

Abstract
Radiation combined injury is defined as an ionizing radiation exposure received in combination with other trauma or physiological insults. The range of radiation threats we face today includes everything from individual radiation exposures to mass casualties resulting from a terrorist nuclear incident, and many of these exposure scenarios include the likelihood of additional traumatic injury. Radiation combined injury sensitizes target organs and cells and exacerbates acute radiation syndrome. Organs and cells with high sensitivity to combined injury are the skin, the hematopoietic system, the gastrointestinal tract, spermatogenic cells, and the vascular system. Among its many effects, radiation combined injury results in decreases in lymphocytes, macrophages, neutrophils, platelets, stem cells, and tissue integrity; activation of the iNOS/NF-κB/NF-IL6 and p53/Bax pathways; and increases in DNA single and double strand breaks, TLR signaling, cytokine concentrations, bacterial infection, and cytochrome c release from mitochondria to cytoplasm. These alterations lead to apoptosis and autophagy and, as a result, increased mortality. There is a pressing need to understand more about the body’s response to combined injury in order to be able to develop effective countermeasures, since few currently exist. In this review, we summarize what is known about how combined injury modifies the radiation response, with a special emphasis on DNA damage/repair, signal transduction pathways, apoptosis, and autophagy. We also describe current and prospective countermeasures relevant to the treatment and prevention of combined injury.
Keywords: DNA damage, apoptosis, autophagy, radiation injury, combined injury, free radical
Introduction
The potential for harmful radiation exposure has increased dramatically since the development of nuclear weapons during World War II. The number of nations with the capability to produce nuclear weapons is ever-increasing. The potential for nuclear accidents and accidental exposures will become greater with the expected proliferation of nuclear power plant construction to meet growing demands for energy more friendly to the environment. The widespread use of radioisotopes in medicine increases the dissemination of radioactive materials and patient exposures. And of course, the frighteningly real possibility that terrorist groups could use nuclear weapons or other radiological weapons poses a serious risk of mass casualties. The fact that more than 50% of cancer patients receive radiotherapy at some point during the course of their disease (5) represents another significant source of exposure as normal tissues are subjected to radiation injury.
Those charged with responding to such threats have modeled many of these potential exposure scenarios, but for the most part they have assumed radiation exposures alone. It is unrealistic however to assume that accidental radiation injury will occur in the absence of other injuries—especially when considering terrorist incidents. It has become abundantly clear that radiation exposure combined with many kinds of other injuries, ranging from trauma to infection, often results in a negative synergistic response more harmful than the sum of the individual injuries. We have only recently begun to appreciate the practical consequences of combined injury and to understand that the body’s response to combined injury may be different from the responses to radiation or physical injury alone.
In this review we aim to summarize our current understanding of how the physiological response to radiation is modified when other injuries are present. We focus on responses especially relevant to health effects: DNA damage and repair; signal transduction processes; free radical-mediated apoptosis and autophagy; and bacterial infection. We also discuss the potential effectiveness of current radiation response-altering drugs that could also be used to treat or prevent combined injury exposures as well as the potential for new drug development.
Radiation Injury
Ionizing radiation is defined as any type of electromagnetic radiation (e.g., X-rays or gamma rays) or particulate radiation (e.g., neutrons or alpha particles) that has sufficient energy to ionize atoms or molecules; that is, to eject electrons from their outer orbits. In considering the effects of radiation on biological systems, it is important to distinguish the different types of ionizing radiation in terms of their linear energy transfer (LET), a measure of the amount of energy transferred to a substance as the radiation passes through it. It is classified to two types of radiation: low linear energy transfer (low-LET) radiations and high linear energy transfer (high-LET) radiations. Table 1 summarizes the types and basic physical characteristics of radiation in these categories. Low-LET radiations include gamma rays, X-rays, beta particles; high-LET radiations include neutrons, alpha particles, and heavy-particle cosmic rays (21). Radiation exposures of concern to health officials cover the full LET spectrum, and exposure could come from external sources as well as internalized radioactive substances (via inhalation, ingestion, or wound contamination).
Table 1.
Characteristics of nuclear radiations (52)
| Name | Relative Mass | Electric Charge | Emitted by | Range in Air | Tissue Penetration | Radiation Stopped by |
|---|---|---|---|---|---|---|
| Alpha | 7,300 | +2 | Unfissioned uranium and plutonium | 5 cm | First layer of skin | Clothing paper |
| Beta | 1 | −1 | Fission products | 12 m | Several layers of skin | Clothing |
| Gamma* | 0 | 0 | Fission products | 100 m | Total body | Several feet of concrete or earth |
| Neutron | 1,830 | 0 | Emitted only during fission | 100 m | Total body | Several feet of concrete or earth |
For the purpose of this presentation X-rays are considered along with gamma rays. X-ray wavelength bands largely overlap those of gamma rays, and they interact at least mechanistically like gamma rays. They are now usually distinguished only by their origin.
The most radiation-sensitive organs include the hematopoietic system (16), the gastrointestinal (GI) system (53), skin (56, 58), and the vascular system (63, 68). A dose range (1–7 Gy in human) of ionizing radiation poses a risk of damage to the hematopoietic system, leading to decreases in blood cells and platelet counts and increases in susceptibility to infection and hemorrhage (8, 76) while high-dose whole-body irradiation (≥8 Gy in humans) causes acute GI syndrome in addition to hematopoietic complications. The GI effects manifest as loss of intestinal crypts and breakdown of the GI mucosal barrier. High doses can also induce GI hemorrhage, endotoxemia, bacteremia, anorexia, nausea, vomiting, diarrhea, and loss of electrolytes and fluid (73). There is no clear demarcation between the hematopoietic and GI syndromes; they represent a continuum of damage. There is hematopoietic damage that influences GI damage at higher radiation and there is likely some GI damage even at lower radiation doses.
Skin injury from radiation burns is characterized by loss of epidermis and dermis (3, 31), reduction of skin stem cells, and impairment of cell communication and cutaneous integrity, a factor that may trigger the failure of other organ systems (55). Vascular endothelium is also damaged (63). Concomitant and interdependent injuries to various organ systems can lead to multi-organ dysfunction (MOD) and multi-organ failure (MOF) and death can occur as a result.
Combined Injury
Large-scale radiation exposure events in history have shown that irradiated victims are also often subjected to other trauma such as wounds or burns. Combined injuries were observed at Hiroshima and Nagasaki, Japan, where 60–70% of victims received thermal burns concurrent with radiation injury, (26, 35). At the Chernobyl reactor meltdown, 10% of 237 victims exposed to radiation received thermal burns as well (3). In animal models of combined injury including mice (31, 43), rats (1, 71, 75), guinea pigs (37), dogs (6, 66), and swine (4), burns, wounds, and infections usually increase mortality after otherwise non-lethal radiation exposures. In rodents, radiation exposure combined with burns, wounds, or infections decreases survival compared to radiation exposure alone (27, 31, 44, 71). Radiation injury also delays wound closure times (31, 47). Consequences of combined injury include acute myelosuppression, immune system inhibition, fluid imbalance, macro/microcirculation failure, massive cellular damage, and disruption of vital organ functions, which, as is the case with radiation exposure alone, can lead to MOD and MOF, the most frequent causes of death after combined injury (36, 42, 78). Although the mode of combined injury death is fairly clear, the molecular events that lead to combined injury-enhanced mortality remain poorly understood.
