

Abstract
Nonalcoholic fatty liver disease (NAFLD) has become the most prevalent cause of liver disease, increasingly contributing to the burden of liver transplantation. In search for effective treatments, novel strategies addressing metabolic dysregulation, inflammation, and fibrosis are continuously emerging. Disturbed bile acid (BA) homeostasis and microcholestasis via hepatocellular retention of potentially toxic BAs may be an underappreciated factor in the pathogenesis of NAFLD and nonalcoholic steatohepatitis (NASH) as its progressive variant. In addition to their detergent properties, BAs act as signaling molecules regulating cellular homeostasis through interaction with BA receptors such as the Farnesoid X receptor (FXR). Apart from being a key regulator of BA metabolism and enterohepatic circulation, FXR regulates metabolic homeostasis and has immune-modulatory effects, making it an attractive therapeutic target in NAFLD/NASH. In this review, the molecular basis and therapeutic potential of targeting FXR with a specific focus on restoring BA and metabolic homeostasis in NASH is summarized.
Keywords: NAFLD, FXR, bile acids, microcholestasis
Nonalcoholic fatty liver disease (NAFLD) has reached a global prevalence of approximately 25% and comprises a disease spectrum in the absence of secondary causes such as consumption of relevant amounts of alcohol. 1 2 3 4 5 The spectrum ranges from isolated hepatic steatosis, defined as lipid droplets present in the cytoplasm of more than 5% of hepatocytes, 6 over progression to nonalcoholic steatohepatitis (NASH) in about one out of four patients with additional signature histopathological changes including hepatocyte ballooning and inflammation driving fibrogenesis, to liver cirrhosis and hepatocellular cancer (HCC). 7 8 Its close relationship with central obesity, type 2 diabetes mellitus, insulin resistance, and dyslipidemia has contributed to the concept of NAFLD being the hepatic manifestation of the metabolic syndrome 9 and resulted in the recent proposal of an overarching description termed metabolic (dysfunction)-associated fatty liver disease (MAFLD). 10 11 This also reflects efforts to make a “positive” diagnosis similar to other liver diseases which does not rely on exclusion of other causes. In line with the metabolic association, several studies have found a strong correlation between MAFLD/NAFLD and cardiovascular disease (CVD), chronic kidney disease (CKD), as well as other extrahepatic metabolic manifestations. For instance, presence of hepatic steatosis has been found to increase the risk for type 2 diabetes mellitus by approximately twofold and purports to be an independent driver for CVD additive to other risk factors. 5 12 13 Moreover, a significant association with CKD has been reported, persisting even after correcting for other known CKD risk factors. 12 Last but not least, NAFLD also significantly impairs quality of life, in particular including fatigue and even occurrence of pruritus, 14 15 16 a fact which may be underappreciated compared with liver disease of other etiology. Importantly, NAFLD/NASH has become a major cause of HCC and to date, the second leading cause for liver transplantation, further emphasizing the need for effective and sustained treatment options. 17 18 19 20 21
Recently elevated bile acid (BA) levels have received increasing attention as prognostic signature in NASH, 22 23 24 25 although the mechanisms underlying this observation are still poorly understood. In addition to their traditional perception as chemical detergents which facilitate dietary fat absorption, BAs have important signaling functions via their nuclear receptor Farnesoid X receptor (FXR) and other G-protein coupled receptors (e.g., Takeda G-protein coupled receptor [TGR5]) on their own as well as lipid/glucose metabolism and inflammation. 24 The molecular alterations that occur in lipid metabolism of hepatocytes during NAFLD/NASH development are summarized in Fig. 1 (further reviewed in Arab et al 26 ). Given the central role of BAs and their receptors in controlling key aspects of NASH pathogenesis, modulation of BA-related pathways holds promise in therapeutically managing NASH. 22 23 25 This review provides a summary of the pathophysiological rationale and therapeutic opportunities of targeting FXR NASH.
Fig. 1.
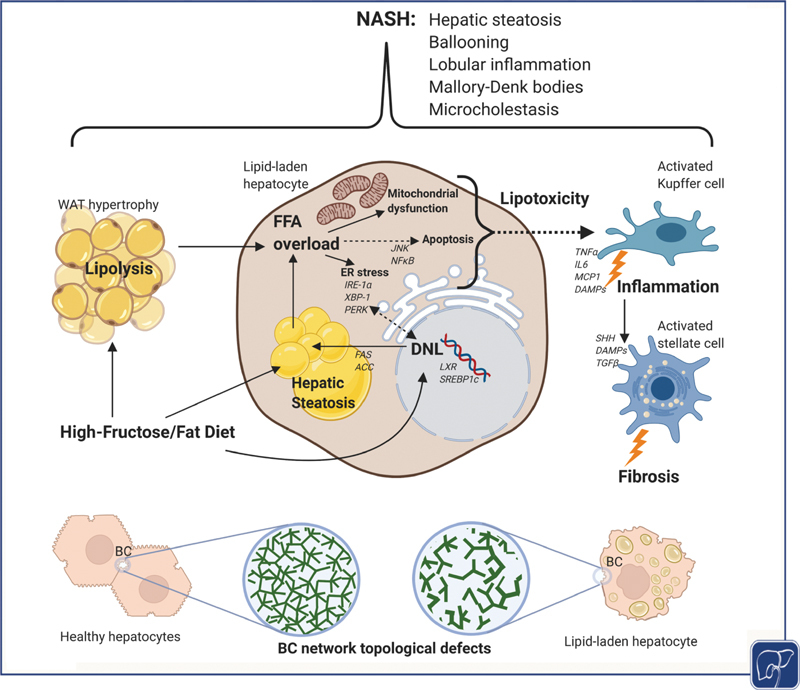
Molecular alterations in liver microenvironment during NASH development. Hepatic steatosis results from overabundance of flux of nonesterified free fatty acids (FFAs) from hypertrophic white adipose tissue (WAT) as a result of increased lipolysis due to insulin resistance. Hepatic de novo lipogenesis (DNL) and dietary fat consumption further contribute, although to a lesser extent, to the development of hepatic steatosis. Due to its peculiar chemical structure, dietary fructose acts directly as a lipogenic substrate in the liver therefore driving DNL through sterol-responsive element-binding protein 1c (SREBP-1c) and liver X receptor (LXR) via acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) as key enzymatic steps. Upon exceeding the hepatic lipid storage capacity, this triggers a FFA overload leading to mitochondrial dysfunction, ER stress via inositol-requiring enzyme 1 α (IRE-1α ), X-box binding protein 1 (XBP-1), and protein kinase RNA-like endoplasmic reticulum kinase (PERK) as well as apoptosis through c-Jun N-terminal kinase (JNK) and nuclear factor kappa-B (NF-kB) signaling. This lipotoxic stress response subsequently activates Kupffer cells involving damage-associated molecular patterns (DAMPs) leading to upregulation of proinflammatory markers such as tumor necrosis factor-α (TNF-α), interleukin 6 (IL6), and monocyte chemotactic protein 1 (MCP-1) as well as activation of hepatic stellate cells (HSCs) through transforming growth factor β (TGFb) and Sonic hedgehog (SHH) signaling, leading to development of lobular inflammation and fibrosis, respectively. These metabolic perturbations are associated with profound changes in BA metabolism in NASH patients where microcholestasis encompassing molecular but also structural alterations as a result of increased pericentral biliary pressure leading to dilatation and reduced connectivity of the bile canalicular (BC) network 112 (lower panel). Figure created with BioRender. BA, bile acid; NASH, nonalcoholic steatohepatitis.
