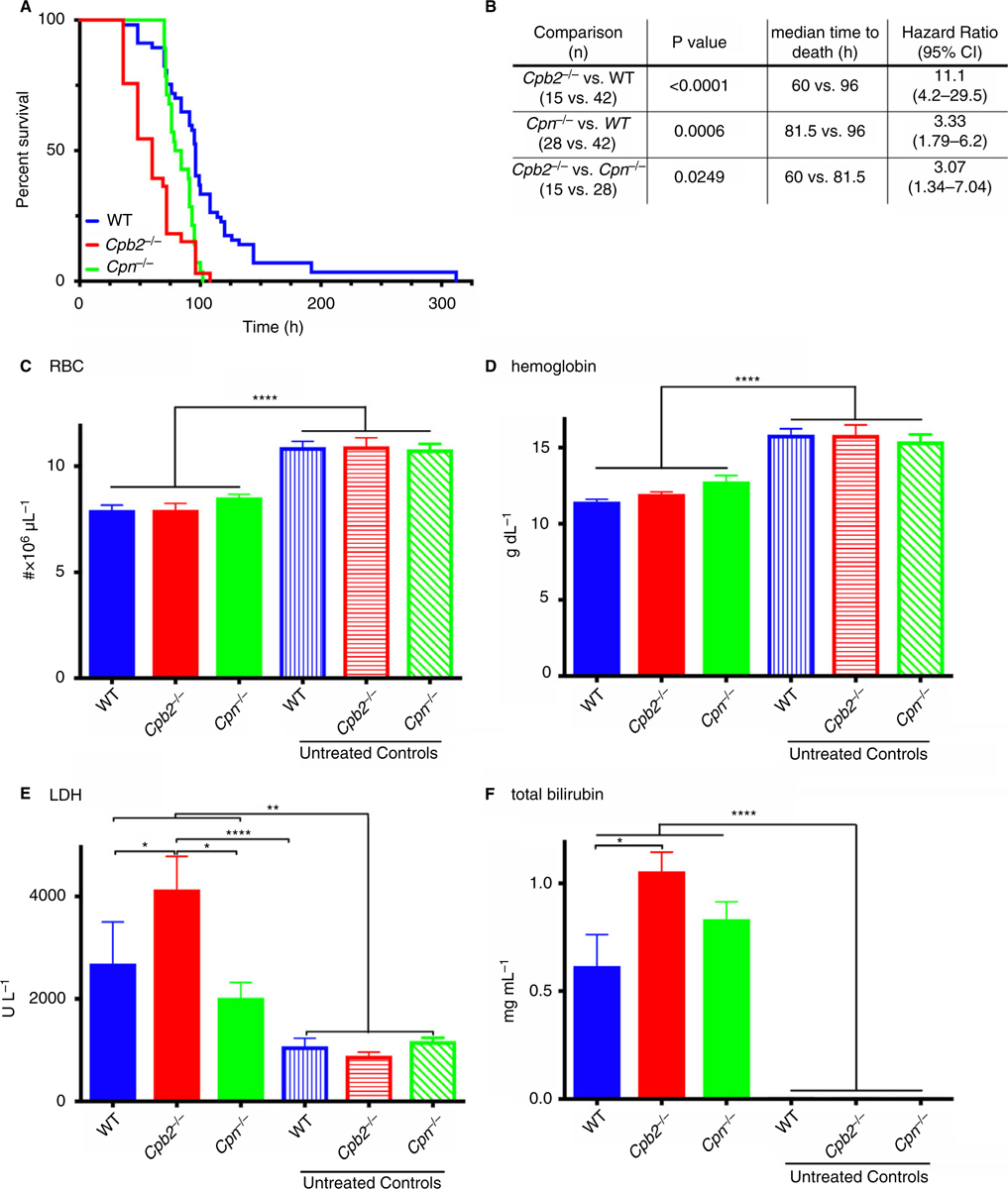
Cpb2−/− mice have exacerbated hemolytic-uremic syndrome (HUS) compared with wild-type (WT) or Cpn−/− mice. Disease was induced by treatment with Stx2 and lipopolysaccharide (LPS) together as described in Methods, and in C-F the mice were sacrificed at 48 h for blood collection (n = 10) for complete blood count (CBC) and clinical chemistry analysis. (A) Cpb2−/−, Cpn−/− and WT mice were followed until death (n = 15, 28 and 42, respectively). (B) Data analyzed by the log rank method with Bonferroni correction for multiple testing. Data analyzed by the log rank method (P = 0.0708). (C) red blood cell (RBC) count, (D) hemoglobin levels, (E) plasma lactate dehydrogenase (LDH) and (F) plasma total bilirubin. *P < 0.05, **P < 0.01, ****P < 0.0001.
Cpb2−/− mice had greater changes in markers of the HUS clinical triad than WT or Cpn−/− mice
To investigate differences between mice with different genotypes following administration of Stx2 and LPS, we assayed markers of hemolysis, thrombocytopenia and renal insufficiency 48 h after induction of disease. In these experiments no mice died before sacrifice, so there was no survivor bias. Stx2 and LPS treatment induced hemolysis in treated mice, as shown by a reduction in red blood cell (RBC) count (Fig. 1C) and hemoglobin levels (Fig. 1D), and more elevated LDH (Fig. 1E) and total bilirubin levels (Fig. 1F). Cpb2−/− mice had a larger increase in LDH and total bilirubin compared with WT or Cpn−/− mice or untreated mice from all three genotypes. Cpb2−/−, Cpn−/− and WT mice all developed thrombocytopenia with platelet counts < 20% of baseline (Fig. 2A). Fewer platelets were present in treated Cpb2−/− mice than in treated Cpn−/− or WT mice. Renal function abnormalities at 48 h with elevated levels of BUN (Fig. 2B) and creatinine (Fig. 2C) were observed only in Cpb2−/− mice. Additionally, there was more liver damage in treated Cpb2−/− mice than in WT and Cpn−/− mice, shown by elevated ALT (Fig. 2D) and AST levels (Fig. 2E).
Fig. 2.