It has been well-characterized that a large radiation dose received over a short period of time can trigger a complicated pattern of physiological responses referred to as acute radiation syndrome (ARS). When an ionizing radiation dose sufficient to induce ARS is combined with concurrent additional physical trauma, the response induced by ionizing radiation is sensitized and a non-lethal radiation dose can be transformed to a lethal one. Concurrent trauma exacerbates radiation-induced white blood cell depletion, activates signal transduction pathways, increases cytokine and chemokine production, and increases susceptibility to bacterial infection (31). The changes observed after radiation combined injury appear at various levels—nucleus, cytoplasm, tissues, organs, and system—and at various time after injury. Whether cells survive or die after ionizing radiation alone or ionizing radiation in combination with other trauma depends on the number and severity of lesions, which determines the extent to which signal transduction pathways responsible for triggering cell death by apoptosis and autophagy are activated.
Recent research has identified key molecular intermediaries involved in radiation injury. Among the many molecules activated by radiation injury, inducible nitric oxide synthase (iNOS) and nitric oxide (NO) play important roles in radiation injury-induced apoptosis (32) and autophagy (18). The promoter region of the iNOS gene contains motifs of many transcriptional factors (33). Radiation injury increases iNOS and its transcription factors such as nuclear factor-κB (NF-κB) and Kruppel-like factor 6 (KLF-6) resulting in increased NO production that leads to caspase-mediated apoptosis (32) and protein nitration-mediated autophagy (18). Radiation injury increases concentrations of interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and interferon-γ (IFN-γ) in human blood (23); IL-1β, IL-3, IL-6, and G-CSF in mouse blood (31, 64, 72); and IL-6 and IL-8 in CNS of non-human primates (19). Cytokines are responsible for stimulating nuclear factor-IL6 (NF-IL6), which subsequently binds to the consensus motif within the iNOS promoter (ranging from +10 to −300 bp upstream of the TATA box) to activate iNOS gene expression (12). In addition, overproduction of IL-6, NO, or nitrogen reactive species can cause dysfunction of the GI barrier (22, 54, 77), which can allow bacteria to enter systemic organs. Radiation combined injury amplifies these changes (31).
The order in which radiation and wounding is received can affect the lethality of combined exposure. Survival from radiation injury improves when wounding occurs prior to radiation exposure; while wounding that occurs after radiation injury results in decreased survival compared to radiation injury alone (45, 46). However, Reid et al. (66) observed similar lethality regardless of order in a model combining radiation exposure with burn trauma.
DNA Damage and Repair
The degree of chromosomal damage is proportional to the absorbed dose of radiation, and high- and low-LET ionizing radiation produce different types of DNA damage. Low-LET ionizing radiation (gamma and X-rays) causes DNA damage mostly indirectly via formation of free radicals, while high-LET ionizing radiation (neutrons, alpha particles, cosmic ray heavy particles) is more likely to cause direct DNA damage that is more complex and difficult to repair than damage from low-LET radiation (65). Acute exposure to ionizing radiation causes damage to macromolecules as well as increased mitochondria-dependent generation of reactive oxygen species and reactive nitrogen species, with subsequent cell cycle checkpoint arrest, apoptosis, and autophagy (18, 32).
Ionizing radiation induces base damage, single strand breaks (SSBs), double strand breaks (DSBs), and DNA crosslinks. DSBs are the primary lethal lesion (7, 60). Two repair pathways, homologous recombination (HR) and non-homologous end joining (NHEJ) efficiently repair DSBs. The majority (80%-90%) of DSB repair involves NHEJ (38, 67).
It is not clear if ionizing radiation combined with wound trauma causes a greater amount of DNA damage than ionizing radiation alone. Studies in this area are ongoing in our laboratory to fill in this data gap.
Signal Transduction Pathway Activation in Response to DNA Damage
DNA repair proteins such as RAD50, MRE11, NBS1, RAD17, RAD1, RAD9, and HUS1 bind to ionizing radiation-induced DSBs to form complexes that are detected by ataxia telangiectasia mutated (ATM) kinases. DSBs stimulate ATM phosphorylation within minutes and the phosphorylated ATM is stable for many hours. MDC1, 53BP, BRCA1, and TopBP1 mediate the phosphorylation of CHK2 by ATM and related kinases. The phosphorylated CHK2 then phosphorylates p53 and CDC25. Phosphorylated p53 arrests the cell cycle at G1/S and phosphorylated CDC25 arrests the cell cycle at both S and G2/M to allow DNA repair [see review (25) and Fig. 1].
Fig. 1. Simplified representation of the DNA-damage-induced checkpoint response.
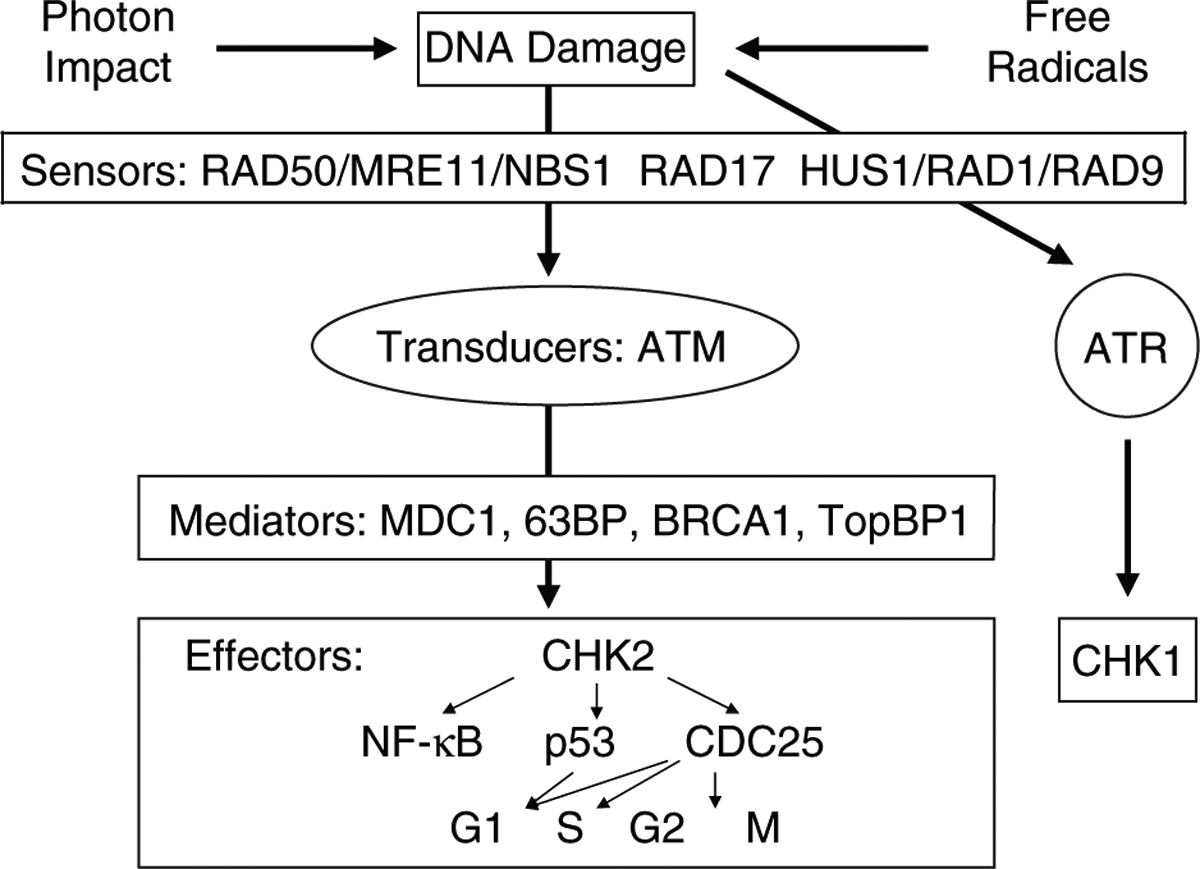
Ionizing radiation induces DNA breaks. After the detection of a given damage by sensor proteins, this signal is transduced to the effector protein CHK2 via the tranducer protein ATM. Depending on the phase of the cell cycle the cell is in, this can lead to activation of p53 and inactivation of CDC25, which eventually leads to cell cycle arrest. Mediator proteins mostly are cell cycle specific and associate with damage sensors, signal transducers, or effectors at particular phases of the cell cycle and, thus, help provide signal transduction specificity. The effect of UV light is via the transducer protein ATR and the effector protein CHK1. MRE11: meiotic recombination 11; NBS1: Nijmegen breakage syndrome 1; ATM: ataxia telangiectasa mutated; ATR: ataxia telangiectasa related; MDC1: mediator of DNA damage checkpoint 1; 63BP: p63 binding protein; BRCA1: breast cancer 1; TopBP1: topoisomerase binding protein 1; CHK1: check 1; CHK2: check 2; CDC25: cell division cycle 25; G1: gap 1; S: synthesis; G2: gap 2; M: mitosis.