Nuclear Receptor FXR as Central Mediator of BA Signaling in Regulation of Metabolism, Inflammation, and Fibrosis
In the liver, cholesterol from plasma or endogenous synthesis is enzymatically processed to primary BAs such as cholic acid (CA) or chenodeoxycholic acid (CDCA) which form glycine or taurine conjugates followed by active excretion at the canalicular membrane via bile salt export pump (BSEP/ABCB11) into bile and storage in the gallbladder. 27 Upon food digestion, BAs are delivered after contraction of the gallbladder to the duodenum where one of their major functions entails micelle formation for facilitated absorption of fat-soluble substances (e.g., dietary lipids, fat soluble vitamins A, D, E). The majority of BAs (95%) is actively reabsorbed involving absorption through apical sodium-dependent BA transporter (ASBT/SLC10A2), transport across the enterocyte via ileal BA-binding protein (IBABP) and followed by basolateral excretion by organic solute transporter (OSTα/β) into portal blood. The remaining BAs are metabolized by resident gut microbiota in the colon resulting in deconjugation and dehydroxylation with formation of secondary BAs such as deoxycholic acid (DCA) and lithocholic acid which can then passively re-enter the enterohepatic circulation so that only a minor fraction of approximately 0.2–0.6 g/d (∼3–5% of daily synthesis) is lost in the feces. 28 To complete the enterohepatic cycle, BAs are very efficiently removed from portal blood at the basolateral membrane of hepatocytes by Na + -taurocholate cotransporting polypeptide (NTCP/SLC10A1) and organic-anion-transporting polypeptide (OATP/SLCO) family members.
In addition to its function as a facilitator of intestinal lipid absorption, bile is a major way of clearance for various exo- and endogenous substances. From their chemical structure, BAs are amphipathic detergents and therefore potentially cytotoxic, thus requiring a highly efficient molecular machinery to maintain intracellular BA concentrations in a nontoxic range. BA homeostasis is maintained through a negative feedback loop encompassing inhibition of BA uptake and synthesis as well as promotion of BA excretion and detoxification (reviewed in Hofmann 29 ). Toxicity from intracellular BAs involves induction of apoptosis, activation of proinflammatory pathways, and necrotic changes in a concentration-dependent manner. 30 BAs are able to directly induce an inflammatory signal in hepatocytes reflected by increased cytokine expression as well as upregulation of adhesion molecules. 31 In response to secretion of these proinflammatory mediators, immune cells such as neutrophils or macrophages are recruited and activated, further aggravating the inflammatory response in the liver. 31 32
Over the recent years, BAs in nontoxic concentrations have been shown to exert signaling functions for key metabolic and homeostatic processes such as self-regulation of BA metabolism, energy balance, and liver injury mechanisms including inflammation, fibrosis, apoptosis, and tumor development. 27 33 34 35 These regulatory properties of BAs derive from their intrinsic ability to act as ligands for a range of receptors including nuclear receptors FXR, constitutive androstane receptor, pregnane X receptor, and vitamin D receptor as well as membranous receptors TGR5, α5β1 integrin, epidermal growth factor receptor, and sphingosine-1-phosphate receptor 2. 36 37 38 39 Upon binding of BAs to FXR, the FXR–retinoid X receptor heterodimer complex is activated resulting in induction of the transcriptional repressor small heterodimer partner (SHP) which in turn downregulates the BA synthetic key enzymes cytochrome P450 7A1 (CYP7A1) and cytochrome P450 8B1 (CYP8B1) by inhibiting their transactivators liver receptor homolog-1 and hepatocyte nuclear factor 4, molecular changes which collectively result in repression of BA synthesis. 40 Apart from controlling BA synthesis, FXR activation also increases biliary BA excretion by upregulating expression of canalicular transporters BSEP as well as multidrug resistance protein 3 (MDR3/ABCB4) and ATP-binding cassette subfamily G members 5 and 8 (ABCG5/G8) for accompanying phospholipid and cholesterol efflux, respectively. 41 42 Concurrently, FXR activation results in repression of the uptake system NTCP via induction of SHP. 43 In enterocytes, resorption of BAs leads to increased expression of FXR downstream targets including SHP, OSTα/β, fibroblast growth factor 19 (FGF19) and IBABP. Elevated levels of IBABP and OSTα/β result in higher transport efficiency and excretion of BAs into the portal venous system, while SHP directly downregulates ASBT expression thereby restricting intestinal BA reabsorption. 44 45 Furthermore, induction of intestinal FGF19 (Fgf15 in mice) leads to activation of hepatic fibroblast growth factor receptor 4 (FGFR4)/β-Klotho downregulating CYP7A1 gene expression through the c-Jun N-terminal kinase signaling pathway. 46 Taken together, FXR functions as an intestinal and hepatic BA sensor and provides elaborate negative feedback and feedforward mechanisms to uphold physiological BA homeostasis while preventing toxic BA concentrations.
