

Summary
Background
Lactobacillus was described as a keystone bacterial taxon in the human vagina over 100 years ago. Using metagenomics, we and others have characterized lactobacilli and other vaginal taxa across health and disease states, including pregnancy. While shifts in community membership have been resolved at the genus/species level, strain dynamics remain poorly characterized.
Methods
We performed a metagenomic analysis of the complex ecology of the vaginal econiche during and after pregnancy in a large U.S. based longitudinal cohort of women who were initially sampled in the third trimester of pregnancy, then validated key findings in a second cohort of women initially sampled in the second trimester of pregnancy.
Findings
First, we resolved microbial species and strains, interrogated their co-occurrence patterns, and probed the relationship between keystone species and preterm birth outcomes. Second, to determine the role of human heredity in shaping vaginal microbial ecology in relation to preterm birth, we performed a mtDNA-bacterial species association analysis. Finally, we explored the clinical utility of metagenomics in detection and co-occurrence patterns for the pathobiont Group B Streptococcus (causative bacterium of invasive neonatal sepsis).
Conclusions
Our highly refined resolutions of the vaginal ecology during and post-pregnancy provide insights into not only structural and functional community dynamics, but highlight the capacity of metagenomics to reveal finer aspects of the vaginal microbial ecologic framework.
Funding
NIH-NINR R01NR014792, NIH-NICHD R01HD091731, NIH National Children’s Study Formative Research, Burroughs Wellcome Fund Preterm Birth Initiative, March of Dimes Preterm Birth Research Initiative, NIH-NIGMS (K12GM084897, T32GM007330, T32GM088129).
Keywords: 16S rRNA, bacteria, GBS, Lactobacillus, metagenomics, microbiome, preterm birth, strains, streptococci, vagina
eTOC blurb
Pace et al. present a highly refined resolution of the vaginal microbiome during after pregnancy that provides insights into structural and functional community dynamics, also highlighting the capacity of metagenomics to reveal finer aspects of the ecology of the vaginal microbiome.
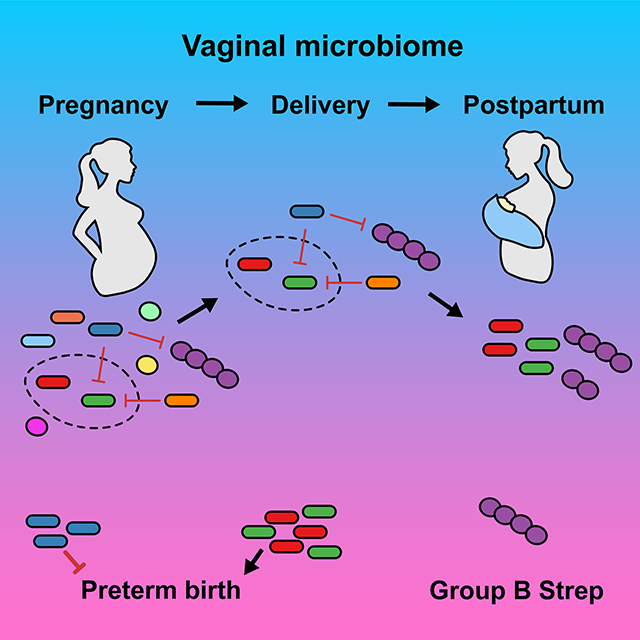
Graphical Abstract
Introduction
In 1892, Gustav Doderlein described his discovery that the vagina was dominantly populated with Lactobacillus spp. Since this time, the notion that lactic acid and hydrogen peroxide-producing lactobacilli are the keystone genera in a healthy vagina has led to the commonly accepted notion that Lactobacillus spp. stability and dominance are hallmarks of a “healthy” vagina and are central to reproductive health. Conversely, it has been largely assumed that vaginal communities where lactobacilli are either unstable or not dominant are dysbiotic and render overgrowth of pathobionts implicated in a number of female reproductive tract disorders (e.g., bacterial vaginosis [BV]1). This has led to over a century of interrogations seeking to specifically identify which non-lactobacilli bacteria are causal pathobionts.
In multiple population-based and case-control studies, BV is associated with an increased risk of occurrence of symptomatic vaginitis, preterm birth, intra-amniotic infections, cervical dysplasia, sexual acquisition and shedding of HIV, and susceptibility to ascending genital infection.1 Since BV is asymptomatic in at least 50% of cases, it is referred to as a vaginosis and not a vaginitis, and given a prevalence as high as 60%, is arguably a community variant rather than a true dysbiosis.2 Moreover, since numerous culture-independent microbial studies have sought and failed to identify precisely which bacterial clades, species, or strains cause BV-associated vaginal or reproductive disease, it is unclear which pathobionts are harbored under the largely clinically defined BV community umbrella.3–17 Of interest, one recent publication from Malawi has found that only minority of recently pregnant women in this sub-Saharan country are Lactobacillus spp. dominated, suggesting that there may be vast regional variation in what constitutes a “healthy” microbiome signature.18
In addition to lack of understanding regarding which species of vaginal bacteria are beneficial and which are potentially harmful from one population to the next, there is poor concordance between vaginal microbial profiling studies and whether the presence or absence of vaginal species or shifts in taxonomic profiles are reliably predictive of preterm birth (i.e., birth before 37 weeks of pregnancy). Studies on the microbial etiology of preterm birth have both found an association9,11,13,19,20 or no association12 with BV and its treatment, an association with an increased abundance of Gardnerella vaginalis and lack of Lactobacillus at the genus level8,11 or no association with either taxa,6,12 as well as an association8,9,19,21 or no association11,12 with L. iners abundance. Interestingly, in one study only the combination of a reduced relative abundance of L. crispatus and an increased abundance of Prevotella were found to associate with preterm birth.11 Of interest, utilizing 16S rRNA gene amplicon sequence variants (ASVs)11 identified multiple sequence variants (i.e., potential strains) of G. vaginalis, of which a single ASV with a potentially different functional capacity was found to drive the observed preterm birth association, indicating that even species level resolution may be insufficient in predicting preterm birth. These data highlight the difficulties in not only reproducing associations between vaginal taxa and preterm birth based on 16S rRNA data, but that the very definition of a “healthy”, a “variant”, or a “dysbiotic” vaginal microbiome varies significantly, further emphasizing the need for high resolution studies in prospective cohorts.20,22
Given the public health and clinical importance of these questions for both maternal and infant health, there is an evident need to (i) reliably resolve the vaginal community membership and its function to the species and strain level; and (ii) model the high-resolution community dynamics during the perinatal period (e.g., during pregnancy and the post-partum interval). With this in mind, we undertook a large, prospective study employing metagenomics sequencing with advanced analytic approaches. In this study we sought to first assess differences in WGS metagenomics and targeted 16S rRNA gene amplicon sequencing (V1V3 hypervariable region) in parallel samples from different vaginal subsites. We then used metagenomics data to determine community transitions during pregnancy, at delivery, and into the postpartum interval. Armed with the resultant WGS reference data resolved to the species and strain level, we quantified ecological interactions within the vaginal microbiome in order to test for associations between species and their strains and host genetic background (as measured by mitochondrial DNA at a genome wide significance) among term and preterm births. Finally, we aimed to explore the potential clinical utility and significance of WGS metagenomics by first determining the concordance between metagenomics-based identification of a clinically relevant pathobiont (Group B streptococci, or Streptococcus agalactiae) and clinical cultivation data, then determined patterns of species and strain co-occurrence and exclusion. The net outcome of these comprehensive analyses is a metagenomics-based model identifying reliable and predictable signatures of vaginal microbial ecology during pregnancy, resolved to the strain level, and resultant implications on two clinically relevant conditions (preterm birth and vaginal GBS), their diagnosis, and potential for innovative therapies.
Results
Vaginal microbial community composition and structure
WGS metagenomics has been held as the gold standard in profiling microbiomes resolved to the species and strain levels. However, since previous studies of the vaginal microbiome have alternately utilized multiple regions of the 16S hypervariable region resulting in discrepancies in results, we sought to determine whether metagenomics could be utilized to more accurately profile the vaginal microbiome. Altogether, we identified 229 taxa resolved to species level from n=182 participants’ vaginal samples (243 samples subjected to WGS, 248 samples subjected to targeted 16S rRNA gene amplicon sequencing; Figure S1). When we examined the overall community species composition of vaginal samples submitted for WGS metagenomic sequencing we found five clusters as the optimum (k-means, average silhouette width of 0.53) (Figure 1A). Three of the clusters are dominated by a single species – L. iners, L. crispatus, and G. vaginalis, the fourth cluster is dominated by two species – L. jensenii and L. iners, and the fifth cluster that contains a diverse assemblage of bacterial species. In comparison, when we examined the overall community composition of samples submitted for 16S rRNA gene amplicon sequencing we found ten clusters (k-means, average silhouette width of 0.56) dominated by seven taxa including multiple Lactobacillus species - L. iners, L. crispatus, L. jensenii, L. acidophilus, and L. gasseri, as well as Atopobium vaginae, and Sneathia sanguinegens (Figure 1B). The remaining three clusters identified within the 16S data consisted of various taxa, including a L. jensenii/L. iners cluster, a L. iners/mixed taxa cluster, and a cluster that contains an assembly of taxa with no single predominant species.
Figure 1.

Characterization of the vaginal microbiome using WGS metagenomics and targeted 16S rRNA gene amplicon sequencing. (A-B) Relative abundance of the top twenty most prevalent microbial species of the vaginal microbiome based on WGS metagenomics and targeted 16S rRNA gene amplicon analysis. (A) WGS metagenomics derived species relative abundances. The 20 most prevalent taxa contribute 94 percent of the total abundance. (B) 16S rRNA ASV derived species level relative abundances. The 20 most prevalent taxa contribute 96 percent of the total abundance. Samples/columns are grouped according to k-means cluster membership and rows are clustered hierarchically using complete linkage. Column annotations indicate race/ethnicity, time point that sample was acquired, vaginal subsite (for WGS metagenomics), and term/preterm outcome; NA indicates not available. (C-D) Vaginal microbiome community structure. (C) Multidimensional scaling (MDS) ordination of Bray-Curtis distance of WGS vaginal samples supports the k-means clustering of five distinct communities (PERMANOVA, p = 0.001; pairwise PERMANOVA, all p = 0.001). (D) MDS ordination of Bray-Curtis distance of 16S rRNA vaginal samples at the species level supports the k-means clustering of ten clusters (PERMANOVA, p = 0.001; pairwise PERMANOVA, all p < 0.05). Samples are color coded according to cluster membership. Arrows/black dots denote landmark samples – samples with the highest relative abundance (in parentheses) of the indicated species. The percentage of variation explained by each axis is shown in parentheses on the x- and y-axis. Ellipses represent 95% confidence intervals.
Multidimensional scaling (MDS) ordination using Bray-Curtis distance of vaginal samples submitted for metagenomic sequencing supported the k-means clustering (PERMANOVA, p=0.001) (Figure 1C). The landmark samples (i.e., samples with the highest observed relative abundance) for L. iners, L. crispatus, and G. vaginalis are positioned near the vertices of the ordination, demonstrating their association with variation in the overall community structure (Figure 1C). We did not strictly observe differences in beta diversity by virtue of vaginal subsite overall or vaginal subsite at the time of sampling (PERMANOVA, p>0.05) (Table 1, Figure 1E). Of note, the PERMANOVA test for vaginal site included samples from the same individual but at different time points. Although we and others have shown diminished diversity and richness in the same individual at the same site across gestation, these cannot be considered independent obsevations and caution with interpretation of PERMANOVA is warranted. However, we did find a significant difference by PERMANOVA in the community structure that corresponded to the sampling time point, driven primarily by the transition from pregnancy to postpartum; these community structure distinctions were observed at both the posterior fornix (p=0.009) and vaginal introitus (p=0.005) subsites (Figure 1E). Comparison of the number of observed taxa revealed a difference by virtue of sampling time point for all samples (Kruskal-Wallis, H=7.491, p=0.0236), that reflected an increase at postpartum compared to 3rd trimester (Dunn’s, p≤0.05) (Figure S2A). However, while the trend towards an increased number of species over time held true, these increases among individual subsites were not significant. In addition, we observed an increase in the number of detected species in the vaginal introitus compared to the posterior fornix for all samples (Mann-Whitney, U=3895, p=0.0048) (Figure S2A). When samples were stratified by sampling time point, we found an increase in the number of observed species in the vaginal introitus at the 3rd trimester time point (Mann-Whitney, U=927, p=0.0363) that was also observed at delivery and postpartum but failed to reach statistical significance (p>0.05) (Figure S2A). When we alternately examined the number of observed taxa on the basis of k-means cluster membership across time points, we found an increase in the number of taxa within the G. vaginalis and mixed community clusters compared to the Lactobacillus dominated clusters (Dunn’s, p ≤ 0.05) (Figure S2B). Differences in taxonomic associations were then tested for using linear discriminate analysis using LEfSe. The Lactobacillus clusters were found to be enriched primarily with their respective representative species, with the sole exception of the L. iners cluster, which was also enriched for Ureaplasma spp. (Figure S2C,D). In contrast, the G. vaginalis cluster was found to be enriched for Megasphaera and Prevotella spp., whereas the mixed cluster was found to be enriched for Atopobium vaginae and other BV-associated taxa (Figure S2C,D).
