

SUMMARY
Loss-of-function mutations in the progranulin gene (GRN), which encodes progranulin (PGRN), are a major cause of frontotemporal dementia (FTD). GRN-associated FTD is characterized by TDP-43 inclusions and neuroinflammation, but how PGRN loss causes disease remains elusive. We show that Grn knockout (KO) mice have increased microgliosis in white matter and an accumulation of myelin debris in microglial lysosomes in the same regions. Accumulation of myelin debris is also observed in white matter of patients with GRN-associated FTD. In addition, our findings also suggest that PGRN insufficiency in microglia leads to impaired lysosomal-mediated clearance of myelin debris. Finally, Grn KO mice that are deficient in cathepsin D (Ctsd), a key lysosomal enzyme, have augmented myelin debris and increased neuronal TDP-43 pathology. Together, our data strongly imply that PGRN loss affects microglial activation and lysosomal function, resulting in the accumulation of myelin debris and contributing to TDP-43 pathology.
Graphical abstract
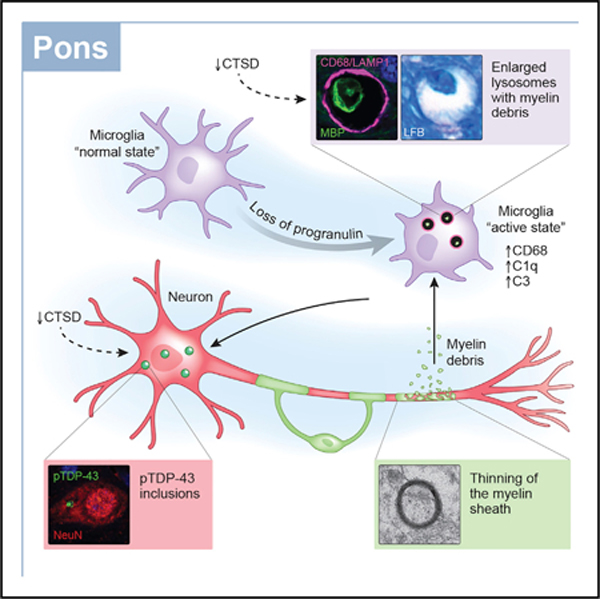
In brief
Wu et al. show increased microgliosis in white matter of Grn knockout mice. Microglial lysosomes accumulate myelin debris in both Grn knockout mice and patients with GRN-associated FTD, and reducing cathespin D levels exacerbates both myelin debris accumulation and pTdp-43 aggregation. Thus, lysosomal dysfunction affects these pathologies in GRN-related FTD.
INTRODUCTION
Frontotemporal dementia (FTD) refers to a group of brain disorders characterized by changes in personality, social behavior, and language proficiency (Neumann and Mackenzie, 2019). It is considered the most common form of dementia in individuals under age 60, and there is currently no cure or effective treatment available for this devastating disease. Approximately 50% of cases with FTD are characterized by proteinaceous inclusions of TAR DNA-binding protein of 43 kDa (TDP-43) in the central nervous system (CNS), representing the most common pathological subtype of FTD (FTLD-TDP) (Arai et al., 2006; Neumann et al., 2006). Mutations in the progranulin gene (GRN) gene, which result in progranulin (PGRN) haploinsufficiency, are a major genetic cause of FTLD-TDP (Shankaran et al., 2008; van Swieten and Heutink, 2008). In addition to TDP-43 pathology, other neuropathological features of GRN-associated FTD include neuronal loss, dystrophic neurites, and inflammation in the frontal and temporal lobes (Rademakers and Rovelet-Lecrux, 2009).
PGRN is an evolutionarily conserved secreted protein that is expressed in multiple cell types in the CNS (Kao et al., 2017). It was first discovered as a growth factor involved in cell proliferation, wound healing, tumorigenesis, and inflammation (Paushter et al., 2018; Zhu et al., 2002). Recent studies indicate that PGRN can contribute to the pathogenesis of FTLD-TDP through its direct role in regulating lysosomal function, as well as its effects on microglial activation. For instance, we and others demonstrated that PGRN is processed by cathepsin L in lysosomes and regulates multiple lysosomal enzymes, including cathepsin D (Ctsd) and β-glucocerebrosidase (Beel et al., 2017; Holler et al., 2017; Lee et al., 2017; Valdez et al., 2017; Zhou et al., 2017, 2019). Moreover, patients with homozygous GRN mutations develop neuronal ceroid lipofuscinosis (NCL), a lysosomal storage disease (Almeida et al., 2016; Smith et al., 2012). NCL-like pathology is also observed in Grn knockout (KO) mice and induced pluripotent stem cell (iPSC)-derived neurons from patients harboring GRN mutations(Ahmed et al., 2010; Götzl et al., 2014; Valdez et al., 2017). At the same time, the complete loss of PGRN in mice leads to exacerbated microglial activation, which produces elevated pro-inflammatory cytokines, causes increased neuronal loss, and promotes Tdp-43 proteinopathy (Martens et al., 2012; Yin et al., 2010a; Zhang et al., 2020). Importantly, Grn KO mice show upregulation of autophagy and lysosome-related genes in microglia (Lui et al., 2016; Zhang et al., 2020), providing a potential link between PGRN-mediated microglial activation and lysosome dysfunction.
The brains of patients with GRN-associated FTD exhibit white matter atrophy, such as myelin pallor, microglial activation, and dystrophic changes in myelin (Caroppo et al., 2014; Kelley et al., 2009; Rohrer et al., 2010; Spina et al., 2007; Sudre et al., 2017; Woollacott et al., 2018), but the connection between the underlying GRN mutations and white matter pathology remains unclear. Myelin, which is synthesized by oligodendrocytes in the CNS, is a lipid-rich substance that encompasses axons and works to increase signal conduction (Nave and Werner, 2014). The myelin membrane is a metabolically stable long-lived structure (Toyama et al., 2013), but it also slowly undergoes turnover and replacement to maintain its compaction and integrity, facilitating axonal conduction. During aging, axonal internode loss and myelin degeneration, including the formation of myelin spheroids and an accumulation of myelin debris, are observed (Hill et al., 2018). It has been well documented that microglial lysosomes play critical roles in the clearance of this myelin debris (Cantuti-Castelvetri et al., 2018; Hill et al., 2018; Safaiyan et al., 2016). In fact, a recently identified subtype of microglia known as white-matter-associated microglia (WAMs) show an upregulation of phagocytosis and lysosome-related genes in response to degenerated myelin in aged mice (Safaiyan et al., 2021). Furthermore, white matter abnormalities have been described in juvenile-onset NCL, including CLN2 (Dyke et al., 2013) and CLN3 (Roine et al., 2018), suggesting that lysosomal impairment could contribute to this phenotype. Although such a phenotype has not been investigated in patients with NCL and GRN mutations, it is tempting to speculate that PGRN-mediated lysosomal dysfunction in microglia reduces their capacity for degradation and contributes to the white matter pathology observed in patients with GRN-associated FTD. If this were the case, we would expect that Grn KO mice would have an increased accumulation of myelin debris, but this has yet to be explored. Similarly, accumulation of myelin debris has not yet been reported in patients with FTLD-TDP. Therefore, we used Grn KO mice as a model to investigate the effect of PGRN loss on lysosomal function and the associated effects on myelin debris accumulation and white matter pathology.
RESULTS
An age-dependent increase in microglial activation is observed in the white matter of Grn KO mice
To comprehensively characterize microglial abnormalities in Grn KO mice, we evaluated the immunoreactivity of microglial markers in the brains of 12- and 21-month-old wild-type (WT) and Grn KO animals. In the brains of 21-month-old Grn KO mice, compared to WT mice, we observed increased immunoreactivity for ionized calcium binding adaptor molecule 1 (Iba1; Figures S1A and S1C), which is constitutively expressed in microglia, and cluster differentiation 68 (CD68; Figures S1B and S1D), which is highly expressed in active phagocytic microglia and shown to be associated with white matter lesions (Waller et al., 2019; Zotova et al., 2013). In particular, microgliosis was observed in white matter regions of the thalamus, cerebellum, corpus callosum, and pons, with the latter being the more severely affected region (Figures S1A–S1D). As elevated CD68 expression indicates active phagocytosis, we asked whether the complement system cascade was activated in Grn KO mice. This protein signaling cascade is involved in the process by which microglia engulf a number of targets, including apoptotic cells, synapses, amyloid beta, and myelin (Kopper and Gensel, 2018; Peterson et al., 2015; Stevens et al., 2007; Taylor et al., 2000; Webster et al., 2000). We observed that the RNA levels of two complement components, namely, C1q and C3b, were significantly upregulated in 21-month-old Grn KO mice compared to those of the WT. This upregulation was most robust in the pons (Figures S1G–S1J). Both the increase in microglial activation and the upregulation of the complement pathway in the pons of Grn KO mice were age dependent (Figures S1E, S1F, S1I, and S1J).