Apart from this regulatory function in BA homeostasis, FXR signaling is involved in lipid and glucose metabolism (reviewed in Chávez-Talavera et al 47 ). Mice deficient of FXR present with significantly increased serum and hepatic triglyceride, cholesterol, and free fatty acid (FA) levels. 48 In line, upon administration of an FXR agonist, wild-type (WT) mice but not FXR-knockout (FXR-KO) mice exhibit decreased serum cholesterol, triglyceride, free FA, and lipoprotein levels. 49 Activation of FXR and subsequent upregulation of SHP result in repressing sterol-regulatory element-binding protein 1c (SREBP-1c) which limits hepatic de novo lipogenesis (DNL). 50 FXR-dependent elimination of serum lipoproteins results from modulation of expression of target genes such as scavenger receptor B1, very low density lipoprotein (LDL) receptor, Syndecan-1, ApoCII/III as coactivators/inhibitors of lipoprotein lipase, and FA translocase (FAT/CD36). 51 On the other hand, FXR promotes β oxidation by activation of peroxisome-proliferator-activated receptor (PPARα). 52 Correspondingly, FXR-mediated FGF15/19 signaling was shown to induce β oxidation in mitochondria 53 via reduction of malonyl-CoA by repression of acetyl-CoA carboxylase (ACC2). Regarding systemic effects, it has been shown that treatment with primary BA CDCA improves hypertriglyceridemia through an FXR- and SHP-dependent mechanism. 50 54 In line, disruption of BA reabsorption through use of BA sequestrants leads to increased biosynthesis of BAs from cholesterol thereby efficiently counteracting hypercholesterinemia. 55 Additionally, reduced plasma glucose levels and a trend toward lower HbA1c have been observed in cholestyramine-treated diabetic patients. 56 At the same time, a common adverse effect of resin treatment lies in aggravation of hypertriglyceridemia, further underlining the role of BAs in triglyceride and cholesterol metabolism. 57 The signaling role of BAs in enterohepatic circulation with systemic metabolic effects of FXR is delineated in Fig. 2 .
Fig. 2.
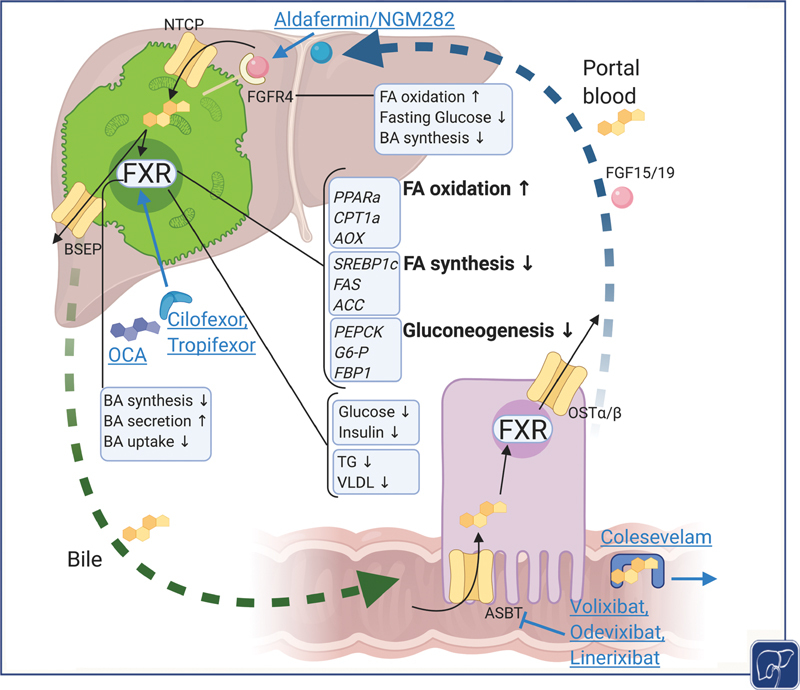
BAs in enterohepatic circulation with systemic metabolic effects of FXR signaling and potentially druggable targets in NAFLD. BAs produced intrahepatically are actively excreted via BSEP into bile. Enterocytic reuptake and basolateral export to portal blood is mediated through ASBT and OSTα/β, respectively. The enterohepatic circle is closed through hepatocellular BA reuptake via NTCP from portal blood. BAs activate the nuclear receptor FXR which leads to increased biliary secretion and reduced synthesis and uptake maintaining BA homeostasis. The systemic effects of FXR encompass metabolic optimization as seen in upregulated fatty acid (FA) oxidation and decreased FA synthesis as well as downregulated gluconeogenesis, contributing to improvement of insulin resistance. Similarly, intestinal FXR leads to elevated FGF 19 levels which mediates increased FA oxidation, decreased fasting glucose via increased glycogen synthesis, and decreased BA synthesis. Modulating BA-related pathways in treatment of NAFLD include direct activation of FXR through steroidal (obeticholic acid, OCA) and nonsteroidal FXR agonists (Cilofexor, Tropifexor), biliary reuptake inhibition with ASBT inhibitors (Volixibat, Odevixibat, Linerixibat) or resins (Colesevelam), and FGF19 analogues (NGM282/Aldafermin). Figure created with BioRender. ASBT, apical sodium-dependent bile acid transporter; BA, bile acid; BSEP, bile salt export pump; FXR, Farnesoid X receptor; NAFLD, nonalcoholic fatty liver disease; NTCP, Na + -taurocholate cotransporting polypeptide.