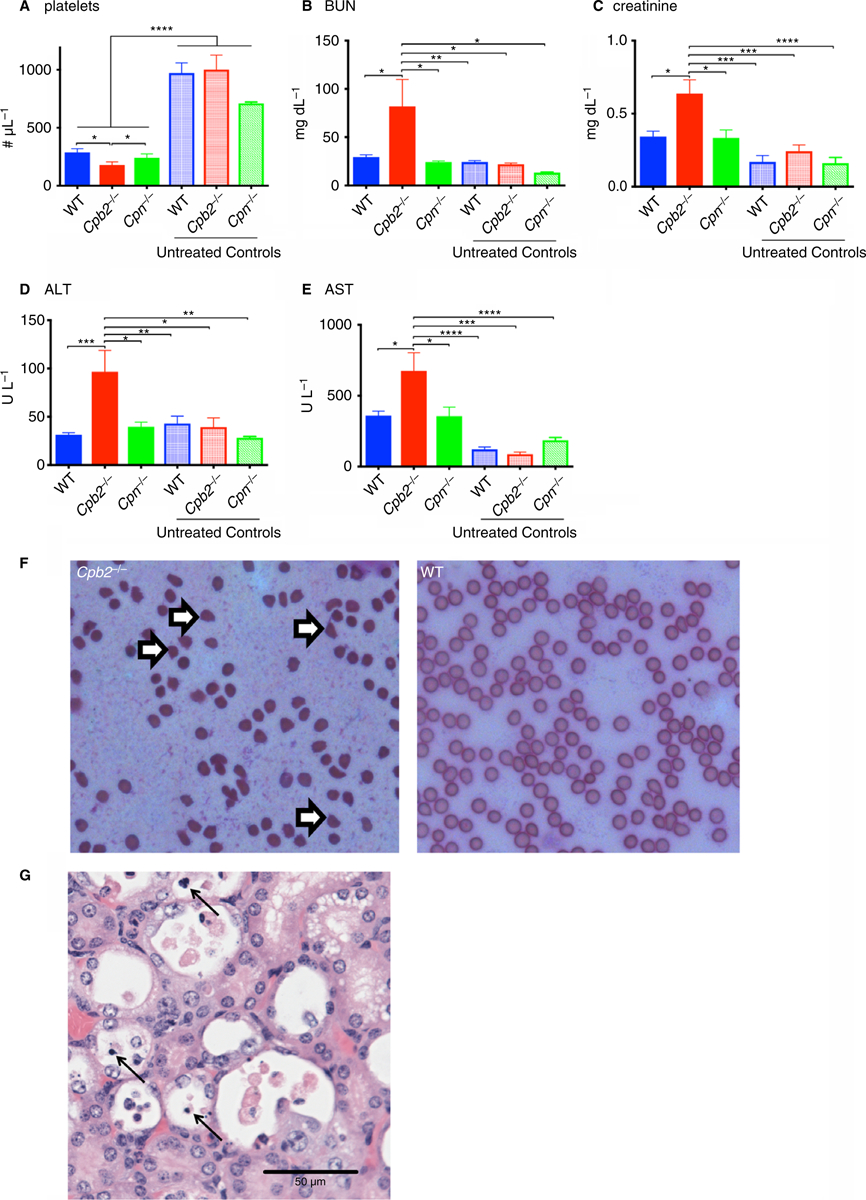
Cpb2−/− mice have worse liver and kidney damage in hemolytic-uremic syndrome (HUS) compared with wild-type (WT) mice. Disease was induced by treatment with Stx2 and lipopolysaccharide (LPS) together as described in Methods and mice were sacrificed at 48 h for blood collection (n = 10). (A) Platelet count, (B) blood urea nitrogen (BUN), (C) creatinine, (D) alanine transaminase (ALT) and (E) aspartate transaminase (AST). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (F) Blood smear showing schistocytes (open arrows) in a sample from Cpb2−/− mice but not WT mice. (G) Kidney section harvested ante-mortem stained with H&E. Arrows point to necrotic tubular epithelial cells. The sections were examined under a Nikon 40×/0.95 objective lens using a Nikon Eclipse E1000M and images captured with a Diagnostic Instruments 15.2 Mp Shifting Pixel using Spot Imaging solutions Version 5.3 acquisition software.
As the Cpn−/− mice had an intermediate phenotype between Cpb2−/− and WT mice, in subsequent HUS experiments we evaluated only those two genotypes. Microangiopathy, as demonstrated by schistocytes on peripheral blood smears taken at 48 h after disease induction, was present in Cpb2−/− mice but not in WT mice (Fig. 2F). Thus, the administration of Stx2 and LPS induces a pathological condition that recapitulates the clinical triad of human HUS. Taken together, these data demonstrate that Cpb2−/− mice are more susceptible to HUS than Cpn−/− or WT mice, supporting our hypothesis that CPB2 plays an important role in inactivating C5a in vivo.
Kidney pathology in mice with HUS
In mice sacrificed 48 h after HUS induction, no kidney pathology was observed, irrespective of genotype (data not shown). Some mice were sacrificed ante-mortem because their health score had reached a value of greater than 23, and kidney pathology was investigated. Irrespective of genotype, necrotic tubular epithelial cells were observed, consistent with previous reports (Fig. 2G) [17,28].
Hematologic parameters in mice with HUS
In addition to the development of hemolytic anemia, there was significant lymphopenia and neutrophilia observed in all three genotypes, which is commonly seen upon acute bacterial infection [29], whereas there was no change in the total leukocyte count (Fig. 3).
Fig. 3.
