

Abstract
Organoids are self-organizing, expanding 3D cultures derived from stem cells. Using tissue derived from patients, these miniaturized models recapitulate various aspects of patient physiology and disease phenotypes including genetic profiles and drug sensitivities. As such, patient-derived organoid (PDO) platforms provide an unprecedented opportunity for improving preclinical drug discovery, clinical trial validation, and ultimately patient care. This article reviews the evolution and scope of organoid technology, highlights recent encouraging results using PDOs as potential patient “avatars” to predict drug response and outcomes, and discusses critical parameters for widespread clinical adoption. These include improvements in assay speed, reproducibility, standardization, and automation which are necessary to realize the translational potential of PDOs as clinical tools. The multiple entry points where PDOs may contribute valuable insights in drug discovery and lessen the risks associated with clinical trials are also discussed.
eTOC Blurb
The development of patient-derived organoids (PDOs) represents a major opportunity for precision medicine. In this review, Bose et al. discuss the evolution and scope of organoid technology and the challenges and potential solutions to clinical adoption for personalized medicine.
Introduction
The promise of personalized medicine has long offered a viable solution to a major hurdle in the path to successful drug discovery and development—namely, the challenge of predicting for whom specific therapies will be most effective. To this end, significant efforts have been undertaken to better characterize the heterogeneity of patient backgrounds and disease subtypes. As our collective understanding has grown, it has also underscored the crucial need for platforms that can recapitulate this complexity in model systems. To this end, immortalized cell lines and genetically engineered mouse models (GEMMs) have formed the cornerstone of scientific inquiry, yielding important insights into human biology, particularly in the interrogation of signaling pathways. However, using either to model human disease can be challenging. The process of immortalizing cell lines from primary tissues is very inefficient and marred by extensive genetic shifts during adaptation to 2D culture and repeated propagations thereafter1. Likewise, the mutations induced in GEMMS to produce disease phenotypes do not capture the diversity of human disease phenotypes or subtypes very well. In addition to being labor-, time- and cost-intensive, GEMMS are thus generally seen as poor predictors of clinical success. As such, the search for a cost-effective, high-throughput model which can capture patient heterogeneity and individual disease phenotypes remains ongoing.
Key advances in this search stemmed from the exploration of cell culture conditions that simulate the in vivo microenvironments. The first and most widely adopted material for this purpose is extracellular matrix (ECM) isolated from Engelbreth-Holm-Swarm (EHS) mouse sarcoma cells—a substance that has since been commercially adopted as Matrigel®. In 1987, breast epithelial cells grown on this material were seen to self-organize into 3D ducts with lumen before beginning to synthesize and secrete milk protein2. The subsequent development of induced pluripotent stem cells (iPSCs) and the growing understanding of how morphogens could be used to choreograph differentiation events paved the way for stem cells isolated from adult tissue (ASCs) to be cultured3, 4. In 2009, single leucine-rich repeat containing G protein-coupled receptor 5 (Lgr5)-expressing adult intestinal stem cells grown in Matrigel® were observed developing into 3D crypt-villus structures—the first reported derivation of organoids from ASCs5. Since this study, organoids have rapidly emerged at the forefront of disease modelling as a valuable, relatively inexpensive, and convenient method to study human disease. In fact, because organoids recapitulate the architecture and behavior of their host tissue in many ways, they have been used to study cancers, hereditary and infectious diseases, and other pathophysiology. The insights derived from using human organoids in these contexts can be used to drive clinical care. As such, the role of organoids in guiding personalized medicine decisions is currently expanding, as the number of human diseases being modeled using these mini-organs is actively growing6-8.
As an important note, organoids have been derived from both iPSCs and ASCs, and the choice of which model to use requires careful consideration of desired experimental outcomes9. Because organoids take advantage of self-renewal and differentiation capabilities of stem cells, both of these organoid types follow the same basic paradigm for initial formation: stem cell populations are isolated using commercial morphogens to block differentiation, terminal differentiation for desired tissue types is initiated using precisely defined media formulations, and the cells are plated into an 3D growth setting which recapitulates the environment in vivo10, 11. In this workflow, iPSCs require additional steps of differentiation to tissue-specific stem cells prior to organoid formation. Despite this, iPSC-derived organoids have been valuable tools to simulate the epithelial-mesenchymal interaction and to establish organoids from tissues with negligible self-renewal capacity, such as parts of the central nervous system12, 13, heart muscle14, 15, or the glomeruli of kidneys16. In the context of precision oncology, the ready availability of tumor specimens allows patient-derived organoids (PDOs) to be directly developed from clinical tumor resection or biopsies without having to resort to the more complex and indirect route of using iPSCs.
Successes of Organoid-Guided Personalized Medicine
Cancers are highly variable in terms of stage, genetic background, and molecular behaviors—a clinical heterogeneity which PDOs are uniquely well-suited to capture. This pursuit has led to the development of large scale biobanks of breast 17, colorectal 18, 19, ovarian 20, pancreatic21, brain22, kidney23, neuroendocrine24, gastric25, 26, cervical27, head and neck28, and liver cancers29. These biorepositories of tissue which reflect the histopathological, genetic profiles of host tissue can be propagated for research purposes, making them powerful avatars to interrogate the efficacies of treatments for different cancer subtypes and patient populations.
Tailoring individual therapies to a patient’s genetic profile has been a principal avenue for the incorporation of personalized medicine into clinical practice. However, conservative estimates place the percentage of cancer patients who benefit from genome-guided therapies at only 7%30. The reason for this limited scope is multifold—narrow patient eligibility, prohibitive costs, differences between predicted and actual clinical response, etc.