Further studies investigating the role of FXR in glucose homeostasis showed that in line with its key role in BA and lipid metabolism, FXR-KO rodents exhibit features of hepatic and systemic insulin resistance as seen in increased fasting and fed glucose levels, decreased peripheral glucose disposal assessed by hyperinsulinemic clamp, and impaired gluconeogenesis. 58 59 Appertaining to this, insulin resistance in ob/ob and db/db but not KO mice could effectively be mitigated by feeding them with an FXR agonist such as CA/GW4064. 60 A potential mechanism by which FXR activation modulates glucose metabolism appears to be repression of key enzymes necessary for gluconeogenesis such as phosphoenolpyruvate carboxykinase, glucose-6-phosphatase, and fructose-1,6-bisphosphatase. 61 By triggering FGF15/19, activation of FXR stimulates glycogen synthesis and therefore decreased hepatic glucose output and plasma levels. 53 62 Furthermore, while activation of FXR in the terminal ileum inhibits glucagon-like peptide (GLP-1) secretion through decreased expression of carbohydrate-responsive element-binding protein and impaired glycolysis, BA signaling via TGR5 plays an important role in glucose homeostasis by inducing secretion of GLP-1 from enteroendocrine L-cells in the gastrointestinal tract, reflective of its therapeutic potential in NAFLD and obesity (reviewed in Chávez-Talavera et al 47 ). These potentially opposing effects might explain differential actions of BA-based therapies and FXR ligands in glucose homeostasis and need to be addressed in more mechanistic detail. In addition to its qualities as a controller of enterohepatic circulation, FXR plays a major role in modification of inflammation. Anti-inflammatory mechanisms of FXR can be attributed to stabilization of the nuclear corepressor NCoR leading to repression of nuclear factor kappa B and downstream inflammatory genes such as tumor necrosis factor-α, interleukin 6, inducible nitric oxide synthase, and cyclooxygenase 1. 63 INT767, a dual agonist for both FXR and TGR5, has been shown to improve liver histology and reduce proinflammatory cytokine expression in db/db mice as well as inducing a gene expression shift toward alternatively activated macrophages. 64
In addition to inflammation, FXR has important impact on fibrogenesis as another key step in the progression of NASH. Although FXR is not expressed to significant amounts in hepatic stellate cells (HSCs), pharmacological activation of FXR was able to ameliorate thioacetamide-induced fibrosis and portal hypertension by counteracting inflammation and reconstitution of endothelial nitric oxide synthase activity in sinusoidal endothelial cells. 65 66 67 68 69 Since inflammation drives activation of HSC and therefore hepatic fibrosis, administration of FXR agonist obeticholic acid (OCA) may counteract fibrosis in rodents by blunting expression of proinflammatory cytokines in hepatocytes and Kupffer cells. 67 Furthermore, nonsteroidal FXR agonist PX20606 (precursor of clinically tested compound Cilofexor) ameliorated liver fibrosis and sinusoidal dysfunction in experimental models of noncirrhotic and cirrhotic portal hypertension. 69 Although these findings provide at least in part the mechanistic basis for clinical efficacy of FXR agonists in NASH fibrosis, the antifibrotic effects of OCA in recent clinical trials appear to be less pronounced (discussed below). Apart from obvious reasons such as species differences and variable duration of the fibrogenic insults, another possible explanation may be SUMOylation of FXR. This phenomenon leads to poor responsiveness to FXR agonist OCA, while upon co-treatment with SUMOylation inhibitors, the antifibrotic effect of FXR activation could be recovered via stabilizing of HSC lipid droplets by inducing Perilipin 1. 70
In addition to their pleiotropic effects on the liver, BAs undergoing enterohepatic circulation also affect the gut microbiome and vice versa. As such, BAs through their detergent properties act as antimicrobial agents by disrupting bacterial membrane integrity as well as by inducing genes involved in antibacterial defense through FXR, therefore inhibiting bacterial translocation. 71 72 73 For instance, treatment of bile duct-ligated mice with naturally occurring BAs or OCA led to improved intestinal barrier, intestinal inflammation, and decreased bacterial translocation. 74 75 Accordingly, in murine models of chemically induced colitis, FXR agonistic treatment led to reduced intestinal inflammation and improved epithelial membrane integrity. 76
On the other hand, microbiota metabolize BAs through introducing chemical modifications such as deconjugation and dehydroxylation, therefore altering their signaling properties. As such microbial generation of secondary BA isoDCA through modulation of intestinal FXR leads to increased differentiation of regulatory T cells, therefore underlining the notion of BA as immunomodulatory mediators. 77 Microbial processing of BAs not only involves activation but also inactivation of FXR. 78 79 For instance, in germ-free mice, accumulation of taurine-conjugated muricholic acid (Tα/βMCA) inhibits intestinal FXR and downstream Fgf15 expression. 80 In line, treatment of rodents with Tempol, which is an antioxidant reducing bile salt hydrolase activity, improves diet-induced obesity potentially through accumulation of intestinal TßMCA, similar to what is observed in intestine-specific FXR-deficiency models. 81 Mechanistically, intestinal inhibition of FXR leads to reduced biosynthesis of ceramides which have been linked to insulin resistance and inflammation in NASH. 82 Furthermore, ceramides induce lipogenesis through upregulation of SREBP1c and have therefore been implied as causal factors in NAFLD. 83 A direct comparison of germ-free and conventionally raised WT and FXR-KO mice revealed that diet-induced obesity and the accompanying systemic phenotype are promoted and transferable by gut microbiota through FXR signaling. 84 In line, in a study investigating gut microbiota of twins discordant for obesity, it was shown from transplantation of fecal microbiota to germ-free mice that lean twins had an FXR antagonistic microbial signature, supporting the concept of targeting the gut microbiome and intestinal FXR for treatment of NAFLD. 85
Changes in BA Homeostasis and Microcholestasis in NASH
