

Abstract
Rhipicephalus microplus, a vector that can transmit many pathogens to humans and domestic animals, is widely distributed in Yunnan province, China. However, few reports on the prevalence of tick-borne pathogens (TBPs) in Rh. microplus in Yunnan are available. The aim of this study was to detect TBPs in Rh. microplus in Yunnan and to analyze the phylogenetic characterization of TBPs detected in these ticks. The adult Rh. microplus (n = 516) feeding on cattle were collected. The pooled DNA samples of these ticks were evaluated using metagenomic next-generation sequencing (mNGS) and then TBPs in individual ticks were identified using genus- or group-specific nested polymerase chain reaction (PCR) combined with DNA sequencing assay. As a result, Candidatus Rickettsia jingxinensis (24.61%, 127/516), Anaplasma marginale (13.18%, 68/516), Coxiella burnetii (3.10%, 16/516), and Coxiella-like endosymbiont (CLE) (8.33%, 43/516) were detected. The dual coinfection with Ca. R. jingxinensis and A. marginale and the triple coinfection with Ca. R. jingxinensis, A. marginale, and CLE were most frequent and detected in 3.68% (19/516) and 3.10% (16/516) of these ticks, respectively. The results provide insight into the diversity of TBPs and their coinfections in Rh. microplus in Yunnan province of China, reporting for the first time that C. burnetii had been found in Rh. microplus in China. Multilocus variable number tandem repeat analysis with 6 loci (MLVA-6) discriminated the C. burnetii detected in Rh. microplus in Yunnan into MLVA genotype 1, which is closely related to previously described genotypes found primarily in tick and human samples from different regions of the globe, indicating a potential public health threat posed by C. burnetii in Rh. microplus in Yunnan.
Keywords: Rhipicephalus microplus, Candidatus Rickettsia jingxinensis, Anaplasma marginale, Coxiella burnetii, Coxiella-like endosymbiont, MLVA, Yunnan province
Introduction
Ticks are distributed widely across the world, and approximately 10% of the currently known 867 tick species act as arthropod vectors in the transmission of human and animal pathogens (Zhang et al., 2019; Yang et al., 2021). Likely due to human movement into tick habitats combined with climate changes, tick-borne diseases are increasing in prevalence and present an increasing global concern (Ostfeld and Brunner, 2015). As tick-borne diseases become more prevalent, the likelihood of coinfection with more than one tick-borne pathogen (TBP) in ticks is increasing and such coinfections have important repercussions on human or animal health which can alter clinical presentation, disease severity, and treatment response in tick-borne diseases (Vaumourin et al., 2015) and may play a role in incidence, distribution, and possible control of tick-borne diseases (Andersson et al., 2017).
Risk of human infection is contingent on the geographic distribution of the tick species as well as the prevalence of TBPs carried by ticks in a given region (Wilson and Elston, 2018). Rhipicephalus microplus is a common vector for the transmission of a great variety of microorganisms including bacteria, viruses, protozoa, fungi, or toxins. Among the TBPs in Rh. microplus, Coxiella burnetii, an obligate intracellular bacterium and the pathogenic agent of Q fever, is worldwide distributed. Q fever is typically an acute febrile illness with non-specific clinical signs in humans, but Q fever may manifest in human as an acute hepatitis and pneumonia or as chronic diseases that are seen in severe cases or life-threatening diseases such as valvular endocarditis (Sulyok et al., 2014).
Rhipicephalus microplus have been proved to be one of six most frequently reported tick species in China and mostly distributed in the southeast part of China (Zhang et al., 2019). Anaplasma spp., Theileria spp., Ehrlichia spp., Hepatozoon canis, and the viral community (Wen et al., 2003; Chen et al., 2014; Guo et al., 2019b; Li et al., 2020a; Shi et al., 2021) have been reported to be found in Rh. microplus in the region, indicating a high risk of exposure to these pathogens for humans and animals here.
Yunnan province is located in the southwestern part of China with wide distribution of Rh. microplus; it is of great importance for epidemiologists and physicians to be aware of Rh. microplus here for evaluating their potential for spread of the tick-borne diseases. In the present study, we investigated the potential TBPs in Rh. microplus collected from cattle in Yunnan. The results in the present study might provide a better understanding of TBPs carried by Rh. microplus in Yunnan, thereby strengthening programs to prevent and control the potential infections caused by these TBPs.
Materials and Methods
Tick Collection and Identification
An investigation was conducted from June to August in 2020, and ticks collected were fed on cattle in Lincang city and Weishan city in Yunnan province (Figure 1). Tick species were identified based on morphological characterization and by molecular biology methods based on the sequences of species-specific 16S rRNA and mitochondrial cytochrome c oxidase I (COI) genes, as previously described (Chitimia et al., 2010). Following identification, the ticks were stored at −80°C for further analysis.
FIGURE 1.

Map of the sampling sites in Yunnan Province, China. Ticks collected were fed on cattle in Lincang city (sampling sites 1–8) and Weishan city (sampling sites 9–12). The colored circles indicate the two cities where the ticks were collected and the dots indicate the sampling regions in this study.
DNA Extraction
To remove environmental contaminants, ticks were individually surface sterilized by 75% ethanol twice, followed by phosphate-buffered saline (PBS) twice. Ticks were then individually homogenized in 300 μl of PBS using the MagNA Lyser Green Beads (Roche, Mannheim, Germany), and DNA extraction was performed on 200 μl of each tick homogenate using QIAamp® Fast DNA Tissue Kit (Qiagen, Düsseldorf, Germany) according to the manufacturer’s instructions. The extracted genomic DNA was dissolved in 100 μl ultrapure water and stored at −20°C for further analysis. Individual DNA samples were mixed in an equal volume (10 μl) to prepare pooled DNA samples for full microbial genome sequencing using mNGS.