Since PDOs recapitulate more features of human disease than genetics alone, PDO-guided therapeutic decision-making may outperform genome-guided therapies clinically in the future. In fact, PDO biobanking efforts and broad-based drug screens have reproduced known associations between genetic mutations and sensitivity to targeted therapies31-33. These results suggest that the genetically tailored approach of current precision oncology is integrated into the PDO-guided therapeutic decisions. Future studies will likely identify such genome-specific therapeutic targets more robustly and with greater clinical relevance than other models19, 29, 34.
Cancer PDOs Predict Patient Response
Overall, a key promise of organoid technology in clinical applications has been its ability to predict patient outcome—specifically, that drugs with antitumor activity in PDOs would have an analogous effect when treating the donor patients. While a number of recent studies have correlated the sensitivity of PDOs treated with chemotherapies to the clinical outcomes of corresponding patients, these studies have often lacked sample sizes to lend their conclusions statistical rigor. Nevertheless, initial results that have been reported suggest the predictive potential of organoids is a promising avenue of exploration. As the sample size and statistical rigor of these investigations become more robust in coming years, the advent of these patient “avatars” as clinical tools to guide therapeutic choice will likely be of significant consequence.
In a landmark study on metastatic gastrointestinal cancers, tumor organoids derived from 21 patients were reported to have a positive predictive value of 88% and a negative predictive value of 100% in forecasting patient response to a library of chemotherapies35. A larger follow-up, the TUMOROID study on metastatic colorectal cancer, reported similar results from 35 PDOs which were used to evaluate combinations of 5-FU, oxaliplatin, and irinotecan as first- and second-line therapies. In this study, a subset of 12 PDOs with available clinical follow-up data was shown to predict patient 5-FU/irinotecan response with an accuracy of 83.3%, and 50% of PDOs most sensitive to the combination were also associated with higher progression-free survival (PFS)36. The correlation between degree of PDO response and length of patient PFS was also reported for 7 patients in a study of rectal cancer patients which also demonstrated that chemoradiation response variability could be simulated using PDOs in culture37, 38. A similar study in head and neck squamous cell carcinoma (HNSCC) organoids also showed that PDO radiosensitivity closely resembled the clinical outcomes of the patients28.
Several of these clinical correlative studies have been undertaken collectively with large-scale biobanking efforts. Despite the large sample sizes of these biobanks, the lack of accessible clinical follow-up data has still limited the statistical power of these studies. For example, a large-scale gastric cancer biobanking effort which demonstrated the broader feasibility of large-scale screening, reported PDO sensitivities to 5-FU and cisplatin congruent with three representative patients26. A biobanking study using a subset of 12 breast cancer PDOs revealed that, while ER+ PDOs responded most robustly to the estrogen receptor (ER) antagonist tamoxifen, PDOs overexpressing HER2 did not reflect corresponding HER2-targeting drug sensitivity17. In ovarian cancer, as a part of the characterization of a large-scale biobank, PDOs from 21 patients were treated with standard platinum/taxane therapies, with PDO response closely predicting patient chemotherapy responsiveness20. In fact, for one patient, the development of chemoresistance was also longitudinally captured in the isolation of organoids derived from different points in the disease course.
Other studies on small cohorts of patients with gastrointestinal and colorectal39-42 or pancreatic21, 43 cancers have similarly reported concordance between PDO drug sensitivity and patient treatment responses. In a separate smaller study of 5 patients with gastric cancer, PDOs derived from one patient exhibited sensitivity to 5-fluorouracil (5-FU), which was used in conjunction with radiotherapy to clinically eradicate the patient’s tumor44. A similar study of 5 patients with esophageal adenocarcinoma revealed overlap between poor clinical response and PDO resistance to chemotherapeutic agents45.
Case studies using organoids derived from patients with glioblastoma46, treatment-refractory peritoneal colorectal cancer47, and liver cancer48 to guide personalized therapies with variable success have also been reported, sometimes yielding novel insights regarding disease mechanisms. For example, after observing improved carboplatin response in castration-resistant prostate cancer PDOs deficient in chromodomain helicase DNA-binding protein 1 (CHD1), a patient with this genetic mutation was subsequently treated with the drug successfully49.
As these studies continue to expand our understanding of how organoids can be used to guide personalized therapies, new technologies for screening and interpreting PDO responses are expanding as well. A recent study using optical metabolic imaging (OMI) of the redox state of pancreatic cancer organoids during treatment enabled accurate stratification of 7 patients as non-responders who exhibited clinical recurrence within a year and as responders who remained tumor-free50. Given that rapid changes in metabolic dynamics in this assay was a valuable predictor of patient response, cell viability in response to drug therapy may be only one of the many metrics of PDO behavior that can be used to predict patient behavior. Further work on using novel cell-based assays and computational pipelines51 may better define the precise features of PDOs that contribute to their predictive value.
PDOs as Functional Biomarkers for Treatment Stratification
Because large-scale PDO libraries can be expanded to include large patient populations, the clinical heterogeneity of various human diseases can be captured in a robust way. As such PDO biobanks can serve as representative microcosms of the disease landscape as a whole. PDOs of rare disease subtypes can thus be selected and used for drug-sensitivity screening. While clinical trials and individualized therapies would be otherwise inaccessible to this small population, the sheer sample sizes of these libraries can enable effective targeted therapies to be identified for specific subsets of patients. In this context, characterization of PDOs—whether by genetic profiling or drug sensitivity screening—may serve as a functional assay that can be used to stratify patients into treatment groups that will optimize clinical outcomes.