Across their disease spectrum, patients with NAFLD exhibit alterations in BA homeostasis and their BA metabolome. A range of studies has revealed that serum BA concentrations are increased in NASH patients compared with healthy controls. 24 86 87 88 Higher serum BA concentrations in NASH were observed in both fasting and postprandial states. 89 90 The elevations of BA levels could be largely attributed to increased primary BAs and BA levels correlated with increasing NAFLD activity score (NAS) and fibrosis stage. 87 In line, 7α-hydroxy-4-cholesten-3-one (C4) and CYP7A1 as markers of BA synthesis as well as serum BA levels correlated with disease severity in NASH patients. 88 91
In addition to serum BA levels, also their composition changes in NASH. More specifically, the ratio of conjugated to unconjugated BAs was elevated in NASH patients and an increased proportion of CA compared with CDCA could be detected which further contributes to mitigation of FXR signaling since CA is a weaker ligand than CDCA. 87 92 In line, gas–liquid chromatography of liver tissue from NASH patients revealed an increase in total BAs with CA and the ratio of trihydroxylated to dihydroxylated BAs being strongly correlated to inflammatory activity. 93
However, it is not clear whether these changes can be attributed to underlying metabolic changes (i.e., insulin resistance and obesity) rather than NASH per se. 94 For instance, insulin-resistant patients were shown to have a shift in BA profile toward 12α-hydroxylated BAs, which correlated with plasma triglyceride levels. 95 Moreover, comparison of obese patients either with biopsy-proven NASH or with healthy livers revealed that BA alterations in these patients are associated with concomitant insulin resistance but did not depend on presence of NASH. 94
The exact pathogenetic mechanism underlying these alterations of BA levels and composition in NASH patients still remains unclear and several options can be considered. In line with a mechanistic correlation to obesity and insulin resistance, multiple rodent models of diabetes exhibit altered BA metabolism (reviewed in Watkins and Sanders 96 ). As such, in streptozotocin-treated rats, decreased bile flow despite increased secretion of BAs together with increased BA levels in serum 97 98 99 support the concept that alterations of BA levels in NASH may reflect underlying insulin resistance rather than the associated severity of liver disease. 94
Paradoxically, FXR-FGF19 signaling—normally repressing BA synthesis—is impaired despite elevated BA levels, possibly reflective of underlying “FXR and/or FGF19 resistance” in NASH. Indeed, preclinical NASH models revealed partial resistance to FXR activation, reflected by transcriptional downregulation of SHP with consequent upregulation of CYP7A1. 100 Furthermore, as a reflection of impaired hepatic FXR activation, increased CYP7A1 expression could be observed in NASH patients while controversial results on BSEP and SHP expression have been reported. 86 87 NASH patients exhibit elevated cholesterol levels and increased activity of HMG-CoA reductase (HMGCR) correlating with histologic features of NASH, integrating cholesterol levels to the concept of lipotoxicity. 101 In line, upregulated synthesis of free cholesterol may contribute to increased BA synthesis resulting from dysregulated FXR signaling. 101 In accordance with these findings, free FAs have been reported to repress FXR-induced SHP-signaling, therefore leading to increased BA synthesis and uptake potentially contributing to hepatocellular injury. 88 Similarly, expression of downstream target of intestinal FXR activation FGF19 is reduced in patients with NAFLD and NASH 102 103 and impaired hepatic repression of BA synthesis in insulin-resistant patients was observed. 104 This inverse relationship was correlated with severity of liver histology in NAFLD patients, therefore highlighting the potential of serum FGF19 levels as a biomarker for NAFLD. 105 Hepatic FXR activation can in part be assessed by measuring C4, an intermediate step in BA synthesis from cholesterol. As such, C4 is robustly elevated in NAFLD patients, supporting the notion of increased BA synthesis due to lower FXR activity. 87 106 In line, increased C4 levels have been associated with fibrosis score in NASH. 107
Moreover, changes in BA levels and composition may also reflect intestinal dysbiosis in NASH. Human fecal analyses revealed increased levels of CA, CDCA, and total BA in stool of NASH patients compared with healthy controls. 91 Furthermore, the obesity-induced BA and gut microbial metabolite DCA have been found to induce a senescence-associated secretory phenotype in HSC thereby facilitating development of liver cancer. 108 Changes in blood microbiota profiles as a reflection of dysbiosis and decreased ratios of secondary to primary BAs in serum suggesting an impaired ability to convert primary BAs have been reported to correlate with fibrosis in obese patients. 109
In addition to increased BA synthesis and altered intestinal/microbial BA metabolism, impaired excretion of BAs and other biliary constituents may contribute to the pathogenesis of NAFLD, consistent with the concept of (micro)cholestasis in NASH. Mechanistically, biliary BA excretion can be characterized by determining bile flow and intrahepatic canalicular bile pressure. Biliary pressure constitutes a hydrostatic force which was shown to disrupt bile canaliculi integrity, therefore contributing to hepatocyte necrosis and bile infarcts. 110 In addition, mechanosensing in the bile canaliculi network either directly through pressure sensors localized on cholangiocytic primary cilia or indirectly through biliary hydrostatic pressure opposing the osmotic gradient has been proposed as a mechanism to induce a Ca 2+ and cAMP-dependent signaling response leading to adaptative changes in the ductular network. 30 111 Albeit cholestasis not generally considered a feature of NASH, elevated gamma-glutamyl transferase (GGT) levels—which have thus far been largely attributed to oxidative stress—in concert with elevated BA levels and altered BA composition point to the fact that there may be a cholestatic component in the pathogenesis of NASH. Impaired BA excretion and transport could result in altered signaling as well as accumulation of potentially toxic BAs and other biliary constituents, serving as a potential additional hit to NASH progression. Indeed, three-dimensional spatially resolved imaging of human NASH livers revealed profound topological defects in the bile canaliculi network, consistent with an underlying microcholestatic state (see Fig. 1 ). 112 Computational modeling of bile flow in these patients predicted higher pericentral biliary pressure which was correlated with GGT levels as a typical serum biochemical finding in NAFLD/NASH, underscoring the concept of microcholestasis. 