Loss of PGRN contributes to the accumulation of myelin debris in microglial lysosomes
Recent studies show that the loss of PGRN leads to dramatic changes in the expression of lysosomal genes in microglia (Lui et al., 2016; Telpoukhovskaia et al., 2020; Zhang et al., 2020). Moreover, abnormally enlarged lysosomes have been observed in the brains of patients with GRN-associated FTD and in Grn KO mice (Lui et al., 2016; Paushter et al., 2018; Zhou et al., 2020). We observed that Lamp1-positive lysosomes were abnormally enlarged in the pons of 21-month-old Grn KO mice compared to those of WT controls (Figure 1A). Co-labeling of Lamp1 with cell-type-specific markers demonstrated that the abnormally enlarged lysosomes were localized within microglia (Iba1) (Figure 1A) but not neurons (microtubule associated protein 2 [Map2]), astrocytes (glial fibrillary acidic protein [Gfap]), or oligodendrocytes (myelin basic protein [Mbp]) (Figures S2A and S2B). Of note, we observed a significant accumulation of Mbp-immunoreactive structures in lysosomes in 21-month-old Grn KO mice (Figure S2B). These Mbp-immunoreactive structures co-localized with lysosomes in microglia (Figure 1B) but not in neurons (Figure S2C) or astrocytes (Figure S2D). Electron microscopy (EM) analysis confirmed that these structures were localized within microglia (Figure 1C). Furthermore, quantitative analysis revealed that Mbp-positive structures specifically accumulated in the white matter of aged (21-month-old) Grn KO mice but not in WT mice (Figures 1D and 1E). Notably, these Mbp-positive structures were abundant in areas with substantial microglial activation. To determine whether the Mbp-positive structures represented myelin debris, we used Luxol fast blue (LFB), a stain commonly used to detect myelin (Kluver and Barrera, 1953). By using this approach, we observed a significant accumulation of myelin debris in the pons of 21-month-old Grn KO mice but not in WT mice (Figures 2A and 2B).
Figure 1. Grn KO mice show enlarged lysosomes and an accumulation of myelin debris in microglial lysosomes.
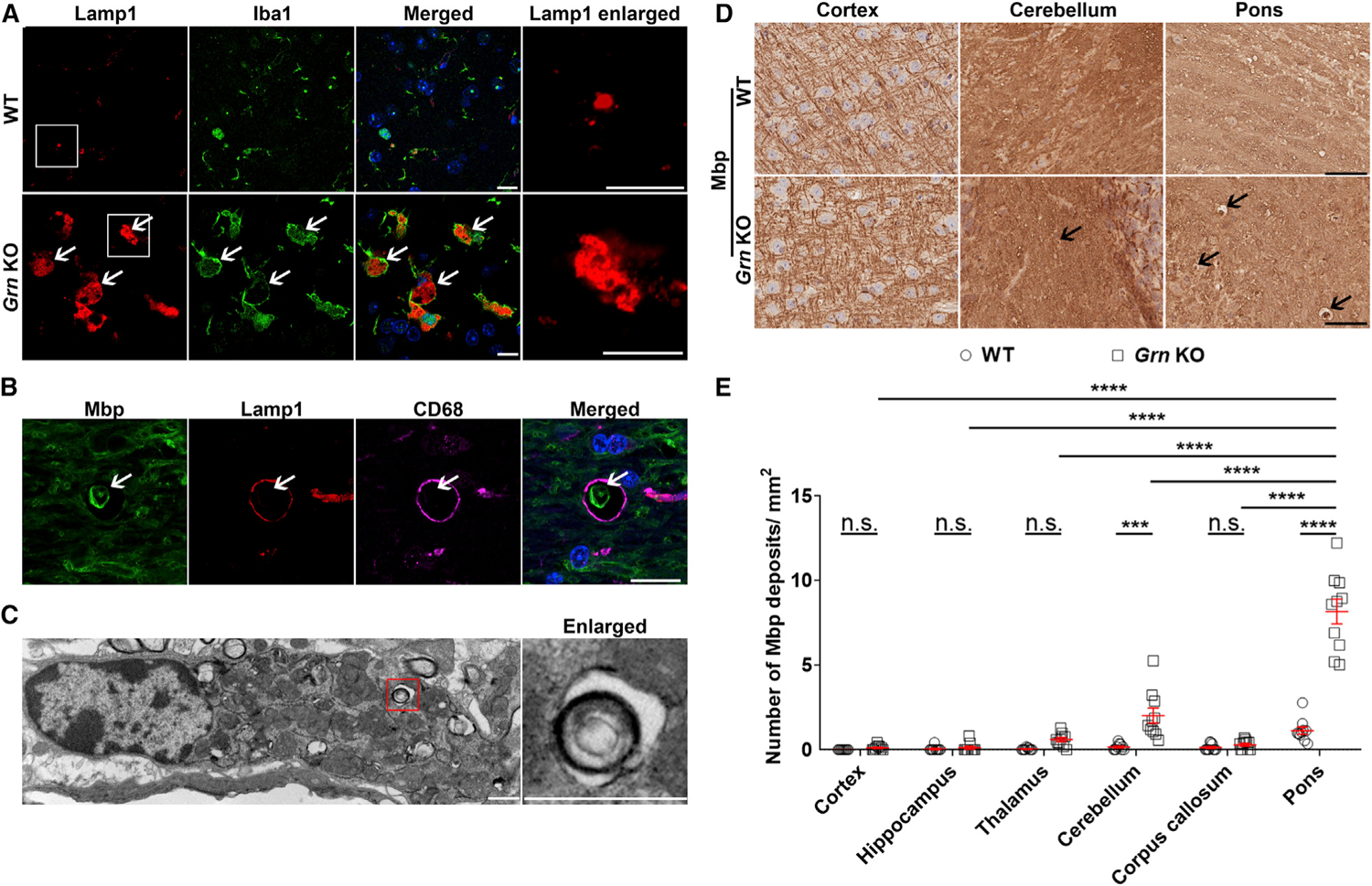
(A) Representative images of the pons of 21-month-old wild-type (WT) and Grn KO mice co-stained for lysosome (Lamp1) and microglia (Iba1) markers. Arrows indicate enlarged lysosomes. Boxes indicate the region enlarged in the enlarged panel.
(B) Representative images of myelin (Mbp), lysosome (Lamp1) and microglia marker (CD68) in the pons of 21-month-old Grn KO mice.
(C) EM image of myelin debris engulfed by microglia in pons of 12-month-old Grn KO mice. Box indicates the region enlarged in the subsequent panel.
(D) Representative images of Mbp staining in different brain regions of 21-month-old WT and Grn KO mice. Arrows indicate myelin debris. Scale bars, 10 μm (A–C) and 50 μm (D).
(E) Quantitative analyses of Mbp staining in different brain regions of 21-month-old WT and Grn KO mice (n = 10 per group). Data are presented as mean ± SEM. ***p < 0.001, ****p < 0.0001, two-way ANOVA, Tukey’s multiple-comparison test. n.s., not significant.
Figure 2. Myelin debris accumulates in Grn KO mice and in patients with GRN-associated FTD.