Table 1.
PERMANOVA p values for WGS and 16S rRNA MDS using Bray Curtis distance.
| Site | 3rd trimester by subsite | Delivery by subsite | Postpartum by subsite | Vaginal introitus by time point | Posterior fornix by time point | |
|---|---|---|---|---|---|---|
| WGS | 0.781 | 0.939 | 0.931 | 0.842 | 0.005 | 0.009 |
| 16S | - | - | - | - | - | - |
Footnote: Timepoint by subsite and subsite by timepoint p-values were generated from data subset to individual timepoints and subsites, respectively.
The marked discrepancy in the relative abundance of G. vaginalis between the metagenomic and 16S data led us to further interrogate which community members were driving these variations. When we examined paired WGS/16S samples, we observed significant differences with respect to G. vaginalis (increased in WGS), Lactobacillus at the genus and species levels (increased in 16S), and limited other taxa that are present in relatively low abundance (Figure S3A,B). Taken together, these data confirm previous reports suggesting that the V1V3 hypervariable region underrepresents G. vaginalis.11
Major transitions in community structure occur from pregnancy to postpartum
In a majority of cases, we found that within participants, the predominant species, with the exception of G. vaginalis, identified via WGS was concordant with that identified via 16S rRNA gene amplicon sequencing across vaginal subsites and from the third trimester to delivery. However, at postpartum, the predominant species differed dramatically from those at delivery (Figure S3C,D). We thus sought to model the temporal dynamics of the vaginal community through discrete time Markov chains (DTMC) of k-means cluster membership using the maximum likelihood estimate for transitions. DTMC analysis revealed that cluster membership was maintained in the 3rd trimester to delivery interval (average self-transition probability of 0.72), with limited transitions occurring between clusters (Figure 2A). Within the 3rd trimester to delivery interval, our model indicated that after mixed clusters (P=1.0), the G. vaginalis dominant cluster had the highest self-transition probability (P=0.83). Interestingly, cluster membership was observed to change considerably from delivery to postpartum, with a majority of Lactobacillus-dominated clusters transitioning to the mixed community cluster (average transition probability of 0.92), while self-transitions within either the mixed or G. vaginalis clusters remained high (P=1.0 and P=0.77, respectively). These patterns of transitions held across vaginal subsite (Figure 2B).
Figure 2.

Transitions between and within predominant species clusters during pregnancy and at postpartum. (A) Transitions from third trimester to delivery. Limited transitions occurred from the third trimester to delivery between the predominant species (k-means) clusters. (B) Transitions from delivery to postpartum. A majority of Lactobacillus dominated participants transition to the mixed community from delivery to postpartum. (C) Transitions between and within predominant species clusters during the perinatal period (during pregnancy and at postpartum) within vaginal subsites. Edge widths and colors represent transition probabilities. The mixed L. jensenii/L. iners cluster is denoted here as L. jensenii.
Microbial species associations within the vaginal econiche
To understand the patterns of species association, we utilized probabilistic modeling to determine significant positive and negative co-occurrences based on our WGS species abundance data.23 A majority of species co-occurrences (10,671 pairs) were omitted based on species pairs expected to have at least one co-occurrence. Of the remaining 1,419 species pairs, 347 (24.5%) significant co-occurrences were identified that corresponded to 294 (85%) positive co-occurrences and 53 (15%) negative co-occurrences. Lactobacillus species, including L. crispatus (85% negative co-occurrences; Fisher’s exact test, p<0.0001), L. jensenii (94% negative co-occurrences; Fisher’s exact test, p<0.0001), and L. iners (65% negative co-occurrences; Fisher’s exact test, p <0.0001) were exclusionary. In contrast, G. vaginalis was relatively permissive (18% negative co-occurrences; Fisher’s exact test, p=0.7317). Consistent with long held microbial characterizations of BV communities, L. crispatus and L. jensenii both negatively co-occurred with G. vaginalis, while the co-occurrence of L. iners and G. vaginalis was random. However, L. iners was found to have both positive (Megasphaera spp. and Ureaplasma spp.) and negative (Anaerococcus lactolyticus and Pophyromonas anaerobius) co-occurrences with prior BV-associated species (Figure 3). The majority of species that L. crispatus and L. jensenii negatively co-occurred with are species previously associated with clinically diagnosed BV (e.g., species from Mycoplasma, Megasphaera, Mobiluncus, Dialister, Pophyromonas, Prevotella, and Atopobium) (Figures 3 and 4).
Figure 3.

Probabilistic model of species co-occurrence demonstrates Lactobacillus spp. are relatively exclusionary. (A) Global network of significant positive and negative co-occurrences for species from the vagina. (B) Significant pair-wise co-occurrence networks for select keystone/predominant vaginal bacterial species. Orange dashed lines represent significant negative co-occurrences (p ≤ 0.05), solid blue lines represent significant positive co-occurrences (p ≤ 0.05), line widths represent the strength of the co-occurrence, with node sizes scaled within each subplot according to the average relative abundance across all samples.
Figure 4.

Species co-occurrences during pregnancy (A, left panel) and at postpartum (A, right panel). (B) Taxonomic associations with preterm birth. Subjects delivering preterm were enriched for G. vaginalis, A. vaginae, and other BV-associated taxa, whereas subjects delivering at term were enriched for Lactobacillus spp.
(C) The effect size for differentially enriched taxa based on preterm birth outcomes was calculated from the average relative abundance derived from WGS metagenomics during pregnancy (3rd trimester and at delivery) via LEfSe.
Inability to robustly predict preterm birth based on vaginal ecology resolved to species
Intrigued by our observations of co-occurrences and exclusions during pregnancy, we hypothesized that different WGS-assigned species co-occurrence patterns might be observed at different gestational age intervals. As an initial step, we first sought to characterize broadly the pregnancy and postpartum intervals (Figure 4A). When samples were stratified to pregnancy (Figure 4A, left panel) or postpartum (Figure 4A, right panel), we found that the pattern of significant co-occurrences during pregnancy comprised a network of positive (18.7%, 190/1012 analyzed pairs) and negative co-occurrences (3.2%, 32/1012 analyzed pairs), i.e., a signature microbiome of pregnancy classifying as “exclusionary”. At postpartum sampling, there was an increase in the number of positive co-occurrences (19.3%, 129/667 analyzed pairs) and decreased in the number of negative co-occurrences (0.7%, 5/667 analyzed pairs) (Fisher’s exact test, p=0.0011) (Figure 4A), thereby classifying the postpartum period as “permissive”.
Given these distinctions between pregnancy and the post-partum period, as well as heterogeneity of prior findings as to whether G. vaginalis or Lactobacillus species reliably predict preterm birth,9,11,13,19 we next sought to determine whether higher resolution vaginal community profiling could more reliably predict preterm birth. Using WGS metagenomics we found that the average relative abundance of G. vaginalis was increased in preterm participants during pregnancy compared to term participants (Mann-Whitney, U=45, p=0.0136) (Figure 4B). When resolved to genus level, Lactobacillus was decreased in preterm participants during pregnancy compared to term participants (Mann-Whitney, U=44.5, p=0.0125), although there was no difference in the relative abundance of L. crispatus or L. iners species (Figure 4B). Similarly, when we examined the metagenomic data for the differential enrichment of taxa via LEfSe, Lactobacillus spp. were found to be enriched in participants with term deliveries, whereas G. vaginalis and other species associated with BV were enriched in participants with preterm deliveries (Figure 4C). When alternately analyzed by ANCOM, only L. gasseri is observed to be differentially abundant when comparing term and preterm.
Stratifying the WGS data by sample time point and vaginal subsite, we again observed a significant increase in the relative abundance of G. vaginalis in preterm participants during the early 3rd trimester (vaginal introitus, U=48, p=0.0481) and an increase in the relative abundance of Lactobacillus in term participants at delivery (vaginal introitus, U=21, p=0.0350; posterior fornix, U=4, p=0.0484) (Figure S4). This is certainly consistent with the observation by us and others that Lactobacllus maybe generally protective against preterm birth by virtue of its association with term birth. However, when we performed a Fisher’s exact test on the presence of G. vaginalis and prediction of preterm birth, we failed to observe a significant association (p>0.99) with an odds ratio of 1.274 (0.06 to 26.29, 95% CI). Taken together, these analyses indicate that the lone presence of G. vaginalis cannot predict nor be considered to attribute to preterm birth.
The relationship between human mtDNA variants, vaginal microbes, and occurrence of preterm birth
Given the inability to consistently replicate studies linking G. vaginalis and Lactobacillus spp. to preterm birth,9,11 other investigators have posited that inherent differences in risk-disparate cohorts may be masking underlying true associations. Since we and others have published an association between genetic polymorphisms of host mitochondria and the microbiome, including the gut and vagina,24 we next sought to evaluate the association of the vaginal microbiome with mitochondrial polymorphisms as risk-modifiers of preterm birth. PLINK, a toolset for linkage analysis, was used to identify significant associations between mitochondrial DNA (mtDNA) single nucleotide polymorphisms (SNPs) and the average abundance of individual taxa during pregnancy. Although a number of significant taxa-SNP associations were identified in WGS (n=1,588) (Figure 5A), these associations were all in relatively minor taxa and did not include the major keystone species driving the vaginal community, including L. crispatus, L. iners, L. jensenii and G. vaginalis. With respect to preterm birth, five SNP-species associations identified by WGS metagenomics were significantly different between term and preterm participants (Figure 5B, Table S1). However, post-hoc comparisons revealed these to be minor taxa present at low abundance and frequency (e.g., Propionibacterium acnes, Haemophilus haemolytica, Veillonella atypica, Veillonella parvum, and Lactobacillus mucosa) (Figure 5C).
Figure 5.

Associations of the vaginal microbiome (WGS metagenomics) with mitochondrial DNA polymorphisms in the context of preterm birth. (A) Manhattan plot demonstrating significant associations between identified taxa and mtSNPs as determined by PLINK associations. (B) Identification of taxa-SNP associations significantly different in the context of preterm birth. Q-values for taxa-SNP associations are plotted on the x-axis, while q-values for quantitative trait interaction (taxa-SNP-preterm birth) are plotted on the y-axis. (C) Relative abundance of vaginal species identified as significantly different between subjects with term and preterm deliveries. Horizontal red bars represent group means.
Strain-level profiling of keystone vaginal microbiota
We next performed strain-level profiling of our metagenomic samples for G. vaginalis, L. crispatus, L. iners, and L. jensenii via pangenome-based phylogenomic analysis (PanPhlAn) to determine whether variations in the presence and function of strains might associate with differences in pregnancy outcomes (term/preterm birth). Altogether, we were able to classify 29 (across 74 samples), 16 (37 samples), 35 (92 samples), and 15 (40 samples) participants at the strain level for G. vaginalis, L. crispatus, L. iners, and L. jensenii, respectively (Figure S5). We found G. vaginalis to cluster into five distinct clades – Gv1a, Gv1b, Gv2a, Gv2b, and Gv3 (PERMANOVA, p=0.001) (Figure 6A). Previously, G. vaginalis reference genomes that belong to the Gv1a/Gv1b groups and Gv2a/Gv2b have been assigned to G1 and G2 clades, respectively, except for reference genome 1400E which was assigned to a third clade that was nested within the G2 clade.11 Here, we found Gardnerella vaginalis strain 1400E to group firmly group within the Gv2a/Gv2 clades and instead found that another reference genome (CMW7778B) represented a different, but distinct Gv3 clade. Within the lactobacilli, we found L. crispatus, L. iners, and L. jensenii to each cluster into two distinct clades (e.g., Lc1 and Lc2, Li 1 and Li2, Lj1 and Lj2, respectively) (PERMANOVA, p=0.001) (Figure 6A).