Perhaps the most illustrative example of this paradigm in recent years has come from treatment of a genetic disease, namely cystic fibrosis (CF). Caused by mutations in the CFTR gene (encoding a chloride channel), this genetic disorder causes significant lung and digestive system damage in affected children. Specific CFTR mutations can be treated with different pharmacological agents, with CFTR-modulating drugs like ivacaftor, lumacaftor, and tezacaftor currently authorized for treatment for only specific mutations52. While CF is a rare disease with 1 in 3000 children affected, the rarity of the precise mutations treated by these drugs is even more pronounced. Thus, clinical trials evaluating the efficacy of these drugs for particular mutational subtypes are often not feasible. Organoids offer a unique opportunity to mitigate this challenge, using rectal biopsies from CF patients as avatars for drug testing to identify whether the CFTR-modulators may be effective for patient treatment. Using a forskolin-induced swelling assay, organoids derived from rectal biopsies of two patients with rare CFTR mutations reflected response to Kalydeco (ivacaftor) treatment, an insight that was translated to clinical care and resulted in significant patient improvement52. Using organoids derived from patient rectal biopsies and assayed in the lab to determine optimal CF treatment for the patient is now much more widely used. In fact, since this approach bypasses the need for expensive genotyping assays and the cost of clinical trials on small patient cohorts with drugs that posed minimal chances of success, it has now defined a key model for clinical translation. In fact, since Kalydeco was previously not approved for use in patients with the identified G1249R mutation, it is highly unlikely that this breakthrough would have otherwise been possible. Thus, a key question emerges—do other clinical opportunities for translational impact with the organoid approach exist? In the drug development and clinical trial pipelines, the potential roles for organoids in guiding personalized medicine are extensive.
While clear milestones around using organoids as avatars for therapeutic decision making exist, more avenues for organoids in clinical care are yet to be explored. For example, establishing rare tumor organoids will likely meet unmet needs, as the lack of alternative models and the challenges of conducting such clinical trials often prevent new therapeutic development for such diseases53. Furthermore, the pathological changes seen in rare disease organoids may improve our understanding of the underlying drivers24. With the role of organoids expanding, the clinical opportunities for patient-derived organoid tissues will likely extend beyond CF to include conditions for which novel technologies like genetically modified PDOs for personalized transplantation may yield therapeutic benefit.
The Advantages of Organoids to Model Disease
As PDOs continue to be explored and characterized for their utility in personalized medicine, unique advantages to this 3D culture method have emerged. The key successes highlighted above capitalize on the ability of organoids to recapitulate aspects of human disease. However, disease phenotypes often emerge from the confluence of various genetic, metabolic, and microenvironmental effects that converge to contribute to an overall pathophysiology. To adequately model the complexity of disease behaviors, organoids must faithfully recapitulate all of these features to serve as valuable tools in determining optimal clinical course—a challenge which they often meet more readily than other in vivo or in vitro counterparts (Figure 1).
Figure 1: Organoids derived from healthy and diseased tissue can be used to model various aspects of human physiology.

These include (clockwise from top left) modelling mutational cascades involved in carcinogenesis, features of pathogenic viral and bacterial infections, capturing the heterogeneity of tumor genetic subtypes, and cell-cell interactions that promote malignant cellular characteristics.
Organoids Reflect Cellular Characteristics
Among these different features, perhaps one of the best studied remains the genetic underpinnings of human disease—with the advent of large-scale genomic profiling technologies paving the way for advances in our collective understanding of mutational events that contribute to aberrant biology. Particularly in the context of cancer, the preservation of this genetic diversity has been well-validated in large-scale PDO biobanks 17, 20, 22, 23. These biorepositories of tissue provide not only a record of interpatient genetic heterogeneity but also present patient-derived samples that can be propagated for research purposes, making them invaluable basic research tools.
In addition to the macroperspective of cancer genetics to which biobanks contribute, recent studies have also shown that the mutational progression of carcinogenesis can be replicated in organoid cultures. In fact, the well-described sequence of adenoma-carcinoma mutations that lead to colorectal cancer (CRC) was simulated in healthy colon organoids genetically engineered using CRISPR-Cas9 to harbor APC, TP53, SMAD4, and activating KRASG12D mutations54, 55. These quadruple-mutant organoids lost Wnt/R-spondin, EGF, and Noggin dependence and reliably formed invasive adenocarcinomas when xenografted. Similar modeling of other mutational cascades has provided insights into the development of cholangiocarcinoma56, breast57, pancreatic58, and gastric59, 60 cancers. Similarly, using CRISPR-Cas9 to precisely edit the genome of healthy organoids and induce particular disease phenotypes has yielded important insights into the causes of both polygenic and monogenic diseases61. Induced frameshift mutations in Hermansky-Pudlak syndrome (HPS) genes in healthy lung organoids resulted in the progressive fibrotic changes characteristic of idiopathic pulmonary fibrosis (IPF)62, and mutations of the DGAT1 in healthy intestinal organoids recapitulated the protein-losing enteropathy and fat intolerance seen in the genetic disorder, DGAT1 deficiency63. Other studies have used intestinal organoids derived from affected patients to study multiple intestinal atresia (MIA)64 and variant microvillus inclusion disease (MVID)65, 66, yielding important insights into the genetic etiologies and aberrant signaling that underlie these disorders.
In addition to these genetic changes that are recapitulated in organoids, the epigenetic landscape of human disease, as a key regulator of gene expression, is of particular interest. Derived organoids appear to preserve these epigenetic markers from patients, reflecting the DNA methylation profile and gene expression profiles of the respective patients with ulcerative colitis67 and Crohn’s Disease68 from whom they were isolated. The epigenetic landscape of primary cancer types was also more closely modeled in tissue-derived organoids than in corresponding 2D cell lines, suggesting that these 3D cultures are well suited to study the role of DNA methylation in cancers69. By more closely preserving the genomic and epigenetic background of tumors, gene expression as profiled using either bulk, or more recently single-cell RNA sequencing and deep proteomics analysis has also revealed significant parity between donor tissue and derived organoids in multiple cancer types70. Notably, while genetic, epigenetic, transcriptomic, and proteomic profiles have been relatively better characterized, the metabolic landscape of organoids remains poorly understood. Future investigations into organoid metabolism may offer another avenue to better understand the effect of individual patient metabolism, shaped by environment and diet, on disease progression and drug response.