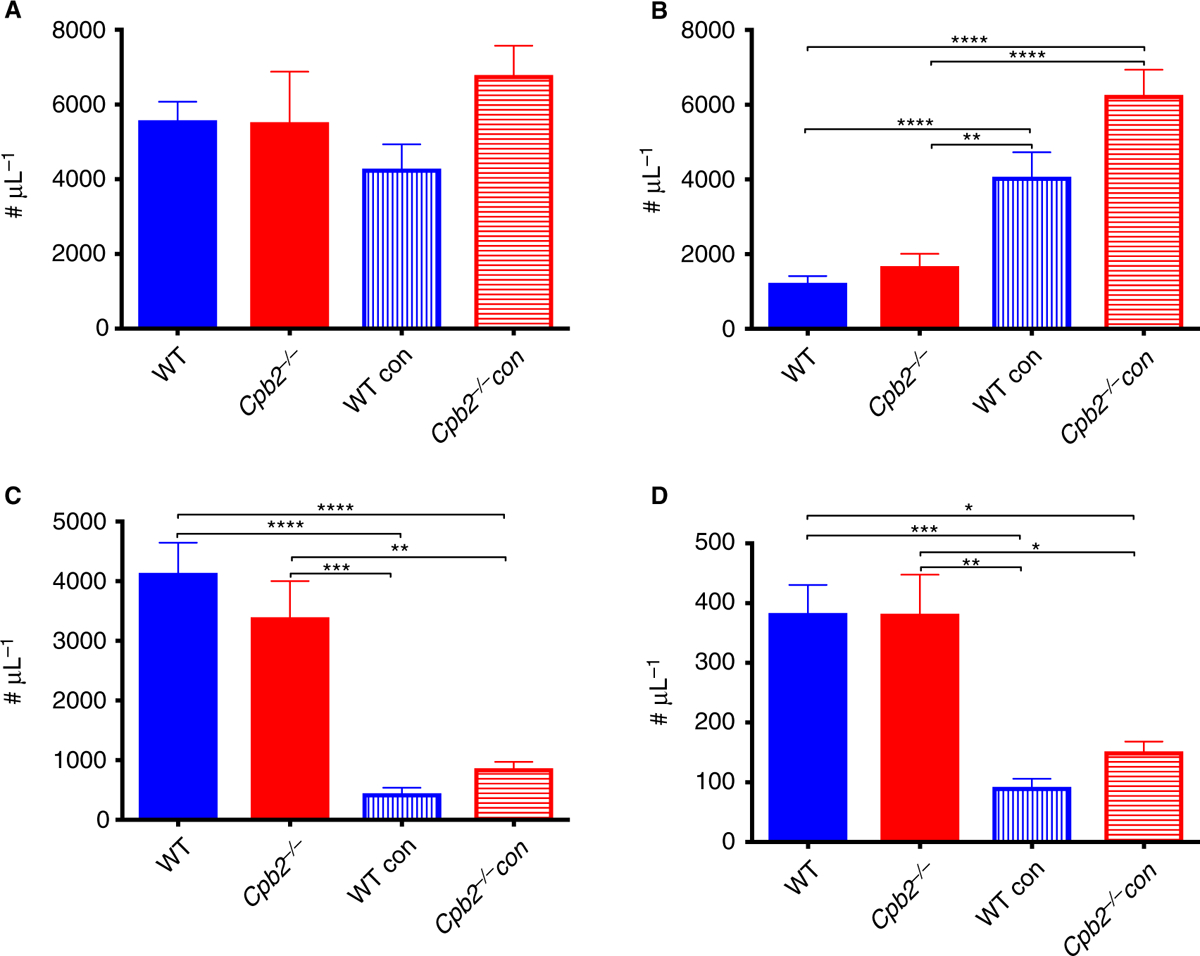
Complete blood count changes in hemolytic-uremic syndrome (HUS) model. Disease was induced by treatment with Stx2 and lipopolysaccharide (LPS) together as described in Methods and mice were sacrificed at 48 h for blood collection (n = 10). (A) White blood cell count, (B) lymphocytes, (C) monocytes and (D) neutrophils. **P < 0.01, ***P < 0.001.
Coagulation and fibrinolysis markers
Plasma fibrinogen levels measured by ELISA were increased upon Stx2 and LPS challenge, similar to the increase we previously observed in a polymicrobial sepsis model, as fibrinogen is an acute phase protein [29], but no difference was detected between WT and Cpb2−/− animals (Fig. 4A). Because the fibrinogen ELISA used here detects both intact and degraded forms of fibrinogen, we also measured D-dimer, a marker of fibrinolysis. There were higher D-dimer levels in WT, Cpb2−/− and Cpn−/− animals than in control animals, but only in Cpb2−/− mice did the increase reach significance (Fig. 4B). This is consistent with the expected increase in fibrinolysis in Cpb2−/− mice compared with WT mice [30]. There was no difference in levels of TAT observed in animals treated with the toxins in either genotype, suggesting that there was no increase in thrombin generation in Cpb2−/− mice despite their exacerbated disease (Figure S1).
Fig. 4.