Metagenomic Profiling
All pooled DNA samples were paired-end sequenced on the Illumina HiSeq platform (insert size 350 bp, read length 150 bp) by The Beijing Genomics Institute (BGI) (Beijing, China), and the depth of sequencing for all pooled DNA samples was 10×. The reads with more than 40-nt low-quality bases (quality value ≤38) were removed. Meanwhile, the reads with more than 10-nt “N” bases were filtered out of the datasets. Lastly, the reads overlapping more than 15-nt bases with the adapters were removed. Reads that aligned to tick genes were also removed using Bowtie 2 (v2.2.4) (Karlsson et al., 2012, 2013). Accordingly, the high-quality data were obtained.
Then, the high-quality reads were mapped against scaffolds using SOAPdenovo (v2.04) (Luo et al., 2012). The unused reads from each sample were then assembled. The scaffolds were broken at N into the scaftigs (Nielsen et al., 2014), and the scaftigs with the length of ≥500 nt were used for further analysis (Li et al., 2014). Open reading frames (ORFs) in the scaftigs (≥500 bp) were predicted by MetaGeneMark (v2.10) (Karlsson et al., 2012; Qin et al., 2014). A non-redundant gene catalog was obtained after processing by using the CD-HIT (v4.5.8) (Li and Godzik, 2006; Fu et al., 2012). To determine the gene abundances, the reads were realigned with the gene catalog using Bowtie 2. Relative abundance of genes was calculated based on the number of reads mapped to the genes and the length of the genes, as previously described (Cotillard et al., 2013; Le Chatelier et al., 2013; Villar et al., 2015).
To access the taxonomic assignments of genes, genes were aligned to the integrated NR database (Version: 2018-01-02) of NCBI using DIAMOND (v0.9.9) (Buchfink et al., 2015). Then, the taxonomical level of each gene was determined by using the lowest common ancestor (LCA)-based algorithm implemented in MEGAN (Huson et al., 2007). The results containing the number of genes and the abundance information of each sample and the relative abundances of each taxonomic group were calculated by adding the relative abundances of genes annotated to the same feature (Karlsson et al., 2012; Li et al., 2014; Feng et al., 2015).
Polymerase Chain Reaction
Based on the results of mNGS, genus/group-specific PCR was performed to confirm the presence of TBPs in individual ticks. For nested PCR, 1 μl of each individual DNA sample was used as template for the first round and 1 μl of the primary PCR production was used as template for the second round. The target genes and specific primers for the spotted fever group rickettsia (SFGR; Cheng et al., 2016), Anaplasma spp. and Ehrlichia spp. (Qin et al., 2018), and Coxiella spp. (Duron et al., 2015) are listed in Table 1. All PCR amplifications were carried out using the PrimeSTAR® HS (Premix) (TaKaRa, Beijing, China) and performed on the PCR System 9700 (Applied Biosystems, GeneAmp®, United States). Amplified products were then electrophoresed in 1.5% agarose gel, and the positive amplicons were sent to TSINGKE Biological Technology (Beijing, China) for sequencing.
TABLE 1.
Target genes and primer sequences used for nested PCR.
| Pathogen | Target gene | Primer name | Sequence (5′–3′) | Tm (T/C) |
| SFGR | gltA | CS2d | ATGACCAATGAAAATAATAAT | 50 |
| CSEndr | CTTATACTCTCTATGTACA | |||
| RpCS.877p | GGGGACCTGCTCACGGCGG | 48 | ||
| RpCS.1258n | ATTGCAAAAAGTACAGTGAACA | |||
| Anaplasma spp. | 16S rRNA | Eh-out1 | TTGAGAGTTTGATCCTGGCTCAGAACG | 55 |
| Ehrlichia spp. | Eh-out2 | CACCTCTACACTAGGAATTCCGCTATC | ||
| Eh-gs1 | GTAATAACTGTATAATCCCTG | 55 | ||
| Eh-gs2 | GTACCGTCATTATCTTCCCTA | |||
| Coxiella spp. | 16S rRNA | Cox16SF1 | CGTAGGAATCTACCTTRTAGWGG | 55 |
| Cox16SR2 | GCCTACCCGCTTCTGGTACAATT | |||
| Cox16SF1 | CGTAGGAATCTACCTTRTAGWGG | 55 | ||
| Cox16SR1 | ACTYYCCAACAGCTAGTTCTCA |
Phylogenetic Analysis
The obtained DNA sequences were compared with those available in GenBank using the National Center for Biotechnology Information (NCBI; Bethesda, MD, United States) Basic Local Alignment Search Tool (BLAST) search engine1, and multiple-sequence alignment was performed using the ClustalW multiple alignment tool with the default parameters in the MEGA X. The phylogenetic analysis of gltA for SFGR, 16S rRNA for Anaplasma spp., or 16S rRNA for Coxiella spp. was performed using the maximum likelihood method based in MEGA X. Bootstrap values were estimated for 1,000 replicates (Hall, 2013; Kumar et al., 2016).
Multilocus Variable Number Tandem Repeat Analysis
Multilocus variable number tandem repeat analysis was performed in PCR targeting six highly variable loci, including ms23, ms24, ms27, ms28, ms33, and ms34 (Arricau-Bouvery et al., 2006). The forward and reverse primer sequences and PCR conditions were applied as described previously (Klaassen et al., 2009; Tilburg et al., 2012; Gonzalez-Barrio et al., 2016). A C. burnetii strain (Nine Mile) which was considered 9–27–4–6–9–5 for loci ms23–ms24–ms27–ms28–ms33–ms34 was used as the reference for normalization and for comparing the MLVA profiles obtained. The MLVA pattern of the isolates was compared in the database of MLVABank2 to check similarities with other isolates. Clustering of obtained MLVA profiles was performed with BioNumerics v.7.6 software (Applied Maths, Belgium).