Phosphorylated ATM can also induce phosphorylation of the histone variant H2AX at serine 139, generating γ-H2AX (15). Immunocytochemical assays with antibodies recognizing γ-H2AX have become the gold standard for detection of DSBs because there is close to a 1:1 relationship between the numbers of DSBs and γ-H2AX foci formed. Furthermore, the rate of DSB repair correlates with the rate of loss of γ-H2AX foci (69). γ-H2AX triggers the CHK2 signal transduction pathway that activates p53 and CDC25. It should be noted that phosphorylated ATM also directly phosphorylates p53, which transcriptionally activates the CDK inhibitor p21 and arrests the cell cycle at G1/S (41).
Recent evidence demonstrates DSB-dependent ATM phosphorylation activates NF-κB (20, 28). Phosphorylated ATM binds to and phosphorylates IKKγ in the nucleus. The complex exits the nucleus and associates with IKKα and IKKβ. The IKK complex releases NF-κB from its inhibitors, IκBα and IκBβ, and unbound NF-κB is then free to move into the nucleus and regulate target genes. The NF-κB signaling network includes DNA repair, cell cycle check regulation, mitochondrial antioxidants, survival and apoptosis, and cytokine and chemokine expression in response to ionizing radiation-induced damage (31).
Additional trauma such as wounding potentiates gene expression induced by ionizing radiation. Table 2 shows that 60Co γ-irradiated mice display increases in expression of p21, Bax, DDB2, and Gadd45α genes. Mice treated with 60Co γ-irradiation and wound trauma exhibit further increases in p21, Bax, and DDB2, but not Gadd45α. The mechanisms underlying this enhancement in radiation combined-injured mice remain unclear. Studies of DSBs, ATM, γ-H2AX, and p53 in radiation combined-injured mice should help define the mechanisms involved.
Table 2.
Gene expression in bone marrow after radiation injury and radiation combined injury
| Gene | Relative to Sham | |||
|---|---|---|---|---|
| Sham | Wound | RI | CI | |
| p21 | 1.0 | 0.4a | 19.7b | 35.9c |
| Bax | 1.0 | 0.5a | 8.6b | 17.5c |
| Bcl-2 | 1.0 | 1.4 | 1.6 | 2.0 |
| Bax/Bcl-2 | 1.0 | 0.4a | 5.5b | 8.6c |
| DDB2 | 1.0 | 1.1 | 5.7b | 7.9c |
| Gadd45α | 1.0 | 1.1 | 5.2b | 4.6b |
| TERT | 1.0 | 0.1a | 0.7b | 0.3c |
B6F2D1/J female mice received 8.5 Gy 60Co gamma (RI) or 8.5 Gy followed by 15% total body surface area skin wound trauma 1 h after radiation (CI). The skin wound was to remove panniculus carnosus muscle and overlying skin (23.5 ± 1.1 mm in length and 14.9 ± 0.7 mm in width; see ref. 31). Gene expression in bone marrow 24 h after RI or CI was measured using real-time PCR. Each group contained 6 mice.
P < 0.05 vs. Sham, RI, and CI;
P < 0.05 vs. Sham, Wound, and CI;
P < 0.05 vs. Sham, Wound, and RI; determined by Chi-square test.
DDB2: DNA damage-binding protein 2; Gadd45α: Growth arrest and DNA-inducible protein 45α; TERT: Telomerase reverse transcriptase
Free Radical-Mediated Apoptosis
In mammalian cells, low-LET ionizing radiation but not high-LET ionizing radiation generates free radicals, including reactive oxygen species (ROS) and reactive nitrogen species (RNS), via mitochondrial mechanisms (13, 24). Consistent with this observation, free radical scavengers or hypoxia treatment can help prevent low-LET ionizing radiation injury. Free radicals are required for the physiological function of cells, but overproduction of free radicals damages cellular components (Fig. 2). ROS are formed from hydrolysis of water in the nucleus and the cytoplasm. ROS in the nucleus cause DNA damage while ROS in the cytoplasm activate multiple signal transduction pathways involved in growth, apoptosis, and autophagy (18, 31, 32, 57). These injuries can lead to cell-cycle arrest, transformation, and cell death.
Fig. 2. Simple representation of the multi-organ dysfunction and multi-organ failure and resultant mortality.
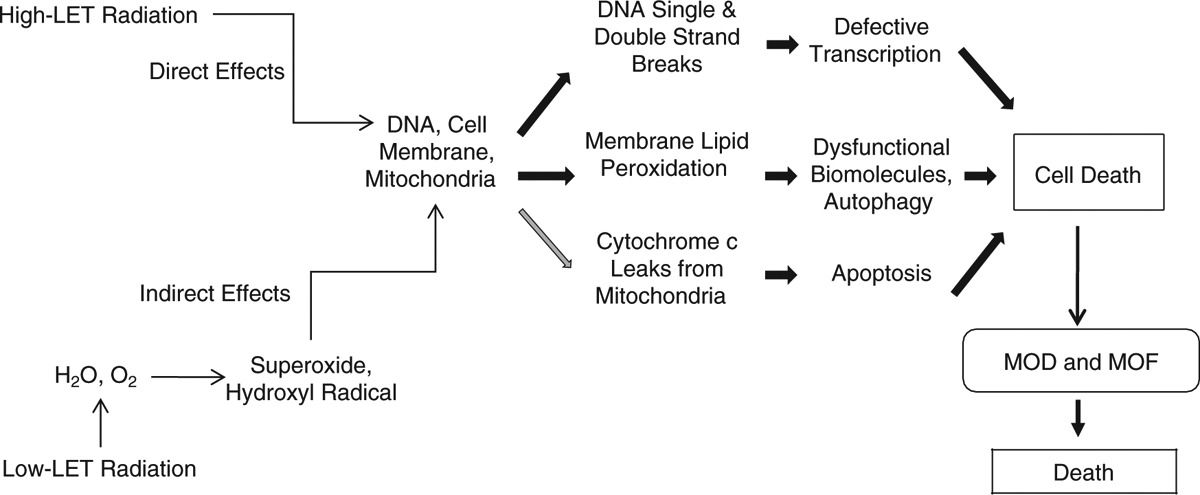
LET: linear energy transfer; MOD: multi-organ dysfunction; MOF: multi-organ failure.