Organoids Recapitulate Cell-Cell Interactions
Multicellularity in organoid cultures enables studies of cell-cell interactions which are otherwise difficult to perform. As such, they offer a unique opportunity to better understand the multicellular signaling networks and behaviors, whether tumor and stroma or host and pathogen, as they interact in the human body during disease71-74. For example, similar to human pancreatic cancers, murine pancreatic stellate cells grown with pancreatic ductal adenocarcinoma cells underwent transformation into cancer-associated fibroblasts and began producing desmoplastic stroma75.
A particular area of interest in recent years has been modeling the interplay of the immune system with either exogenous or endogenous pathogens73, 76. Interestingly, while immunological research has long relied on animal models as the cornerstone of modeling immune response, viral and bacterial infection of healthy organoids reflect morphological changes associated with patient infection12. A common etiology of stomach ulcers in patients, Heliobacter Pylori bacteria was used to infect healthy gastric organoids, leading to strong inflammatory responses that reflected the degrees of inflammation of corresponding donor patients77, 78. In cancer, co-cultures of immune cell and tumor organoids have provided a novel avenue to understand the role of the immune system in cancer progression. In fact, initial studies using microfluidic co-culture models of bladder cancer have revealed macrophages with upregulated Arg-1 migrating towards neoplastic cancer cells79, 80. Similar applications of organoid and microfluidic technologies may be used to develop CAR-T cells and other immunotherapeutic approaches in the futures71.
Organoids Recapitulate Unique Aspects of Human Pathophysiology
Particularly, in contexts like the lung and the brain, where infection dynamics of in vivo murine models do not reflect the human physiology and specialized animal models are required, organoid cultures have been proven to be of unique value. As an illustrative example, understanding the precise mechanisms by which the Zika virus (ZIKV) causes microencephaly is currently not possible in murine models, as mice lack the outer subventricular zone (OSVZ). Thus, the relevance of the model is unknown. Instead, infecting hESC-derived cerebral organoids with ZIKV recapitulated first-trimester brain development and allowed in depth investigation into the processes of disrupted neurogenesis that lead to microcephaly81. In modeling pulmonary infection, exposing healthy lung organoids to respiratory syncytial virus (RSV) led to infection and sloughing of Type 2 pneumocytes82. Infection of small intestine and lung organoids by Cryptosporidium, a protozoan parasite, similarly yielded in vitro behaviors physiologically relevant to in vivo infections83. Most recently, the complex distal lung bronchioles and alveoli most affected in SARS-CoV-2 infection were derived from iPSCs in organoid culture, and faithfully recapitulated features of COVID-19 associated pneumonia84.
In the context of microenvironmental influences, organoids offer a particularly robust model to manipulate morphogen exposure and extracellular scaffolding with a level of precision not offered with animal models. In fact, combining bioprinting85 and so-called designer matrices86, creating artificial extracellular matrices and vasculature for organoids that closely mimic the in vivo environment has become possible. Using these controlled environments, further insights into the precise microenvironmental makeup (growth factors, stromal and niche cells, and external factors) that contribute to specific disease conditions will undoubtedly be generated.
Parameters to Improve for Clinical Translation
Historically, a number of in vivo and in vitro models have been used to model disease and predict treatment response in translational research. Models that do not use patient-derived tissue, like established cell lines and genetically engineered mouse models (GEMMs), have been cornerstones of this research, yielding important insights into drug mechanisms, signaling cascades, and core aspects of cellular biology. However, these models are not ideal tools for personalized medicine, as cell lines have exhibited significant genetic variability with repeated passaging and questions around the translatability of GEMM tumor physiology to human patients exist. As such, these models remain most useful to define and interrogate broader biological paradigms in cancer.
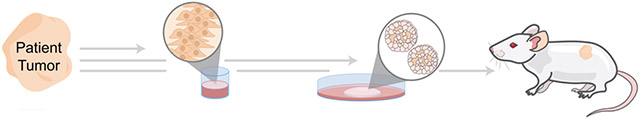
In cancer drug discovery and personalized medicine, patient-derived xenografts (PDXs) have remained the gold-standard for translational models over past decades—with tumor tissue isolated from patients engrafted into immune-compromised mice. These models provide a number of benefits: they retain intra-tumoral clonal architecture after repeated passaging87, faithful recapitulate of patient drug response88, 89, and capture genetic diversity of tumor types across patients. However, PDXs are quite time consuming and expensive. In comparison, PDOs offer better cost, higher rates of successful establishment, and higher throughput (Table 1). As larger-scale studies on PDOs establish their potential to predict drug response and capture patient heterogeneity, organoids as a whole are quickly becoming one of the most powerful tools in our arsenal to model human physiology35-37.
Table 1:
Patient-derived cell lines, organoids, and xenografts are important tools to model patient tumor behaviors.
| |||
|---|---|---|---|
| Patient Derived Cell Lines |
Patient Derived Organoids (PDOs) |
Patient Derived Xenografts (PDXs) |
|
| Development Time | 1 week | 4-6 weeks | 6-8 months |
| Initiation Success | Minimal | Moderate | Moderate |
| Cost | $ | $$ | $$$$ |
| Throughput | High | Moderate | Low |
| Standardization | High | Low | Moderate |
The ability of PDOs to mimic patient response to treatments have made these models valuable tools at multiple points in the entire pipeline of drug development and clinical medicine (Figure 2). By capturing patient diversity, PDOs can be used as medium-throughput pre-clinical models to validate drug safety and efficacy, complementing 2D cell lines, GEMMs, and PDX models to bridge the gap between drug discovery and clinical trials. As the aforementioned studies show, PDOs correlate well with patient drug response, suggesting that PDOs might have an important role as a companion diagnostic assay. PDO treatment at the bench can thus serve as a functional biomarker to direct treatment choices at the bedside, both during clinical trials and after the treatment is approved. The extent of these roles is currently evolving and exploring the potential role of PDOs in guiding personalized therapies has become a pivotal undertaking of several individual companies and academic institutions.