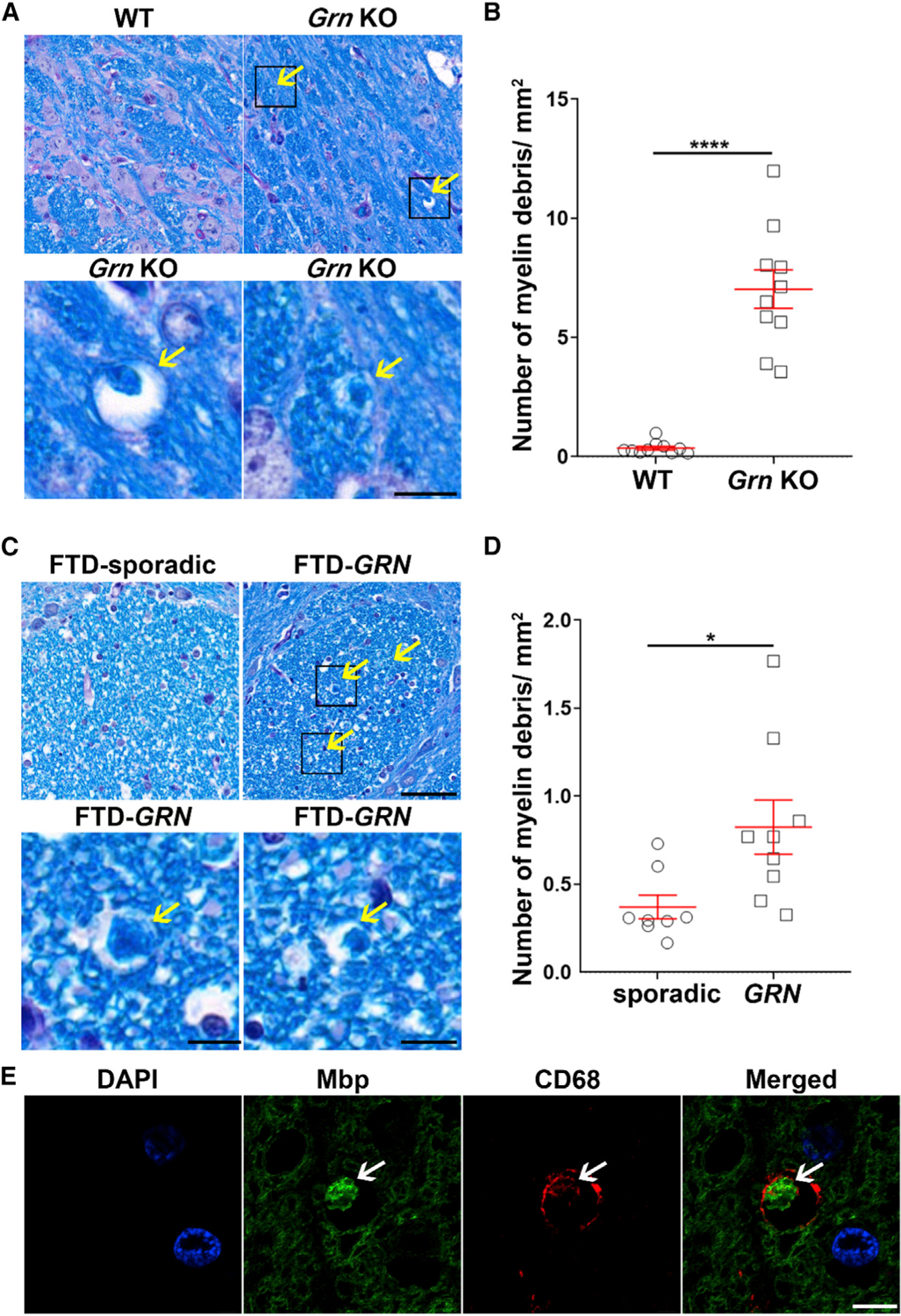
(A–D) Representative images (A and C) and quantitative analysis (B and D) of myelin staining (LFB) in the pons of 21-month-old WT or Grn KO mice (n = 10 per group) (A and B) and in the pons of patients with sporadic FTD and patients with GRN-associated FTD (n = 8–9 per group) (C and D). Arrows indicate myelin debris. Scale bar, 50 μm. Boxes in (A) and (C) indicate the region enlarged below. Data in (B) and (D) are presented as mean ± SEM. *p < 0.05, ****p < 0.0001, unpaired Student’s t tests.
(E) Representative images of myelin (MBP) and microglial lysosome (CD68) co-staining in pons of patients with GRN-associated FTD. Arrow indicates myelin debris. Scale bar, 5 μm.
To determine whether myelin debris may also be detected in white matter of patients with GRN-associated FTD, we stained the pons of patients with sporadic FTD and patients harboring GRN mutations with LFB (patient information is listed in Table S1). Indeed, the brains of individuals with GRN mutations showed a significant accumulation of myelin debris compared to patients with sporadic FTD (Figures 2C and 2D). Furthermore, myelin debris accumulated within microglial lysosomes (Figure 2E) but not in astrocytes or neurons (Figures S2E and S2F), similar to what we observed in Grn KO mice (Figures S2C and S2D). Together, our data suggest that the loss of Grn causes microglial lysosomal dysfunction, leading to the accumulation of myelin debris.
Accumulation of myelin debris in microglia is exacerbated by lysosomal dysfunction
The accumulation of myelin debris may result from increased phagocytosis and/or decreased lysosomal degradation. To determine the contribution of each of these events, we performed both in vitro phagocytosis and degradation assays by using cultured microglia. First, to evaluate the phagocytic activity, we treated purified WT and Grn KO microglia from postnatal day 0 (P0) mice with myelin conjugated to a pH-sensitive dye (pHrodo) and measured the fluorescence intensity of pHrodo-myelin inside microglia over time. We found that Grn KO microglia have similar phagocytosis activity as WT (Figures 3A and 3B). To evaluate the ability of cultured microglia to degrade myelin, we treated WT and Grn KO microglia with purified myelin for 3 h, washed away any extra myelin that had not be internalized by microglia, and then measured myelin levels by quantifying Mbp fluorescent intensity after 24 h. Of interest, Mbp intensity was significantly reduced by 94% in WT microglia, but Grn KO microglia still accumulated 68% of myelin (Figure 3C), strongly suggesting that myelin degradation is severely impaired in Grn KO microglia.
Figure 3. Accumulation of myelin debris in Grn KO microglia is exacerbated by the partial loss of Ctsd.
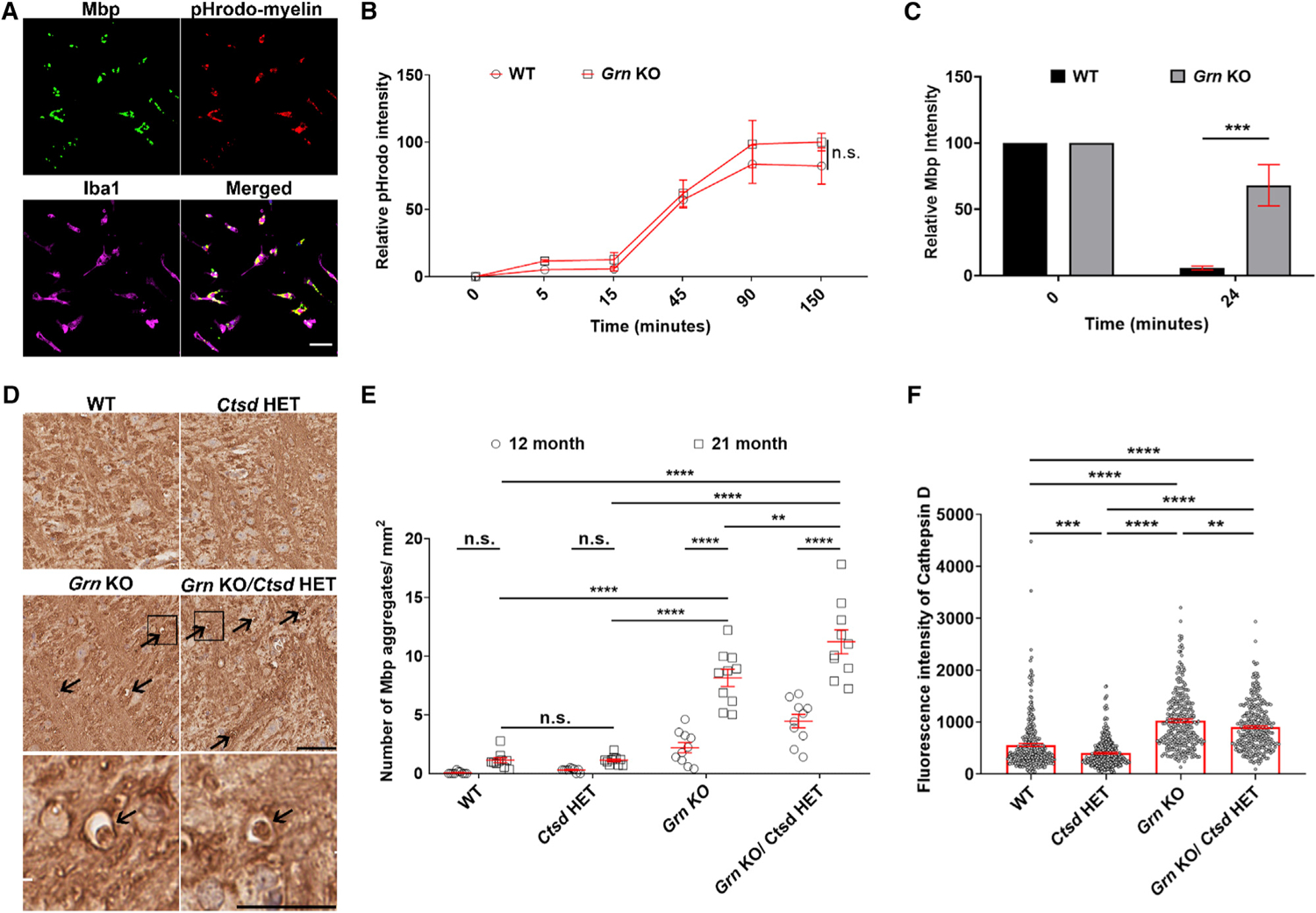
(A–C) Representative images of myelin (Mbp), pHrodo-myelin, and microglia marker (Iba1) co-staining in WT microglia fed with pHrodo-myelin (A), and quantitative analyses of relative phagocytosed pHrodo intensity (B) and Mbp intensity (C) in a time-dependent manner after adding pHrodo-myelin into medium (B) or after washing away extra myelin after incubation with microglia (C).