Results
Taxonomic Classification
All adult hard ticks collected were identified as Rh. microplus (n = 516) based on morphological identifications and confirmed by species-specific PCR and sequencing assay. Fifteen pools of Rh. microplus DNA samples were finally analyzed using mNGS. Sequencing yielded between 6,166 and 7,273 million reads per pool, while all were of high quality (Clean_Q20 > 96%) (Supplementary Table 1).
Then, the presence of Rickettsia and Anaplasma in the pooled tick samples was identified by the taxonomic profiles at the genus level (Supplementary Table 2) and the 10 most abundant bacterial genera in pooled tick samples are as shown in Figure 2. Rickettsia spp. and Anaplasma spp. were abundant in all the sample pools. In addition, Pseudomonas spp. was most abundant in pool 4.
FIGURE 2.

Relative abundances of potential top 10 pathogens at the genus level in pooled Rhipicephalus microplus samples. The x-axis indicates pooled DNA sample names of Rh. microplus for metagenomic next-generation sequencing; the y-axis indicates relative abundance. “Others” indicates the sum of the relative abundance of all genera except the top 10.
Tick-Borne Pathogen Identification
By mNGS, the important pathogenic bacterial genera Rickettsia and Anaplasma were found in the pooled tick samples, and thus each tick sample was analyzed by the genus/group-specific PCR combined with sequencing in order to identify TBPs carried by it. In addition, Coxiella spp. was commonly found in ticks, and thus each tick sample was also analyzed by Coxiella-specific PCR.
As a result, 34.69% (179/516) of ticks were tested positive for at least one of the four pathogens/endosymbionts [Candidatus Rickettsia jingxinensis, Anaplasma marginale, C. burnetii, and Coxiella-like endosymbiont (CLE)] (Table 2). Ca. R. jingxinensis was the highest prevalence in these ticks (24.61%, 127/516), while the prevalence of A. marginale and C. burnetii were 13.18 and 3.10% in these ticks, respectively (Table 2).
TABLE 2.
Prevalence of tick-borne pathogens in individual tick.
| Pathogen |
Number of individual tick positive for single and coinfections
|
| Rhipicephalus microplus (n = 516) | |
| Single | |
| Candidatus Rickettsia jingxinensis | 74 (14.34%) |
| Anaplasma marginale | 27 (5.23%) |
| Coxiella burnetii | 14 (2.71%) |
| Coxiella-like endosymbiont | 5 (0.97%) |
| Double | |
| Ca. R. jingxinensis, A. marginale | 19 (3.68%) |
| A. marginale, C. burnetii | 2 (0.39%) |
| A. marginale, Coxiella-like endosymbiont | 4 (0.78%) |
| Ca. R. jingxinensis, Coxiella-like endosymbiont | 18 (3.49%) |
| Triple | |
| Ca. R. jingxinensis, A. marginale, Coxiella-like endosymbiont | 16 (3.10%) |
| Total | 179 (34.69%) |
Coinfections
In 179 TBP-positive ticks in the present study, 59 ticks (32.96%) were found to be coinfected with more than one TBP identified (Table 2). The dual coinfections with Ca. R. jingxinensis and A. marginale (8.33%, 43/516) were most frequent in Rh. microplus, while the dual coinfections with A. marginale and C. burnetii, the dual coinfection with A. marginale and CLE, and the dual coinfection with Ca. R. jingxinensis and CLE were detected in 0.39% (2/516), 0.78% (4/516), and 3.49% (18/516) of these ticks, respectively. The triple coinfections with Ca. R. jingxinensis, A. marginale, and CLE were detected in 3.10% (16/516) of these ticks (Table 2).
Phylogenetic Analysis
The gltA sequences of Ca. R. jingxinensis and 16S rRNA of A. marginale and C. burnetii in this study were 100% identical to those in GenBank, while CLE detected in the Rh. microplus collected in Weishan showed 97.57–99.20% nucleotide sequence identity to the known CLE strains and was most similar to the CLE strain (JQ480818.1) detected in Rhipicephalus turanicus in the 16S rRNA comparison. By phylogenetic analysis, Ca. R. jingxinensis detected in the present study were placed in a clade with Ca. R. jingxinensis (MH500217, MW114882, and MW114883) and an uncultured Rickettsia sp. clone NKGT-UR (MN842268) (Figure 3). The A. marginale identified was shown to be clustered with A. marginale (CP006847, CP023731, NC022784, and CP001079) and Anaplasma centrale str. Israel (CP001759) (Figure 4). C. burnetii was most close to C. burnetii CbuK (NC011528) and C. burnetii Dugway 5J108-111 (NC009727), while CLE detected in the present study was placed in a separated clade with the known CLE strains (Figure 5).
FIGURE 3.

Phylogenetic tree of Candidatus Rickettsia jingxinensis in ticks based on partial gltA gene sequence similarity. The sequences obtained in this study are indicated with a colored dot. Sequences were aligned using the MEGA X (Version 10.2.5) software package. Phylogenetic analysis was performed by the maximum likelihood method, and bootstrap values were estimated for 1,000 replicates.
FIGURE 4.

Phylogenetic tree of Anaplasma marginale in ticks based on partial 16S rRNA gene sequence similarity. The sequences obtained in this study are indicated with a colored dot. Sequences were aligned using the MEGA X (Version 10.2.5) software package. Phylogenetic analysis was performed by the maximum likelihood method, and bootstrap values were estimated for 1,000 replicates.
FIGURE 5.

Phylogenetic tree of Coxiella spp. in ticks based on partial 16S rRNA gene sequence similarity. The sequences obtained in this study are indicated with a colored dot. Sequences were aligned using the MEGA X (Version 10.2.5) software package. Phylogenetic analysis was performed by the maximum likelihood method and bootstrap values were estimated for 1,000 replicates.