While ROS are short-lived and extremely reactive, RNS are longer-lived and more specific in the reactions they undergo (57). NO reacts with superoxide to form the peroxynitrite anion, resulting in oxidative stress (31). This results in the release of cytochrome c from the mitochondria to the cytoplasm and the subsequent conjugation of the cytochrome c with caspase-9 and Apaf-1 to form apoptosomes that activate caspase-3 and caspase-7. Activated caspase-3 then activates caspase-2, -6, -8, and -10, resulting in apoptosis (33).
Because exposure to ionizing radiation combined with wound trauma enhances iNOS gene expression and iNOS protein levels, due to activation of both NF-κB and NF-IL6 and increases in serum cytokines (31), greater production of peroxynitrite anion and more protein nitration is anticipated relative to that seen after radiation exposure alone. Apoptosis can thus be expected to occur to a greater extent after radiation combined injury. Peroxynitrite anion also leads to more LC3-mediated autophagy (see below).
Ionizing radiation activates PI3K/AKT and mitogen-activated protein kinase (MAPK) pathways (10). The PI3K/AKT pathway activates anti-apoptotic proteins (17, 30). The MAPK pathways include extra-cellular signal-regulated kinase 1/2 (ERK1/2) activity (2), JNK (48), and p38 (34). The former is anti-apoptosis, whereas the latter two are pro-apoptosis. It is not clear if radiation combined with wound trauma enhances these pathways.
Free Radical-Mediated Autophagy
A growing body of evidence suggests that ionizing radiation induces programmed cell death mediated not only by the Bcl-2 family of proteins and caspase proteases (type I cell death) but also autophagy-dependent programmed cell death type 2 (PCDT2) (50). The role of ionizing radiation-induced autophagy in normal cells, especially in the cells of dose-sensitive tissues such as small intestine, is a subject that requires attention.
Autophagy (or autophagocytosis) is a lysosomal mechanism of degradation of self-constituents that is evolutionary conserved and occurs in various eukaryotic cells (14, 40, 70). Three forms of autophagy have been distinguished, based on how intracellular material is delivered to lysosomes: chaperone-mediated autophagy, microautophagy, and macroautophagy (49). Macroautophagy (mAG) is the most common form of autophagy; under normal conditions mAG is responsible for the routine bulk degradation of redundant or defective organelles, long-lived proteins, large macromolecules, and pathogens. mAG thus provides a homeostatic balance between biosynthetic and biodegradative activities and innate immunity. mAG is characterized by the formation of autophagosomes (phagophores), in which portions of cytoplasm are sequestered, cargo packaged within a double membrane-enclosed vacuole, and transported to lysosomes or late endosomes for biodegradation (29, 59).
One of the crucial steps of this multistage process is conversion of light chain protein 3 type I (LC3-I) (also known as ubiqitin-like protein Atg8) to type II (LC3-II) either by a redox sensitive Atg4 serine pro-tease or by E-1 and E-2 like enzymes Atg7 and Atg3 (9, 62, 74). LC3 protein is considered a marker for autophagosomes (9, 62).
mAG is induced in response to certain conditions including exposure to ionizing radiation. Induction of mAG in response to cytotoxic stress can be either protective or detrimental. It has been recently shown that PCDT2 is related to the damage-regulated autophagy modulator (DRAM), the death associated protein kinase (DAPK), autophagic massive elimination of apoptotic mitochondria, and oxidative activation of Atg4 serine protease, which can occur via free radical mechanisms activated by ionizing radiation. Although the free-radical species produced by ionizing radiation have short-term effects, the subsequent activation of pro-oxidant pathways, such as the iNOS cascade, can potentiate and prolong oxidation and thus extend up-regulation of mAG.
LC3-II is identified in host small intestine-defense cells such as Paneth cells, which are considered to be relatively resistant to radiation and can therefore help maintain the GI barrier after otherwise lethal insults. We assessed the dynamics of LC3 protein to track mAG in ileal crypt cells after ionizing radiation or radiation combined injury. We found that there is a larger increase in LC3-II in CD15-positive Paneth cells at day 7 after radiation combined injury than after radiation injury alone (Figs. 3 and 4). The increase is correlated with iNOS activation, NO production, lipid peroxidation, and protein nitration. The up-regulation of autophagy is accompanied by a decrease in protein-protein interaction between LC3, heat shock protein 70 kDa, and Bcl-2-associated anthanogene-1 (18).
Fig. 3. Radiation combined injury induces more LC3-II than radiation in mouse ileum.
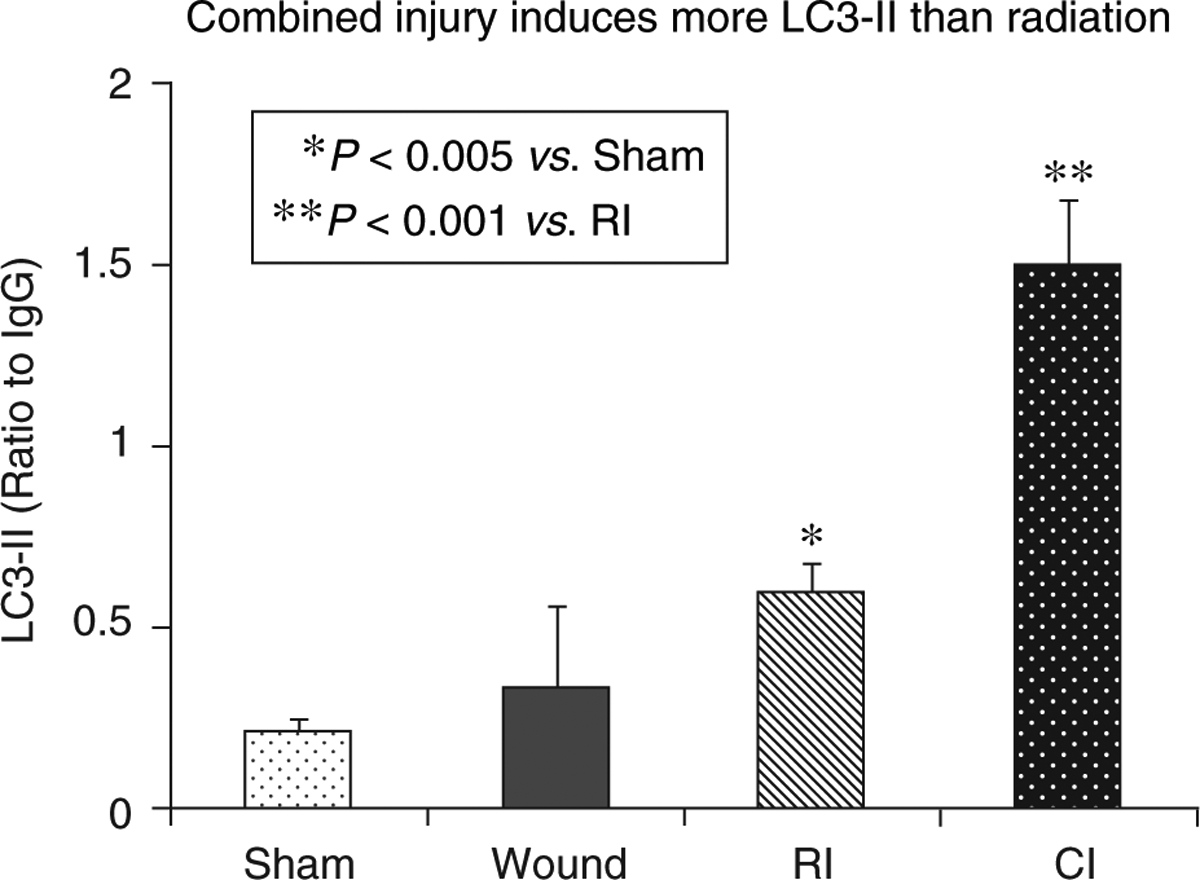
B6D2F1/J female mice received 60Co gamma 9.75 Gy followed immediately by 15% total body surface skin-wound trauma. Ileal lysates were prepared 7 d after sham-treatment (Sham), wounding (Wound), radiation-injury (RI), and radiation combined injury (CI). Each groups had 6 mice. Western blots were performed to quantitate the LC3-II level.