112 Hepatocyte morphometry revealed increased number and size of lipid droplets pericentrally correlating with disease progression and potentially contributing to apical protein trafficking disruption. 112 More specifically, dipeptidyl peptidase 4 which follows a transcytotic route for apical expression was mislocalized while multidrug resistance-associated protein (MRP2) and BSEP localization remained intact. Contrasting this finding and in line with preclinical studies in rodent models, 113 114 altered protein trafficking and mislocalization of MRP2 and MRP3 have been found in NASH patients, further substantiating the concept of microcholestasis in NASH. 115 116 Under cholestatic conditions, hepatocellular transport systems undergo adaptive changes mainly through FXR activation, resulting in downregulation of the BA uptake transporter NTCP, whereas compensatory basolateral efflux increases through OSTα/β, MRP3, and MRP4 bypassing impaired canalicular excretion. 117 118 Zucker rats, a rodent model of NAFLD, show impaired hepatobiliary transport compatible with an underlying cholestatic component. 113 More precisely, decreased BA-independent bile flow coinciding with downregulation of hepatic MRP2 and OATP2 has been reported while BSEP and NTCP expression remained unchanged, reflecting an impaired hepatobiliary transport capacity. 113 114 Interestingly, mice lacking the canalicular BA export pump BSEP display impaired mitochondrial FA oxidation as well as reduced white adipose tissue mass. 119 In line, challenging these mice with dietary models of hepatic steatosis leads to attenuation of hepatic lipid accumulation despite worsened inflammation, potentially through impaired FXR and PPARα signaling. 120 121 Additionally, genetic polymorphisms of ABCB11, the gene encoding for human BSEP, have been correlated to higher triglyceride and cholesterol levels as well as body mass index. 122 123 However, the exact mechanism of microcholestasis as well as addressing the topological and molecular alterations of the hepatobiliary network in NASH is still poorly understood and requires further interrogation. Even so, these findings support the concept to apply empirically used or novel anticholestatic therapies such as ursodeoxycholic acid (UDCA) and norUDCA or FXR agonists, respectively, in NASH (see the following section).
Targeting FXR in Microcholestasis of NASH and Beyond
Due to the key role of FXR in regulating not only BA homeostasis, but also glucose and lipid metabolism, as well as inflammation and fibrosis, pharmacological targeting of FXR has become a promising strategy for treating NAFLD/NASH. 24 25 124 125 In line with the key role of FXR in maintaining biliary homeostasis, FXR ligands have first been developed as therapy for cholestatic liver diseases. 27 Since BAs are increasingly conceived as important biomarkers of NAFLD and microcholestasis has emerged as a feature of NASH, modulating FXR activity as a key regulator of BA homeostasis may be a key asset or at least important added benefit for treating NASH (see Fig. 2 ).
In multiple preclinical models, FXR agonism led to resolution of features of steatohepatitis through its metabolic and anti-inflammatory effects. 53 Steroidal FXR agonists such as OCA but also natural ligands such as CA and CDCA have been shown to improve insulin resistance and steatosis in obese rats 59 as well as ameliorate inflammation and fibrosis in mouse models of NASH. 126 As another example, administration of nonsteroidal FXR agonist WAY-362450 ameliorated steatohepatitis and fibrosis in mice fed a methionine-choline-deficient diet in an FXR-dependent fashion. 127 Additionally, FXR agonism reduces vascular inflammation and plaque formation in rodent models of atherosclerosis, highlighting its therapeutic potential on CVD associated with NAFLD/NASH. 128 129 130 131 Moreover, administration of OCA in rodent models of CKD led to attenuation of renal injury and counteracted diabetic nephropathy. 132 133 Taken together, FXR agonism may not only be a promising target for treatment of NASH, but also for associated cardiometabolic comorbidity, warranting further investigations.
Several clinical trials currently investigate the effects of FXR ligands in NAFLD/NASH and most progress has so far been made for OCA as first in class steroidal FXR agonist. 134 In a small proof-of-concept phase II study testing OCA in diabetic patients with NAFLD, administration of either 25 or 50 mg OCA once daily over 6 weeks improved insulin sensitivity assessed by hyperinsulinemic-euglycemic clamp as well as liver enzymes (GGT, alanine aminotransferase [ALAT], aspartate aminotransferase [ASAT]), findings which were associated with a modest reduction in noninvasive fibrosis parameters (Enhanced Liver Fibrosis [ELF] score) in the 25-mg OCA group compared with placebo. 135 Additionally, OCA treatment increased FGF19 levels in a dose-dependent manner as well as decreasing C4 levels and levels of total endogenous BAs, reflecting target engagement and giving mechanistic insight into the regulation of insulin resistance in NASH patients. 135 A subsequent large multicenter phase IIb study (FLINT—FXR Ligand Obeticholic Acid in NASH Treatment) enrolling 283 patients revealed effectiveness of 25 mg OCA treatment in NASH patients compared with placebo after 72 weeks. OCA treatment resulted in a significant improvement in the primary histological endpoint, defined as a decrease in NAS of at least two points with no worsening of fibrosis, as compared with placebo, while no significant impact on overall resolution of NASH was observed. 136 Again, levels of ALAT, ASAT, and GGT significantly improved upon OCA treatment while serum levels of alkaline phosphatase (known to be upregulated by FXR) increased. However, OCA treatment led to an unfavorable lipid profile as reflected by increased levels of serum LDL and decreased levels of serum high-density lipoprotein (HDL). In addition, development of moderate to severe pruritus was more common in the OCA-treated group compared with placebo. Surprisingly, measurement of hepatic insulin resistance using the homeostasis model of assessment (HOMA-IR) showed an increase in insulin resistance despite weight loss during OCA treatment, both of which reverted back after treatment discontinuation. This effect on insulin resistance contrasting the findings from the previous proof-of-concept phase II study in diabetics 135 might be in part attributed to different methodology in assessing insulin resistance as well as decentralized insulin measurements.