Multilocus Variable Number Tandem Repeat Analysis Typing
In total, 14 tick DNA samples positive for C. burnetii and 10 DNA samples were characterized by a complete MLVA analysis, and not all C. burnetii DNA-positive samples could be characterized probably due to the poor quality and quantity of DNA. The allele codes of the identified MLVA type were 9–27–4–6–9–5 for loci ms23–ms24–ms27–ms28–ms33–ms34 and recognized as MLVA genotype 1, suggesting that these 10 strains belong to the same genotype and were closely related to tick Coxiella strains isolated from United States and human Coxiella strains isolated from United States, France, and Canada (Figure 6). A minimum spanning tree based on host origin of the MLVA analysis showed that the C. burnetii strains detected in the present study were clustered to the previously described genotypes found primarily in ticks and patients of Q fever from different regions of the globe (Figure 7).
FIGURE 6.

UPGMA cluster analysis of Coxiella burnetii genotypes using a six-multiple locus variable number tandem repeat analysis. All 34 selected samples are based on the MLVA-6 database (http://mlva.i2bc.paris-saclay.fr/mlvav4/genotyping/). The reference strain included in the tree is Nine Mile RSA493. Strain, source, geographical origin, and MLVA-6 type are indicated. The same genotype was coded with the same color. The hollow dots indicate the sample genotype obtained in this study.
FIGURE 7.
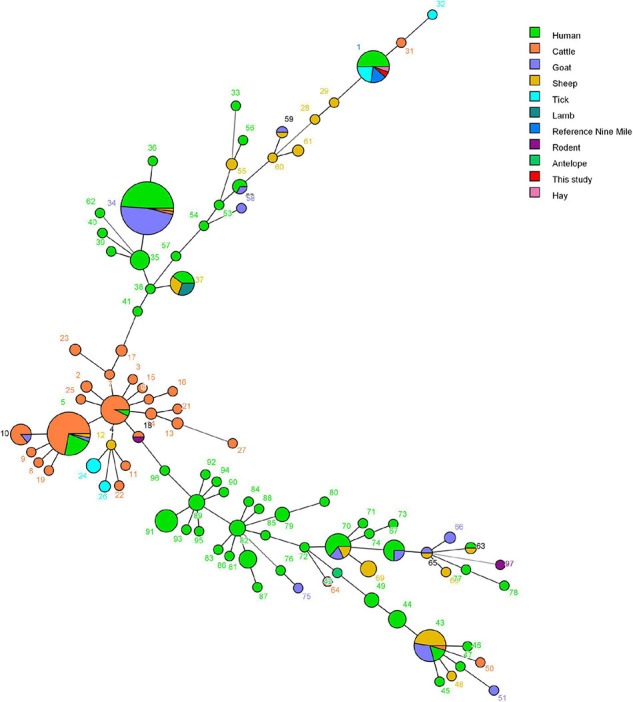
Minimum spanning tree of Coxiella burnetii strains using a six-multiple locus variable number tandem repeat analysis. The obtained MLVA genotypes identified in this study and data of 304 strains based on the MLVA-6 database (http://mlva.i2bc.paris-saclay.fr/mlvav4/genotyping/) were used. Each circle represents a unique genotype (1–97). The minimum spanning tree of the analyzed isolates provides information on the proportion of hosts of each identified genotype (see color index), and the size of the pie charts represents the number of isolates of the corresponding genotype.
Discussion
Although a variety of pathogens have been identified in ticks, the single infection and coinfection with multiple pathogens in Rh. microplus have rarely been investigated in China. In this study, we applied mNGS combined with nested PCR to survey TBPs in Rh. microplus feeding on cattle in Yunnan province and to analyze the phylogenetic characterization of TBPs detected in these ticks.
The pooled DNA samples of Rh. microplus collected were assayed by mNGS. Although each tick was surface sterilized and ultrapure water, sterile tubes, and filter tips were used and all operations were carried out in a biological safety cabinet during DNA extraction, bacteria like Pseudomonas, Enterobacter, and Rhizophagus were abundant in the pooled DNA samples of Rh. microplus when analyzed using mNGS. Pathogenic, environmental, and skin-associated bacteria have also been reported as highly abundant (Greay et al., 2018), and it is one of the possibilities that bacteria like Pseudomonas were predominant in the present study. Rickettsia spp. and Anaplasma spp. were also revealed at the genus level in pooled DNA samples. Moreover, Ca. R. jingxinensis, A. marginale, C. burnetii, and CLE were further identified in individual tick DNA by sequencing of PCR-amplified DNA fragments. Coxiella spp. was not identified in the pooled DNA sample via mNGS, and one of the possibilities is the degradation of nucleic acids during sequencing.
Spotted fever group rickettsias are devastating human infections, and no licensed vaccine is available (Liu et al., 2021). More than 20 species of Rickettsia are associated with SFGR, of which 16 are considered as human pathogens (Cohen et al., 2021). Ca. R. jingxinensis, one uncultured SFGR species, was identified in 24.61% of these ticks. The sequence of Ca. R. jingxinensis was first described in Haemaphysalis longicornis in Japan (Ishikura et al., 2003), and its presence in Ha. longicornis or Rh. microplus has been reported in China (Zou et al., 2011; Dong et al., 2014) and then was named based on its geographical origin in 2016 (Liu et al., 2016). Many Ca. R. jingxinensis-specific DNA sequences have been deposited in GenBank. Of these, a gltA sequence (KU853023) was recovered from a patient, suggesting its potential pathogenicity to humans (Guo et al., 2019a). Our analysis showed that the gltA of Ca. R. jingxinensis in Rh. microplus was 100% identical to that of Candidatus Rickettsia longicornii, suggesting that the two organisms should be recognized as one species, which is consistent with the previous report (Jiang et al., 2018). These results revealed that Ca. R. jingxinensis is widely distributed in China even in the world and its pathogenicity remains to be determined.