Fig. 4. Radiation combined injury induces more LC3-II than radiation in crypt cells of mouse ileum.
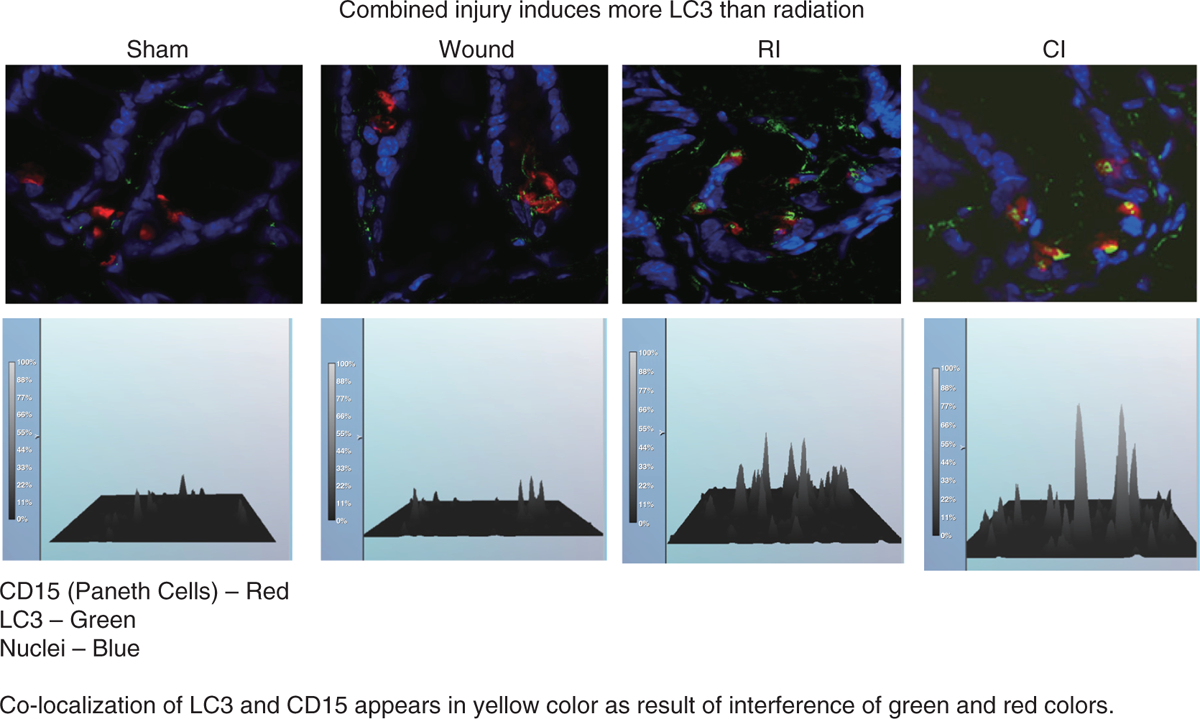
B6D2F1/J female mice received 60Co gamma 9.75 Gy followed immediately by 15% total body surface skin-wound trauma. Ileum was prepared 7 d after sham-treatment (Sham), wounding (Wound), radiation-injury (RI), and radiation combined injury (CI). Each groups had 6 mice. Immunofluorescent staining was performed to identify and quantitate LC3-II in CD15-positive Paneth cells.
Bacterial Infection Activates Signal Transduction Pathways
It is evident that overproduction of IL-6, NO, or nitrogen reactive species can cause dysfunction of the GI barrier (22, 54, 77), resulting in bacterial entry into the systemic organs. In our laboratory, we collected heart blood and liver tissue from recently deceased or euthanized sham, wounded, radiation-injured, or radiation combined-injured mice and cultured the tissue to determine if facultative bacteria had entered the circulation. Since tissues from healthy animals are normally sterile (except for occasional, transient bacteremia), the presence of bacteria in detectable numbers is indicative of systemic infection.
In sham-treated mice no bacteria were found in the tissues tested. In wounded and radiation-injured mice Enterococcus sp. and Staphylococcus sp. were only occasionally detected. However, in radiation combined-injured mice, Enterococcus sp., Staphylococcus sp., Bacillus sp., and Lactobacillus sp. were common, and the same bacterial species were also isolated from ileum. Bacteremia in mice receiving wounds alone was transient and present only until day 3 after wounding. On the other hand, systemic infection was demonstrated in radiation combined-injured mice through day 17 and sporadically in radiation-injured mice through day 25. In radiation combined injured-mice, Bacillus and Lactobacillus were isolated within the first 8 d after radiation combined injury. The data imply that mice receiving wounds alone were able to resist infection. While systemic infection occurred in both radiation combined-injured mice and mice receiving radiation alone, it occurred several days sooner in the radiation combined-injured mice.
Bacteremia induced increases in serum cytokine concentrations, which further promoted iNOS over-expression and activation in radiation combined-injured mice (31). It is important to note when interpreting these data that luminal microbiota composition may influence the host’s intestinal response to radiation and may change in those developing postirradiation diarrhea (61). For this reason, it is not surprising to observe variations in the intestinal response either to radiation or radiation combined with wound trauma.
Radiation Combined-Injury Countermeasures
A synergistic effect between radiation and traumatic injury has been reported in mice (31, 43), rats (1, 71, 75), guinea pigs (37), dogs (6, 66), and swine (4). Key features of radiation combined injury include: (a) shock, which occurs earlier and is more frequent and severe compared to simple radiation injury, often becoming the main cause of death at times soon after injury; (b) dramatic suppression of hematopoiesis and the immune system, which negatively affects prognosis after radiation combined injury; (c) extensive and severe GI damage, such as mechanical and immune barrier breakdown, which leads to dysfunction in absorption and secretion and increased risk of infection; and (d) delayed wound healing—often double the healing time of wounding alone.
Since the mechanisms of radiation combined injury appear to be more complicated than the mechanisms of the individual injuries alone, it can be expected that the treatments are also not as straightforward. DiCarlo et al. (11) suggests that the complexity of the response makes them pessimistic that any effective treatments amenable for use in a mass casualty scenario can be found. However, the search for pharmacological countermeasures for radiation combined injury has shown some promise.