(D and E) Representative images (D) and quantitative analysis (E) of Mbp staining in pons of 21-month-old WT, Ctsd HET, Grn KO, or Grn KO/Ctsd HET mice (n = 10 per group). Box indicates the region enlarged in the panels below. Arrows indicate Mbp deposits. Scale bar, 20 μm (A) and 50 μm (D).
(F) Quantitative analysis of Ctsd staining in microglia of pons of 21-month-old WT, Ctsd HET, Grn KO, or Grn KO/Ctsd HET mice (n = 300 per group). Data are presented as mean ± SEM. **p < 0.01, ***p < 0.001, ****p < 0.0001, two-way ANOVA (B, C, and E) or one-way ANOVA (F), Tukey’s multiple-comparison test.
To further investigate the role of lysosomes in the accumulation of myelin debris, we generated double-KO mice that lacked both Grn and Ctsd. Ctsd encodes Ctsd, a lysosomal protease that can digest MBP in vitro (Pritzker et al., 2000). Because homozygous Ctsd KO mice die within a month from birth from neurological and systemic defects (Saftig et al., 1995), we used Ctsd heterozygous KO mice (Ctsd HET). Of interest, we did not observe increased Mbp immunoreactivity (Figures 3D and 3E) or an accumulation of myelin debris (Figures S3A and S3B) in 21-month-old Ctsd HET mice, despite the widespread decrease in microglial Ctsd levels in these animals (Figures 3F and S3C). Consistent with previous studies showing that lysosomal genes are upregulated in Grn KO microglia (Lui et al., 2016; Telpoukhovskaia et al., 2020), Ctsd levels were significantly upregulated in microglia of Grn KO mice (Figures 3F, S3C, and S3D). This increase is likely a compensatory response aimed at degrading cargo (including myelin debris) that accumulates inside lysosomes or a reflection of the global upregulation of lysosomal genes that results from Grn loss. Nevertheless, Ctsd levels were significantly lower in Grn KO/Ctsd HET mice than those in Grn KO mice (Figures 3F and S3C), and there was a corresponding increase in Mbp (Figures 3D and 3E) and myelin debris (Figures S3A and S3B) in these animals. There was no apparent change in microglial activation in Grn KO/Ctsd HET mice compared with that of Grn KO mice (Figures S3E and S3F), suggesting that the observed increase in myelin debris accumulation was not due to an overall change in microgliosis. Thus, our data support the hypothesis that lysosomal dysfunction in microglia of Grn KO mice leads to increased myelin debris accumulation and indicate that Ctsd may be involved in this process.
PGRN loss induces demyelination as well as neuronal Tdp-43 inclusion formation in white matter regions, and the latter is exacerbated by a partial loss of Ctsd
Given the effects of Pgrn on myelin debris clearance, we wondered whether loss of Pgrn would result in compromised myelination in Grn KO mice. To address this question, we used EM to analyze the myelin sheath of axons in the pons for each of our mouse lines and quantified the relative thickness of the myelin sheath using the G-ratio (ratio of the inner-to-outer diameter of a myelinated axon) (Hildebrand and Hahn, 1978). We observed a reduced myelin sheath thickness in Grn KO and Grn KO/Ctsd Het mice compared to that of WT and Ctsd mice (Figures S4A and S4B), suggesting that Pgrn loss results in demyelination.
Patients with GRN-associated FTD and Grn KO mice have an aberrant accumulation of TDP-43 in cellular inclusions (Mackenzie et al., 2011). The presence of Tdp-43 pathology in the white matter regions of Grn KO mice is poorly described, with few groups reporting inclusions of phosphorylated Tdp-43 (pTdp-43) in the thalamus of Grn KO mice (Tanaka et al., 2014; Yin et al., 2010a; Zhang et al., 2020). We also observed some pTdp-43 inclusions in the thalamus of Grn KO mice, but they were scarce (Figures S4C and S4D). On the other hand, pTdp-43 inclusions were abundant in the pons of 21-month-old Grn KO mice (Figures 4A and 4B), and these inclusions were ubiquitin positive (Figure 4C). Although there were no pTdp-43 inclusions observed in Ctsd HET mice (Figures 4A and 4B), Grn KO/Ctsd HET mice had more pTDP-43 inclusions than Grn KO mice in both the pons (Figures 4A and 4B) and thalamus (Figure S4C). Importantly, pTdp-43 inclusions were detected in neurons (Figures 4D, S4E, and S4F) but not in microglia (Figures 4E and S4F). This observation is consistent with previous findings (Zhang et al., 2020) and with relative levels of Ctsd, as immunofluorescence analyses revealed that Ctsd levels were reduced in the neurons of Grn KO mice compared to those in WT animals and further reduced in Grn KO/Ctsd HET mice (Figures S4G–S4I).
Figure 4. pTdp-43 inclusions are observed in the pons of Grn KO mice and are increased in response to a partial loss of Ctsd.
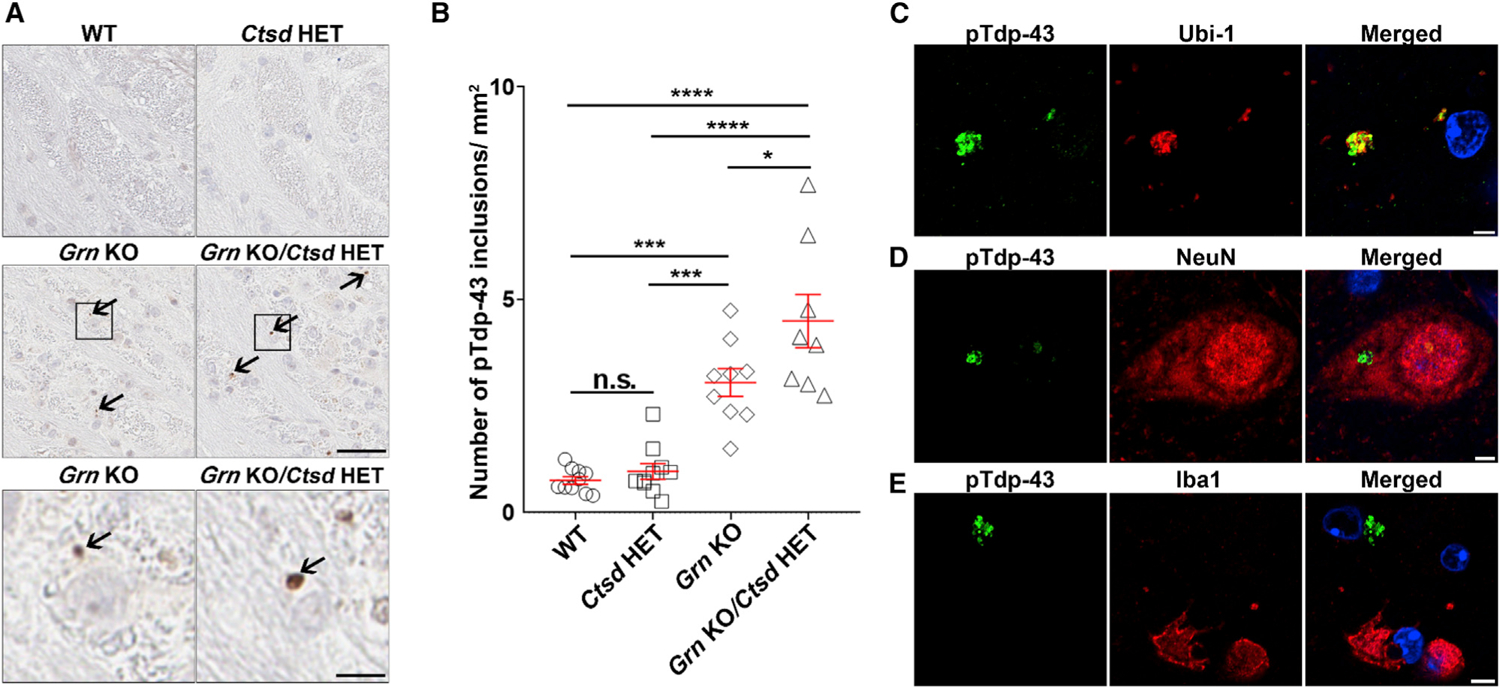
(A and B) Representative images (A) and quantitative analysis (B) of pTdp-43 staining in pons of 21-month-old WT, Ctsd HET, Grn KO, or Grn KO/Ctsd HET mice (n = 8–10 per group). Arrows indicate pTdp-43 inclusions. Scale bar, 50 μm. Boxes in (A) represent regions enlarged below. Data in (B) are presented as mean ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001, one-way ANOVA, Tukey’s multiple-comparison test.