Figure 6.

Keystone species are present as multiple strains. (A) Principal coordinate analysis (PCoA) of the binary Jaccard distances of pangenome centroids for vaginal samples and reference strains of keystone species. Bacterial reference strains are colored by their respective cluster. Vaginal samples are represented as filled black circles. The percentage of variation explained by each axis is shown in parentheses. Ellipses show the 95% confidence intervals for the reference strains. (B) Species and strain level co-occurrences for pairwise complete observations. (C) Strain-specific functional capacity in vaginal samples during the perinatal interval. Relative proportions of strain-specific metabolism, cellular processes, and environmental information processing KEGG pathways for G. vaginalis, L. crispatus, and L. jensenii.
Within our samples, nearly half of participants with G. vaginalis strain profiles contained strains from multiple G. vaginalis clades (13/29, 45%) (Figure S5A). Participants classified with possessing a single G. vaginalis strain, however, were found to be stably maintained over time. In contrast, participants with strain profiles for L. crispatus, L. iners, and L. jensenii were found to contain strains from single clades (Figure S5B–C), with the exception of one subject that was found to possess two different L. iners strains at different time points – Li2 at third trimester and delivery and Li 1 at postpartum (Figure S5D). Although we identified two distinct L. crispatus clades among the reference genomes, only a single subject with a term delivery was found to possess a strain from the Lc2 clade and the remaining participants possessed strains from the Lc1 clade (15/16 participants) (Figure S5C).
As G. vaginalis variants or strains have previously been shown to associate with preterm birth, specifically a strain belonging to the Gv2 clade,11 we next tested whether differences in strain frequencies were associated with term or preterm birth, and then determined whether exclusionary or permissive interactions between strains exist. We did not find a significant difference in the frequency of multiple G. vaginalis strains during pregnancy on a per subject basis (preterm: 40%, 2/5; term: 40%, 8/20; Fisher’s exact test, p>0.99), which also did not hold up as significant when alternately analyzed by attribution (OR=1.0; 0.1352–7.396 95% CI). When we examined the frequency of individual G. vaginalis and lactobacilli strains, we also failed to identify any clear associations with preterm birth (Fisher’s exact test, p>0.05; Table S2). When we then examined the patterns of co-occurrence resolved to the strain level, we found L. crispatus strains belonging to Lc1 and L. jensenii strains belonging to Lj 1 negatively co-occurred with G. vaginalis strains belonging to the Gv1 and Gv2 clades, respectively (Figure 6B). Similarly, L. jensenii negatively co-occurred with G. vaginalis Gv2b strains. While no L. iners strains were found to positively or negatively co-occur with G. vaginalis, at the species level L. iners negatively co-occurred with G. vaginalis Gv2b strains and positively co-occurred with G. vaginalis Gv3 strains.
Given that differences in the metagenomics-determined functional capacity of G. vaginalis (Gv1 and Gv2) have been previously described,11 we next determined whether clade-specific differences existed. We found that the majority of strain-specific functions were largely redundant (Figure 6C). One notable exception was Gv2b, whereby the Gv2b clade demonstrated enrichment for transport and catabolism, as well as lipid and xenobiotics metabolism. Similarly, the functional capacity of the keystone Lactobacillus spp., L. crispatus strains in the Lc1 clade could be differentiated from Lc2 by virtue of enrichment for metabolism of carbohydrates, lipids, xenobiotics, cofactors and vitamins, alongside signal transduction and membrane transport (Figure 6C). For L. iners, we found that strains from Li2 were functionally distinct in their capacity for glycan biosynthesis, specifically peptidoglycan biosynthesis. L. jensenii strains from Lj 1 were functionally dissimilar from Lj2 in their capacity for metabolism of carbohydrates, glycans, lipids, xenobiotics, and membrane transport (Figure 6C).
The vaginal ecology of the pathobiont Group B Streptococcus
We first determined the concordance between metagenomic detection of group B Streptococcus (GBS) and results from clinical cultivation tests meeting current U.S. guidelines.25–27 To reliably identify GBS, we utilized two tools that differ in their approach for classifying microbial metagenomes, MetaPhlAn2 and Centrifuge. MetaPhlAn2 utilizes species and clade-specific marker genes, whereas Centrifuge relies on alignments to compressed pan-genomes. We found Centrifuge to perform better than MetaPhlAn2 at detecting GBS when benchmarked to positive clinical cultivation samples (Figure S6A). Of participants with a positive GBS clinical culture, MetaPhlAn2 identified 1/5 participants as having GBS, whereas Centrifuge identified 4/5 participants. For participants with a negative GBS culture, MetaPhlAn2 identified 2/55 participants with a greater than zero relative abundance of GBS, whereas Centrifuge identified 50/55 participants. We did not detect a significant difference in the relative abundance of GBS based on clinical culture status overall (Mann-Whitney, p=0.196, U=1201; Figure S6B), maximum relative abundance per subject (Mann-Whitney, p=0.3595, U=102; Figure S6C), or when data was stratified by vaginal subsite or sampling time point (Mann-Whitney, p>0.05; Figure S6D). Furthermore, we did not detect a significant difference in the relative abundance of GBS over time in participants, including those with a positive clinical culture and subsequent administration of intrapartum antibiotics at delivery. When we set clinical culture as the benchmark “gold standard” for the detection of GBS, we found metagenomics to be an equally sensitive predictor of GBS carrier status in the vagina, and ability to detect with WGS was not impeded following intrapartum antibiotics (Supplemental Methods, S1).
To further corroborate the accuracy of our metagenomic GBS prediction, we mapped to the reference genome 2603V/R. On average, samples with a GBS relative abundance of zero had a 0.23-fold coverage and percent coverage of 2.12%, compared to samples with a greater than zero relative abundance that had a 0.40-fold coverage and percent coverage of 3.69%. At >1% relative metagenomic abundance, the fold coverage marginally increased to 0.47-fold with a percent coverage of 8.06%. Interestingly, nine of the top ten samples with the highest percent coverage came from participants with negative GBS clinical cultures (Figure 7A) and the sample with the highest percent coverage (81.5%, average 2.73-fold coverage, 70.2% relative abundance) came from a subject with a negative clinical culture (Figure 7B). In comparison, the sample with the highest relative abundance of GBS from a subject with a positive clinical culture (2.7% relative abundance) had a percent coverage of 4.8% and average 1.89-fold coverage (Figure 7B). These data suggest that in at least once case, clinical cultivation missed GBS carriage while metagenomics detected the organism at 2.73X and high (70%) relative abundance.
Figure 7.

Metagenomic identification of GBS. (A) GBS reference genome coverage. Samples from participants with positive GBS clinical cultures are shaded red and negative GBS clinical cultures are shaded green, circle size represents GBS relative abundance. Representative samples annotated i-iii indicate the i) sample with the highest relative abundance of GBS that had a positive clinical culture, ii) sample with a zero-relative abundance of GBS that had a negative clinical culture, iii) sample with the highest relative abundance of GBS that had a negative clinical culture. (B) Genomic coverage of reference genome 2603V/R binned at 1 kb for representative samples i-iii from panel A. Positions of 16S genes (16S) are highlighted by the dashed red lines and the CAMP factor gene (cfb) is highlighted by the dashed purple line. (C) GBS positively co-occurs with bacterial vaginosis associated species and differentially co-occurs with Lactobacillus spp. Positive co-occurrence with L. iners, but a negative co-occurrence (exclusion) with L. crispatus.
We next sought to determine whether an initial positive diagnosis of GBS via metagenomics sample might be predictive of future detection, and whether there was a difference in species abundance based on GBS status. Markov chain modeling of GBS status as defined by WGS indicated GBS positive participants are more likely to remain GBS positive at subsequent time points (Supplemental Methods, S1). We analyzed the differential enrichment of species during pregnancy using LEfSe based on positive GBS clinical cultivation and WGS-assigned GBS status (Supplemental Methods, S1). As assessed by positive GBS clinical cultivation, we found a limited number of differentially abundant taxa, including an increased enrichment of Ureaplasma urealyticum, Corynebacterium glucuronolyticum, Propionibacterium acnes, and Haemophilus haemolyticus (Figure S6E). When samples were instead classified by WGS-assigned GBS status (at least one sample of a given subject with an observed relative abundance during pregnancy), we identified an increased enrichment of Veillonella parvula, Peptoniphilus harei, and decrease of Akkermansia muciniphila (Figure S6F). When WGS-assigned GBS status was modified to a relative abundance >1%, we identified an increased enrichment of Megasphaera sp. UPII 199 6, S. agalactiae, Varibaculum cambriense, Jonquetella anthropi, Propionibacterium avidum, Lactobacillus iners, Staphylococcus aureus, Acinetobacter baumannii, Corynebacterium glucuronolyticum, and Fusobacterium gonidiaformans (Figure S6G).
When we imputed the Centrifuge GBS presence/absence calls into our previous species co-occurrence model we found 21 significant associations that were not previously identified in the initial clade-specific marker classification (Figure 7C). The majority of the significant associations represented positive co-occurrences (n=19), including a positive co-occurrence with L. iners. The two negative co-occurrences identified were with an unclassified Neisseria spp. and, interestingly, L. crispatus. When we examined the patterns of co-occurrence over time, we found significant co-occurrences for GBS during pregnancy, with a shift towards random co-occurrences postpartum. A notable exception of a positive and negative co-occurrence was observed with Prevotella buccalis and Prevotella copri, respectively (Figure S6H). Prevotella are among the members of the microbial consortium that define BV, including P. buccalis.
Second trimester cohort study
To determine whether the vaginal microbiome trends associated with preterm birth outcomes observed in our initial cohort (“third trimester cohort”) might be observed earlier in pregnancy, we compared a subset of cases and controls from a prospectively enrolled cohort. Specifically, we performed metagenomic sequencing on posterior fornix samples collected during the second trimester and at delivery from a case-control nested cohort (“second trimester cohort”) of 23 participants. Of the 23 preterm birth cases and controls utilized for this current nested analysis, 11 went on to deliver preterm (average GA of 33.3±3.5 weeks) and 12 had term deliveries (average GA of 38.1 ±1.3 weeks) The average GA (weeks) for those that went on to deliver term and preterm at the second trimester sampling was 22.7±2.6 and 22.9±2.6, respectively; Mann-Whitney, U=61.5, p=0.79). In contrast with the results from our initial third trimester cohort, when we compared the relative abundance of keystone species based on term versus preterm birth outcomes in this subset of cases and controls sampled in the second trimester, we found only L. jensenii to differ at the second trimester time-point (average abundance in term: 0%, in PTB: 7.3%; no difference at delivery), with no significant difference in the average relative abundance of G. vaginalis, Lactobacillus species (genus-level), L. crispatus, or L. iners species nor strains at either the second trimester or delivery time points (Figure S7). We found no significant difference in the relative abundance of keystone species between preterm and term deliveries (Figure S7) but acknowledge the risk of underpowering by comparing n= 11 cases and n=12 controls.
Similarly, an evaluation of the abundance of species identified via SNP-species associations with the occurrence of preterm birth identified in the late third trimester cohort was inconclusive in the second third trimester cohort. Only two of the five species originally identified were sparsely present - P. acnes (i.e., Cutibacterium acnes, present in 14 samples; relative abundance 2.5±15.4%, mean±standard deviation) and V. atypica (1 sample at 0.11%). There was no significant difference in the relative abundance of P. acnes based on preterm birth occurrence.
Finally, we examined how strain profiles of the vaginal keystone taxa in this second trimester cohort might also be associated with birth outcomes. We identified distinct strain profiles of G. vaginalis in 11 participants (16 samples) at least once and in 5 participants at both time points; L. iners in 15 participants (20 samples) and in 3 participants at both time points; L. crispatus in 6 participants (9 samples) at least once and in 3 participants at both time points; and L. jensenii in 6 participants (7 samples) at least once, and in a single participant at both time points. As observed in the initial cohort, we found no statistical support for the presence of any individual keystone strain and birth outcomes (Table S3).