Zou et al. (78) reports that cervical sympathetic nerve block once a day for 14 d after radiation combined injury significantly decreases mortality (51). Ledney and Elliott (43) reported that the nonspecific immunomodulator S-TDCM given i.p. immediately after radiation combined injury, along with systemic and topical application of gentamicin, improves survival. They also reported that syngeneic bone marrow transplantation increases the survival of mice with combined injury. Shah et al. (71) reported that human ghrelin attenuated organ injury and improves survival in a rat model of radiation combined with sepsis.
The medical response to a mass casualty scenario will likely always be different from how a small number of first responders to a radiation-contaminated area or radiation therapy patients are treated. It is clearly unrealistic in mass casualty situations to undertake cervical sympathetic ganglia blocks, bone marrow transplants, or even the intravenous administrations of drugs. Intramuscular injections, orally administered drugs, and perhaps subcutaneous injections (39) may be the most complex treatments available to mass casualty victims. Countermeasures for radiation attacks or nuclear accidents that must be given prior to radiation exposure are impractical since it is unlikely such events will occur with adequate warning; however, they could prove useful if radiation exposures are likely or if they are planned, as for radiation therapy. Successful countermeasure development must therefore address the requirements of a variety of very different scenarios.
Conclusion
Radiation combined with wound trauma results in decreases in lymphocytes, macrophages, neutrophils, platelets, cell adhesion molecules, tissue integrity, and stem cells, but increases in activity of the iNOS/NF-κB/NF-IL6 and p53/Bax pathways, TLR signaling, cytokine concentrations, bacterial infection, cytochrome c release from mitochondria to cytoplasm, and DNA single and double strand breaks. These alterations lead to apoptosis and autophagy, resulting in mortality. Radiation injury combined with burns, infection, or fractures may be mediated by mechanisms similar to those observed after radiation injury combined with wound trauma. Countermeasures available for radiation combined injury are currently very limited (Fig. 5), so the development of agents for prevention, mitigation, and treatment remains a pressing need.
Fig. 5. Radiation combined injury attenuates the normal defenses.
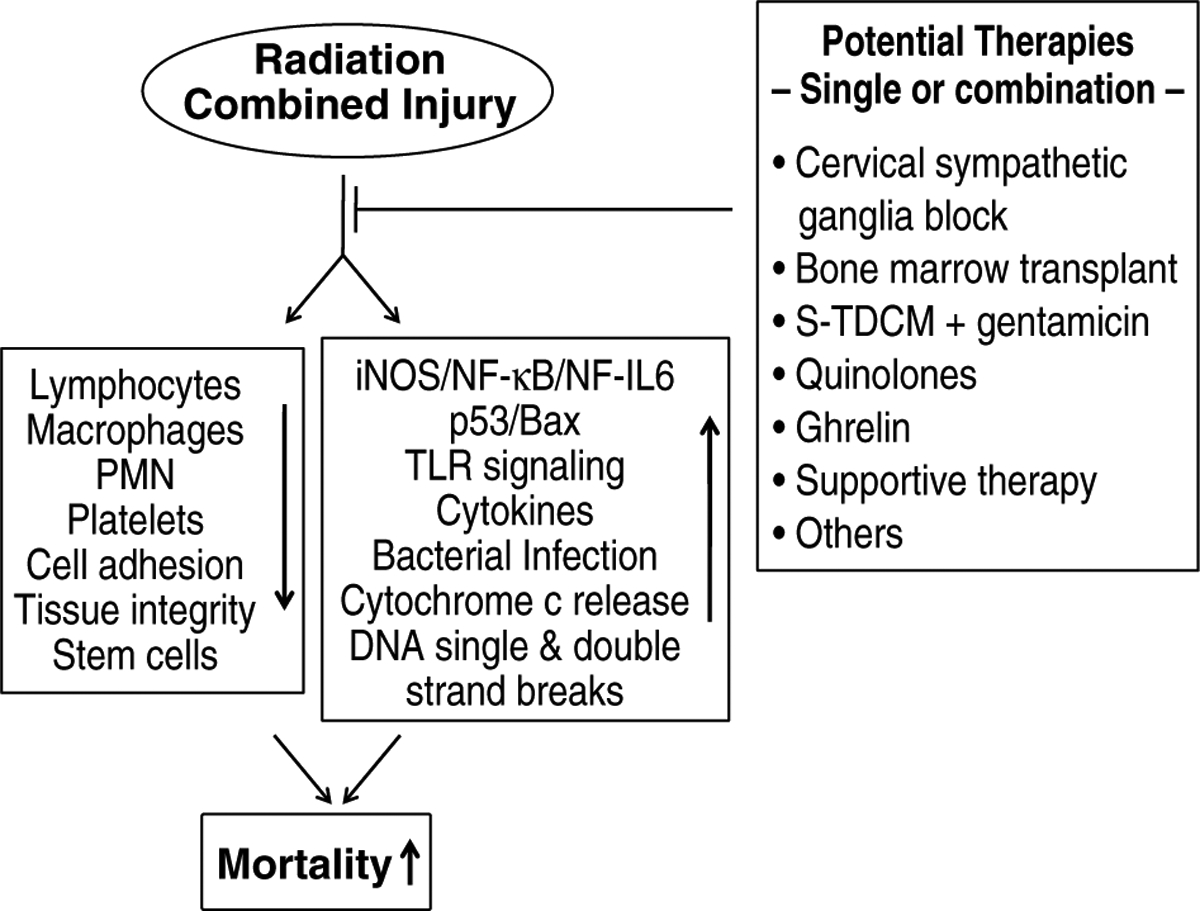
Various interventions to treat radiation combined injury may be used alone or in combination to improve the chance of survival in severely injured patients.
Acknowledgments
The authors thank Dr. David E. McClain for his editorial assistance. The opinions or assertions contained herein are the authors’ private views and are not to be construed as official or reflecting the views of the Uniformed Services University of the Health Sciences or the US Department of Defense.