(C–E) Representative images of pTdp-43 inclusions co-stained with ubiquitin (Ubi-1) (C) neuronal (NeuN) (D), and microglial (Iba1) (E) markers in the pons of Grn KO mice. Scale bar, 10 μm.
DISCUSSION
FTD is a devastating disease for which there is no cure. Mutations in GRN lead to haploinsufficiency and are a common cause of familial FTD (Shankaran et al., 2008; van Swieten and Heutink, 2008). Patients with GRN-associated FTD have neuronal loss and inflammation in the temporal and frontal lobes (Rademakers and Rovelet-Lecrux, 2009), as well as white matter lesions, such as myelin staining pallor and gliosis (Caroppo et al., 2014; Kelley et al., 2009; Rohrer et al., 2010; Spina et al., 2007; Woollacott et al., 2018). The mechanisms by which loss-of-function Grn mutations cause white matter pathology remain unclear. Grn KO mice have a progressive neurodegenerative phenotype that mimics human disease, including gliosis and pTdp-43 inclusions (Yin et al., 2010a, 2010b). Our findings show that microgliosis is especially abundant in white matter of Grn KO mice. In particular, the pons, a white-matter-rich region that relays signals from the forebrain to the cerebellum, was most severely affected. We observed an accumulation of myelin debris in enlarged microglial lysosomes in the pons of Grn KO mice. Of importance, we also observed myelin debris within enlarged microglial lysosomes in the pons of cases with GRN-associated FTD. Finally, we found that degradation of myelin is defective in Grn KO microglia and that enhancing lysosomal dysfunction through a partial loss of the lysosomal enzyme Ctsd exacerbated both myelin debris accumulation and pTdp-43 aggregation in Grn KO mice. Together, these results strongly suggest that microglial lysosomal dysfunction induced by PGRN deficiency leads to an accumulation of myelin debris and contributes to the accumulation of pTDP-43 aggregates in neurons.
Microglial abnormalities have been observed throughout the brain of Grn KO mice, including the thalamus (Ahmed et al., 2010), a white-matter-rich region that relays sensory and motor signals into the cortex. Our studies show that an age-dependent increase in microglial activation is also observed in additional white-matter-rich regions such as the cerebellum, corpus callosum, and pons. In fact, the pons showed the highest levels of activated microglia. This region contains several nerve tracts that connect the forebrain and cerebellum, as well as nuclei that control and regulate vital functions. We observed microgliosis throughout several regions of the pons, as well as pTDP-43 inclusions, particularly near the cortico-spinal tracts. It will be interesting to determine whether either of these pathologies correlates with clinical features in patients harboring GRN mutations. In addition, although white matter abnormalities have been previously reported in patients with FTD (Caroppo et al., 2014; Kelley et al., 2009; Rohrer et al., 2010; Spina et al., 2007; Sudre et al., 2017; Woollacott et al., 2018), we have demonstrated that microglial lysosomes in the pons of patients with GRN-associated FTD significantly accumulate myelin debris, a result consistent with our findings in Pgrn-deficient mice. In contrast, myelin debris was not as frequent in patients with sporadic FTD. Thus, our findings add myelin debris pathology to the list of defects specifically associated with loss-of-function GRN mutations.
Furthermore, we found that degradation of myelin, but not the engulfment of myelin, is impaired in Grn KO microglia. Meanwhile, we also observed that a partial loss of Ctsd was associated with enhanced accumulation of myelin debris in Grn KO mice. The pons of Grn KO mice showed significantly higher levels of microglial Ctsd than WT mice, consistent with previous studies showing upregulation of several lysosomal genes, including Ctsd, in Grn KO microglia (Lui et al., 2016; Telpoukhovskaia et al., 2020). These changes may result from a compensatory mechanism induced in response to lysosomal dysfunction in Pgrn-deficient mice. Alternatively, they could result from an activation of the ‘‘CLEAR’’ network, as PGRN-deficient microglia showed increased nuclear localization of TFEB, a transcription factor that regulates lysosomal genes (Tanaka et al., 2013). Nevertheless, Ctsd levels were decreased in microglia of Grn KO/Ctsd HET mice compared to those of Grn KO mice, and there was a concomitant increase in myelin debris in these animals. Interestingly, accumulation of myelin debris was also observed in the spinal cords of Tmem106b KO/Grn KO mice (Feng et al., 2020; Zhou et al., 2020). Tmem106b is also a lysosomal protein, further supporting our findings that myelin debris accumulation is enhanced by the downregulation of lysosomal proteins and the associated increase in lysosomal dysfunction.
Additionally, we observed a significant accumulation of phosphorylated and ubiquitinated neuronal Tdp-43 inclusions in the pons of Grn KO mice, and inclusion levels were further increased by the partial loss of Ctsd. Based on this finding, we predict that lysosomal dysfunction can modulate pTDP-43 inclusion formation in cases harboring GRN mutations, perhaps by negatively affecting the direct clearance of TDP-43 in neurons. Indeed, neuronal Ctsd levels are further decreased in Grn KO/Ctsd HET mice compared to those in Grn KO animals, and these mice also show a corresponding increase in pTdp-43 inclusions. At the same time, despite a significant decrease in Ctsd, Ctsd HET mice did not accumulate pTdp-43 inclusions; thus, it seems unlikely that decreasing Ctsd in neurons alone is sufficient to trigger pTdp-43 pathology. Instead, PGRN loss must induce pTDP-43 aggregation through a separate mechanism. To this end, our studies point toward an important association between microglia dysfunction and pTDP-43 inclusion formation. Although pTdp-43 inclusions were found in neurons in Grn KO and Grn KO/Ctsd HET mice, neuronal inclusions accumulated in the pons, a region that also had severe microgliosis. Furthermore, although the loss of Pgrn clearly induces microgliosis, the concomitant loss of Ctsd did not synergistically affect microglial activation, supporting our hypothesis that lysosomal dysfunction modulates Tdp-43 aggregation in Pgrn-deficient mice, whereas microgliosis influences its induction. Damage to microglia can negatively affect the microenvironment in the brain. For instance, an excess amount of myelin debris in microglia has been shown to rupture microglial phagolysosomal membranes and stimulate an inflammasome response in aged mice and in myelin-treated human iPSC-derived microglia (Cantuti-Castelvetri et al., 2018; Nugent et al., 2020). Accumulation of myelin debris can also negatively affect the overall myelination process (Kotter et al., 2006), as indicated by the thinner myelin sheath in the pons of Grn KO and Grn KO/Ctsd Het mice. Importantly, we found that the complement pathway was activated in the pons of Grn KO mice in an age-dependent manner, and activation of this pathway was recently associated with enhanced pTDP-43 proteinopathy in the thalamus (Zhang et al., 2020). Our data show that components of the complement pathway are more strongly upregulated in the pons than the thalamus, and we predict that this robust activation could be detrimental to other cell types in the CNS. Finally, recent studies suggest Grn-deficient microglia can secrete factors that promote formation of neuronal Tdp-43 inclusions in vitro (Zhang et al., 2020). Our studies provide additional evidence to support the hypothesis that dysfunctional microglia can non-cell-autonomously influence neuronal TDP-43 aggregation.