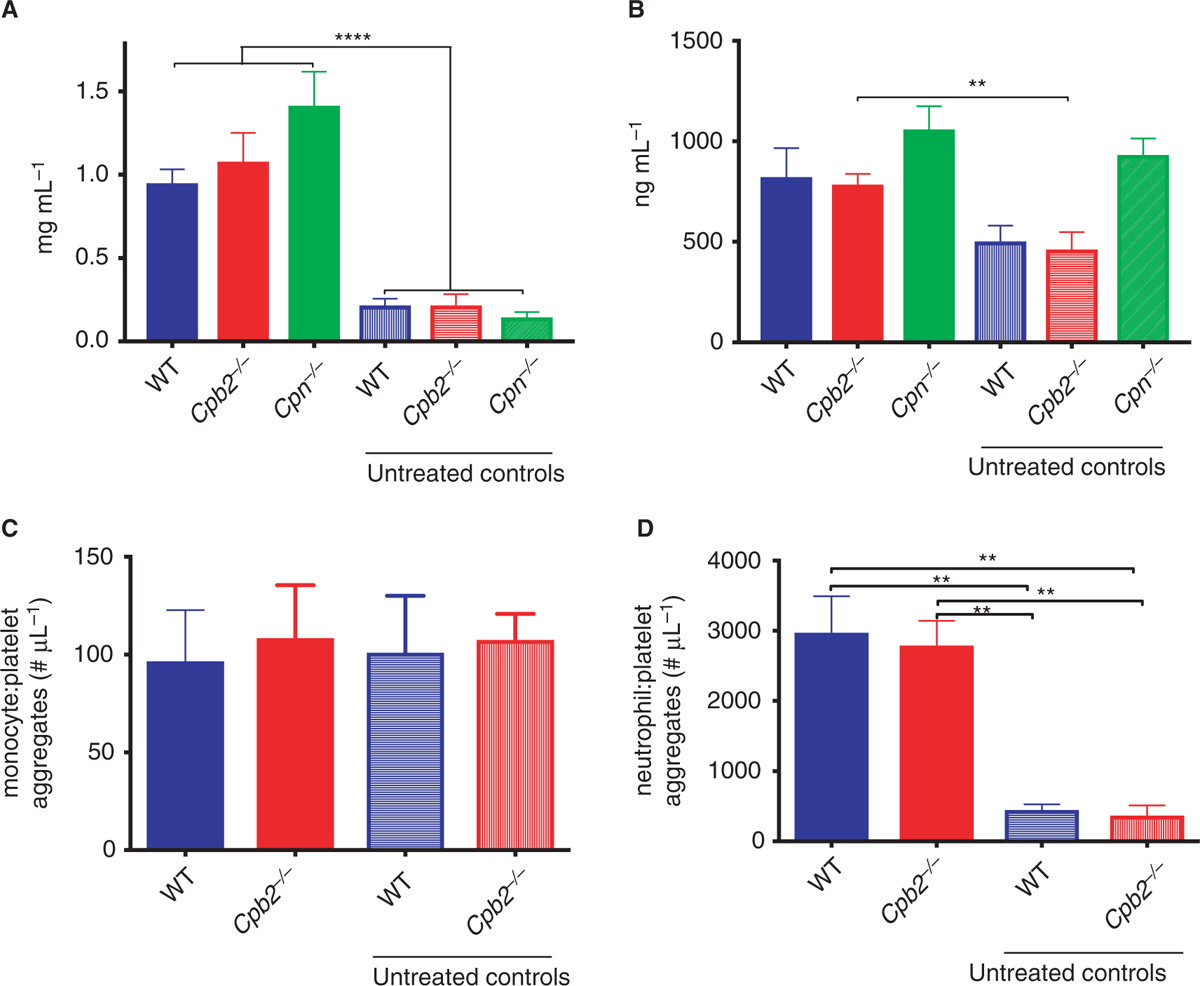
Hemolytic-uremic syndrome (HUS) causes changes in coagulation and fibrinolysis markers and platelet–leukocyte aggregates. Disease was induced by treatment with Stx2 and lipopolysaccharide (LPS) together as described in Methods and mice were sacrificed at 48 h for blood collection (n > 8). ELISAs were run for (A) fibrinogen and (B) D-dimer. Platelets were identified by staining with anti-CD41, neutrophils were defined as CD11bhighLy6Ghigh and monocytes were defined as CD11b+Ly6G−. Events in neutrophil and monocyte gates that were also CD41 positive were leukocyte–platelet aggregates. N = 6. (C) Monocyte–platelet aggregates and (D) neutrophil–platelet aggregates. *P < 0.05, **P < 0.01.
Leukocyte–platelet aggregates were decreased more in Cpb2−/− mice than in WT mice
Leukocyte–platelet aggregates contribute to the pathogenesis of HUS [31]. We measured the absolute number of neutrophil–platelet and monocyte–platelet aggregates in mouse whole blood and showed that in Cpb2−/− and WT mice the number of neutrophil–platelet aggregates was markedly increased with Stx/LPS treatment (Fig. 4D) but the number of monocyte–platelet aggregates was unaltered (Fig. 4C). The percentage of neutrophils and monocytes aggregated with platelets was reduced in animals with HUS as the platelet number dropped in both genotypes (Fig. 2A), but in Cpb2−/− mice there was less reduction in the proportion of neutrophils and monocytes in aggregates than in WT mice (Figure S2).
Anti-C5 treatment attenuated Stx2 and LPS-induced HUS in Cpb2−/− and WT mice
CPB2 has several substrates, including fibrin, C3a and C5a, of which C5a is known to be involved in HUS. We tested if C5a was the relevant CPB2 substrate in our model by administering an antibody that prevents C5 cleavage, thereby preventing generation of C5a, a known CPB2 substrate, and C5b-9 (MAC) [26,32]. Both WT and Cpb2−/− mice had prolonged survival with treatment (Fig. 5; n = 11 in each group). Cpb2−/− mice had increased survival (P < 0.0001), with a median survival of 60 h vs. 168 h with anti-C5 treatment (hazard ratio = 0.078; 95% CI, 0.023–0.264), and in WT mice survival was also improved (P = 0.003), with median survival of 96 h vs. 168 h in the treatment group (hazard ratio = 0.129; 95% CI, 0.022–0.755). In this experiment, untreated Cpb2−/− mice had worse survival than untreated WT mice (P = 0.0253; hazard ratio = 3.53; 95% CI, 1.17–10.6), in agreement with the earlier data. Interestingly the difference in survival between WT and Cpb2−/− mice was not observed in animals treated with anti-C5 antibody, as median survival was 168 h in both genotypes and the hazard ratio is 1.18 (95% CI, 0.431–3.22).
Fig. 5.