Figure 2: Using organoids can have impacts at multiple stages of the drug development pipeline, including during drug candidate screening, target validation, and clinical trials.

As the current paradigm results in only a few drugs introduced to market, patient derived organoids (PDOs) can reflect a greater range of patient backgrounds, thus identifying therapeutics which may be effective but had been rejected in prior models.
However, the translation of organoid model systems for use in clinical settings to drive patient care will require the removal of several hurdles, including the speed of development, success rates of organoid establishment, cost, throughput and reproducibility. Automated, microfluidics-driven approaches of culturing organoids, potentially combined with novel bioengineered matrices, appear to present the most promising road to clinical implementation of PDOs. The adoptability of this promising technology in the clinic is reliant on advances in these key areas (Table 1).
Speed and Throughput of Organoid Development
In the context of cancer therapeutics, speed of development is a crucial parameter that determines applicability of organoid technologies as personalized diagnostic tools. In current clinical practice, the time between a diagnosis of metastatic cancers via imaging and/or biopsy and the start of treatment, whether via chemotherapy, radiation, or initial resection surgery, ranges optimistically between 12-14 days in the United States. However, in cases of aggressive and fast-growing cancers, delays in this diagnostic interval on the timescale of days can lead to drastically different patient prognoses, motivating the need for diagnostic tools that can be implemented quickly.
The time required to develop robust patient-derived organoids, especially from low volume tissue samples such as 18-gauge biopsies, represent a significant limitation in their utility in guiding clinical decision-making. Currently, while PDOs may be able to guide second-line or adjuvant therapy choices, the 4-6 weeks necessary to develop PDOs disqualify them from being used to define first line therapies. During this time, patients being treated with first-line therapies may also develop resistance which may not be reflected in PDOs derived at a treatment-naïve stage. Thus, acceleration of the PDO development and testing timeline to under 14 days is necessary for these personalized medicine tools to used clinically. The current time course for PDO development is still significantly shorter than the 6-8 months required to establish useful PDX models89. While academic and industrial groups currently are developing technologies to address this need, the relatively slow speed of PDO establishment remains a key hurdle in the application of PDOs to clinical practice.
Success of Establishing Organoids
This lengthy development course is further complicated by the fact that establishing organoids from patient derived tissues is not always successful. In fact, the establishment of organoid cultures is often highly dependent on culture and maintenance processes used. The method of harvesting tissues that are used for organoid development can also be particularly important.
For larger solid tumors, resected tissue has also been used to establish various cancer organoids with considerable success, with large biobanking efforts reporting establishment rates of 70-95% 21, 23, 90. However, establishment rates for some types of cancers remain low91. More challenging still are smaller biopsies which can also be taken at earlier stages of disease, better simulating premalignant conditions and metastatic growths. However, using resected tissues or even different biopsy sites in the pursuit of higher cell numbers which may yield better establishment rates is not always a valid approach. Variability in the biopsy site can have significant consequences, as demonstrated by a study of ovarian cancer PDOs derived from different tumor biopsy sites from 7 patients which exhibited differences in drug sensitivities92. This inter- and intra-patient heterogeneity suggests that different methods of harvesting tissue for organoids can yield different phenotypes.
An area of current exploration focuses on using circulating tumor cells (CTCs) to develop robust organoid cultures. This approach, while still in its infancy, will represent a step forward in using liquid biopsies—peripheral blood samples, urine samples, or bronchoalveolar lavage—as sources for organoids in diagnosis and clinical care31, 93. Adaptations to protocols used to establish organoids to accommodate relatively sparse stem cell populations will be necessary to develop these methods further.
Another complication which may limit organoid establishment involves ‘contamination’ of the tumor samples by normal epithelial cells, which may overgrow the PDO culture. This phenomenon has been described in both prostate and lung cancers91. This complication can be overcome by using biopsies of ‘cancer-only’ metastatic outgrowths, or by applying growth factor conditions which selectively allow outgrowth only of tumor cells. For example, this outcome be achieved by removing Wnt from the growth factor cocktail when growing colorectal carcinoma PDOs94 or pancreatic tumor PDOs58.
Cost of Organoid Culture
PDOs must be competitive in terms of cost for a path forward into clinical use. Currently, the cost of current organoid culturing is largely determined by: (1) the time- and labor-consuming process of establishing and subsequently passaging PDOs, as well as by (2) the materials required to develop and maintain the cultures. These reagents include an expensive cocktail of growth factors and animal-based matrix extracts like Matrigel®. The development of synthetic matrices has been a key endeavor in minimizing the cost of establishing and maintaining organoids. Efforts to develop polyethylene glycol (PEG) hydrogels as a cost-reducing alternative to Matrigel® have been underway in recent years95-97. However, such gels need to be ‘functionalized’ by including extracellular matrix signals such as short peptides motifs derived from collagen or laminin; to date Matrigel® has remained superior to any of the synthetic matrices 98. Ongoing development of these synthetic scaffolds will likely yield benefits for both cost and reproducibility in the future. A potential overarching solution involves using microfluidics approaches. These would offer ways to miniaturize organoid assays, bypassing the need to expand PDOs by passaging and allowing rapid screening, possibly even within 7-10 days.
Standardization of Organoid Cultures
The complexity of the processes required to generate PDOs poses not only a considerable challenge for clinical adoption but also for hypothesis-generating insights derived from PDO studies. Further standardization and automation of these procedures – from the materials used in culture conditions to the devices that yield uniform organoid generation – will enhance reproducibility and form an important foundation for the advent of PDO technology to market.