Anaplasma marginale, the causative agent of bovine anaplasmosis, can be transmitted by at least 20 species of ticks mainly in the genera Dermacentor and Rhipicephalus (Duan et al., 2020). The infection rate of A. marginale is determined by the level of rickettsemia in the host and the ability to infect the midgut of tick vector and undergo successful biological replication (Ueti et al., 2007). A. marginale has been detected in ticks from Ningxia, Hubei, and Henan provinces and the Qinghai-Tibet Plateau in China (Lu et al., 2017; Cui et al., 2018; Han et al., 2019; Duan et al., 2020). The presence of A. marginale has been detected in 13.18% of Rh. microplus in the present study, suggesting its wider geographical distribution in China.
Coxiella burnetii is the causative agent of Q fever, and this bacterium is highly infectious and classified as a category B biological weapon (Madariaga et al., 2003). In the 1950s, Q fever was first reported in China and then C. burnetii has been isolated from patients, livestock, wild mice, and ticks (Ni et al., 2020). The sporadic human Q fever cases and several small outbreaks of Q fever that occurred in leather factories or goat/sheep farms were reported in China (Yu, 2000). In 2018–2019, an epidemic of human Q fever in Zhuhai city of China was determined by mNGS (Huang et al., 2021). In the present study, C. burnetii were detected in 3.10% of Rh. microplus (14/516), while the previous reports show that it was detected in 12.50% of Dermacentor nuttalli (7/56), 2.79% of Dermacentor silvarum (11/394), 14.75% of Dermacentor niveus (9/61), and 22.65% of Hyalomma asiaticum (41/181) in China (El-Mahallawy et al., 2015; Li et al., 2020b,c; Ni et al., 2020), strongly demonstrating its wide distribution in China. C. burnetii has been detected in Rh. microplus in the Philippines, Thailand, and Mali before (Muramatsu et al., 2014; Diarra et al., 2017; Galay et al., 2020), and this is the first time that C. burnetii has been detected in Rh. microplus in China.
Genotyping C. burnetii from wildlife will help in tracing back clinical cases in humans directly exposed to wildlife. MLVA is nowadays the first-choice method to compare C. burnetii genotypes due to its powerful method to type C. burnetii from a diversity of hosts and geographic origins (Klaassen et al., 2009; Roest et al., 2011). In the present study, C. burnetii genotypes 1 were obtained using the MLVA-6-marker and genotype 1 was mainly found in strains from both patients and ticks. C. burnetii genotypes from ticks in the present study clustered mainly with C. burnetii genotypes from human Q fever cases in France, Canada, and United States (Figures 6, 7), suggesting that these strains identified in the ticks in Yunnan province were phylogenetically closely related to the strains from ticks and patients in different regions of the world. Interestingly, Q fever in humans in Yunnan province where our samples were collected has been reported (Fang et al., 2015). C. burnetii is a zoonotic pathogen transmitted from infected vertebrate animals to humans via contaminated aerosols (Graves et al., 2016). Although direct transmission of C. burnetii to humans through ticks has never been properly documented (Pacheco et al., 2013), tick bite cannot be ruled out and a previous report has suggested that Q fever may have been transmitted by tick bite (Beaman and Hung, 1989), and ticks may play a critical role in the transmission of C. burnetii among vertebrate animals (both domestic and native). To the best of our knowledge, this is the first time that C. burnetii found in China has been genotyped using MLVA.
A common characteristic of the various tick species is the presence of bacterial endosymbionts, typically bacterial members of Coxiella, Rickettsia, and Francisella genera, some of which are closely related to vertebrate pathogens (Tsementzi et al., 2018). CLEs are uncultured and relatively common in the microbiota of various tick species around the world and affect the development, nutrition, chemical defense, or reproduction of the hosts (Ben-Yosef et al., 2020). CLEs form multiple subclusters in the cluster of the genus Coxiella in phylogenetic analysis (Duron et al., 2015). CLE has not been reported in Rh. microplus before, and in the present study, CLEs were detected in 8.33% of Rh. microplus, similar to the prevalence level of CLE in Rhipicephalus sanguineus in North America and Europe (Ueti et al., 2007). More importantly, CLE detected in Rh. microplus collected from Weishan shared 97.57–99.20% of 16S rRNA sequence identity with the known CLE strains, forming a separated clade in phylogenetic analysis.
Conclusion
This study provides a better understanding of TBPs in Rh. microplus in Yunnan and the presence of TBP coinfections in Rh. microplus, reporting for the first time that C. burnetii had been found in Rh. microplus in China. MLVA data-based phylogenetic analysis showed that the C. burnetii strains detected in Rh. microplus in Yunnan belonged to MLVA genotype 1, which is closely related to previously described genotypes found primarily in ticks and patients from different regions in the world, and suggesting a potential public health threat to the people living close to the natural foci of C. burnetii. It is important to plan future surveys, applying molecular methods for investigation of these TBPs in livestocks and farmers closely related to Rh. microplus in Yunnan province of China.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/, ERS6645636.