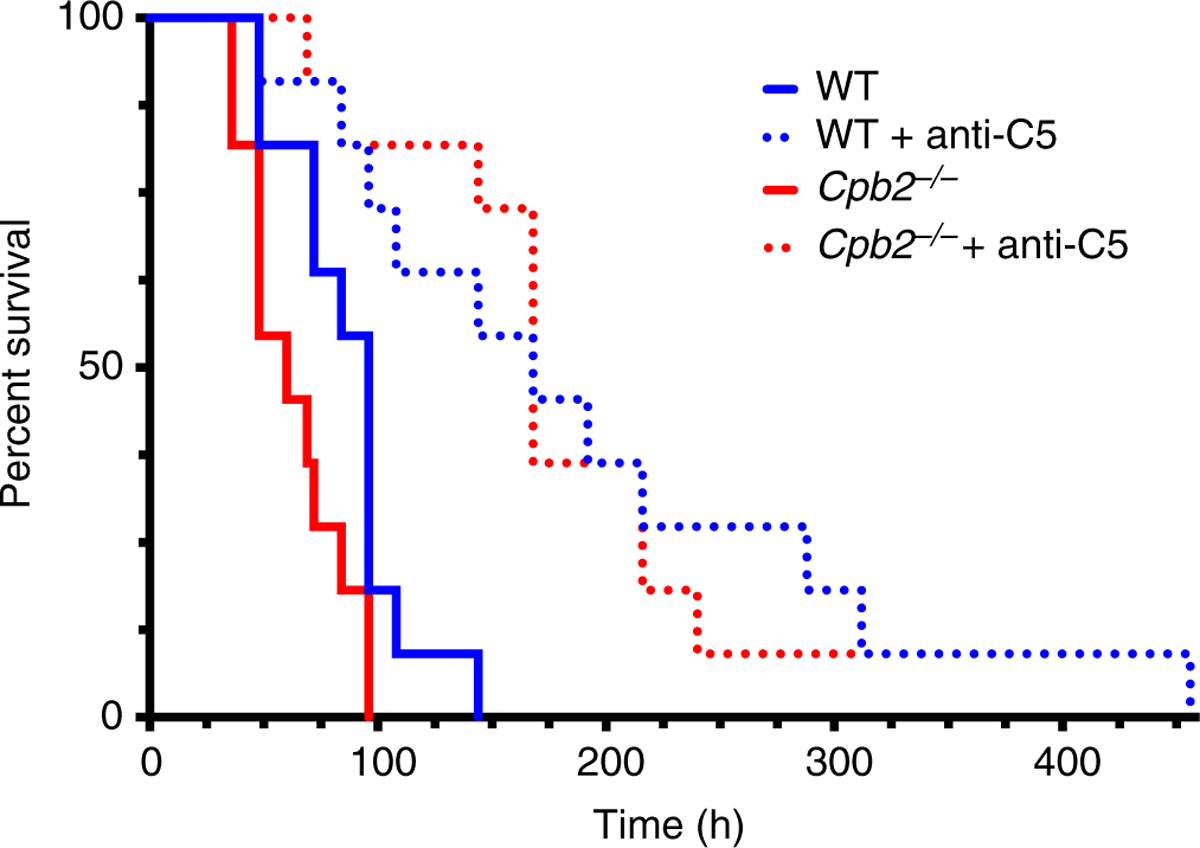
Anti-C5 antibody prolongs survival in the hemolytic-uremic syndrome (HUS) model in both Cpb2−/− and wild-type (WT) mice. Disease was induced by treatment with Stx2 and lipopolysaccharide (LPS) together as described in Methods and mice were followed until death (n = 11). Anti-C5 antibody was administered intraperitoneally at 3 h before disease induction and then every 24 h. Data were analyzed by the log rank method with the Bonferroni correction for multiple tests.
Susceptibility to CVF was increased in Cpn−/− but not Cpb2−/− mice
Administration of CVF at 7.5 μg i.v. per mouse to Cpn−/− mice caused 100% mortality within 1 h, with no deaths occurring in WT mice [8]. To test susceptibility to C5a, we treated WT, Cpb2−/−, Cpn−/− and Cpb2−/−/Cpn−/− mice with 7.5 or 15 μg per mouse of C5a i.v., but no responses were observed in any genotype, even at the higher dose.
CVF is a C3b homolog that forms a C3/C5 convertase that activates complement systemically, generating C3a, C5a and the membrane attack complex, C5b-9 [33]. In preliminary studies, we treated WT and Cpb2−/−/Cpn−/− mice with increasing doses of CVF and found that 300 ug per mouse was the lowest dose at which some Cpb2−/−/Cpn−/− mice, but no WT mice, died. We treated WT, Cpb2−/−, Cpn−/− and Cpb2−/−/Cpn−/− mice (n = 9, except Cpn−/− mice where n = 10) with this dose and found that deaths only occurred in the Cpb2−/−/Cpn−/− mice between 30 min and 1 h after administration of CVF (44% of Cpb2−/−/Cpn−/− mice died; deaths in Cpb2−/−/Cpn−/− mice different from the other three genotypes, P < 0.0001). Cpb2−/−, Cpn−/− and Cpb2−/−/Cpn−/− mice all had a significant deterioration in their health score at 30 min compared with WT mice, with the Cpb2−/−/Cpn−/− mice worst, then Cpn−/− mice, whereas the Cpb2−/− mice had relatively mild disease (Fig. 6A). All mice alive at 1 h subsequently recovered. Thus, in this acute challenge model, CPN plays the dominant role, whereas CPB2 played a supplementary role in inactivating C5a, as the absence of CPN alone exacerbates the disease, which is compounded when CPB2 is also missing.
Fig. 6.