Taken together, our findings reveal myelin defects in the white matter of Grn-deficient mice and patients with GRN-associated FTD and highlight the importance of microglial lysosome dysfunction in disease. Furthermore, our studies support a model in which PGRN loss induces microglial activation, which influences the formation of pTDP-43 pathology. TDP-43 aggregation levels can then be further exacerbated by lysosomal dysfunction in PGRN-deficient neurons. Overall, these studies underscore the need to further understand the role of microglia in disease, which may uncover targets for therapeutic intervention.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Leonard Petrucelli (petrucelli.leonard@mayo.edu).
Materials availability
All unique reagents generated in this study are available upon reasonable request from the Lead Contact.
Data and code availability
Data reported in this paper will be shared by the Lead Contact upon request
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All procedures in this study using mice were performed in accordance with the National Institutes of Health Guide for Care and Use of Experimental Animals and approved by the Mayo Clinic Institutional Animal Care and Use Committee (Protocol number A34315–15-R17). Grn KO (Kayasuga et al., 2007) and Ctsd HET mice (Saftig et al., 1995) were maintained in the animal facilities at Mayo Clinic Florida on a 12-hour light/dark cycle in standard housing. When in their home cage, animals had access to standard mouse chow and water ad libitum. All mice were of the same genetic background (C57BL/6J) and were assigned to experimental groups based on genotype. WT, Grn KO, Ctsd Het and Grn KO/Ctsd Het mice were littermates and generated via Grn HET x Grn HET matings. For the 12-month-old experimental cohort, 5 male and 5 female WT, 6 male and 4 female Ctsd Het, 6 male and 4 female Grn KO, and 4 male and 6 female Grn KO/Ctsd Het mice were included. For the 21-month-old experimental cohort, 5 male and 5 female mice of each genotype were included. No differences between sexes were observed. Mice were aged to 12 or 21 months as indicated in the text and then sacrificed using CO2 inhalation. Brains were harvested and fixed as described in the Method details section below.
Post-Mortem Human Samples
Post-mortem tissues from patients with neuropathologically confirmed frontotemporal dementia (FTD) were obtained from the Brain Bank for Neurodegenerative Disorders at Mayo Clinic Florida. Information on human cases is provided in Table S1. Autopsies were performed after consent by the next-of-kin or someone with legal authority to grant permission. The brain bank operates under protocols approved by the Mayo Clinic Institutional Review Board (IRB).
METHOD DETAILS
Tissue processing
Mouse brains were harvested without previously perfusing the animals. Sagittal half brains and human brains were immersion fixed in 10% formalin, embedded in paraffin, sectioned (5 μm thick), and then mounted on positively-charged glass slides. Sections were dried overnight and then used for immunofluorescence or immunohistochemistry staining as indicated.
Immunohistochemistry staining
Paraffin-embedded brain sections were deparaffinized in xylene, and rehydrated through a series of ethanol solutions, followed by washing in deionized H2O. Antigen retrieval was performed by steaming slides in deionized H2O or Tris-EDTA (DAKO), pH 9.0 for 30 minutes followed by a 5-minute incubation in DAKO Peroxidase Block (S2001, DAKO) to block endogenous peroxidase activity. To detect Iba1, CD68 and Mbp, slides were blocked with DAKO Protein Block Serum-Free (X0909, DAKO) for 1 hour, and incubated with primary antibodies (working dilutions are listed in Table S2) for 45 minutes. After washing, sections were incubated for 30 minutes in DAKO Envision-Plus anti-rabbit (K4003, DAKO) or anti-mouse (K4001, DAKO) labeled HRP polymer, respectively. Peroxidase labeling was visualized with the Liquid DAB + Substrate Chromogen System (K3468, DAKO).
To detect pTdp-43 in mice, slides were deparaffinized and rehydrated as described above, and antigen retrieval was performed by steaming in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween-20, pH 6.0) for 30 minutes. After cooling and washing with distilled H2O, slides were incubated with Dako Dual Endogenous Enzyme Block (DAKO), and subsequently washed in PBS. Sections were then blocked with 2% normal goat serum for 1 hour, followed by an overnight incubation with primary pTDP-43 antibody (Table S2) at 4°C. The next day, slides were washed with PBS, incubated with biotinylated goat anti-rabbit or rabbit anti-rat secondary (ThermoFisher) (1:200) for 2 hours, and again washed in PBS. Slides were then incubated with avidin-biotin complex solution for 30 minutes, washed in PBS, and reacted with 3,3′-diaminobenzidine (Acros Organics) activated with hydrogen peroxide. The reaction was stopped by rinsing the slides in dH2O.
Following labeling, all sections were counterstained with hematoxylin (Thermo Fisher Scientific), dehydrated through a series of ethanol and xylene washes, and coverslipped with Cytoseal mounting medium (Thermo Fisher Scientific). Slides were scanned with a ScanScope AT2 (Leica Biosystems), and representative images were taken with ImageScope software (v12.1; Leica Biosystems).
Luxol fast blue-periodic acid–Schiff staining
Paraffin-embedded mouse brain sections were deparaffinized and hydrated as described above. Slides were incubated in Luxol fast blue solution (10% Luxol fast blue dissolved in 95% ethanol and 5% acetic acid) overnight at room temperature. Slides were then washed with 95% ethanol and deionized water followed by quick immersion in a 0.05% lithium carbonate solution and a wash in deionized water. Then slides were incubated in 2% periodic acid solution for 5 minutes and rinsed in deionized water, followed by incubation in Schiff’s solution for 15 minutes. After a 5-minute wash in tap water, the slides were counterstained in Gill-1 hematoxylin for 3–4 s and rinsed in tap water. Slides were then dehydrated in ethanol and xylene and mounted with permanent mounting media. Slides were scanned with a ScanScope AT2 (Leica Biosystems) and representative images were taken with ImageScope software (v12.1; Leica Biosystems).
Digital pathology
Quantitative analysis of myelin debris and pTdp-43 pathology was carried out using ImageScope software (v12.1; Leica Biosystems). The entire pons from one section for each mouse was analyzed from mid-sagittal serial sections. Mbp deposits, myelin debris and pTdp-43 inclusions were quantified as the number of deposits/debris/inclusions per area. These analyses were performed in a blinded fashion. Iba1- and CD68-positive microglia were quantified in a blinded fashion using custom-designed Positive Pixel Count algorithms. The number of positively stained pixels as a proportion of all pixels in the annotated area was used as the output parameter.
Immunofluorescence staining in mouse and human brains
Paraffin sections of mouse and human brain tissues were deparaffinized, rehydrated, steamed for 30 minutes in DAKO antigen retrieval solution, blocked with DAKO All Purpose Blocker for 1 hour, and incubated with each primary antibody of interest (working dilutions are listed in Table S2). After washing, sections were incubated with corresponding Alexa Fluor 488-, 568- or 647-conjugated donkey anti-species secondary antibodies (1:200, Molecular Probes) for 2 hours. Hoechst 33258 (1 μg/ml, Thermo Fisher Scientific) was used to stain cellular nuclei. Images were obtained on a Zeiss LSM 880 laser scanning confocal microscope.
Quantification of fluorescence intensity of cathepsin D
Cathepsin D fluorescence intensity was measured from Iba1- or NeuN-positive cells from each mouse model using Zeiss Zen 2.3. After collecting images, thirty Iba1 or NeuN positive cells per brain were randomly picked. Then, cathepsin D fluorescence intensity was evaluated for those cells. Measurements from all ten animals from each group were included, and the average fluorescence intensity from a total of 300 cells is reported as the overall cathespin D level for each study group. We also included quantifications of cathepsin D per mouse level in addition to the per cell level. All analyses were done in a blinded manner and all stains and images were obtained in the same batch and using the same imaging parameters.
RNA extraction and qRT-PCR
Frozen pons were homogenized in Trizol LS (300 ul), and RNA were extracted using the Direct-zol MicroPrep kit (Zymo Research) according to manufacturer’s instructions. cDNA was then obtained using the High Capacity cDNA Transcription Kit (Applied Biosciences). qRT-PCR was performed in triplicate for all samples using the SYBR green assay (Life Technologies) on an ABI Prism 7900HT Fast Real-Time PCR system (Applied Biosystems). The primers used were: C1q: 5′-AAAGGCAATCCAGGCAATATCA-3′ and 5′-TGGTTCTGGTATGGACTCTCC-3.; C3b: 5′-CCAGCTCCCCATTAGCTCTG-3′ and 5′-GCACTTGCCTCTTTAGGAAGTC-3′(Lui et al., 2016).; Gapdh: 5′-CATGGCCTTCCGTGTTCCTA-3′ and 5′-CCTGCTTCA CCACCTTCTTGAT-3. Relative RNA expression of C1q and C3b was normalized to Gapdh values.