Discussion
In this study we have performed a robust and high-resolution taxonomic profiling of the vaginal microbiome during and after pregnancy using WGS metagenomics to illustrate the importance in resolving constituent members to the strain level. We identified multiple and specific pitfalls in relying solely on 16S targeted amplicon profiling, notably an underrepresentation of G. vaginalis and overrepresentation of specific Lactobacillus spp. We find that although a majority of species and strain interactions are random, significant and predictable co-occurrences are generally exclusionary in pregnancy (‘non-permissive”), whereas postpartum is classified by an increase in positive co-occurrences (“permissive”). These observations held true for strain level profiling of Lactobacillus spp., G. vaginalis, and the often neglected pathobiont Group B Streptococcus. Taken together, these data demonstrate that the ecology of the vaginal microbiome is mainly driven by the abundance of four keystone species, that both community profiling and functional differences exist within these species to the strain-level, and that there is considerable value in evaluating the contribution of individual strains when examining for associations with disease risk. These findings are consistent with principles of microbial ecology, and meaningfully advance our initial observations29 to show for the first time that in a diverse U.S. based cohort, its unique vaginal microbiome signature during pregnancy arises as a result of exclusionary co-occurrences of species, strains, and clades.
WGS metagenomics robustly captures the vaginal microbial community structure at the species level
Although WGS metagenomic sequencing has been held as the gold-standard for studies of microbial communities, targeted 16S rRNA gene amplicon sequencing has disproportionally been utilized in studies of the vaginal microbiome due to a comparatively lower cost and fewer computational demands. We found that WGS metagenomics more accurately captures the diversity and dynamic ecology of the vaginal microbiome compared to targeted 16S rRNA gene amplicon analysis. Specifically, we found that V1V3 greatly underrepresents the abundance of Gardnerella vaginalis (Figure 1). Therefore, we propose that the V1V3 primer set should be used with attention to this point, and/or in combination with another primer set, when profiling the vaginal microbiome. Overall, the vaginal community structure in pregnancy is largely structured by the abundance of four species – L. crispatus, L. iners, L. jensenii, and G. vaginalis, or their relative absence (Figure 1).
The vaginal microbiome has complex community dynamics that comprise a signature profile during pregnancy resulting from exclusionary co-occurrences
Consistent with previous data,28 when we examined transitions within and between cluster membership, indicative of the predominant species during pregnancy, we found the vaginal microbiome to have a distinct and mostly stable signature. Cluster membership was unlikely to change during pregnancy, which is consistent with previous observations27 that stability of the vaginal microbiome tends to increase with increasing gestational age, with Lactobacillus species predominating. However, as previously reported,29 in the time period from delivery to postpartum we observed a dramatic shift for each Lactobacillus-dominated cluster towards the mixed community cluster. In contrast, prior membership in either G. vaginalis-dominated or mixed community clusters was maintained. These changes are likely a consequence of parturition, though the shift toward a mixed community cluster also occurred in women who delivered by an unlabored Cesarean surgery. This would suggest that there are inherent changes to the vaginal niche preceding and independent from labor and descent of the newborn through the vaginal canal, which precipitate these microbial community shifts. Furthermore, it is unclear how long this postpartum signature lasts and if and when the vaginal microbiome transitions back to a Lactobacillus dominant state.
Lack of robust association between any single vaginal microbe and preterm birth
Despite the challenges in reliably demonstrating that there is a predictive vaginal microbial signature for preterm birth which is replicative across racially and ethnically distinct cohorts in different regions of the U.S., the importance of doing so is evident. Worldwide, 12–15 million neonates are delivered preterm annually.30 This is accompanied by as high as a 27.5% mortality rate,31 and significant morbidity among those that survive. Currently our clinical prediction tools are limited and of poor prognostic value, resulting in both unnecessary interventions or ineffective therapies to millions of pregnant women annually.32,33 The circumstantial evidence for culpability of members of the vaginal ecologic community has long been present, including decades of data associating BV with preterm birth.34 Since level I evidence (randomized controlled trial) has shown that antibiotic treatment for asymptomatic vaginal dysbiosis can result in a higher rate of preterm birth when compared to placebo,35–38 metagenomics studies which resolve species and strain community functional ecology are needed prior to undertaking further interventions.
Host genetics must also be taken into consideration, as previous research has failed to reach a consensus on whether the vaginal microbiome has a characteristic profile that reliably predicts PTB. Romero et al. reported that the bacterial composition and abundance did not differ between mothers who delivered preterm compared to those who delivered at term in a primarily African-American cohort.6 Conversely, DiGiulio et al. reported that reduced Lactobacillus and increased Gardnerella or Ureaplasma was associated with increased risk of PTB in a largely Caucasian cohort.9 Callahan et al. also identified an association of Gardnerella with preterm birth, but only within one cohort of primarily Caucasian women, and not within a larger African-American cohort.11 Notably, the ethnic and racial demographics of these studies varied significantly, suggesting that what may be considered vaginal dysbiosis may differ depending on host factors.39,40 Within this study, we attempted to separate social determinants of health from race or ethnicity, using mitochondrial DNA as a genetic measure; however, while we identified taxa by mtDNA SNP associations within the vagina, none were strongly predictive of preterm birth.
However, we did identify that an increased abundance of Gardnerella vaginalis and decreased abundance of Lactobacillus at the genus level was associated with preterm birth, but absolute presence/absence of any given strain was not associated with preterm birth. Prior studies have indicated that the V1V3 hypervariable region best discriminates Lactobacillus spp., leading to its nearly uniform adoption for vaginal microbiome studies. However, using matched samples sequenced in parallel by both 16S (V1V3) and WGS methodologies, we have shown that 16S leads to an underrepresentation of Gardnerella. This has been alluded to previously by Callahan et al,11 but is more definitively evidenced by the current study data. However, only a small minority of participants had clinically apparent BV, whereas many more carried Gardnerella vaginalis without overt symptoms and without preterm birth.
Strain-level ecology of the predominant vaginal species
Although we did not identify a robust association between any single microbe and preterm birth, we did observe several significant patterns of strain co-occurrence when assessed at the strain level (Figure 8B). The major co-occurrence patterns differ significantly between L. crispatus and L. iners, where L. crispatus demonstrates a strong negative co-occurrence with BV-associated taxa, while L. iners is permissive or positively co-occurs (potentially facilitates or enhances its presence) to BV-associated taxa. Genomic comparisons between L. crispatus and L. iners may potentially explain these differences. L. crispatus, in contrast to L. iners, produces the D-isomer of lactic acid, which has been shown to potently inhibit infection by Chlamydia trachomatis in vitro. Indeed, co-colonization studies using clinical isolates have shown L. crispatus strains to inhibit the growth or adhesion of G. vaginalis strains.41–43 Conversely, G. vaginalis strains (e.g., 101-a BV-isolate classified in our work as a member of the Gv1b clade, and 5–1- a healthy isolate classified as a Gv1a member) are also capable of differential adhesion inhibition of Lactobacillus spp. G. vaginalis strains from the Gv1b clade, but not the Gv1a clade, have been shown to displace L. crispatus.43 Therefore, these observed co-occurrence patterns can be a consequence of reciprocal inhibitory actions by both L. crispatus and Gardnerella vaginalis. Interestingly, L. iners produces inerolysin, a pore-forming cytolysin, that is similar in structure and function to vaginolysin produced by G. vaginalis, which can facilitate host cell lysis for resource liberation44. Interestingly, when G. vaginalis is grown in the presence of L. iners (ATCC 55195 - classified as a member of the Li1 clade), there is an enhanced adhesion of the Gv1b strain, but not the Gv1a strain.43 This specific feature of L. iners may facilitate G. vaginalis colonization, or may confer a selective advantage in resource poor conditions that similarly promote G. vaginalis. Our co-occurrence observations cannot distinguish the true driver of the associations, and further studies are needed to explain these interactions.
We found participants colonized by the major Lactobacillus species are typically colonized predominantly by single strains. Interestingly, L. crispatus CTV-05 is a vaginally derived and isolated strain that has been suggested to be efficacious in the treatment of BV, and has been shown to decrease the abundance of G. vaginalis in limited trials.45–47 A review of our strain level data shows that L. crispatus CTV-05 belongs to the Lc1 clade, which we found to negatively co-occur with G. vaginalis Gv1 strains. Furthermore, colonization of CTV-05 when used as a vaginal probiotic in women already colonized by L. crispatus is found to be reduced or minimized compared to women lacking endogenous L. crispatus.45,47 Although the exact strains present in the women already colonized with L. crispatus at enrollment were unknown, our findings of only a single strain for each Lactobacillus species within participants is also consistent with prior data,48 and suggest that once a single Lactobacillus strain has colonized, exclusionary interactions with other strains of the same species may occur. Alternatively, the presence of a single strain may be due to founder effects, and the opportunity to be colonized by an additional strain has not yet occurred. This could have profound implications for potential therapies that seek to utilize antibiotics and/or probiotics in the treatment of vaginal dysbiosis and reproductive health.49 Furthermore, as associations with vaginal health can be confounded at the level of genus and even species,11 our findings highlight that strain-level profiling can provide an even more accurate and highly specific definition of what constitutes a normal or dysbiotic vaginal microbiome.
Conversely, we found participants colonized by G. vaginalis can be found to contain multiple strains. Prior work has demonstrated that colonization of multiple G. vaginalis is associated with BV.50 Additionally, our data suggest that G. vaginalis strains belonging to the Gv1 clade, specifically Gv1a variants, and Gv2b variants, might moderate risk occurrence of preterm birth. This is partially consistent with prior results showing G. vaginalis strains belonging to the Gv2 clade were significantly associated with preterm birth in a Caucasian, but not African-American, cohort.11 Furthermore, different strains of the same species have been found to elicit different host immune responses.51 However, further studies focusing on participants with vaginal microbiomes containing an appreciable abundance of G. vaginalis are needed since our data failed to reach strong significance in either prediction or attribution.
Potential clinical implications of our findings with respect to Group B streptococci ecology
Although a large number of studies over the years have focused on identifying which pathobionts drive BV-associated disease and PTB in particular, much less attention has been focused on understanding the ecology of other pathobionts, such as vaginal Group B Streptococcus (GBS or Streptococcus agalactiae). GBS is a Gram-positive alpha-hemolytic bacterium that can cause invasive GBS disease in the early newborn (<6 days of age), characterized primarily by neonatal sepsis and pneumonia. In contrast to the early neonate, GBS rarely causes morbidity in the pregnant women who carry it, although it may be associated with urinary tract infections, amnionitis, endometritis, or sepsis/meningitis either during pregnancy or in the postpartum interval.25 With GBS colonization of the vagina or rectum occurring in an estimated ten to thirty percent of pregnant women,25 GBS may be considered a pathobiont member of the human microbiome. In an effort to eliminate neonatal mortality due to early invasive GBS disease, the current U.S. standard for maternal GBS detection during pregnancy is universal screening by vaginal/rectal culture at 35–37 weeks gestation, or with preterm labor or preterm premature rupture of membranes.25–27 Since 2011, U.S. guidelines have provided a permissive statement for a limited role of nucleic acid amplification tests for intrapartum testing for GBS.25–27 The current U.S. recommendation for a positive GBS culture test result (or with history of previous infant with GBS septicemia, positive maternal GBS bacteriuria during pregnancy) is intrapartum antibiotic prophylaxis, resulting in as many as 1 million U.S. women receiving multiple doses of antibiotics in labor annually.25 However, other developed countries with similar rates of asymptomatic maternal GBS colonization during pregnancy instead take a risk-based approach to GBS screening and treatment.52 Irrespective of the method used to determine who receives prophylaxis for the prevention of perinatal GBS disease, given a current case prevalence of invasive early newborn GBS disease of less than 0.4 cases/1000 live births (and a pre-national guidelines prevalence of 1.7 cases/1000 live births25–27), thousands of women will be exposed to multiple antibiotic courses in order to prevent a single neonatal case. As expected, the current guidelines on either continent have had no effect on late-onset GBS disease (defined as occurring in neonates older than 6 days).25–27 If viewed through the lens of a vaginal microbial ecologist, given the great disparity between maternal prevalence (10–30%) and early invasive newborn disease (<0.02% at baseline, currently <0.004%), studies that detail the exclusion or co-occurrence of other microbes with vaginal GBS might provide a refined definition of who is at risk for transmission and invasive newborn GBS disease. The overarching goal of such work would be to provide an evidence-based rationale for subsequent clinical trials which could potentially refine and reduce the need for intrapartum antibiotics prophylaxis. This is clinically important, since although the rate of early onset GBS sepsis in both low and normal weight birthweight neonates have declined, the rate of ampicillin resistant E. coli sepsis has concomitantly increased.53,54 As a result, the overall rate of early-onset sepsis has not significantly changed but the prevalence of resistant organisms has significantly risen.53,54
Moving away from identification of pathobionts associated with BV, we sought to apply the same analytic principles to the pathobiont Group B streptococci. In the current study, we have shown that WGS metagenomics is a sensitive predictor of vaginal GBS colonization status and confirm decades of clinical microbiology demonstrating that GBS is a member of the vaginal microbial community in as great as 28% of women, although marginal in relative abundance. Within this study, we further demonstrate via Markov modeling of GBS positivity high probability of becoming or staying GBS positive. Consistent with our finding that L. crispatus negatively co-occurs with GBS, previous data have shown Lactobacillus spp. are capable of inhibiting GBS.55–57 Our findings are of marked significance, as they demonstrate that GBS is a keystone and landmark commensal of the vaginal niche with highly predictable co-occurrence ecology. Although we had no cases of neonatal GBS disease in our cohort and thus cannot comment on the implications of our observations for risk modification, future studies leveraging our novel observations are indicated. The goal of such studies would be to refine screening and prediction to both further reduce neonatal morbidity and mortality and reduce the current prevalence of use of multiple courses of intrapartum antibiotic prophylaxis.