References
- 1.Alpen EL and Sheline GE The combined effects of thermal burns and whole body X irradiation on survival time and mortality. Ann. Surg 140: 113–118, 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Astsaturov I, Cohen RB and Harari P Targeting epidermal growth factor receptor signaling in the treatment of head and neck cancer. Expert Rev. Anticancer Ther 6: 1179–1193, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Barabanova AV Significance of beta-radiation skin burns in Chernobyl patients for the theory and practice of radiopathology. Vojnosanit Pregl. 63: 477–480, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Baxter H, Drummond JA, Stephens-Newsham LG and Randall RG Studies on acute total body ionizing radiation in animals. I. Effect of streptomycin following exposure to a thermal burn and ionizing radiation. Plast. Reconstr. Surg 12: 439–445, 1953. [DOI] [PubMed] [Google Scholar]
- 5.Bentzen SM Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat. Rev. Cancer 6: 702–713, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Brooks JW, Evans EI, Ham WT Jr. and Reid JD The influence of external body radiation on mortality from thermal burns. Ann. Surg 136: 533–545, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burma S, Chen BP and Chen DJ Role of non-homologous and end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst.) 5: 1042–1048, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Coleman CN, Stone HB, Moulder JE and Pellmar TC Medicine. Modulation of radiation injury. Science 304: 693–694, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T and Ryan KM DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126: 121–134, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Dent P, Reardon DB, Park JS, Bowers G, Logsdon C, Valerie K and Schmidt-Ullrich R Radiation-induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation-induced cell death. Mol. Biol. Cell 10: 2493–2506, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiCarlo AL, Hatchett RJ, Kaminski JM, Ledney GD, Pellmar TC, Okunieff P and Ramakrishnan N Medical countermeasures for radiation combined injury: radiation with burn, blast, trauma and/or sepsis. Report of an NIAID Workshop, March 26–27, 2007. Radiat. Res 169: 712–721, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dlaska M and Weiss G Central role of transcription factor NF-IL6 for cytokines and iron-mediated regulation of murine inducible nitric oxide synthase expression. J. Immunol 162: 6171–6177, 1999. [PubMed] [Google Scholar]
- 13.Epperly MW, Sikora CA, DeFilippi SJ, Gretton JA, Zhan Q, Kufe DW and Greenberger JS Managanese superoxide dismutase (SOD2) inhibits radiation-induced apoptosis by stabilization of the mitochondrial membrane. Radiat. Res 157: 568–577, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Fengsrud M, Sneve ML, Overbye A and Seglen PO Structural aspects of mammalian autophagy. In: Autophagy, edited by Klionsky DJ, Georgetown, TX: Landes Bioscience, 2004, pp. 11–25. [Google Scholar]
- 15.Fernandez-Capetillo O, Lee A, Nussenzweig M and Nussenzweig A H2AX: the histone guardian of the genome. DNA Repair (Amst.). 3: 959–967, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Fliedner TM, Graessel D, Meineke V and Dorr H Pathological principles underlying the blood cell concentration responses used to assess the severity of effect after accidental whole-body radiation exposure: an essential basis for an evidence-based clinical triage. Exp. Hematol 35: 8–16, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Gomez-Vicente V, Doonan F, Donovan M and Cotter TG Induction of BIM(EL) following growth factor withdrawal is a key event in caspase-dependent apoptosis of 661W photoreceptor cells. Eur. J. Neurosci 24: 981–990, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Gorbunov NV and Kiang JG Up-regulation of autophagy in the small intestine Paneth cell in response to total body γ-irradiation. J. Pathol 219: 242–252, 2009. [DOI] [PubMed] [Google Scholar]
- 19.Gourmelon P, Marquette C, Agay D, Mathieu J and Clarencon D Involvement of the central nervous system in radiation-induced multi-organ dysfunction and/or failure. Brit. Inst. Radiol 27 (suppl): 62–68, 2005. [Google Scholar]
- 20.Habraken Y and Piette J NF-kappaB activation by double-strand breaks. Biochem. Pharmacol 72: 1132–1141, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Hall EJ and Giacca AJ In: Radiobiology for the Radiologist. Sixth Ed., Philadelphia, PA: Lippincott Williams & Wilkins, pp. 252–268, 2006. [Google Scholar]
- 22.Han X, Fink MP and Delude RL Proinflammatory cytokines cause NO*-dependent and -independent changes in expression and localization of tight junction proteins in intestinal epithelial cells. Shock 19: 229–237, 2003. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi T, Morishita Y, Kudo Y, Kusunoki Y, Hayashi I, Kasagi F, Hakoda M, Kyoizumi S and Nakachi K Long-term effects of radiation dose on inflammatory markers in atomic bomb survivors. Am. J. Med 118: 83–86, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi T, Hayashi I, Shinohara T, Morishita Y, Nagamura H, Kusunoki Y, Kyoizumi S, Seyama T and Nakachi K Radiation-induced apoptosis of stem/progenitor cells in human umbilical cord blood is associated with alterations in reactive oxygen and intracellular pH. Mutat. Res 556: 83–91, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Houtgraff JH, Versmissen J and van der Giessen WJ A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc. Revasc. Med 7: 165–172, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Iijima S Pathology of atomic bomb casualties. Acta Pathol. Jpn 32(Suppl. 2): 237–270, 1982. [PubMed] [Google Scholar]
- 27.Jacob A, Shah KG, Wu R and Wang P Ghrelin as a novel therapy for radiation combined injury. Mol. Med 2010. January 24. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janssens S and Tschopp J Signals from within: the DNA-damage-induced NF-kB response. Cell Death Differ. 13: 773–784, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19: 5720–5728, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kao GD, Jiang Z, Fernandes AM, Gupta AK and Maity A Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem 282: 21206–21212, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiang JG, Jiao W, Cary LH, Mog SR, Elliott TB, Pellmar TC and Ledney GD Wound trauma increases radiation-induced mortality by activation of iNOS pathway and elevation of cytokine concentrations and bacterial infection. Radiat. Res 173: 319–332, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kiang JG, Smith JT and Agravante NG Geldanamycin analog 17-DMAG inhibits iNOS and caspases in gamma irradiated human T cells. Radiat, Res. 172: 321–330, 2009. [DOI] [PubMed] [Google Scholar]
- 33.Kiang JG and Tsen K-T Biology of hypoxia. Chinese J. Physiol 49: 223–233, 2006. [PubMed] [Google Scholar]
- 34.Kim BJ, Ryu SW and Song BJ JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem 281: 21256–21265, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Kishi HS Effects of the “special bomb”: recollection of a neurosurgeon in Hiroshima. Neurosurgery 47: 441–446, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Koenig KL, Goans RE, Hatchett RJ, Mettler FA Jr., Schumacher TA, Noji EK and Jarrett DG Medical treatment of radiological casualties: current concepts. Ann. Emerg. Med 45: 643–652, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Korlof B Infection of burns, I. A bacteriological and clinical study of 99 cases. II. Animal experiments: burns and total body x-ray radiation. Acta Chir. Scand Suppl 209: 1–144, 1956. [PubMed] [Google Scholar]
- 38.Kuhne M, Riballo E, Rief N, Rothkamm K, Jeggo PA and Lobrich M A double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Res. 64: 500–508, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Kumar KS, Kiang JG, Whitnall MH and Hauer-Jensen M Perspectives in radiological and nuclear countermeasures. In: Medical Consequences of Nuclear Warfare. In press, 2010. [Google Scholar]