Author Contributions
JJ and XX conceived and designed the study and drafted the manuscript. JZ, PH, YS, and QY performed the sample collection, tick species identification, and laboratory work. JJ, XO, and YY performed the experimental data analysis. BW and QY edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
Funding
This work was supported by the Beijing Natural Science Foundation (grant number 5204039), the National Key Research and Development Program of China (No. 2019YFC1200500), and the National Natural Science Foundation of China (grant numbers 31970178 and 32000139).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.736484/full#supplementary-material
References
- Andersson M. O., Vichova B., Tolf C., Krzyzanowska S., Waldenstrom J., Karlsson M. E. (2017). Co-infection with Babesia divergens and Anaplasma phagocytophilum in cattle (Bos taurus), Sweden. Ticks Tick Borne Dis. 8 933–935. 10.1016/j.ttbdis.2017.08.005 [DOI] [PubMed] [Google Scholar]
- Arricau-Bouvery N., Hauck Y., Bejaoui A., Frangoulidis D., Bodier C. C., Souriau A., et al. (2006). Molecular characterization of Coxiella burnetii isolates by infrequent restriction site-PCR and MLVA typing. BMC Microbiol. 6:38. 10.1186/1471-2180-6-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaman M. H., Hung J. (1989). Pericarditis associated with tick-borne Q fever. Aust. N. Z. J. Med. 19 254–256. 10.1111/j.1445-5994.1989.tb00258.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yosef M., Rot A., Mahagna M., Kapri E., Behar A., Gottlieb Y. (2020). Coxiella-like endosymbiont of Rhipicephalus sanguineus is required for physiological processes during ontogeny. Front. Microbiol. 11:493. 10.3389/fmicb.2020.00493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Xie C., Huson D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12 59–60. 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- Chen Z., Liu Q., Liu J. Q., Xu B. L., Lv S., Xia S., et al. (2014). Tick-borne pathogens and associated co-infections in ticks collected from domestic animals in central China. Parasit. Vectors 7:237. 10.1186/1756-3305-7-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C., Fu W., Ju W., Yang L., Xu N., Wang Y. M., et al. (2016). Diversity of spotted fever group Rickettsia infection in hard ticks from Suifenhe, Chinese-Russian border. Ticks Tick Borne Dis. 7 715–719. 10.1016/j.ttbdis.2016.02.023 [DOI] [PubMed] [Google Scholar]
- Chitimia L., Lin R. Q., Cosoroaba I., Wu X. Y., Song H. Q., Yuan Z. G., et al. (2010). Genetic characterization of ticks from southwestern Romania by sequences of mitochondrial cox1 and nad5 genes. Exp. Appl. Acarol. 52 305–311. 10.1007/s10493-010-9365-9 [DOI] [PubMed] [Google Scholar]
- Cohen R., Finn T., Babushkin F., Paran Y., Ben Ami R., Atamna A., et al. (2021). Spotted fever group Rickettsioses in Israel, 2010-2019. Emerg. Infect. Dis. 27 2117–2126. 10.3201/eid2708.203661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotillard A., Kennedy S. P., Kong L. C., Prifti E., Pons N., Le Chatelier E., et al. (2013). Dietary intervention impact on gut microbial gene richness. Nature 500 585–588. 10.1038/nature12480 [DOI] [PubMed] [Google Scholar]
- Cui Y., Wang X., Zhang Y., Yan Y., Dong H., Jian F., et al. (2018). First confirmed report of outbreak of theileriosis/anaplasmosis in a cattle farm in Henan, China. Acta Trop. 177 207–210. 10.1016/j.actatropica.2017.08.029 [DOI] [PubMed] [Google Scholar]
- Diarra A. Z., Almeras L., Laroche M., Berenger J. M., Kone A. K., Bocoum Z., et al. (2017). Molecular and MALDI-TOF identification of ticks and tick-associated bacteria in Mali. PLoS Negl. Trop. Dis. 11:e0005762. 10.1371/journal.pntd.0005762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Chen X. P., Liu N., Dumler S. J., Zhang Y. Z. (2014). Co-circulation of multiple species of Rickettsiales bacteria in one single species of hard ticks in Shenyang, China. Ticks Tick Borne Dis. 5 727–733. 10.1016/j.ttbdis.2014.05.011 [DOI] [PubMed] [Google Scholar]
- Duan D. Y., Liu G. H., Cheng T. Y. (2020). Microbiome analysis of the saliva and midgut from partially or fully engorged female adult Dermacentor silvarum ticks in China. Exp. Appl. Acarol. 80 543–558. 10.1007/s10493-020-00478-2 [DOI] [PubMed] [Google Scholar]
- Duron O., Noel V., Mccoy K. D., Bonazzi M., Sidi-Boumedine K., Morel O., et al. (2015). The recent evolution of a maternally-inherited endosymbiont of ticks led to the emergence of the Q fever pathogen, Coxiella burnetii. PLoS Pathog. 11:e1004892. 10.1371/journal.ppat.1004892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mahallawy H. S., Lu G., Kelly P., Xu D., Li Y., Fan W., et al. (2015). Q fever in China: a systematic review, 1989-2013. Epidemiol. Infect. 143 673–681. 10.1017/s0950268814002593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L. Q., Liu K., Li X. L., Liang S., Yang Y., Yao H. W., et al. (2015). Emerging tick-borne infections in mainland China: an increasing public health threat. Lancet Infect. Dis. 15 1467–1479. 10.1016/s1473-3099(15)00177-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q., Liang S., Jia H., Stadlmayr A., Tang L., Lan Z., et al. (2015). Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 6:6528. 10.1038/ncomms7528 [DOI] [PubMed] [Google Scholar]