Limitations of Study
The primary limitation of our study is the few subjects who were enrolled prospectively in the second trimester, and went onto have a preterm birth. However, that is also the studies primary strength as such prospective samples enable one to compare findings by gestational age, naïve to eventual term vs preterm birth. We are currently analyzing the entirety of this prospectively acquired cohort which will be the focus of future work. Secondary limitations include those inherent to any large translational cohort, inclusive of depth of sequencing, bias of birth outcome by other secondary contributing co-factors, and being prone to limitations of the analytic approach and computational methodology chosen. We attempted to address and overcome these limitations, but have undoubtedly fallen short by others estimates.
Conclusions
To our knowledge, to date this is the largest metagenomic interrogation of the vaginal microbiome during pregnancy and postpartum using whole genome shotgun sequencing outside of the MOMS-PI study20. Parallel sequencing of identical samples using the typical 16S rRNA targeted amplicon-based approach with primers against the V1V3 hypervariable regions revealed a notable underrepresentation of Gardnerella vaginalis when compared to WGS. Microbial communities characterized by 16S rRNA gene sequencing have been previously demonstrated to have inherent biases depending on the hypervariable regions sequenced.58–61 Our study further corroborates this notion, revealing that Gardnerella vaginalis is significantly underrepresented with V1V3 sequencing, which has been suggested by others previously.11 Therefore, it is likely that previous studies on the vaginal microbiome using V1V3 primer sets are likely to miss Gardnerella representation in their study, thereby biasing conclusions pertaining to the association between the vaginal microbiome and PTB. Alternatively, the use of additional primer sets, like V4, in conjunction with V1V3 (or phased V1V3 primer sets62) may help to overcome primer biases and provide more accurate characterization of the vaginal microbiota.11 In our hands, WGS metagenomic sequencing, although costlier and more computationally intensive, provides a more sensitive detection of the major and minor constituents of the vaginal microbiome, including Gardnerella vaginalis, Lactobacillus species, and Group B Streptococcus. In addition, WGS metagenomic sequencing enables strain-level differentiation, which may be of importance when attempting to understand the complex ecology of the vaginal niche.
From a translational perspective, our study is significant for our observations showing that although a majority of species and strain interactions are random, significant co-occurrences are generally exclusionary in pregnancy, whereas postpartum is classified by an increase in positive co-occurrences. These observations held true for both strain level profiling of Lactobacillus spp., G. vaginalis, and the pathobiont Group B Streptococcus. Taken together, these data demonstrate that the ecology of the vaginal microbiome is mainly driven by the abundance of four keystone species, that both community profiling and functional differences exist within these species to the strain-level, and that more attention should be focused on the contribution of individual strains with respect to potential differences in health outcomes such as preterm birth and likely invasive GBS disease. These findings are consistent with the essential principles of microbial ecology and show for the first time that the unique vaginal microbiome signature of pregnancy arises as a result of exclusionary co-occurrences of bacterial species, strains, and clades.
STAR★Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kjersti Aagaard (aagaardt@bcm.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The WGS metagenomic and targeted 16S rRNA gene amplicon sequence data generated from this study have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject PRJNA451212.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
As shown in Figure S1, this was a prospective cohort study of healthy pregnant women enrolled in the third trimester and followed to delivery and early postpartum (referred to as the “third trimester cohort”). A second prospective cohort of pregnant women at risk for preterm birth were enrolled during the second trimester (referred to as the “second trimester cohort”). In all cases the vaginal introitus and/or posterior fornix were swabbed by a trained individual for each subject at each time point as described previously.63,64 An overview of the study design and samples collected is shown in Figure S1. Demographics of both cohorts are reported in Tables S4 and S5.
This study was reviewed and approved by the Baylor College of Medicine Institutional Review Board (IRB) under protocols H-27393 and H-34056. Participants in both cohorts were included if they had a viable pregnancy, were 18 years age or older and were willing to consent to all aspects of the study. Exclusion criteria included known HIV or Hepatitis C infection, immunosuppressive disease, use of cytokines or immunosuppressive agents within the last 6 months, a history of cancer squamous or basal cell carcinoma of the skin managed by local excision, treatment of suspicion of ever having toxic shock syndrome, or major surgery of the GI tract except cholecystectomy or appendectomy in the past five years. Participants were informed and consented to the potential risks of participation, including minimal physical discomfort associated with specimen collection and the possibility of accidental release of protected health information. Participants consented to having data from de-identified, human DNA scrubbed data uploaded to public repositories.
Clinical metadata was queried and abstracted from subject electronic medical records, including gestational age at delivery (weeks and days), mode of delivery at birth (Cesarean or vaginal), and Group B Streptococcus (GBS) clinical culture status obtained during the course of care. Participants were sampled for clinical GBS cultivation during the third trimester (~35–37 weeks of gestation) or with symptoms consistent with preterm labor and according to the American College of Obstetricians and Gynecologists (ACOG) guidelines.26 Participants were classified as having delivered preterm if gestational age at delivery was <37 weeks.
METHOD DETAILS
Sample Processing and DNA Extraction.
After collection, vaginal swabs were immediately dounced for 30 seconds in MoBio collection tubes with PowerSoil Garnet 0.70 mm beads. Samples were collected in duplicate and stored at 4°C in preparation for DNA extraction within 24 hours, or long-term storage at −80°C. DNA extraction was performed using the MoBio PowerSoil extraction kits according to the manufacturer’s recommended protocol. Samples were heated at 65C for 5 minutes then 95C for 5 minutes. 60 ul of C1 was added to each sample and vortexed for 20 minutes on max speed using a radial tube adaptor. Samples were centrifuged at 10,000 rpm for 30 seconds and the supernatant was transferred to a new tube containing 200 ul C2. Samples were vortexed briefly and chilled at 4°C for 5 minutes. Samples were centrifuged at 10,000 rpm for 30 seconds and the supernatant was transferred to a new tube containing 250 ul C3. Samples were vortexed briefly and chilled at 4°C for 5 minutes. Samples were centrifuged at 10,000 rpm for 30 seconds and the supernatant was transferred to a new tube containing 1250 ul C4. Samples were vortexed vigorously and 675 ul of the mixture was applied to individual columns. Samples were centrifuged briefly and the flow-through discarded. The previous step was repeated until all the sample/C4 mixture was applied to the column. 500 ul of C5 was applied to the column and centrifuged at 10,000 rpm for 30 seconds, discarding the flow-through. The columns were centrifuged empty at 10,000 rpm for 30 seconds to eliminate residual buffer. The columns were transferred to a clean 1.5 ul collection tube and 100 ul of water was applied directly to the column and incubated at room temperature for 1 minute. The samples were centrifuged at 10,000 rpm for 1 minute and the flow-through was saved containing the extracted DNA.
16S (V1V3 hypervariable region) rRNA gene sequencing and data processing.
The V1V3 region of the 16S rRNA gene was amplified by PCR using barcoded universal primers (reverse primer, 5’-CAAGCAGAAGACGGCATACGAGATxrefxrefGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT- 3’, where X denotes the index region of the adapter; forward primer, 5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’). 16S rRNA amplicon data was processed through the DADA265 pipeline (v1.6) in R (v3.4.4). Sequences were manually examined for drop off of sequencing quality. Subsequently the forward and reverse reads were quality filtered and uniformly trimmed to 230 bp using the filterAndTrim() command with the following parameters: truncLen=c(230,230), maxN=0-, truncQ=11, maxEE=c(2,2), rmphix=TRUE). Error rates for both the forward and reverse reads were learned using 2 million subsampled reads. Amplicon Sequence Variants (ASVs) were identified per sample after sequence de-replication. Chimeric ASVs were identified using the command removeBimeraDenovo() with the consensus method. ASVs were assigned taxonomy using the RDP classifier against the GreenGenes database (v13.8)66,67. For species level assignments, the representative sequence for the GreenGene ID assigned to each ASV was blasted (blastn 2.2.29+) against the NCBI 16S Microbial blast database (ftp://ftp.ncbi.nih.gov/blast/db). The top blast hit was subsequently assigned as the taxonomy for each ASV. A resultant ASV table was constructed consisting of the abundance of each ASV in every sample and imported into the R package phyloseq (v1.20.0) for downstream analysis.68
Whole genome shotgun metagenomic sequencing and data processing.
For the third trimester cohort, raw sequence reads generated from Illumina HiSeq 2500 (2×150bp) sequencing were trimmed and filtered for host reads with Trimmomatic69 and BMTagger70 using KneadData (https://bitbucket.org/biobakery/kneaddata/wiki/Home). Trimmomatic was used to remove Illumina adapters and quality filter. Default parameters were used for BMTagger mapping and filtering of hosts reads. Further quality filtering was successively performed using BBDuk as implemented in BBTools version 37.33 (http://jgi.doe.gov/data-and-tools/bbtools/) to remove PhiX viral reads derived from Illumina HiSeq quality control and partial adapter sequences. This resulted in a total of 232,971,950 high quality reads, with an average of 1,159,064 reads per sample (median of 428,816 reads per sample). For the second trimester cohort, raw sequence reads generated from Illumina Hiseq X (2×150bp) sequencing (1,703,909,984 reads) were trimmed and filtered for host reads using the same KneadData/BBDuk pipeline, resulting in a total of 35,267,854 high quality reads, with an average of 1,396,729 reads per sample (median of 722,534 reads per sample).
Microbial taxonomic classification.
Microbial taxonomic classification of host-filtered sequence reads was performed using MetaPhlAn2 (Metagenomic Phylogenetic Analysis 2, v2.6.0)71 using default parameters and Centrifuge (v1.0.3)72 against the Centrifuge prokaryote, human, and viral database, and excluding the following tax-ids – 9606 (human), 374840 (PhiX), 32630 (synthetic constructs), and 10239 (viral sequences). Strain-level profiling was performed using PanPhlAn (Pangenome-based Phylogenomic Analysis, v1.2.3)22 using custom pangenome centroid databases generated for Gardnerella vaginalis (43 reference genomes, 10,964 gene families), Lactobacillus crispatus (51 reference genomes, 7,923 gene families), Lactobacillus iners (20 reference genomes, 2,161 gene families), and Lactobacillus jensenii (17 reference genomes, 3,618 gene families) from reference genomes downloaded from NCBI (November 2017 and March 2018)(Supp. Table 7). Briefly, PanPhlAn identifies the strain specific gene sets present in samples by screening for all prospective genes from the species pangenome. Default PanPhlAn parameters were used, including clustering of pangenome centroids at a gene similarity threshold of 95%, except strain detection thresholds were adjusted to the following during profiling: --min_coverage 1 --left_max 1.70 --right_min 0.30. Strain-level functional modules/pathways were profiled using the PanPhlAn-generated pangenome centroids. Indicator values73 were calculated and used to identify strain-specific pangenome centroids, that were then submitted to UBLAST74 against the prokaryotic KEGG database (e-value, 1e-9) and filtered for the top hit. KEGG gene IDs were mapped to KEGG KOs and used to retrieve the KEGG functional pathway hierarchy. Metagenomic samples were assigned to strain clades based on clustering of reference strains using binary Jaccard distance and the presence and abundance of strain-specific pangenome centroids determined by significant indicator values (p ≤ 0.05).
Mapping of metagenomic reads to Streptococcus agalactiae reference genome.
Mapping of metagenomic reads to the representative Streptococcus agalactiae reference genome 2603V/R (NC_004116) was performed with the Burrows-Wheeler Alignment Tool (BWA, v0.7.15-r1140)75 using the BWA-MEM algorithm with default parameters. Genomic coverage was calculated using the BBTools pileup.sh script.