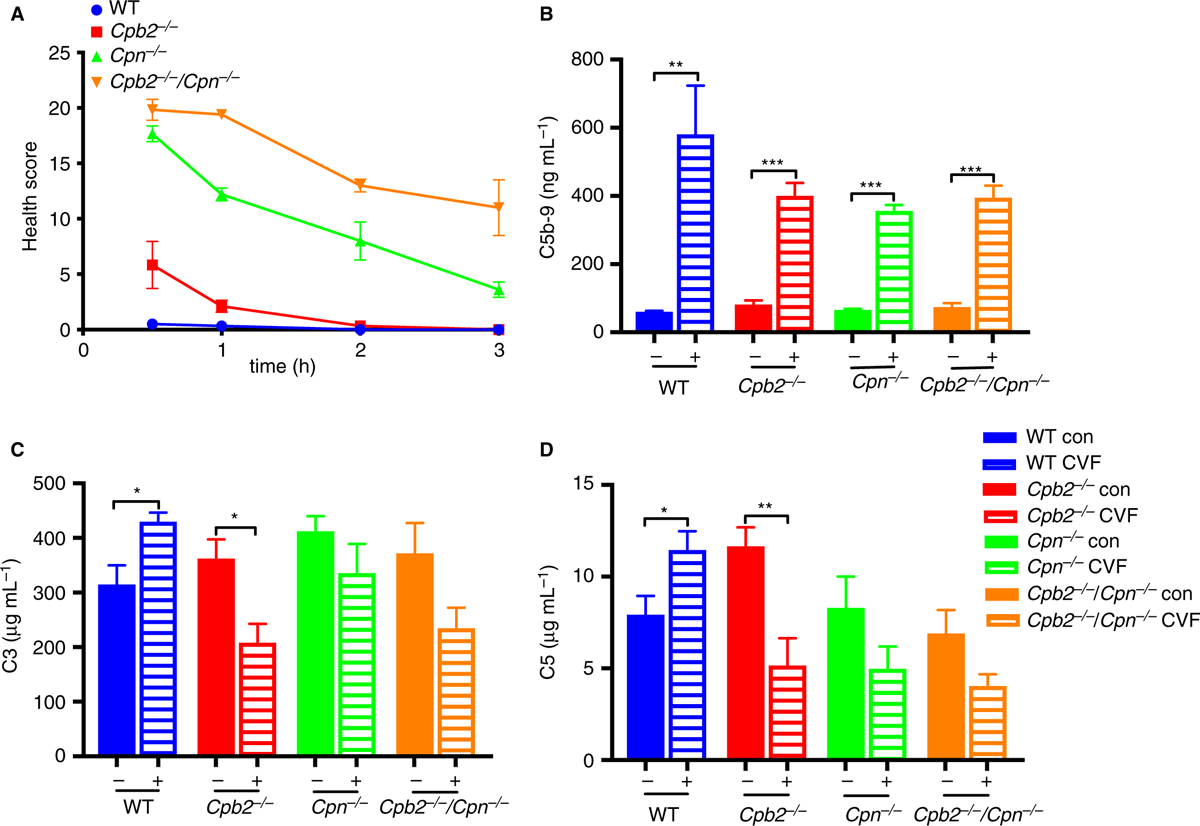
Cpb2−/−/Cpn−/− mice have worse disease after cobra venom factor (CVF) treatment than Cpb2−/−, Cpn−/− or wild-type (WT) mice. Mice were treated with 300 μg/mouse CVF and their health score was monitored. (A) Four Cpb2−/−/Cpn−/− mice died between 30 min and 1 h. The health scores for all genotypes were different from each other (Cpb2−/−/Cpn−/−, Cpb2−/− and WT, n = 9; Cpn−/−, n = 10; P < 0.0001). Plasma was collected before and 20 min after CVF administration and ELISAs were run for (B) C5b-9, (C) C3 and (D) C5 (n = 6). *P < 0.05, **P < 0.01.
There was no difference in baseline levels of C3, C5 and C5B-9 before CVF challenge (Fig. 6B–D). In control mice there was a significant increase in C5b-9 in all four genotypes, whereas C3 and C5 were consumed in Cpb2−/−, Cpn−/− and Cpb2−/−/Cpn−/− mice but not WT mice.
Discussion
HUS has been described, in part, as a disease of complement dysregulation [34]. Although CPB2 has several substrates described in vitro, only three have been validated as physiological substrates, fibrin, C3a and C5a. Based on our previous data showing CPB2 inactivation of C3a and C5a is relevant pathophysiologically in a murine polymicrobial sepsis model [7], we hypothesized that in an HUS model, Cpb2−/− mice would have worse disease than WT mice due to excessive levels of the anaphylatoxins. We employed a published model and found the expected changes in renal insufficiency, microangiopathic hemolysis and thrombocytopenia [25]. Cpb2−/− mice clearly had worse disease than WT mice, consistent with our hypothesis. CPB2 is activated from proCPB2 by either the thrombin/TM complex or plasmin bound to glycosaminoglycans, and evidence of activation of fibrinolysis was found, as documented by elevated D-dimer levels, consistent with enhanced plasmin generation. Increased plasmin has paradoxical effects on the complement system, both causing an increase in activation of C5 [35] but also inhibiting complement by cleaving C3b into inactive fragments [36,37].
The lack of an observable phenotype in the Cpb2−/−/Cpn−/− mice shows that inactivation of the complement anaphylatoxins is not required for fertility, development or survival. No differences were observed in baseline levels of several markers related to coagulation and complement activation between the four genotypes, showing that lack of one or both of the plasma basic carboxypeptidases does not result in a phenotype in the absence of challenge.
Treatment of Cpb2−/− mice with LPS, using 30 or 40 mg kg−1 LPS, the LD50 for LPS in C57Bl/6 mice, did not result in any observable differences with WT mice in survival experiments [23]. Here we used 75 μg kg−1, which is ~0.1% of the dose used previously. The level of endotoxin in the Stx2 lots used here would increase the exposure by < 1% of the LPS administered, as the model requires both toxins (data not shown). Thus, the disease phenotype observed in this model was not mediated by a direct toxic effect of LPS.
Treatment with an antibody that prevents cleavage of C5, and hence formation of C5a as well as the MAC, protected both Cpb2−/− and WT mice, showing that the cleavage products of C5 play an important role in exacerbation of the disease. C5a is a good substrate for CPB2 in vitro [38], whereas to our knowledge there are no reports of CPB2 affecting the formation of the MAC. Thus, C5a is the probable CPB2 substrate in this HUS model responsible for the difference in phenotype to WT mice. This study does not, however, eliminate a role for other substrates of CPB2 in this model. Blockade of the C3a receptor protects mice against HUS induced by Stx2 and LPS [19] and it is plausible that increased levels of C3a could also play a role here. On the other hand, enhanced plasmin generation and fibrinolysis, as expected in the Cpb2−/− mice and supported by the higher levels of D-dimer, should lead to a decreased clot burden and a beneficial effect in these mice, rather than increased disease susceptibility; thus, fibrin is probably not a key substrate in this model. Eculizumab is an anti-human C5 antibody with an equivalent mode of action to the anti-mouse C5 antibody used in our studies and has been shown to have beneficial effects in Shiga toxin-producing E. coli infections, especially when administered early [39–41].