Myelin purification, phagocytosis, and degradation assay
Myelin was purified from 8-week-old WT C57BL/6 mouse brain using previously described methods (Safaiyan et al., 2016). Ten whole brains were dissected and placed in lysis buffer (1.2 mL lysis buffer per mouse, 10 mM HEPES, 5 mM EDTA, 0.3 M sucrose, cOmplete protease inhibitor tablet (11873580001, Roche) and chopped into fine pieces, followed by Dounce homogenization. Homogenates were then added on top of equal volumes of 0.32 M sucrose and 0.85 M sucrose in an Ultra-Clear tube (344060, Beckman). Samples were spun at 75,000 g, 4◦C, for 30 min in a SW41Ti rotor in an Optima XPN-90 Ultracentrifuge (Beckman) with Level 1 acceleration and deceleration. Then crude myelin at the 0.32/0.85 M sucrose interface were transferred to a new ultracentrifuge tube and resuspended in distilled water, followed by 12,000 g, 4°C, 15 min centrifugation with maximum acceleration and deceleration. This step was repeated once and then the pellet was resuspended in lysis buffer. All steps from sucrose layer preparation were repeated once more time to obtain pure myelin. Finally, purified myelin was resuspended in 1X PBS and adjusted to 1mg/ml protein concentration. For phagocytosis assay, fractions of myelin were labeled using the pHrodo Red Microscale Labeling Kit (P35363, ThermoFisher) following manufacturer’s instructions. Cultured microglia in Opti-MEM (51985–034, GIBCO) with 10% FBS were seeded into 24-well plate with PDL-coated coverslips at a density of 300,000 cells per well. After 24 hours, pHrodo-myelin was diluted using cell culture medium at a concentration of 5 μg/ml and sonicated for 1 min and then added to microglia. Cells were fixed with 4% PFA after 5/15/45/90/150 min and subjected to immunofluorescence staining. For degradation assay, microglia were incubated with 5 μg/ml myelin for 3 hours then subjected to fixation right away or 24 hours after washing away extra myelin prior to immunofluorescence staining.
Immunofluorescence staining and quantification in cultured microglia
Fixed cells were permeabilized with 0.5% Triton X-100 for 10 mins, blocked with 5% nonfat dry milk in PBS for 1 hour, then incubated with primary antibody (working dilutions are listed in Table S2) overnight at 4°C. After washing with PBS for 3 times, cells were incubated with corresponding Alexa Fluor 488-, 568-, or 647-conjugated donkey/goat anti-species antibodies (1:500, Molecular Probes) for 2 hours. Hoechst 33258 (1 μg/ml, H3569, Thermo Fisher Scientific) was used to stain cellular nuclei. Images were obtained on a Zeiss LSM 880 laser scanning confocal microscope. To quantify the level of pHrodo-myelin or Mbp, fluorescence intensity of both markers for each cell was measured in a blinded fashion (50–70 cells were measured for each group from 3 independent experiments).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical information for each experiment, including the total n number analyzed and the specific tests performed, is reported in the figure legends. In general, data are presented as mean ± standard error of mean (SEM) and analyzed with one-way or 2-way ANOVA followed by Tukey’s post hoc analysis, or by unpaired Student’s t tests (GraphPad Prism, version 8.0.1). All data with p < 0.05 were considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Cathespin D | ProteinTech | Cat#21327–1-AP; RRID:AB_10733646 |
| Rabbit anti-CD68 | Abcam | Cat#ab125212; RRID:AB_10975465 |
| Rabbit anti-Gfap | Biogenex | Cat#PU020-UP |
| Rabbit anti-Iba1 | Wako Chemicals | Cat#019–19741; RRID:AB_839504 |
| Rat anti-Lamp1 | Invitrogen | Cat#14–1071-82; RRID:AB_657531 |
| Mouse anti-Map2 | Sigma | Cat#M1406; RRID:AB_477171 |
| Mouse anti-Mbp | R&D Systems | Cat#MAB42282 |
| Mouse anti-NeuN | Chemicon | Cat#MAB377; RRID:AB_2298772 |
| Rabbit anti-pTdp-43 | Rb3655 | |
| Mouse anti-Ubi1 | EMD Millipore | Cat#MAB1510; RRID:AB_2180556 |
|
| ||
| Biological samples | ||
| Patient-derived pons tissue | Mayo Clinic Florida Brain Bank | http://www.mayo.edu/research/departments-divisions/department-neuroscience-florida/brain-banks/mayo-clinic-brain-bank |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
| pHrodo Red, succinimidyl ester (pHrodo Red, SE) | ThermoFisher | P36600 |
| cOmplete Protease Inhibitor Tablets | Roche | 11873580001 |
|
| ||
| Experimental models: Organisms/strains | ||
| Mouse: Grn KO | Kayasuga et al., 2007 | N/A |
| Mouse: Ctsd KO | Saftig et al., 1995 | N/A |
|
| ||
| Software and algorithms | ||
| Aperio eSlide Manager v12.1 | Leica Biosystems | https://www.leicabiosystems.com/aperio-eslide-manager/ |
| Aperio ImageScope v12.1 | Leica Biosystems | https://www.leicabiosystems.com/digital-pathology/manage/aperio-imagescope/ |
| Graphpad Prism v8.1.1 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Zeiss Zen 2.3. | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| SDS v2.2.2 | ThermoFisher | https://www.thermofisher.com/us/en/home/technical-resources/software-downloads/applied-biosystems-7900ht-fast-real-timespcr-system.html |
Highlights.
Progranulin loss induces microgliosis in white matter of Grn knockout (KO) mice
Microglial lysosomes accumulate myelin debris in Grn KO mice and GRN-related FTD
Lysosomal degradation of myelin debris is impaired in Grn KO cultured microglia
Reducing Ctsd exacerbates myelin debris and pTDP-43 accumulation in Grn KO mice
ACKNOWLEDGMENTS
We thank all the patients and their families for their contribution to this study, and we are grateful to all individuals who assisted in the procuring of these samples. The authors would also like to acknowledge Virginia Phillips (Mayo Clinic, Jacksonville) for immunohistochemistry (IHC) support, Anneliese Hill (Mayo Clinic, Jacksonville) for IHC imaging support, and Jon E Charlesworth (Mayo Clinic, Microscopy and cell analysis core Rochester) for EM support. We would also like to thank Katie Vicari for the artwork depicted in the graphical abstract. This work was supported by the NINDS, NIH (R35NS097273 to L.P.; P01NS084974 to L.P. and Y.-J.Z.; RF1NS120992 to M.P.), the Mayo Clinic Foundation (to L.P.), the Association of Frontotemporal Dementia (AFTD) (to L.P.), and the Alzheimer’s Association-AD Strategic Fund (to L.P.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109581.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, Yu X, Wuertzer CA, Hou H, Chiba S, et al. (2010). Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol 177, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida MR, Macario MC, Ramos L, Baldeiras I, Ribeiro MH, and Santana I (2016). Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging 41, 200.e201–200.e205. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, and Oda T (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun 351, 602–611. [DOI] [PubMed] [Google Scholar]
- Beel S, Moisse M, Damme M, De Muynck L, Robberecht W, Van Den Bosch L, Saftig P, and Van Damme P (2017). Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum. Mol. Genet 26, 2850–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L, Fitzner D, Bosch-Queralt M, Weil MT, Su M, Sen P, Ruhwedel T, Mitkovski M, Trendelenburg G, Lütjohann D, et al. (2018). Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 359, 684–688. [DOI] [PubMed] [Google Scholar]
- Caroppo P, Le Ber I, Camuzat A, Clot F, Naccache L, Lamari F, De Septenville A, Bertrand A, Belliard S, Hannequin D, et al. (2014). Extensive white matter involvement in patients with frontotemporal lobar degeneration: think progranulin. JAMA Neurol 71, 1562–1566. [DOI] [PubMed] [Google Scholar]
- Dyke JP, Sondhi D, Voss HU, Shungu DC, Mao X, Yohay K, Worgall S, Hackett NR, Hollmann C, Yeotsas ME, et al. (2013). Assessment of disease severity in late infantile neuronal ceroid lipofuscinosis using multiparametric MR imaging. AJNR Am. J. Neuroradiol 34, 884–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T, Mai S, Roscoe JM, Sheng RR, Ullah M, Zhang J, Katz II, Yu H, Xiong W, and Hu F (2020). Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep 21, e50219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, Janssens J, van der Zee J, Lang CM, Kremmer E, et al. (2014). Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol 127, 845–860. [DOI] [PubMed] [Google Scholar]
- Hildebrand C, and Hahn R (1978). Relation between myelin sheath thickness and axon size in spinal cord white matter of some vertebrate species. J. Neurol. Sci 38, 421–434. [DOI] [PubMed] [Google Scholar]
- Hill RA, Li AM, and Grutzendler J (2018). Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci 21, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler CJ, Taylor G, Deng Q, and Kukar T (2017). Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins that Are Haploinsufficient in Patients with Frontotemporal Dementia Caused by GRN Mutations. eNeuro 4, ENEURO.0100–17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao AW, McKay A, Singh PP, Brunet A, and Huang EJ (2017). Progranulin, lysosomal regulation and neurodegenerative disease. Nat. Rev. Neurosci 18, 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayasuga Y, Chiba S, Suzuki M, Kikusui T, Matsuwaki T, Yamanouchi K, Kotaki H, Horai R, Iwakura Y, and Nishihara M (2007). Alteration of behavioural phenotype in mice by targeted disruption of the progranulin gene. Behav. Brain Res 185, 110–118. [DOI] [PubMed] [Google Scholar]