Mitochondrial DNA SNP variant calling.
WGS paired-end reads identified as host by the BMTagger filtering step were aligned to the human mitochondrial reference genome (NC_012920.1) using BWA (v0.7.12-r1039) and variant calls were generated using samtools mpileup.76 Only single nucleotide variants were considered for subsequent analysis.
Taxa by mtSNP associations.
Associations between species and mtSNPs were performed using PLINK (v1.07) with the variants considered a haploid genotype and each taxon considered a quantitative trait. For associations between variants, species and preterm birth, we utilized the Quantitative trait interaction (GxE) algorithm in PLINK using preterm birth as the covariate groups (term versus preterm). Resultant p-values from both analyses were corrected for False Discovery Rates (FDR) using the R command p.adjust. Plots were generated in R using the manhattanly package (v0.2.0).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis.
Except where noted, all statistical analyses were performed using R (version 3.4.3) and/or GraphPad Prism (GraphPad Software Inc., La Jolla, CA). The R packages factoextra (v1.0.5), pheatmap (v1.0.8), vegan (v2.4–6)77, phyloseq (v1.22.3)68, and ggplot2 (v2.2.1)78 were used to perform and visualize cluster analyses and ordinations. Differential taxonomic features were identified via Linear discriminant analysis effect size (LEfSe)79, using an alpha value of 0.05 for the Kruskal-Wallis/Wilcoxon tests and a threshold of 2.0 for the logarithmic linear discriminant analysis (LDA) score for discriminative features and analysis of composition of microbiomes (ANCOM)80. Indicator values (IndVal) were calculated using the labdsv (v1.8–0) R package.73 Spearman correlations were performed with base R. Species co-occurrence analysis was performed using the cooccur (v1.3) R package23 and modeled in Cytoscape.81
Supplementary Material
Table S1. PLINK SNP-species associations, Related to Figure 5.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Biological samples | ||
| Vaginal introitus and posterior fornix swabs | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Critical commercial assays | ||
| PowerSoil DNA isolation kit | MoBio/Qiagen | Cat# 12888 |
| Deposited data | ||
| Human filtered metagenomic sequencing data | This study | NCBI-SRA BioProject: PRJNA451212 |
| Gardnerella vaginalis 409-05 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000025205.1 |
| Gardnerella vaginalis ATCC 14019 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000159155.2 |
| Gardnerella vaginalis 101 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000165615.1 |
| Gardnerella vaginalis 41V | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000165635.1 |
| Gardnerella vaginalis AMD | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000176475.1 |
| Gardnerella vaginalis 44317 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000176495.1 |
| Gardnerella vaginalis ATCC 14018 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000178355.1 |
| Gardnerella vaginalis HMP9231 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000213955.1 |
| Gardnerella vaginalis 315-A | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000214315.1 |
| Gardnerella vaginalis 284V | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263435.1 |
| Gardnerella vaginalis 55152 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263475.1 |
| Gardnerella vaginalis 1400E | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263495.1 |
| Gardnerella vaginalis 00703C2mash | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263515.1 |
| Gardnerella vaginalis 75712 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263535.1 |
| Gardnerella vaginalis 0288E | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263555.1 |
| Gardnerella vaginalis 6420B | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263575.1 |
| Gardnerella vaginalis 1500E | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263595.1 |
| Gardnerella vaginalis 00703Bmash | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263615.1 |
| Gardnerella vaginalis 00703Dmash | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263635.1 |
| Gardnerella vaginalis 6119V5 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000263655.1 |
| Gardnerella vaginalis JCP8522 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414425.1 |
| Gardnerella vaginalis JCP8151B | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414485.1 |
| Gardnerella vaginalis JCP8151A | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414505.1 |
| Gardnerella vaginalis JCP8108 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414525.1 |
| Gardnerella vaginalis JCP8070 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414545.1 |
| Gardnerella vaginalis JCP8066 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414565.1 |
| Gardnerella vaginalis JCP8017B | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414585.1 |
| Gardnerella vaginalis JCP8017A | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414605.1 |
| Gardnerella vaginalis JCP7719 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414625.1 |
| Gardnerella vaginalis JCP7672 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414645.1 |
| Gardnerella vaginalis JCP7659 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414665.1 |
| Gardnerella vaginalis JCP7276 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414685.1 |
| Gardnerella vaginalis JCP7275 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000414705.1 |
| Gardnerella vaginalis JCM 11026 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001042655.1 |
| Gardnerella vaginalis 3549624 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001049785.1 |
| Gardnerella vaginalis 14019_MetR | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001278345.1 |
| Gardnerella vaginalis GED7275B | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001546445.1 |
| Gardnerella vaginalis CMW7778B | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001563665.1 |
| Gardnerella vaginalis 23-12 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001660735.1 |
| Gardnerella vaginalis 18-4 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001660755.1 |
| Gardnerella vaginalis ATCC 49145 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001913835.1 |
| Gardnerella vaginalis GV37 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001953155.1 |
| Gardnerella vaginalis DSM 4944 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_900105405.1 |
| Lactobacillus crispatus ST1 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000091765.1 |
| Lactobacillus crispatus JV.V01 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000160515.1 |
| Lactobacillus crispatus MV.1A.US | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000161915.2 |
| Lactobacillus crispatus 125.2.CHN | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000162255.1 |
| Lactobacillus crispatus MV.3A.US | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000162315.1 |
| Lactobacillus crispatus CTV.05 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000165885.1 |
| Lactobacillus crispatus SJ.3C.US | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000176975.2 |
| Lactobacillus crispatus 214.1 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000177575.1 |
| Lactobacillus crispatus FB049.03 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000301115.1 |
| Lactobacillus crispatus FB077.07 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000301135.1 |
| Lactobacillus crispatus 2029 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000466885.2 |
| Lactobacillus crispatus EM.LC1 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000497065.1 |
| Lactobacillus crispatus JCM 1185 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001311685.1 |
| Lactobacillus crispatus DSM 20584 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001434005.1 |
| Lactobacillus crispatus VMC3 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001541385.1 |
| Lactobacillus crispatus VMC4 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001541405.1 |
| Lactobacillus crispatus VMC6 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001541505.1 |
| Lactobacillus crispatus VMC5 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001541515.1 |
| Lactobacillus crispatus VMC7 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001541535.1 |
| Lactobacillus crispatus VMC8 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001541585.1 |
| Lactobacillus crispatus VMC1 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001546015.1 |
| Lactobacillus crispatus VMC2 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001546025.1 |
| Lactobacillus crispatus PSS7772C | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001563615.1 |
| Lactobacillus crispatus JCM 5810 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001567095.1 |
| Lactobacillus crispatus C037 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001700475.1 |
| Lactobacillus crispatus C25 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001704465.1 |
| Lactobacillus crispatus ATCC 33820 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002088015.1 |
| Lactobacillus crispatus UMNLC2 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218565.1 |
| Lactobacillus crispatus UMNLC1 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218615.1 |
| Lactobacillus crispatus UMNLC3 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218645.1 |
| Lactobacillus crispatus UMNLC4 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218655.1 |
| Lactobacillus crispatus UMNLC8 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218685.1 |
| Lactobacillus crispatus UMNLC6 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218695.1 |
| Lactobacillus crispatus UMNLC9 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218735.1 |
| Lactobacillus crispatus UMNLC5 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218765.1 |
| Lactobacillus crispatus UMNLC7 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218775.1 |
| Lactobacillus crispatus UMNLC10 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218805.1 |
| Lactobacillus crispatus UMNLC11 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218815.1 |
| Lactobacillus crispatus UMNLC13 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218845.1 |
| Lactobacillus crispatus UMNLC12 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218855.1 |
| Lactobacillus crispatus UMNLC14 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218885.1 |
| Lactobacillus crispatus UMNLC15 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218895.1 |
| Lactobacillus crispatus UMNLC16 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218925.1 |
| Lactobacillus crispatus UMNLC18 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218945.1 |
| Lactobacillus crispatus UMNLC19 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218965.1 |
| Lactobacillus crispatus UMNLC20 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002218975.1 |
| Lactobacillus crispatus UMNLC21 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002219005.1 |
| Lactobacillus crispatus UMNLC24 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002219015.1 |
| Lactobacillus crispatus UMNLC22 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002219045.1 |
| Lactobacillus crispatus UMNLC25 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002219055.1 |
| Lactobacillus crispatus UMNLC23 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002219085.1 |
| Lactobacillus iners LactinV 11V1-d | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000149065.1 |
| Lactobacillus iners LactinV 09V1-c | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000149085.1 |
| Lactobacillus iners LactinV 03V1-b | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000149105.1 |
| Lactobacillus iners LactinV 01V1-a | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000149125.1 |
| Lactobacillus iners SPIN 2503V10-D | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000149145.1 |
| Lactobacillus iners DSM 13335 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000160875.1 |
| Lactobacillus iners AB-1 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000177755.1 |
| Lactobacillus iners LEAF 2053A-b | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000179935.1 |
| Lactobacillus iners LEAF 2052A-d | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000179955.1 |
| Lactobacillus iners LEAF 2062A-h1 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000179975.1 |
| Lactobacillus iners LEAF 3008A-a | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000179995.1 |
| Lactobacillus iners ATCC 55195 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000185405.1 |
| Lactobacillus iners UPII 143-D | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000191685.1 |
| Lactobacillus iners UPII 60-B | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000191705.1 |
| Lactobacillus iners SPIN 1401G | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000204435.1 |
| Lactobacillus iners DSM 13335 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001435015.1 |
| Lactobacillus iners UMB0033 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002871595.1 |
| Lactobacillus iners UMB1051 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002884695.1 |
| Lactobacillus iners UMB0030 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002884705.1 |
| Lactobacillus iners KA00186 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002892385.1 |
| Lactobacillus jensenii SNUV360 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001936235.1 |
| Lactobacillus jensenii JV-V16 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000159335.1 |