- 40.Kundu M and Thompson CB Autopagy: Basic principles and relevance to disease. Annu. Rev. Pathol. Mecha. Dis 3: 247–255, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Kurz EU and Lees-Miller SP DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst.) 3: 889–900, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Lausevic Z, Lausevic M, Trbojevic-Stankovic J, Krstic S and Stojimovic B Predicting multiple organ failure in patients with severe trauma. Can. J. Surg 51: 97–102, 2008. [PMC free article] [PubMed] [Google Scholar]
- 43.Ledney GD and Elliott TB Combined injury: factors with potential to impact radiation dose assessments. Health Phys. 98: 145–152, 2010. [DOI] [PubMed] [Google Scholar]
- 44.Ledney GD, Elliott TB and Moore MM Modulations of mortality by tissue trauma and sepsis in mice after radiation injury. In: The Biological Basis of Radiation Protection Practice, edited by Mossman KL and Mills WA Baltimore, MD: Williams and Wilkins, 1992, pp. 202–217. [Google Scholar]
- 45.Ledney GD Exum ED and Sheehy PA Survival enhanced by skin-wound trauma in mice exposed to 60Co radiation. Experientia 37: 193–194, 1981. [DOI] [PubMed] [Google Scholar]
- 46.Ledney GD Exum, E.D., Stewart, D.A., Gelston, H.M. Jr. and Weinberg, S.R. Survival and hematopoietic recovery in mice after wound trauma and whole-body irradiation. Exp. Hematol 10(Suppl 12): 263–278, 1982.6175531 [Google Scholar]
- 47.Ledney GD, Stewart DA, Exum ED and Sheehy PA Skin wound-enhanced survival and myelocytopoiesis in mice after whole-body ionizing radiation. Acta Radiol. Oncol 20: 29–38, 1981. [DOI] [PubMed] [Google Scholar]
- 48.Lee ER, Kim JY, Kang YJ, Kim BW, Choi HY, Jeong MY and Cho SG Interplay between PI3K/Akt and MAPK signaling pathways in DNA-damaging drug-induced apoptosis. Biochim. Biophys. Acta 1763: 958–968, 2006. [DOI] [PubMed] [Google Scholar]
- 49.Levine B and Klionsky DJ Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6: 463–477, 2004. [DOI] [PubMed] [Google Scholar]
- 50.Lockshin RA and Zakeri Z Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol 36: 2405–2419, 2004. [DOI] [PubMed] [Google Scholar]
- 51.Lu J, Shi Z, Su Y, Cheng T and Du Z Effect of cervical sympathetic ganglia block on the mortality of mice with combined radiation and burn injury and its possible mechanism. Chinese J. Clin. Rehabil 10: 177–180, 2006. [Google Scholar]
- 52.Luckett LW and Vesper BE Radiological considerations in medical operations. In: Medical Consequences of Nuclear Warfare, edited by Walker RI and Cerveny TJ Falls Church, VA: TMM publications, 1989, part 1, vol. 2, pp. 227–244. [Google Scholar]
- 53.MacNaughton WK Review article: new insights into the pathologenesis of radiation-induced intestinal dysfunction. Aliment Pharmacol. Ther 14: 523–528, 2000. [DOI] [PubMed] [Google Scholar]
- 54.Mazzon E, De Sarro A, Caputi AP and Cuzzocrea S Role of tight junction derangement in the endothelial dysfunction elicited by exogenous and endogenous peroxynitrite and poly(ADP-ribose) synthetase. Shock 18: 434–439, 2002. [DOI] [PubMed] [Google Scholar]
- 55.Meineke V The role of damage to the cutaneous system in radiation-induced multi organ failure. Brit. J. Radiol. Suppl 27: 85–99, 2005. [Google Scholar]
- 56.Meistrich ML and Kangasniemi M Hormone treatment after irradiation stimulates recovery of rat spermatogenesis from surviving spermatogonia. J. Androl 18: 80–87, 1997. [PubMed] [Google Scholar]
- 57.Mikkelsen RB and Wardman P Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 22: 5734–5754, 2003. [DOI] [PubMed] [Google Scholar]
- 58.Muller K and Meineke V Radiation-induced alterations in cytokine production by skin cells. Exp. Hematol 35: 96–104, 2007. [DOI] [PubMed] [Google Scholar]
- 59.Ohsumi Y Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell. Biol 2: 211–216, 2001. [DOI] [PubMed] [Google Scholar]
- 60.Olive PL Impact of the comet assay in radiobiology. Mutat. Res 681: 13–23, 2007. [DOI] [PubMed] [Google Scholar]
- 61.Packey CD and Ciorba MA Microbial influences on the small intestinal response to radiation injury. Curr. Opin. Gastroenterol 26: 88–94, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D and Yahalom J A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 61: 439–444, 2001. [PubMed] [Google Scholar]
- 63.Pena LA, Fuks Z and Kolesnick RN Radiation-induced apoptosis of endothelial cells in the murine central nervous system: protection by fibroblast growth factor and sphingomyelinase deficiency. Cancer Res. 60: 321–327, 2000. [PubMed] [Google Scholar]
- 64.Peterson VM, Adamovicz JJ, Elliott TB, Moore MM, Madonna GS, Jackson WE 3rd, Ledney GD and Gause WC Gene expression of hematoregulatory cytokines is elevated endogenously after sublethal gamma irradiation and is differentially enhanced by therapeutic administration of biologic response modifiers. J. Immunol 153: 2321–2330, 1994. [PubMed] [Google Scholar]
- 65.Pogozelski WK, Xapsos MA and Blakely WF Quantitative assessment of the contribution of clustered damage to DNA double-strand breaks induced by 60Co gamma rays and fission neutrons. Radiat. Res 151: 442–448, 1999. [PubMed] [Google Scholar]
- 66.Reid JD, Brooks JW, Ham WT and Evans EI The influence of X-radiation on mortality following thermal flash burns: The site of tissue injury as a factor determining the type of invading bacteria. Ann. Surg 142: 844–850, 1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA and Lobrich M A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell 16: 715–724, 2004. [DOI] [PubMed] [Google Scholar]
- 68.Rodemann HP and Blaese MA Responses of normal cells to ionizing radiation. Semin. Radiat. Oncol 17: 81–88, 2007. [DOI] [PubMed] [Google Scholar]
- 69.Rothkamm K and Lobrich M Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl. Acad. Sci. U.S.A 100: 5057–5062, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schmidt D and Munz C Innate and adaptive immunity through autophagy. Immunity 27: 11–21, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shah KG, Wu R, Jacob A, Blau SA, Ji Y, Dong W, Marini CP, Ravikumar TS, Coppa GF and Wang P Human ghrelin ameliorates organ injury and improves survival after radiation injury combined with severe sepsis. Mol. Med 15: 407–414, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singh VK, Grace MB, Jacobsen KO, Chang CM, Parekh VI, Inal CE, Shafran RL, Whitnall AD, Kao TC, Jackson WE 3rd. and Whitnall MH Administration of 5-androstenediol to mice: Pharmacokinetics and cytokine gene expression. Exp. Mol. Pathol 84: 178–188, 2008. [DOI] [PubMed] [Google Scholar]
- 73.Somosy Z, Horvath G, Telbisz A, Rez G and Palfia Z Morphological aspects of ionizing radiation response of small intestine. Micron 33: 167–178, 2002. [DOI] [PubMed] [Google Scholar]
- 74.Tanida I, Ueno T and Kominami E LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol 36: 2503–2518, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Valeriote FA and Baker DG The combined effects of thermal trauma and x-ray radiation on early mortality. Radiat. Res 22: 693–702, 1964. [PubMed] [Google Scholar]
- 76.Waselenko JK, MacVitte TJ, Blakely WF Pesik N, Wiley AL, Dickerson WE, Tsu H, Confer DL, Coleman CN, Seed T, Lowry P, Armitage JO and Dainiak N Medical management of the acute radiation syndrome: recommendations of the Strategic National Stockpile Radiation Working Group. Ann. Intern. Med 140: 1037–1051, 2004. [DOI] [PubMed] [Google Scholar]
- 77.Yang R, Han X, Uchiyama T, Watkins SK, Yaguchi A, Delude RL and Fink MP IL-6 is essential for development of gut barrier dysfunction after hemorrhagic shock and resuscitation in mice. Am. J. Physiol. Gastrointest. Liver Physiol 285: G621–G629, 2003. [DOI] [PubMed] [Google Scholar]
- 78.Zou Z, Sun H, Su Y, Cheng T and Luo C Progress in research on radiation combined injury in China. Radiat. Res 169: 722–729, 2008. [DOI] [PubMed] [Google Scholar]