Significant work has been done to define media and cocktails of growth factors that are best suited for different subtypes of tumor organoids. However, these compositions are not standardized. As such individual studies optimize growth factor concentrations based on cost and availability, results derived from PDO studies can often not be compared across publications because of the variability in microenvironmental factors99, 100. While some media and growth factor cocktails are now commercially available, these media compositions must be standardized for adequate reproducibility. In addition, software pipelines51 that can be used to quantify these results will be essential for PDOs to be used in driving clinical decision making.
Discussion and Future Directions
As large-scale organoid biobanking for diseases like cancer and genetic disorders become more prevalent, the utility of these PDOs as surrogates for clinical trial patients becomes more viable. For this, it will not only be necessary to improve features of PDO use along the parameters described above, but also to extensively validate PDOs as predictors of clinical response through further larger-scale studies. While the ideal paradigm of the randomized controlled trial (RCT) has long dictated the format of clinical studies, novel study designs may allow PDOs to be evaluated with the same degree of clinical rigor without trial constraints; establishment and maintenance of PDOs from treatment-naïve patients can be concurrently performed while medical providers use standards of care and clinical judgements to determine course of treatment. Subsequent screening of organoids may yield a PDO-predicted optimal treatment, at which point, patients may be stratified in treatment and control groups based on whether there is concordance between PDO-predicted treatments and clinician choice (Figure 3). Despite this approach offering the benefit of including patients without impacting treatment (as with an observational or retrospective study) while using treatment-naïve PDOs as in a prospective study, two key drawbacks emerge: (1) this study design is limited clinically to drugs approved for each disease and (2) the current month-long timeline to establish PDOs mean results from these studies cannot be readily adopted.
Figure 3:

Retrospective studies have been used to establish the validity of PDO-guided therapies in clinic.
While the role of organoids as avatars for personalized patient testing are evolving, the potential advantages of these 3D cultures in reshaping the current drug discovery pipeline appear equally evident. Current drug development paradigms rely largely on identifying “hits” from high-throughput drug screens. Only the drugs identified as potentially efficacious are followed up with further validation and mechanistic studies, discarding the vast majority of compounds deemed to be ineffective. Assuming that human, patient-derived PDOs are more representative than cell lines, revisiting the paradigm of high throughput drug screening through the lens of patient-derived organoids will likely yield compounds that were previously discarded but may, in fact, be highly effective for certain patient populations.
In addition, to the role of organoids in drug discovery processes, the subsequent validation of compounds as viable therapeutics poses another node of the drug development pipeline where organoids may provide valuable insights. Following the identification of “effective” compounds from the drug screening process, further validation studies are currently often carried out in murine models of disease and through mechanistic studies in larger panels of cell lines before a drug can proceed to clinical trials. This crucial step again eliminates a large percentage of candidate drugs in the pipeline. PDOs, by providing a human context and the variability of patient background, may hold significant value at this step as well. As an illustrative example, if a metabolic therapy is particularly effective in a subset of PDOs derived from patients with genetic disease, it follows that metabolic predispositions may contribute to the disease pathophysiology. In addition to identifying an effective drug for that subpopulation, further studies can be pursued to understand the exact mechanisms which sensitize some patients.
The application of novel computational technologies to precision medicine using PDOs is another area of interest. PDOs, by virtue of their ability to recapitulate multiple features of disease tissue (metabolic, immune, genetic, etc.) provide multiple inputs which can be computationally deconvoluted using machine learning algorithms currently in development. It is possible to envision that in the coming years, these studies will identify predisposing factors for human disease that are yet unknown.
Finally, even after successful advancing through drug screening, validation, and testing in vivo and in vitro, drugs are often unsuccessful in clinical trials – leading to significant financial and intellectual capital lost. Unfortunately, while some degree of this is unavoidable in translation of drugs from the bench to the bedside, the potential benefits of using PDOs as phenotypic biomarker assays may provide a multifold solution. In addition to allowing much larger populations to be prescreened prior to inclusion in clinical trials, PDOs can also allow clinicians to make informed decisions about drug combinations that will work best for individual patients. In fact, in designing clinical trials, enriching the patient population with predicted responders as defined by a favorable PDO response would minimize cost, and may potentially accelerate the introduction of lifesaving therapeutics on the market. As advances in methods of establishing and maintaining organoids continue to reduce costs and increase reproducibility and throughput, these PDO biomarker assays can continue to expand beyond the realm of clinical trials and into regular clinical practice as well.
Novel technologies are reshaping how organoids are being established and maintained, in ways that will alleviate the constraints that currently limit clinical adoption. These technologies will allow organoid technology to impact the way drugs are developed, diseases are treated, and patient care is personalized.
Acknowledgments
The work was supported by NIH R35GM122465, U01 CA214300, and U01 CA217514.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
S.B. declares no competing interests. X.S. is a co-founder and the CEO of Xilis Inc. H.C. is a member of the board of directors of Roche and a co-founder of Xilis Inc. H.C.’s full disclosure is given at https://www.uu.nl/staff/JCClevers/.