| Lactobacillus jensenii 1153 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000155915.2 |
| Lactobacillus jensenii 27-2-CHN | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000161895.2 |
| Lactobacillus jensenii SJ-7A-US | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000162335.1 |
| Lactobacillus jensenii 115-3-CHN | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000162435.1 |
| Lactobacillus jensenii IM11 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001012655.1 |
| Lactobacillus jensenii IM18-1 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001012665.1 |
| Lactobacillus jensenii IM59 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001012675.1 |
| Lactobacillus jensenii IM18-3 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001012685.1 |
| Lactobacillus jensenii IM1 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001012735.1 |
| Lactobacillus jensenii IM3 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001012745.1 |
| Lactobacillus jensenii 269-3 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000175035.1 |
| Lactobacillus jensenii MD IIE-70(2) | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_000466805.1 |
| Lactobacillus jensenii TL2937 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001742045.1 |
| Lactobacillus jensenii DSM 20557 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_001436455.1 |
| Lactobacillus jensenii UMB0077 | ftp://ftp.ncbi.nlm.nih.gov/genome/ | GCF_002848045.1 |
| Experimental models: cell lines | ||
| Experimental models: organisms/strains | ||
| Oligonucleotides | ||
| 16S rRNA V1V3 reverse primer: 5’-CAAGCAGAAGACGGCATACGAGATXXXXXXXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ | This study | N/A |
| 16S rRNA V1V3 forward primer: 5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’ | This study | N/A |
| Recombinant DNA | ||
| Software and algorithms | ||
| R (versions 3.4.3 and 3.4.4) | https://www.r-project.org/ | |
| GraphPad Prism (version 7) | https://www.graphpad.com/scientific-software/prism/ | |
| DADA2 (version 1.6) | https://benjjneb.github.io/dada2/index.html | |
| GreenGenes (version 13.8) | 66,67 | https://greengenes.secondgenome.com/ |
| blastn (version 2.2.29+) | ||
| phyloseq (versions 1.20.0 and 1.22.3) | 68 | https://joey711.github.io/phyloseq/ |
| Trimmomatic | 69 | http://www.usadellab.org/cms/?page=trimmomatic |
| BMTagger | 70 | ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger/ |
| KneadData | https://bitbucket.org/biobakery/kneaddata/wiki/Home | |
| BBTools (version 37.33) | http://jgi.doe.gov/data-and-tools/bbtools/ | |
| MetaPhlAn2 (Metagenomic Phylogenetic Analysis 2, version 2.6.0) | 71 | https://github.com/biobakery/MetaPhlAn |
| Centrifuge (version 1.0.3) | 72 | https://ccb.jhu.edu/software/centrifuge/ |
| PanPhlAn (Pangenome-based Phylogenomic Analysis, version 1.2.3) | 22 | https://github.com/segatalab/panphlan |
| labdsv (version 1.8-0) | 73 | https://cran.r-project.org/web/packages/labdsv |
| UBLAST | 74 | https://www.drive5.com/usearch/manual/ublast_algo.html |
| Burrows-Wheeler Alignment Tool (BWA, version 0.7.15-r1140 and version 0.7.12-r1039) | 75 | http://bio-bwa.sourceforge.net/ |
| Samtools | 76 | http://www.htslib.org/ |
| PLINK (version 1.07) | https://zzz.bwh.harvard.edu/plink/ | |
| Manhattanly (version 0.2.0) | https://cran.r-project.org/web/packages/manhattanly | |
| factoextra (version 1.0.5) | https://cran.r-project.org/web/packages/factoextra | |
| pheatmap (version 1.0.8) | https://cran.r-project.org/web/packages/pheatmap | |
| PLINK (version 1.07) | https://zzz.bwh.harvard.edu/plink/ | |
| vegan (version 2.4-6) | 77 | https://cran.r-project.org/web/packages/vegan |
| ggplot2 (version 2.2.1) | 78 | https://ggplot2.tidyverse.org/ |
| Linear discriminant analysis effect size (LEfSe) | 79 | https://github.com/biobakery/lefse |
| Analysis of composition of microbiomes (ANCOM) | 80 | |
| cooccur (version 1.3) | 23 | https://cran.r-project.org/web/packages/cooccur |
| Cytoscape | 81 | https://cytoscape.org/ |
| Other | ||
| NCBI 16S Microbial blast database | ftp://ftp.ncbi.nih.gov/blast/db | |
LIFE SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Bacterial and virus strains | ||
| pAAV-hSyn-DIO-hM3D(Gq)-mCherry | Krashes et al., 2011 | Addgene AAV5; 44361-AAV5 |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP | Hope Center Viral Vectors Core | N/A |
| Cowpox virus Brighton Red | BEI Resources | NR-88 |
| Zika-SMGC-1, GENBANK: KX266255 | Isolated from patient (Wang et al., 2016) | N/A |
| Staphylococcus aureus | ATCC | ATCC 29213 |
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC 700294 |
| Biological samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children’s Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, peptides, and recombinant proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical commercial assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | PerkinElmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Nanog STILT inference | This paper; Mendeley Data | http://dx.doi.org/10.17632/wx6s4mj7s8.2 |
| Affinity-based mass spectrometry performed with 57 genes | This paper; Mendeley Data | Table S8; http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Experimental models: cell lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental models: organisms/strains | ||
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl. y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtml.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard, 2000 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Oligonucleotides | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al., 2013 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al., 2013 | https://github.com/ChristophRau/wMICA |
| ICS algorithm | This paper; Mendeley Data | http://dx.doi.org/10.17632/5hvpvspw82.1 |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
PHYSICAL SCIENCE TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| QD605 streptavidin conjugated quantum dot | Thermo Fisher Scientific | Cat#Q10101MP |
| Platinum black | Sigma-Aldrich | Cat#205915 |
| Sodium formate BioUltra, ≥99.0% (NT) | Sigma-Aldrich | Cat#71359 |
| Chloramphenicol | Sigma-Aldrich | Cat#C0378 |
| Carbon dioxide (13C, 99%) (<2% 18O) | Cambridge Isotope Laboratories | CLM-185-5 |
| Poly(vinylidene fluoride-co-hexafluoropropylene) | Sigma-Aldrich | 427179 |
| PTFE Hydrophilic Membrane Filters, 0.22 μm, 90 mm | Scientificfilters.com/TischScientific | SF13842 |
| Critical commercial assays | ||
| Folic Acid (FA) ELISA kit | Alpha Diagnostic International | Cat# 0365-0B9 |
| TMT10plex Isobaric Label Reagent Set | Thermo Fisher | A37725 |
| Surface Plasmon Resonance CM5 kit | GE Healthcare | Cat#29104988 |
| NanoBRET Target Engagement K-5 kit | Promega | Cat#N2500 |
| Deposited data | ||
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Structure of compound 5 | This paper; Cambridge Crystallographic Data Center | CCDC: 2016466 |
| Code for constraints-based modeling and analysis of autotrophic E. coli | This paper | https://gitlab.com/elad.noor/sloppy/tree/master/rubisco |
| Software and algorithms | ||
| Gaussian09 | Frish et al., 2013 | https://gaussian.com |
| Python version 2.7 | Python Software Foundation | https://www.python.org |
| ChemDraw Professional 18.0 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| Weighted Maximal Information Component Analysis v0.9 | Rau et al., 2013 | https://github.com/ChristophRau/wMICA |
| Other | ||
| DASGIP MX4/4 Gas Mixing Module for 4 Vessels with a Mass Flow Controller | Eppendorf | Cat#76DGMX44 |
| Agilent 1200 series HPLC | Agilent Technologies | https://www.agilent.com/en/products/liquid-chromatography |
| PHI Quantera II XPS | ULVAC-PHI, Inc. | https://www.ulvac-phi.com/en/products/xps/phi-quantera-ii/ |
Context and Significance.
There is a lack of knowledge regarding which vaginal bacteria are beneficial (and on the contrary, which are potentially harmful) across populations, especially in relation with the risk of pre-term birth. Here, scientists from the Baylor College in Huston, Texas present a high-resolution investigation of the vaginal microbiome during and after pregnancy. This analysis allows a better understanding of the relationships between the most important vaginal microbes, maternal genetics, and the risk of preterm birth.
Highlights.
The vaginal microbiome differs in its bacterial composition during and after pregnancy.
Preterm birth-vaginal microbiome associations differ at the species/strain level.
Heredity of mitochondrial DNA may play a role in bacterial-preterm birth associations.
Group B Streptococcus is a prevalent, low abundant member of the vaginal microbiome.
Acknowledgements
The authors gratefully acknowledge the support of the NIH-NINR (R01 NR014792, K.M.A.), NIH-NICHD (R01 HD091731, K.M.A.), NIH National Children’s Study Formative Research (N01-HD-80020, K.M.A.), the Burroughs Wellcome Fund Preterm Birth Initiative (K.M.A.), the March of Dimes Preterm Birth Research Initiative (K.M.A.), NIH IRACDA Award (NIGMS K12 GM084897, R.M.P.), the Baylor College of Medicine Medical Scientist Training Program (NIGMS T32 GM007330, D.M.C. and K.M.A.), the National Institute of General Medical Sciences (T32 GM088129, D.M.C.), Baylor Research Advocates for Student Scientists (D.M.C.) and the Human Microbiome Project funded through the NIH Director’s Common Fund at the National Institutes of Health (as part of NIH Roadmap 1.5, K.M.A.). All sequencing and adaptation of protocols for WGS sequencing were performed by the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC), which is funded by direct support from the National Human Genome Research Institute (NHGRI) (U54HG004973 (BCM); R. Gibbs, Principal Investigator).
The authors also thank the staff members who were directly involved in clinical recruitment and specimen processing (B. Boggan, T. Barrett, L. Showalter, C. Shope). The authors are grateful to Drs. Melissa Suter and James Versalovic for critical review of the manuscript.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marrazzo JM, Martin DH, Watts DH, Schulte J, Sobel JD, Hillier SL, Deal C, and Fredricks DN (2010). Bacterial vaginosis: Identifying research gaps proceedings of a workshop sponsored by DHHS/NIH/NIAID November 19–20, 2008. Sex Transm. Dis. 37, 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van de Wijgert JHHM, Borgdorff H, Verhelst R, Crucitti T, Francis S, Verstraelen H, and Jespers V (2014). The vaginal microbiota: What have we learned after a decade of molecular characterization? PLoS ONE 9, e105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, et al. (2011). Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. U S A 108 Suppl 1, 4680–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gajer P, Brotman RM, Bai G, Sakamoto J, Schütte UME, Zhong X, Koenig SSK, Fu L, Ma ZS, Zhou X, et al. (2012). Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 4, 132ra52–132ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romero R, Hassan SS, Gajer P, Tarca AL, Fadrosh DW, Nikita L, Galuppi M, Lamont RF, Chaemsaithong P, Miranda J, et al. (2014). The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romero R, Hassan SS, Gajer P, Tarca AL, Fadrosh DW, Bieda J, Chaemsaithong P, Miranda J, Chaiworapongsa T, and Ravel J (2014). The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datcu R (2014). Characterization of the vaginal microflora in health and disease. Dan. Med. J. 61, B4830. [PubMed] [Google Scholar]
- 8.Brown RG, Marchesi JR, Lee YS, Smith A, Lehne B, Kindinger LM, Terzidou V, Holmes E, Nicholson JK, Bennett PR, et al. (2018). Vaginal dysbiosis increases risk of preterm fetal membrane rupture, neonatal sepsis and is exacerbated by erythromycin. BMC Med. 16, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, Sun CL, Goltsman DSA, Wong RJ, Shaw G, et al. (2015). Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl. Acad. Sci. U S A 112, 11060–11065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dols JAM, Molenaar D, van der Helm JJ, Caspers MPM, de Kat Angelino-Bart A, Schuren FHJ, Speksnijder AGCL, Westerhoff HV, Richardus JH, Boon ME, et al. (2016). Molecular assessment of bacterial vaginosis by Lactobacillus abundance and species diversity. BMC Infect. Dis. 16, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Callahan BJ, DiGiulio DB, Goltsman DSA, Sun CL, Costello EK, Jeganathan P, Biggio JR, Wong RJ, Druzin ML, Shaw GM, et al. (2017). Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc. Natl. Acad. Sci. U S A 114, 9966–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stout MJ, Zhou Y, Wylie KM, Tarr PI, Macones GA, and Tuuli MG (2017). Early pregnancy vaginal microbiome trends and preterm birth. Am. J. Obstet. Gynecol. 217, 356.e1–356.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kindinger LM, Bennett PR, Lee YS, Marchesi JR, Smith A, Cacciatore S, Holmes E, Nicholson JK, Teoh TG, and MacIntyre DA (2017). The interaction between vaginal microbiota, cervical length, and vaginal progesterone treatment for preterm birth risk. Microbiome 5, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brooks JP, Buck GA, Chen G, Diao L, Edwards DJ, Fettweis JM, Huzurbazar S, Rakitin A, Satten GA, Smirnova E, et al. (2017). Changes in vaginal community state types reflect major shifts in the microbiome. Microb. Ecol. Health Dis. 28, 1303265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drell T, Štšepetova J, Simm J, Rull K, Aleksejeva A, Antson A, Tillmann V, Metsis M, Sepp E, Salumets A, et al. (2017). The influence of different maternal microbial communities on the development of infant gut and oral microbiota. Sci. Rep. 7, 9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas-White K, Forster SC, Kumar N, Van Kuiken M, Putonti C, Stares MD, Hilt EE, Price TK, Wolfe AJ, and Lawley TD (2018). Culturing of female bladder bacteria reveals an interconnected urogenital microbiota. Nat. Commun. 9, 1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parolin C, Foschi C, Laghi L, Zhu C, Banzola N, Gaspari V, D’Antuono A, Giordani B, Severgnini M, Consolandi C, et al. (2018). Insights into vaginal bacterial communities and metabolic profiles of chlamydia trachomatis infection: positioning between eubiosis and dysbiosis. Front. Microbiol. 9, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doyle R, Gondwe A, Fan Y-M, Maleta K, Ashorn P, Klein N, and Harris K (2018). A Lactobacillus-deficient vaginal microbiota dominates postpartum women in rural Malawi. Appl. Environ. Microbiol. 84, 4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kindinger LM, MacIntyre DA, Lee YS, Marchesi JR, Smith A, McDonald JAK, Terzidou V, Cook JR, Lees C, Israfil-Bayli F, et al. (2016). Relationship between vaginal microbial dysbiosis, inflammation, and pregnancy outcomes in cervical cerclage. Sci. Transl. Med. 8, 350ra102–350ra102. [DOI] [PubMed] [Google Scholar]