It is notable that although the deficiency of either carboxypeptidase renders the mouse more susceptible in the HUS model, Cpb2−/− mice clearly had worse disease than Cpn−/− mice. In contrast, when challenged acutely with CVF, the reverse phenotype was observed. Cpn−/− mice had markedly worse disease than Cpb2−/− mice, whereas the WT mice were resistant at this dose of CVF. Previous studies have also shown that Cpn−/−, but not Cpb2−/−, mice are susceptible to acute CVF challenge [8,9]. Taken together, the data suggest that CPN and CPB2 play overlapping but non-redundant roles in regulating complement activation in vivo. The constitutively active CPN is key for inactivation of systemic C5a in the blood compartment, whereas CPB2, whose activation from proCPB2 requires either the thrombin/TM complex or plasmin, functions as an on-demand supplementary anaphylatoxin inhibitor in inactivating excessive C5a that is formed locally. Thus, the Cpn−/− mice succumb to the acute systemic CVF challenge, whereas Cpb2−/− mice are more susceptible to the Stx2/LPS-induced HUS, which is a more subacute disease model. It would be interesting to determine if this phenotypic difference between the Cpn−/− and Cpb2−/− mice is observed in other complement-mediated murine disease models.
Supplementary Material
Essentials.
Two basic carboxypeptidases are present in plasma, B2 (CPB2) and N (CPN).
Cpb2−/− and Cpn−/− mice were challenged in a hemolytic uremic syndrome (HUS) model vs. wild type.
Cpb2−/− exacerbates HUS while Cpn−/− exacerbates cobra venom factor challenge vs. wild type mice.
CPB2 and CPN have overlapping but non-redundant roles.
Acknowledgements
This study was supported by NIH grants HL057530 and AI085268. Zhifei Shao was supported by NIH fellowship grant 1T32HL098049. We thank V. M. Holers (University of Colorado, Denver, Colorado, USA) for providing the anti-C5 antibody, BB5.1, and L. Gigliello and P. Castro for helping with observing the mice.
Footnotes
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Plasma TAT levels are unaltered by HUS.
Fig. S2. HUS causes changes in % of leukocytes in platelet-leukocyte aggregates.
References
- 1.Willemse JL, Heylen E, Nesheim ME, Hendriks DF. Carboxypeptidase U (TAFIa): a new drug target for fibrinolytic therapy? J Thromb Haemost 2009; 7: 1962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurewich V, Pannell R. Structure-function relationships in thrombin-activatable fibrinolysis inhibitor: comment. J Thromb Haemost 2016; 14: 1899–900. [DOI] [PubMed] [Google Scholar]
- 3.Mao SS, Cooper CM, Wood T, Shafer JA, Gardell SJ. Characterization of plasmin-mediated activation of plasma procarboxypeptidase B. Modulation by glycosaminoglycans. J Biol Chem 1999; 274: 35046–52. [DOI] [PubMed] [Google Scholar]
- 4.Bajzar L, Morser J, Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem 1996; 271: 16603–8. [DOI] [PubMed] [Google Scholar]
- 5.Myles T, Nishimura T, Yun TH, Nagashima M, Morser J, Patterson AJ, Pearl RG, Leung LL. Thrombin activatable fibrinolysis inhibitor, a potential regulator of vascular inflammation. J Biol Chem 2003; 278: 51059–67. [DOI] [PubMed] [Google Scholar]
- 6.Skidgel RA, Erdos EG. Structure and function of human plasma carboxypeptidase N, the anaphylatoxin inactivator. Int Immunopharmacol 2007; 7: 1888–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morser J, Gabazza EC, Myles T, Leung LL. What has been learnt from the thrombin-activatable fibrinolysis inhibitor-deficient mouse? J Thromb Haemost 2010; 8: 868–76. [DOI] [PubMed] [Google Scholar]
- 8.Mueller-Ortiz SL, Wang D, Morales JE, Li L, Chang JY, Wetsel RA. Targeted disruption of the gene encoding the murine small subunit of carboxypeptidase N (CPN1) causes susceptibility to C5a anaphylatoxin-mediated shock. J Immunol 2009; 182: 6533–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asai S, Sato T, Tada T, Miyamoto T, Kimbara N, Motoyama N, Okada H, Okada N. Absence of procarboxypeptidase R induces complement-mediated lethal inflammation in lipopolysaccharide-primed mice. J Immunol 2004; 173: 4669–74. [DOI] [PubMed] [Google Scholar]
- 10.Song JJ, Hwang I, Cho KH, Garcia MA, Kim AJ, Wang TH, Lindstrom TM, Lee AT, Nishimura T, Zhao L, Morser J, Nesheim M, Goodman SB, Lee DM, Bridges SL Jr, Gregersen PK, Leung LL, Robinson WH. Plasma carboxypeptidase B downregulates inflammatory responses in autoimmune arthritis. J Clin Invest 2011; 121: 3517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shao Z, Nishimura T, Leung LL, Morser J. Carboxypeptidase B2 deficiency reveals opposite effects of complement C3a and C5a in a murine polymicrobial sepsis model. J Thromb Haemost 2015; 13: 1090–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishimura T, Myles T, Piliponsky AM, Kao PN, Berry GJ, Leung LL. Thrombin-activatable procarboxypeptidase B regulates activated complement C5a in vivo. Blood 2007; 109: 1992–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thurman JM, Marians R, Emlen W, Wood S, Smith C, Akana H, Holers VM, Lesser M, Kline M, Hoffman C, Christen E, Trachtman H. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol 2009; 4: 1920–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, Proulx F, Clermont MJ, Le Deist F, Niaudet P, Schaefer F. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med 2011; 364: 2561–3. [DOI] [PubMed] [Google Scholar]
- 15.Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, Eitner F, Feldkamp T, Fouque D, Furman RR, Gaber O, Herthelius M, Hourmant M, Karpman D, Lebranchu Y, Mariat C, et al. Terminal complement inhibitor eculizumab in atypical haemolytic-uremic syndrome. N Engl J Med 2013; 368: 2169–81. [DOI] [PubMed] [Google Scholar]