- Fu L., Niu B., Zhu Z., Wu S., Li W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28 3150–3152. 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galay R. L., Talactac M. R., Ambita-Salem B. V., Chu D. M. M., Costa L., Salangsang C. M. A., et al. (2020). Molecular detection of Rickettsia Spp. and Coxiella Burnetii in cattle, water buffalo, and Rhipicephalus (Boophilus) Microplus ticks in Luzon island of the Philippines. Trop. Med. Infect. Dis. 5:54. 10.3390/tropicalmed5020054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Barrio D., Hagen F., Tilburg J. J., Ruiz-Fons F. (2016). Coxiella burnetii genotypes in Iberian wildlife. Microb. Ecol. 72 890–897. 10.1007/s00248-016-0786-9 [DOI] [PubMed] [Google Scholar]
- Graves S. R., Jackson C., Hussain-Yusuf H., Vincent G., Nguyen C., Stenos J., et al. (2016). Ixodes holocyclus tick-transmitted human pathogens in north-eastern new South Wales, Australia. Trop. Med. Infect. Dis. 1:4. 10.3390/tropicalmed1010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greay T. L., Gofton A. W., Paparini A., Ryan U. M., Oskam C. L., Irwin P. J. (2018). Recent insights into the tick microbiome gained through next-generation sequencing. Parasit. Vectors 11:12. 10.1186/s13071-017-2550-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W. P., Wang Y. H., Lu Q., Xu G., Luo Y., Ni X., et al. (2019a). Molecular detection of spotted fever group rickettsiae in hard ticks, northern China. Transbound. Emerg. Dis. 66 1587–1596. 10.1111/tbed.13184 [DOI] [PubMed] [Google Scholar]
- Guo W. P., Zhang B., Wang Y. H., Xu G., Wang X., Ni X., et al. (2019b). Molecular identification and characterization of Anaplasma capra and Anaplasma platys-like in Rhipicephalus microplus in Ankang, Northwest China. BMC Infect. Dis. 19:434. 10.1186/s12879-019-4075-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B. G. (2013). Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 30 1229–1235. 10.1093/molbev/mst012 [DOI] [PubMed] [Google Scholar]
- Han R., Yang J. F., Mukhtar M. U., Chen Z., Niu Q. L., Lin Y. Q., et al. (2019). Molecular detection of Anaplasma infections in ixodid ticks from the Qinghai-Tibet Plateau. Infect. Dis. Poverty 8:12. 10.1186/s40249-019-0522-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Ma J., Jiao J., Li C., Chen L., Zhu Z., et al. (2021). The epidemic of Q fever in 2018 to 2019 in Zhuhai city of China determined by metagenomic next-generation sequencing. PLoS Negl. Trop. Dis. 15:e0009520. 10.1371/journal.pntd.0009520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D. H., Auch A. F., Qi J., Schuster S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17 377–386. 10.1101/gr.5969107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikura M., Ando S., Shinagawa Y., Matsuura K., Hasegawa S., Nakayama T., et al. (2003). Phylogenetic analysis of spotted fever group rickettsiae based on gltA, 17-kDa, and rOmpA genes amplified by nested PCR from ticks in Japan. Microbiol. Immunol. 47 823–832. 10.1111/j.1348-0421.2003.tb03448.x [DOI] [PubMed] [Google Scholar]
- Jiang J., An H., Lee J. S., O’guinn M. L., Kim H. C., Chong S. T., et al. (2018). Molecular characterization of Haemaphysalis longicornis-borne rickettsiae, Republic of Korea and China. Ticks Tick Borne Dis. 9 1606–1613. 10.1016/j.ttbdis.2018.07.013 [DOI] [PubMed] [Google Scholar]
- Karlsson F. H., Fak F., Nookaew I., Tremaroli V., Fagerberg B., Petranovic D., et al. (2012). Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 3:1245. 10.1038/ncomms2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson F. H., Tremaroli V., Nookaew I., Bergstrom G., Behre C. J., Fagerberg B., et al. (2013). Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498 99–103. 10.1038/nature12198 [DOI] [PubMed] [Google Scholar]
- Klaassen C. H., Nabuurs-Franssen M. H., Tilburg J. J., Hamans M. A., Horrevorts A. M. (2009). Multigenotype Q fever outbreak, the Netherlands. Emerg. Infect. Dis. 15 613–614. 10.3201/eid1504.081612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Tamura K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Chatelier E., Nielsen T., Qin J., Prifti E., Hildebrand F., Falony G., et al. (2013). Richness of human gut microbiome correlates with metabolic markers. Nature 500 541–546. 10.1038/nature12506 [DOI] [PubMed] [Google Scholar]
- Li J., Jia H., Cai X., Zhong H., Feng Q., Sunagawa S., et al. (2014). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 32 834–841. 10.1038/nbt.2942 [DOI] [PubMed] [Google Scholar]
- Li J., Kelly P., Guo W., Zhang J., Yang Y., Liu W., et al. (2020a). Molecular detection of Rickettsia, Hepatozoon, Ehrlichia and SFTSV in goat ticks. Vet. Parasitol. Reg. Stud. Reports 20:100407. 10.1016/j.vprsr.2020.100407 [DOI] [PubMed] [Google Scholar]
- Li J., Li Y., Moumouni P. F. A., Lee S. H., Galon E. M., Tumwebaze M. A., et al. (2020b). First description of Coxiella burnetii and Rickettsia spp. infection and molecular detection of piroplasma co-infecting horses in Xinjiang Uygur Autonomous Region, China. Parasitol. Int. 76:102028. 10.1016/j.parint.2019.102028 [DOI] [PubMed] [Google Scholar]
- Li W., Godzik A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22 1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- Li Y., Li J., Chahan B., Guo Q., Zhang Y., Moumouni P. F. A., et al. (2020c). Molecular investigation of tick-borne infections in cattle from Xinjiang Uygur Autonomous Region, China. Parasitol. Int. 74:101925. 10.1016/j.parint.2019.05.003 [DOI] [PubMed] [Google Scholar]
- Liu H., Li Q., Zhang X., Li Z., Wang Z., Song M., et al. (2016). Characterization of rickettsiae in ticks in northeastern China. Parasit. Vectors 9:498. 10.1186/s13071-016-1764-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhou C., Su Z., Chang Q., Qiu Y., Bei J., et al. (2021). Endothelial exosome plays a functional role during rickettsial infection. mBio 12 e007 69–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M., Tian J. H., Yu B., Guo W. P., Holmes E. C., Zhang Y. Z. (2017). Extensive diversity of rickettsiales bacteria in ticks from Wuhan, China. Ticks Tick Borne Dis. 8 574–580. 10.1016/j.ttbdis.2017.03.006 [DOI] [PubMed] [Google Scholar]
- Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madariaga M. G., Rezai K., Trenholme G. M., Weinstein R. A. (2003). Q fever: a biological weapon in your backyard. Lancet Infect. Dis. 3 709–721. 10.1016/s1473-3099(03)00804-1 [DOI] [PubMed] [Google Scholar]
- Muramatsu Y., Usaki N., Thongchai C., Kramomtong I., Kriengsak P., Tamura Y. (2014). Seroepidemiologic survey in Thailand of Coxiella burnetii infection in cattle and chickens and presence in ticks attached to dairy cattle. Southeast Asian J. Trop. Med. Public Health 45 1167–1172. [PubMed] [Google Scholar]
- Ni J., Lin H., Xu X., Ren Q., Aizezi M., Luo J., et al. (2020). Coxiella burnetii is widespread in ticks (Ixodidae) in the Xinjiang areas of China. BMC Vet. Res. 16:317. 10.1186/s12917-020-02538-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H. B., Almeida M., Juncker A. S., Rasmussen S., Li J., Sunagawa S., et al. (2014). Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32 822–828. [DOI] [PubMed] [Google Scholar]
- Ostfeld R. S., Brunner J. L. (2015). Climate change and Ixodes tick-borne diseases of humans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370:20140051. 10.1098/rstb.2014.0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco R. C., Echaide I. E., Alves R. N., Beletti M. E., Nava S., Labruna M. B. (2013). Coxiella burnetii in ticks, Argentina. Emerg. Infect. Dis. 19 344–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N., Yang F., Li A., Prifti E., Chen Y., Shao L., et al. (2014). Alterations of the human gut microbiome in liver cirrhosis. Nature 513 59–64. [DOI] [PubMed] [Google Scholar]
- Qin X. R., Han F. J., Luo L. M., Zhao F. M., Han H. J., Zhang Z. T., et al. (2018). Anaplasma species detected in Haemaphysalis longicornis tick from China. Ticks Tick Borne Dis. 9 840–843. 10.1016/j.ttbdis.2018.03.014 [DOI] [PubMed] [Google Scholar]
- Roest H. I., Ruuls R. C., Tilburg J. J., Nabuurs-Franssen M. H., Klaassen C. H., Vellema P., et al. (2011). Molecular epidemiology of Coxiella burnetii from ruminants in Q fever outbreak, the Netherlands. Emerg. Infect. Dis. 17 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Shen S., Wu H., Zhang Y., Deng F. (2021). Metagenomic profiling of viruses associated with Rhipicephalus microplus ticks in Yunnan Province, China. Virol. Sin. 36 623–635. 10.1007/s12250-020-00319-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulyok K. M., Hornok S., Abichu G., Erdelyi K., Gyuranecz M. (2014). Identification of novel Coxiella burnetii genotypes from Ethiopian ticks. PLoS One 9:e113213. 10.1371/journal.pone.0113213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilburg J. J., Rossen J. W., Van Hannen E. J., Melchers W. J., Hermans M. H., Van De Bovenkamp J., et al. (2012). Genotypic diversity of Coxiella burnetii in the 2007-2010 Q fever outbreak episodes in the Netherlands. J. Clin. Microbiol. 50 1076–1078. 10.1128/jcm.05497-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsementzi D., Castro Gordillo J., Mahagna M., Gottlieb Y., Konstantinidis K. T. (2018). Comparison of closely related, uncultivated Coxiella tick endosymbiont population genomes reveals clues about the mechanisms of symbiosis. Environ. Microbiol. 20 1751–1764. 10.1111/1462-2920.14104 [DOI] [PubMed] [Google Scholar]
- Ueti M. W., Reagan J. O., Jr., Knowles D. P., Jr., Scoles G. A., Shkap V., Palmer G. H. (2007). Identification of midgut and salivary glands as specific and distinct barriers to efficient tick-borne transmission of Anaplasma marginale. Infect. Immun. 75 2959–2964. 10.1128/iai.00284-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaumourin E., Vourc’h G., Gasqui P., Vayssier-Taussat M. (2015). The importance of multiparasitism: examining the consequences of co-infections for human and animal health. Parasit. Vectors 8:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar E., Farrant G. K., Follows M., Garczarek L., Speich S., Audic S., et al. (2015). Ocean plankton. Environmental characteristics of Agulhas rings affect interocean plankton transport. Science 348:1261447. [DOI] [PubMed] [Google Scholar]
- Wen B., Cao W., Pan H. (2003). Ehrlichiae and ehrlichial diseases in china. Ann. N. Y. Acad. Sci. 990 45–53. 10.1111/j.1749-6632.2003.tb07335.x [DOI] [PubMed] [Google Scholar]
- Wilson K. D., Elston D. M. (2018). What’s eating you? Ixodes tick and related diseases, part 3: coinfection and tick-bite prevention. Cutis 101 328–330. [PubMed] [Google Scholar]
- Yang X., Gao Z., Wang L., Xiao L., Dong N., Wu H., et al. (2021). Projecting the potential distribution of ticks in China under climate and land use change. Int. J. Parasitol. 51 749–759. 10.1016/j.ijpara.2021.01.004 [DOI] [PubMed] [Google Scholar]
- Yu S. (2000). [Progress in the study of Q fever in China]. Zhonghua Liu Xing Bing Xue Za Zhi 21 456–459. [PubMed] [Google Scholar]
- Zhang G., Zheng D., Tian Y., Li S. (2019). A dataset of distribution and diversity of ticks in China. Sci. Data 6:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y., Wang Q., Fu Z., Liu P., Jin H., Yang H., et al. (2011). Detection of spotted fever group Rickettsia in Haemaphysalis longicornis from Hebei Province, China. J. Parasitol. 97 960–962. 10.1645/ge-2751.1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/, ERS6645636.