Furthermore, in a recent interim analysis of the subsequent multicenter phase III study (REGENERATE—Randomized Global Phase 3 Study to Evaluate the Impact on NASH With Fibrosis of Obeticholic Acid Treatment), statistically significant improvement of fibrosis was seen in 12% in patients receiving placebo compared with 18% in patients receiving 10 mg OCA and 23% in patients receiving 25 mg OCA, while the endpoint for resolution of NASH was not met. 137 Side effects of OCA treatment again were increased LDL cholesterol levels at the expense of HDL cholesterol and mild to moderate pruritus in a dose-dependent manner. With reduction of NASH activity and fibrosis on one hand and features of dyslipidemia on the other, the overall clinical benefit of OCA treatment in terms of cardiometabolic comorbidity or all-time mortality is the subject of currently ongoing studies.
With the initial intention to avoid adverse effects such as LDL increase or occurrence of pruritus, various nonsteroidal synthetic FXR agonists have been developed and several compounds are currently tested in phase II clinical trials. 138 Administration of Cilofexor (formerly known as GS-9674, follow-up compound to Px-102/Px-104) in noncirrhotic NASH patients revealed reduction of hepatic steatosis as measured by magnetic resonance imaging (MRI-PDFF) and decreased levels of GGT, C4, and primary BA, while no changes were observed for noninvasive parameters of liver fibrosis using ELF, transient elastography, and magnetic resonance elastography. While the serum lipid profile did not change significantly upon Cilofexor treatment, dose-dependent occurrence of pruritus was observed. 139
In another trial, Cilofexor, ACC inhibitor Firsocostat, and apoptosis-signal regulating kinase 1 inhibitor Selonsertib have been tested alone or in a dual-drug combination strategy in a phase IIb trial in NASH patients for 48 weeks. 140 None of the treatment arms achieved significant changes compared with placebo for the primary endpoint (≥ 1-stage improvement in fibrosis without worsening of NASH). However, significant improvements in biochemical and noninvasive fibrosis parameters, ≥ 2-point NAS reductions and its subcomponents, as well as a significant shift of advanced (F3/F4) to nonadvanced fibrosis stages using a machine learning-based algorithm, were observed with the combination of Cilofexor and Firsocostat compared with placebo. Again, occurrence of pruritus has been reported more often in Cilofexor-treated patients.
Another non-BA FXR agonist called Tropifexor (LJN452) has been successfully tested in multiple preclinical NASH models. 141 142 In healthy volunteers, administration of Tropifexor was well tolerated and did not lead to significant alterations in the plasma lipid profile encouraging further studies. 143 Recent interim results from a currently undergoing phase II clinical trial in NASH patients (FLIGHT-FXR: NCT02855164) revealed robust and dose-dependent reduction of ALAT, GGT, hepatic fat fraction, and body weight in patients receiving either 140 or 200 µg Tropifexor over 12 weeks. 144 Despite a significant reduction of collagen proportional area in the 200-µg-treated group, neither of the Tropifexor groups showed significant difference in terms of histological improvement of NASH or fibrosis stage as a predefined endpoint. 144 Regarding adverse events, a dose-related increase in LDL cholesterol and an association with mild pruritus were reported. Notable changes in LDL cholesterol levels appear to generally increase in therapies that reduce hepatocellular BA levels through accumulation of total cholesterol which impairs SREBP2 signaling, leading to downregulation of LDL receptor (LDLR) expression and therefore, increased serum LDL cholesterol. 145 Collectively, these data also raise the question whether pruritus stems from FXR agonism directly rather than from elevated levels of BAs or BA derivatives and drives the necessity for comprehensive therapeutic strategies to manage adverse effects in FXR agonistic treatment including pruritus. 146 In addition to Cilofexor and Tropifexor, multiple other nonsteroidal FXR agonists are currently under investigation for treatment of NAFLD/NASH (reviewed in Gege et al 138 ).