- Kelley BJ, Haidar W, Boeve BF, Baker M, Graff-Radford NR, Krefft T, Frank AR, Jack CR Jr., Shiung M, Knopman DS, et al. (2009). Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol. Aging 30, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluver H, and Barrera E (1953). A method for the combined staining of cells and fibers in the nervous system. J. Neuropathol. Exp. Neurol 12, 400–403. [DOI] [PubMed] [Google Scholar]
- Kopper TJ, and Gensel JC (2018). Myelin as an inflammatory mediator: Myelin interactions with complement, macrophages, and microglia in spinal cord injury. J. Neurosci. Res 96, 969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter MR, Li WW, Zhao C, and Franklin RJ (2006). Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci 26, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Stankowski JN, Chew J, Cook CN, Lam YW, Almeida S, Carlomagno Y, Lau KF, Prudencio M, Gao FB, et al. (2017). The lysosomal protein cathepsin L is a progranulin protease. Mol. Neurodegener 12, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, Shang Y, Oldham MC, Martens LH, Gao F, et al. (2016). Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 165, 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, and Lee VM (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122, 111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, Min SW, Gan L, Finkbeiner S, Huang EJ, and Farese RV Jr. (2012). Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J. Clin. Invest 122, 3955–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA, and Werner HB (2014). Myelination of the nervous system: mechanisms and functions. Annu. Rev. Cell Dev. Biol 30, 503–533. [DOI] [PubMed] [Google Scholar]
- Neumann M, and Mackenzie IRA (2019). Review: Neuropathology of nontau frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol 45, 19–40. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. [DOI] [PubMed] [Google Scholar]
- Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, et al. (2020). TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 105, 837–854.e9. [DOI] [PubMed] [Google Scholar]
- Paushter DH, Du H, Feng T, and Hu F (2018). The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol 136, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SL, Nguyen HX, Mendez OA, and Anderson AJ (2015). Complement protein C1q modulates neurite outgrowth in vitro and spinal cord axon regeneration in vivo. J. Neurosci 35, 4332–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritzker LB, Joshi S, Gowan JJ, Harauz G, and Moscarello MA (2000). Deimination of myelin basic protein. 1. Effect of deimination of arginyl residues of myelin basic protein on its structure and susceptibility to digestion by cathepsin D. Biochemistry 39, 5374–5381. [DOI] [PubMed] [Google Scholar]
- Rademakers R, and Rovelet-Lecrux A (2009). Recent insights into the molecular genetics of dementia. Trends Neurosci 32, 451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC, Rossor MN, and Warren JD (2010). Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 53, 1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roine U, Roine TJ, Hakkarainen A, Tokola A, Balk MH, Mannerkoski M, Åberg LE, Lönnqvist T, and Autti T (2018). Global and Widespread Local White Matter Abnormalities in Juvenile Neuronal Ceroid Lipofuscinosis. AJNR Am. J. Neuroradiol 39, 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, Edinger AL, Jung S, Rossner MJ, and Simons M (2016). Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci 19, 995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaiyan S, Besson-Girard S, Kaya T, Cantuti-Castelvetri L, Liu L, Ji H, Schifferer M, Gouna G, Usifo F, Kannaiyan N, et al. (2021). White matter aging drives microglial diversity. Neuron 109, 1100–1117.e10. [DOI] [PubMed] [Google Scholar]
- Saftig P, Hetman M, Schmahl W, Weber K, Heine L, Mossmann H, Köster A, Hess B, Evers M, von Figura K, et al. (1995). Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J 14, 3599–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, and Haass C (2008). Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J. Biol. Chem 283, 1744–1753. [DOI] [PubMed] [Google Scholar]
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, et al. (2012). Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet 90, 1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina S, Murrell JR, Huey ED, Wassermann EM, Pietrini P, Baraibar MA, Barbeito AG, Troncoso JC, Vidal R, Ghetti B, and Grafman J (2007). Clinicopathologic features of frontotemporal dementia with progranulin sequence variation. Neurology 68, 820–827. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Sudre CH, Bocchetta M, Cash D, Thomas DL, Woollacott I, Dick KM, van Swieten J, Borroni B, Galimberti D, Masellis M, et al. ; Genetic FTD Initiative, GENFI (2017). White matter hyperintensities are seen only in GRN mutation carriers in the GENFI cohort. Neuroimage Clin 15, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Matsuwaki T, Yamanouchi K, and Nishihara M (2013). Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience 250, 8–19. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Chambers JK, Matsuwaki T, Yamanouchi K, and Nishihara M (2014). Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol. Commun 2, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, and Walport MJ (2000). A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J. Exp. Med 192, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telpoukhovskaia MA, Liu K, Sayed FA, Etchegaray JI, Xie M, Zhan L, Li Y, Zhou Y, Le D, Bahr BA, et al. (2020). Discovery of small molecules that normalize the transcriptome and enhance cysteine cathepsin activity in progranulin-deficient microglia. Sci. Rep 10, 13688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR 3rd, and Hetzer MW (2013). Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez C, Wong YC, Schwake M, Bu G, Wszolek ZK, and Krainc D (2017). Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum. Mol. Genet 26, 4861–4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swieten JC, and Heutink P (2008). Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol 7, 965–974. [DOI] [PubMed] [Google Scholar]
- Waller R, Baxter L, Fillingham DJ, Coelho S, Pozo JM, Mozumder M, Frangi AF, Ince PG, Simpson JE, and Highley JR (2019). Iba-1-/CD68+ microglia are a prominent feature of age-associated deep subcortical white matter lesions. PLoS One 14, e0210888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, and Tenner AJ (2000). Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp. Neurol 161, 127–138. [DOI] [PubMed] [Google Scholar]
- Woollacott IOC, Bocchetta M, Sudre CH, Ridha BH, Strand C, Courtney R, Ourselin S, Cardoso MJ, Warren JD, Rossor MN, et al. (2018). Pathological correlates of white matter hyperintensities in a case of progranulin mutation associated frontotemporal dementia. Neurocase 24, 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, et al. (2010a). Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J. Exp. Med 207, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, Beal MF, Nathan C, Thomas B, and Ding A (2010b). Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J 24, 4639–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Velmeshev D, Hashimoto K, Huang YH, Hofmann JW, Shi X, Chen J, Leidal AM, Dishart JG, Cahill MK, et al. (2020). Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature 588, 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Paushter DH, Feng T, Sun L, Reinheckel T, and Hu F (2017). Lysosomal processing of progranulin. Mol. Neurodegener 12, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Paushter DH, Pagan MD, Kim D, Nunez Santos M, Lieberman RL, Overkleeft HS, Sun Y, Smolka MB, and Hu F (2019). Progranulin deficiency leads to reduced glucocerebrosidase activity. PLoS One 14, e0212382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Brooks M, Jiang P, Koga S, Zuberi AR, Baker MC, Parsons TM, Castanedes-Casey M, Phillips V, Librero AL, et al. (2020). Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin-deficient mice. EMBO Rep 21, e50197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, and Ding A (2002). Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell 111, 867–878. [DOI] [PubMed] [Google Scholar]
- Zotova E, Bharambe V, Cheaveau M, Morgan W, Holmes C, Harris S, Neal JW, Love S, Nicoll JA, and Boche D (2013). Inflammatory components in human Alzheimer’s disease and after active amyloid-b42 immunization. Brain 136, 2677–2696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the Lead Contact upon request
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.