References
- 1.Letai A Functional precision cancer medicine—moving beyond pure genomics. Nature medicine 23, 1028 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Li ML et al. Influence of a reconstituted basement membrane and its components on casein gene expression and secretion in mouse mammary epithelial cells. Proceedings of the National Academy of Sciences 84, 136–140 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi K & Yamanaka S Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. cell 126, 663–676 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Clarke DL et al. Generalized potential of adult neural stem cells. Science 288, 1660–1663 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Sato T et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Van Der Vaart J & Clevers H Airway organoids as models of human disease. Journal of internal medicine (2020). [DOI] [PubMed] [Google Scholar]
- 7.Clevers H Modeling development and disease with organoids. Cell 165, 1586–1597 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Baker LA, Tiriac H, Clevers H & Tuveson DA Modeling pancreatic cancer with organoids. Trends in cancer 2, 176–190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuveson D & Clevers H Cancer modeling meets human organoid technology. Science 364, 952–955 (2019). [DOI] [PubMed] [Google Scholar]
- 10.McCauley HA & Wells JM Pluripotent stem cell-derived organoids: using principles of developmental biology to grow human tissues in a dish. Development 144, 958–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Q, Oost KC & Liberali P Engineering human knock-in organoids. Nature cell biology 22, 261–263 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Sun G et al. Modeling human cytomegalovirus-induced microcephaly in human iPSC-derived brain organoids. Cell Reports Medicine 1, 100002 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anastasaki C et al. Human iPSC-derived neurons and cerebral organoids establish differential effects of germline NF1 gene mutations. Stem cell reports 14, 541–550 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider MD Upstairs, downstairs: atrial and ventricular cardiac myocytes from human pluripotent stem cells. Cell stem cell 21, 151–152 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Lee J et al. In vitro generation of functional murine heart organoids via FGF4 and extracellular matrix. Nature communications 11, 1–18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishinakamura R Human kidney organoids: progress and remaining challenges. Nature Reviews Nephrology 15, 613–624 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Sachs N et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 172, 373–386. e310 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Sato T et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 141, 1762–1772 (2011). [DOI] [PubMed] [Google Scholar]
- 19.van de Wetering M et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopper O et al. An organoid platform for ovarian cancer captures intra-and interpatient heterogeneity. Nature medicine 25, 838–849 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Driehuis E et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proceedings of the National Academy of Sciences 116, 26580–26590 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacob F et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter-and intra-tumoral heterogeneity. Cell 180, 188–204. e122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calandrini C et al. An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nature communications 11, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawasaki K et al. An organoid biobank of neuroendocrine neoplasms enables genotype-phenotype mapping. Cell 183, 1420–1435. e1421 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Seidlitz T, Koo B-K & Stange DE Gastric organoids—an in vitro model system for the study of gastric development and road to personalized medicine. Cell Death & Differentiation 28, 68–83 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan HH et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell stem cell 23, 882–897. e811 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Lõhmussaar K et al. Patient-derived organoids model cervical tissue dynamics and viral oncogenesis in cervical cancer. Cell Stem Cell (2021). [DOI] [PubMed] [Google Scholar]
- 28.Driehuis E et al. Oral mucosal organoids as a potential platform for personalized cancer therapy. Cancer discovery 9, 852–871 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Broutier L et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nature medicine 23, 1424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marquart J, Chen EY & Prasad V Estimation of the percentage of US patients with cancer who benefit from genome-driven oncology. JAMA oncology 4, 1093–1098 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao D et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertotti A et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer discovery 1, 508–523 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Verissimo CS et al. Targeting mutant RAS in patient-derived colorectal cancer organoids by combinatorial drug screening. elife 5, e18489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pauli C et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer discovery 7, 462–477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vlachogiannis G et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ooft SN et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Science translational medicine 11 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Ganesh K et al. A rectal cancer organoid platform to study individual responses to chemoradiation. Nature medicine 25, 1607–1614 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao Y et al. Patient-derived organoids predict chemoradiation responses of locally advanced rectal cancer. Cell stem cell 26, 17–26. el6 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Steele NG et al. An organoid-based preclinical model of human gastric cancer. Cellular and molecular gastroenterology and hepatology 7, 161–184 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J et al. Malignant ascites-derived organoid (MADO) cultures for gastric cancer in vitro modelling and drug screening. Journal of cancer research and clinical oncology 145, 2637–2647 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arena S et al. A subset of colorectal cancers with cross-sensitivity to olaparib and oxaliplatin. Clinical Cancer Research 26, 1372–1384 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Pasch CA et al. Patient-derived cancer organoid cultures to predict sensitivity to chemotherapy and radiation. Clinical Cancer Research 25, 5376–5387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiriac H et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer discovery 8, 1112–1129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao M et al. Development of patient-derived gastric cancer organoids from endoscopic biopsies and surgical tissues. Annals of surgical oncology 25, 2767–2775 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Derouet MF et al. Towards personalized induction therapy for esophageal adenocarcinoma: Organoids derived from endoscopic biopsy recapitulate the pre-treatment tumor. Scientific reports 10, 1–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loong HH et al. Patient-derived tumor organoid predicts drugs response in glioblastoma: A step forward in personalized cancer therapy? Journal of Clinical Neuroscience 78, 400–402 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Narasimhan V et al. Medium-throughput drug screening of patient-derived organoids from colorectal peritoneal metastases to direct personalized therapy. Clinical Cancer Research 26, 3662–3670 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skardal A, Devarasetty M, Rodman C, Atala A & Soker S Liver-tumor hybrid organoids for modeling tumor growth and drug response in vitro. Annals of biomedical engineering 43, 2361–2373 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shenoy T et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Annals of Oncology 28, 1495–1507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharick JT et al. Metabolic heterogeneity in patient tumor-derived organoids by primary site and drug treatment. Frontiers in Oncology 10, 553 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kong J et al. Network-based machine learning in colorectal and bladder organoid models predicts anti-cancer drug efficacy in patients. Nature communications 