- 20.Fettweis JM, Serrano MG, Brooks JP, Edwards DJ, Girerd PH, Parikh HI, Huang B, Arodz TJ, Edupuganti L, Glascock AL, et al. (2019). The vaginal microbiome and preterm birth. Nat. Med. 1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petricevic L, Domig KJ, Nierscher FJ, Sandhofer MJ, Fidesser M, Krondorfer I, Husslein P, Kneifel W, and Kiss H (2014). Characterisation of the vaginal Lactobacillus microbiota associated with preterm delivery. Sci. Rep. 4, 5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scholz M, Ward DV, Pasolli E, Tolio T, Zolfo M, Asnicar F, Truong DT, Tett A, Morrow AL, and Segata N (2016). Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Meth. 13, 435–438. [DOI] [PubMed] [Google Scholar]
- 23.Griffith DM, Veech JA, and Marsh CJ (2016). cooccur: Probabilistic species co-occurrence analysis in R. J. Stat. Softw. 69, 1–17. [Google Scholar]
- 24.Ma J, Coarfa C, Qin X, Bonnen PE, Milosavljevic A, Versalovic J, and Aagaard KM (2014). mtDNA haplogroup and single nucleotide polymorphisms structure human microbiome communities. BMC Genom. 15, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verani JR, McGee L, and Schrag SJ (2010). Prevention of perinatal group B streptococcal disease--revised guidelines from CDC, 2010. [PubMed]
- 26.American College of Obstetricians and Gynecologists (2011). Committee opinion no. 485: Prevention of early-onset Group B Streptococcal disease in newborns. Obstet. Gynecol. 117, 1019–1027. [DOI] [PubMed] [Google Scholar]
- 27.American College of Obstetricians and Gynecologists (2018). Committee opinion no. 485: Prevention of early-onset Group B Streptococcal disease in newborns: correction. Obstet. Gynecol. 131. [DOI] [PubMed] [Google Scholar]
- 28.Aagaard KM, Riehle K, Ma J, Segata N, Mistretta T-A, Coarfa C, Raza S, Rosenbaum S, Van den Veyver I, Milosavljevic A, et al. (2012). A Metagenomic Approach to Characterization of the Vaginal Microbiome Signature in Pregnancy. PLoS One 7, e36466–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacIntyre DA, Chandiramani M, Lee YS, Kindinger L, Smith A, Angelopoulos N, Lehne B, Arulkumaran S, Brown R, Teoh TG, et al. (2015). The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci. Rep. 5, 8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blencowe H, Cousens S, Oestergaard MZ, Chou D, Moller A-B, Narwal R, Adler A, Vera Garcia C, Rohde S, Say L, et al. (2012). National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet 379, 2162–2172. [DOI] [PubMed] [Google Scholar]
- 31.Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu J, Lawn JE, Cousens S, Mathers C, and Black RE (2016). Global, regional, and national causes of under-5 mortality in 2000–15: an updated systematic analysis with implications for the sustainable development goals. Lancet 388, 3027–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muglia LJ, and Katz M (2010). The enigma of spontaneous preterm birth. N. Engl. J. Med. 362, 529–535. [DOI] [PubMed] [Google Scholar]
- 33.American College of Obstetricians and Gynecologists (2016). Practice Bulletin No. 171: Management of Preterm Labor. Obstet. Gynecol. 128, e155–e164. [DOI] [PubMed] [Google Scholar]
- 34.Chu DM, Seferovic M, Pace RM, and Aagaard KM (2018). The microbiome in preterm birth. Best Pract. Res. Clin. Obstet. Gynaecol. 52, 103–113. [DOI] [PubMed] [Google Scholar]
- 35.McGregor JA, French JI, Jones W, Milligan K, McKinney PJ, Patterson E, and Parker R (1994). Bacterial vaginosis is associated with prematurity and vaginal fluid mucinase and sialidase: results of a controlled trial of topical clindamycin cream. Am. J. Obstet. Gynecol. 170, 1048–59–discussion1059–60. [DOI] [PubMed] [Google Scholar]
- 36.Klebanoff MA, Carey JC, Hauth JC, Hillier SL, Nugent RP, Thom EA, Ernest JM, Heine RP, Wapner RJ, Trout W, et al. (2001). Failure of metronidazole to prevent preterm delivery among pregnant women with asymptomatic Trichomonas vaginalis infection. N. Engl. J. Med. 345, 487–493. [DOI] [PubMed] [Google Scholar]
- 37.Kigozi GG, Brahmbhatt H, Wabwire-Mangen F, Wawer MJ, Serwadda D, Sewankambo N, and Gray RH (2003). Treatment of Trichomonas in pregnancy and adverse outcomes of pregnancy: A subanalysis of a randomized trial in Rakai, Uganda. Am. J. Obstet. Gynecol. 189, 1398–1400. [DOI] [PubMed] [Google Scholar]
- 38.Carey JC, and Klebanoff MA (2005). Is a change in the vaginal flora associated with an increased risk of preterm birth? - PubMed - NCBI. Am. J. Obstet. Gynecol. 192, 1341–1346. [DOI] [PubMed] [Google Scholar]
- 39.Mason SM, Kaufman JS, Emch ME, Hogan VK, and Savitz DA (2010). Ethnic density and preterm birth in African-, Caribbean-, and US-born non-Hispanic black populations in New York City. Am. J. Epidemiol. 172, 800–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Craig ED, Mitchell EA, Stewart AW, Mantell CD, and Ekeroma AJ (2004). Ethnicity and birth outcome: New Zealand trends 1980–2001: Part 4. Pregnancy outcomes for European/other women. Aust. N. Z. J. Obstet. Gynaecol. 44, 545–548. [DOI] [PubMed] [Google Scholar]
- 41.Breshears LM, Edwards VL, Ravel J, and Peterson ML (2015). Lactobacillus crispatus inhibits growth of Gardnerella vaginalis and Neisseria gonorrhoeae on a porcine vaginal mucosa model. BMC Microbiol. 15, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ojala T, Kankainen M, Castro J, Cerca N, Edelman S, Westerlund-Wikstrom B, Paulin L, Holm L, and Auvinen P (2014). Comparative genomics of Lactobacillus crispatus suggests novel mechanisms for the competitive exclusion of Gardnerella vaginalis. BMC Genom. 15, 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castro J, Henriques A, Machado A, Henriques M, Jefferson KK, and Cerca N (2013). Reciprocal Interference between Lactobacillus spp. and Gardnerella vaginalis on Initial Adherence to Epithelial Cells. Int.J. Med. Sci. 10, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rampersaud R, Planet PJ, Randis TM, Kulkarni R, Aguilar JL, Lehrer RI, and Ratner AJ (2011). Inerolysin, a cholesterol-dependent cytolysin produced by Lactobacillus iners. J. Bacteriol. 193, 1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ngugi BM, Hemmerling A, Bukusi EA, Kikuvi G, Gikunju J, Shiboski S, Fredricks DN, and Cohen CR (2011). Effects of BV-associated bacteria and sexual intercourse on vaginal colonization with the probiotic Lactobacillus crispatus CTV-05. Sex Transm. Dis. 38, 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mastromarino P, Macchia S, Meggiorini L, Trinchieri V, Mosca L, Perluigi M, and Midulla C (2009). Effectiveness of Lactobacillus-containing vaginal tablets in the treatment of symptomatic bacterial vaginosis. Clin. Microbiol. Infect. 15, 67–74. [DOI] [PubMed] [Google Scholar]
- 47.Antonio MAD, Meyn LA, Murray PJ, Busse B, and Hillier SL (2009). Vaginal colonization by probiotic Lactobacillus crispatus CTV-05 Is decreased by sexual activity and endogenous lactobacilli. J. Infect. Dis. 199, 1506–1513. [DOI] [PubMed] [Google Scholar]
- 48.Marrazzo JM, Antonio M, Agnew K, and Hillier SL (2009). Distribution of genital Lactobacillus strains shared by female sex partners. J. Infect. Dis. 199, 680–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tachedjian G, Aldunate M, Bradshaw CS, and Cone RA (2017). The role of lactic acid production by probiotic Lactobacillus species in vaginal health. Res. Microbiol. 168, 782–792. [DOI] [PubMed] [Google Scholar]
- 50.Balashov SV, Mordechai E, Adelson ME, and Gygax SE (2014). Identification, quantification and subtyping of Gardnerella vaginalis in noncultured clinical vaginal samples by quantitative PCR. J. Med. Microbiol. 63, 162–175. [DOI] [PubMed] [Google Scholar]
- 51.Sela U, Euler CW, da Rosa JC, and Fischetti VA (2018). Strains of bacterial species induce a greatly varied acute adaptive immune response: The contribution of the accessory genome. PLOS Pathog. 14, e1006726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Homer CSE, Scarf V, Catling C, and Davis D (2014). Culture-based versus risk-based screening for the prevention of group B streptococcal disease in newborns: a review of national guidelines. Women Birth 27, 46–51. [DOI] [PubMed] [Google Scholar]
- 53.Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, Lemons JA, Donovan EF, Stark AR, Tyson JE, et al. (2002). Changes in pathogens causing early-onset sepsis in very-low-birth-weight infants. N. Engl. J. Med. 347, 240–247. [DOI] [PubMed] [Google Scholar]
- 54.Bizzarro MJ, Dembry L-M, Baltimore RS, and Gallagher PG (2008). Changing patterns in neonatal Escherichia coli sepsis and ampicillin resistance in the era of intrapartum antibiotic prophylaxis. Pediatrics 121, 689–696. [DOI] [PubMed] [Google Scholar]
- 55.Juárez Tomás MS, Saralegui Duhart CI, De Gregorio PR, Vera Pingitore E, and Nader-Macías ME (2011). Urogenital pathogen inhibition and compatibility between vaginal Lactobacillus strains to be considered as probiotic candidates. Eur. J. Obstet. Gynecol. Reprod. Biol. 159, 399–406. [DOI] [PubMed] [Google Scholar]
- 56.De Gregorio PR, Tomás MSJ, Terraf MCL, and Nader-Macías MEF (2014). In vitro and in vivo effects of beneficial vaginal lactobacilli on pathogens responsible for urogenital tract infections. J. Med. Microbiol. 63, 685–696. [DOI] [PubMed] [Google Scholar]
- 57.Ho M, Chang Y-Y, Chang W-C, Lin H-C, Wang M-H, Lin W-C, and Chiu T-H (2016). Oral Lactobacillus rhamnosus GR-1 and Lactobacillus reuteri RC-14 to reduce Group B Streptococcus colonization in pregnant women: A randomized controlled trial. Taiwan J. Obstet. Gynecol. 55, 515–518. [DOI] [PubMed] [Google Scholar]
- 58.Jumpstart Consortium Human Microbiome Project Data Generation Working Group (2012). Evaluation of 16S rDNA-based community profiling for human microbiome research. PLOS ONE 7, e39315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qunfeng D, and Claudia V (2012). Evaluation of the RDP Classifier accuracy using 16S rRNA gene variable regions. Metagenomics 2012, 1–5. [Google Scholar]
- 60.Huse SM, Ye Y, Zhou Y, and Fodor AA (2012). A core human microbiome as viewed through 16S rRNA sequence clusters. PLOS ONE 7, e34242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fettweis JM, Serrano MG, Sheth NU, Mayer CM, Glascock AL, Brooks JP, Jefferson KK, and Buck GA (2012). Species-level classification of the vaginal microbiome. BMC Genom. 13 Suppl 8, S17–S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, and Olsen GJ (2008). Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 74, 2461–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.The Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aagaard KM, Petrosino J, Keitel W, Watson M, Katancik J, Garcia N, Patel S, Cutting M, Madden T, Hamilton H, et al. (2013). The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J. 27, 1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Meth. 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, and Tiedje JM (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, and Hugenholtz P (2011). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McMurdie PJ, and Holmes S (2013). phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLOS ONE 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rotmistrovsky K, and Agarwala R (2011). BMTagger: Best Match Tagger for removing human reads from metagenomics datasets. Ftp://Ftp.Ncbi.Nlm.Nih.Gov/Pub/Agarwala/Bmtagger/.
- 71.Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Meth. 12, 902–903. [DOI] [PubMed] [Google Scholar]
- 72.Kim D, Song L, Breitwieser FP, and Salzberg SL (2016). Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dufrene M, and Legendre P (1997). Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol. Monogr. 67, 345. [Google Scholar]
- 74.Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- 75.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, and McGlinn D vegan: Community ecology package. R package version 2.4–6. [Google Scholar]
- 78.Wickham H (2009). ggplot2: Elegant Graphics for Data Analysis (Springer; ). [Google Scholar]
- 79.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mandal S, Van Treuren W, White RA, Eggesbo M, Knight R, and Peddada SD (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smoot ME, Ono K, Ruscheinski J, Wang P-L, and Ideker T (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. PLINK SNP-species associations, Related to Figure 5.
Data Availability Statement
The WGS metagenomic and targeted 16S rRNA gene amplicon sequence data generated from this study have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject PRJNA451212.