- 16.Rathbone J, Kaltenthaler E, Richards A, Tappenden P, Bessey A, Cantrell A. A systematic review of eculizumab for atypical haemolytic uraemic syndrome (aHUS). BMJ Open 2013; 3: e003573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keir LS. Shiga toxin associated hemolytic uremic syndrome. Hematol Oncol Clin North Am 2015; 29: 525–39. [DOI] [PubMed] [Google Scholar]
- 18.Cofiell R, Kukreja A, Bedard K, Yan Y, Mickle AP, Ogawa M, Bedrosian CL, Faas SJ. Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood 2015; 125: 3253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morigi M, Galbusera M, Gastoldi S, Locatelli M, Buelli S, Pezzotta A, Pagani C, Noris M, Gobbi M, Stravalaci M, Rottoli D, Tedesco F, Remuzzi G, Zoja C. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J Immunol 2011; 187: 172–80. [DOI] [PubMed] [Google Scholar]
- 20.Orth D, Khan AB, Naim A, Grif K, Brockmeyer J, Karch H, Joannidis M, Clark SJ, Day AJ, Fidanzi S, Stoiber H, Dierich MP, Zimmerhackl LB, Wurzner R. Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol 2009; 182: 6394–400. [DOI] [PubMed] [Google Scholar]
- 21.Poolpol K, Orth-Holler D, Speth C, Zipfel PF, Skerka C, de Cordoba SR, Brockmeyer J, Bielaszewska M, Wurzner R. Interaction of Shiga toxin 2 with complement regulators of the factor H protein family. Mol Immunol 2014; 58: 77–84. [DOI] [PubMed] [Google Scholar]
- 22.Ehrlenbach S, Rosales A, Posch W, Wilflingseder D, Hermann M, Brockmeyer J, Karch H, Satchell SC, Wurzner R, Orth-Holler D. Shiga toxin 2 reduces complement inhibitor CD59 expression on human renal tubular epithelial and glomerular endothelial cells. Infect Immun 2013; 81: 2678–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagashima M, Yin ZF, Zhao L, White K, Zhu Y, Lasky N, Halks-Miller M, Broze GJ Jr, Fay WP, Morser J. Thrombin-activatable fibrinolysis inhibitor (TAFI) deficiency is compatible with murine life. J Clin Invest 2002; 109: 101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shrum B, Anantha RV, Xu SX, Donnelly M, Haeryfar SM, McCormick JK, Mele T. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res Notes 2014; 7: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zoja C, Locatelli M, Pagani C, Corna D, Zanchi C, Isermann B, Remuzzi G, Conway EM, Noris M. Lack of the lectin-like domain of thrombomodulin worsens Shiga toxin-associated hemolytic uremic syndrome in mice. J Immunol 2012; 189: 3661–8. [DOI] [PubMed] [Google Scholar]
- 26.Banda NK, Kraus D, Vondracek A, Huynh LH, Bendele A, Holers VM, Arend WP. Mechanisms of effects of complement inhibition in murine collagen-induced arthritis. Arthritis Rheum 2002; 46: 3065–75. [DOI] [PubMed] [Google Scholar]
- 27.Gerrits AJ, Frelinger AL 3rd, Michelson AD. Whole blood analysis of leukocyte-platelet aggregates. Curr Protoc Cytom 2016; 78: 6.15–6.15.10. [DOI] [PubMed] [Google Scholar]
- 28.Keepers TR, Psotka MA, Gross LK, Obrig TG. A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J Am Soc Nephrol 2006; 17: 3404–14. [DOI] [PubMed] [Google Scholar]
- 29.Fish RJ, Neerman-Arbez M. Fibrinogen gene regulation. Thromb Haemost 2012; 108: 419–26. [DOI] [PubMed] [Google Scholar]
- 30.Schultz G, Tedesco MM, Sho E, Nishimura T, Sharif S, Du X, Myles T, Morser J, Dalman RL, Leung LL. Enhanced abdominal aortic aneurysm formation in thrombin-activatable procarboxypeptidase B-deficient mice. Arterioscler Thromb Vasc Biol 2010; 30: 1363–70. [DOI] [PubMed] [Google Scholar]
- 31.Stahl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 2011; 117: 5503–13. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Rollins SA, Madri JA, Matis LA. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc Natl Acad Sci U S A 1995; 92: 8955–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogel CW, Fritzinger DC. Cobra venom factor: Structure, function, and humanization for therapeutic complement depletion. Toxicon 2010; 56: 1198–222. [DOI] [PubMed] [Google Scholar]
- 34.Jokiranta TS. HUS and atypical HUS. Blood 2017; 129: 2847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foley JH, Walton BL, Aleman MM, O’Byrne AM, Lei V, Harrasser M, Foley KA, Wolberg AS, Conway EM. Complement activation in arterial and venous thrombosis is mediated by plasmin. EBioMedicine 2016; 5: 175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barthel D, Schindler S, Zipfel PF. Plasminogen is a complement inhibitor. J Biol Chem 2012; 287: 18831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foley JH. Plasmin(ogen) at the nexus of fibrinolysis, inflammation, and complement. Semin Thromb Hemost 2017; 43: 135–42. [DOI] [PubMed] [Google Scholar]
- 38.Du XY, Zabel BA, Myles T, Allen SJ, Handel TM, Lee PP, Butcher EC, Leung LL. Regulation of chemerin bioactivity by plasma carboxypeptidase N, carboxypeptidase B (activated thrombin-activable fibrinolysis inhibitor), and platelets. J Biol Chem 2009; 284: 751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delmas Y, Vendrely B, Clouzeau B, Bachir H, Bui HN, Lacraz A, Helou S, Bordes C, Reffet A, Llanas B, Skopinski S, Rolland P, Gruson D, Combe C. Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France: outcome with eculizumab. Nephrol Dial Transplant 2014; 29: 565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dinh A, Anathasayanan A, Rubin LM. Safe and effective use of eculizumab in the treatment of severe Shiga toxin Escherichia coli-associated hemolytic uremic syndrome. Am J Health Syst Pharm 2015; 72: 117–20. [DOI] [PubMed] [Google Scholar]
- 41.Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical Hemolytic Uremic Syndrome (HUS) with neurological involvement. Medicine 2015; 94: e1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.