11, 1–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berkers G et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell reports 26, 1701–1708. e1703 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Puca L et al. Patient derived organoids to model rare prostate cancer phenotypes. Nature communications 9, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drost J et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 521, 43–47 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Matano M et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nature medicine 21, 256–262 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Artegiani B et al. Probing the tumor suppressor function of BAP1 in CRISPR-engineered human liver organoids. Cell Stem Cell 24, 927–943. e926 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Dekkers JF et al. Modeling Breast Cancer Using CRISPR-Cas9-mediated engineering of human breast organoids. JNCI: Journal of the National Cancer Institute 112, 540–544 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seino T et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell stem cell 22, 454–467. e456 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Lo Y-H et al. A CRISPR/Cas9-engineered ARID1A-deficient human gastric cancer organoid model reveals essential and non-essential modes of oncogenic transformation. Cancer Discovery (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nanki K et al. Divergent routes toward Wnt and R-spondin niche independency during human gastric carcinogenesis. Cell 174, 856–869. e817 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Hendriks D, Clevers H & Artegiani B CRISPR-Cas tools and their application in genetic engineering of human stem cells and organoids. Cell Stem Cell 27, 705–731 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Strikoudis A et al. Modeling of fibrotic lung disease using 3D organoids derived from human pluripotent stem cells. Cell reports 27, 3709–3723. e3705 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Rijn JM et al. Intestinal failure and aberrant lipid metabolism in patients with DGAT1 deficiency. Gastroenterology 155, 130–143. e115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bigorgne AE et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. The Journal of clinical investigation 124, 328–337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Müller T et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nature genetics 40, 1163–1165 (2008). [DOI] [PubMed] [Google Scholar]
- 66.Wiegerinck CL et al. Loss of syntaxin 3 causes variant microvillus inclusion disease. Gastroenterology 147, 65–68. e10 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Dotti I et al. Alterations in the epithelial stem cell compartment could contribute to permanent changes in the mucosa of patients with ulcerative colitis. Gut 66, 2069–2079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Howell KJ et al. DNA methylation and transcription patterns in intestinal epithelial cells from pediatric patients with inflammatory bowel diseases differentiate disease subtypes and associate with outcome. Gastroenterology 154, 585–598 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Joshi R et al. The DNA methylation landscape of human cancer organoids available at the American type culture collection. Epigenetics 15, 1167–1177 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cristobal A et al. Personalized proteome profiles of healthy and tumor human colon organoids reveal both individual diversity and basic features of colorectal cancer. Cell reports 18, 263–274 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Ye W, Luo C, Li C, Huang J & Liu F Organoids to study immune functions, immunological diseases and immunotherapy. Cancer letters 477, 31–40 (2020). [DOI] [PubMed] [Google Scholar]
- 72.Cattaneo CM et al. Tumor organoid–T-cell coculture systems. Nature protocols 15, 15–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dutta D & Clevers H Organoid culture systems to study host–pathogen interactions. Current opinion in immunology 48, 15–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qin X et al. Cell-type-specific signaling networks in heterocellular organoids. Nature methods 17, 335–342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Öhlund D et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. Journal of Experimental Medicine 214, 579–596 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bar-Ephraim YE, Kretzschmar K & Clevers H Organoids in immunological research. Nature Reviews Immunology 20, 279–293 (2020). [DOI] [PubMed] [Google Scholar]
- 77.Bartfeld S & Clevers H Organoids as model for infectious diseases: culture of human and murine stomach organoids and microinjection of Helicobacter pylori. Journal of visualized experiments: JoVE (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bartfeld S et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148, 126–136. e126 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yin X et al. Engineering stem cell organoids. Cell stem cell 18, 25–38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu P.-f. et al. A bladder cancer microenvironment simulation system based on a microfluidic co-culture model. Oncotarget 6, 37695 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Garcez PP et al. Zika virus impairs growth in human neurospheres and brain organoids. Science 352, 816–818 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Chen Y-W et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nature cell biology 19, 542–549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Heo I et al. Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nature microbiology 3, 814–823 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Salahudeen AA et al. Progenitor identification and SARS-CoV-2 infection in human distal lung organoids. Nature 588, 670–675 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brassard JA, Nikolaev M, Hübscher T, Hofer M & Lutolf MP Recapitulating macro-scale tissue self-organization through organoid bioprinting. Nature Materials 20, 22–29 (2021). [DOI] [PubMed] [Google Scholar]
- 86.Gjorevski N et al. Designer matrices for intestinal stem cell and organoid culture. Nature 539, 560–564 (2016). [DOI] [PubMed] [Google Scholar]
- 87.Bruna A et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell 167, 260–274. e222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gao H et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nature medicine 21, 1318–1325 (2015). [DOI] [PubMed] [Google Scholar]
- 89.Hidalgo M et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer discovery 4, 998–1013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li X et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nature communications 9, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dijkstra KK et al. Challenges in establishing pure lung cancer organoids limit their utility for personalized medicine. Cell reports 31, 107588 (2020). [DOI] [PubMed] [Google Scholar]
- 92.de Witte CJ et al. Patient-derived ovarian cancer organoids mimic clinical response and exhibit heterogeneous inter-and intrapatient drug responses. Cell reports 31, 107762 (2020). [DOI] [PubMed] [Google Scholar]
- 93.Mout L et al. Generating human prostate cancer organoids from leukapheresis enriched circulating tumour cells. European Journal of Cancer 150, 179–189 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Schwab RH et al. Wnt is necessary for mesenchymal to epithelial transition in colorectal cancer cells. Developmental Dynamics 247, 521–530 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Aisenbrey EA & Murphy WL Synthetic alternatives to Matrigel. Nature Reviews Materials 5, 539–551 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kaur S, Kaur I, Rawal P, Tripathi DM & Vasudevan A Non-matrigel scaffolds for organoid cultures. Cancer Letters (2021). [DOI] [PubMed] [Google Scholar]
- 97.Cruz-Acuña R et al. PEG-4MAL hydrogels for human organoid generation, culture, and in vivo delivery. Nature protocols 13, 2102–2119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Collier JH & Segura T Evolving the use of peptides as biomaterials components. Biomaterials 32, 4198 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xie AW & Murphy WL Engineered biomaterials to mitigate growth factor cost in cell biomanufacturing. Current Opinion in Biomedical Engineering 10, 1–10 (2019). [Google Scholar]
- 100.Weeber F, Ooft SN, Dijkstra KK & Voest EE Tumor organoids as a pre-clinical cancer model for drug discovery. Cell chemical biology 24, 1092–1100 (2017). [DOI] [PubMed] [Google Scholar]