As another strategy in the treatment of NASH, recent studies have turned toward FGF19 analogues. Being a downstream target of intestinal FXR activation, FGF19 has been proposed to reduce hepatic lipid content while potentially sidestepping adverse effects of direct FXR agonism. Notably engineered FGF19 analogues show distinct dissociation of metabolic from proliferative potentially procarcinogenic effects by blunting the signal transducer and activator of transcription 3-activating function of the naturally occurring protein. 147 In line, preclinical models testing a synthetic and nontumorigenic FGF 19 analogue, Aldafermin/NGM282 (formerly known as M70), were even able to prevent tumor formation. 148 Additionally, Aldafermin/NGM282 treatment in humans led to a dose-dependent enrichment of BA-sensitive commensal microbe Veillonella, which correlated to physical performance-enhancement through its ability to degrade exercise-induced lactate to propionate. 149 150
An interim analysis investigating 1 or 3 mg Aldafermin/NGM282 treatment in NASH patients revealed a significant histological response (defined as > 2 point improvement of NAS without worsening of fibrosis or > 1 point improvement in fibrosis without worsening of NASH) in 50 and 68% after 12 weeks treatment, respectively. 151 Although Aldafermin/NGM282 treatment for 24 weeks also showed a robust decrease in liver fat content (measured by MRI-PDFF) as well as serum transaminases and BA and C4 levels, no significant impact on histological assessment of liver fibrosis or NASH resolution could be observed at this extended time point. 152 Interestingly, Aldafermin/NGM282 treatment over 12 weeks also improved noninvasive markers of fibrosis significantly in patients with primary sclerosing cholangitis independent of biochemical improvement of cholestasis, suggesting potential direct antifibrotic effects. 153 Adverse events included diarrhea, nausea, and abdominal pain as well as increased LDL-cholesterol levels which, however, could be effectively and safely counteracted by rosuvastatin administration. 151 152 154 155
Apart from the above-mentioned beneficial effects of systemic treatment to induce hepatic FXR signaling, gut-selective modulation of FXR activity has been proposed as a mechanism to target obesity and development of NAFLD. Intestinal FXR agonism counteracted diet-induced obesity, insulin resistance, and inflammation in mice through induction of FGF15 without altering hepatic FXR signaling. 156 Contrasting this finding, intestinal knockout of FXR in high-fat diet (HFD)-fed mice led to ameliorated hepatic steatosis due to reduced circulation of ceramides. 83 Moreover, dietary modulation of the gut microbiome through accumulation of TßMCA resulting in intestinal FXR antagonism led to decreased ceramide levels and therefore, reduced hepatic gluconeogenesis and insulin resistance. 157 Modulation of gut microbiota in rodents by antibiotics changes intestinal BA composition toward an intestine-specific inhibition of FXR signaling also through accumulating of TßMCA normally degraded by intact gut microbiota and leads to lower levels of circulating ceramides, therefore decreasing DNL and ameliorating hepatic steatosis. 83 Interestingly, UDCA may be seen as a weak FXR antagonist. 78 Indeed, in morbidly obese NAFLD patients, administration of UDCA exerted FXR-antagonistic effects as seen through upregulation of CYP7A1 impacting on BA synthesis as well as increasing SREBP2 and consequently LDLR reflecting cholesterol homeostasis in addition to promoting neutral lipid accumulation in adipose tissue and liver. 158 In summary, opposing effects of FXR modulation in an organ/tissue-specific manner reveal novel therapeutic avenues to treat NAFLD, warranting further studies to resolve these apparent discrepant findings between the effects of FXR agonism versus antagonism.
In addition to the aforementioned effects, UDCA treatment in multiple rodent models of NASH led to resolution of liver steatosis and inflammation. 159 160 Furthermore, TUDCA has been proposed as a chemical chaperone to increase the adaptive capacity of the endoplasmic reticulum (ER), therefore reducing ER stress and leading to normalized insulin sensitivity and resolution of NAFLD in obese and diabetic mice. 161 However, clinical trials elucidating the effect of UDCA in NASH have been inconsistent regarding patient databases, primary endpoints, and therapeutic benefits. In particular, while reduction of transaminases and hepatic steatosis were observed in an initial pilot study, 162 following randomized controlled trials could not show histological benefits or an improvement of liver steatosis or ALAT compared with placebo. 163 164 165 In a subsequent high-dose treatment strategy, improvement of liver enzymes and HOMA-IR could be observed, but liver histology was not assessed. 166 Furthermore, a trial investigating a combination strategy of UDCA with vitamin E revealed improved transaminases and histological regression of liver steatosis in the combination arm while no histologic changes could be seen in UDCA alone. 165 Moreover, treatment of obese patients with TUDCA led to improved insulin sensitivity in liver and muscle, whereas adipose tissue insulin sensitivity and cellular markers of ER stress were unchanged compared with placebo. 167 In summary, based on this evidence UDCA is no longer recommended for treatment of NASH by current guidelines. 1 2
Beyond modulation of FXR signaling, TGR5 is a widely expressed G-protein coupled receptor which upon activation through BAs has been shown to increase glucose homeostasis efficiency and exert potent anti-inflammatory effects in preclinical studies (reviewed in Pols et al 168 ). Activating TGR5 through administration of BAs increases energy expenditure in brown adipose tissue through promoting thyroid hormone metabolism and therefore counteracts diet-induced obesity and insulin resistance. 169 Accordingly, both TGR5-selective agonists such as INT-777 and TGR5/FXR dual agonists such as INT-767 have been reported to counteract the development of obesity, steatosis, and inflammation in multiple experimental models of NASH. 64 170 171 However, clinical trials investigating TGR5 agonists are scarce. To date, there has been only one human study of TGR5 agonist administration in type 2 diabetic patients yielding negative results with even elevation instead of reduction of blood glucose levels. 172 Moreover, TGR5 agonistic treatment approaches are associated with safety concerns. Such being the case, multiple in vitro studies proposed that TGR5 signaling is involved in carcinogenesis. 173 174 Additionally, TGR5 agonism is important in gallbladder physiology through mediating gallbladder distension 175 with preclinical models of TGR5 deficiency exhibiting resistance to gallstone formation. 176 To address these concerns, further mechanistic studies are warranted.
Other possibilities to manipulate lipid metabolism through modulating BA homeostasis include prevention of biliary reuptake from the intestine. The initial intention of using BA sequestrants such as Colesevelam for hypercholesterolemia was to compensate for the loss of BAs through conversion of cholesterol, therefore lowering total and LDL cholesterol levels in patients. At the same time, Colesevelam treatment results in increased HDL cholesterol and triglyceride levels, reflecting partial deactivation of FXR by interrupting the enterohepatic cycle. 177 Furthermore, administering BA sequestrants to mice in dietary models of steatohepatitis led to reduction of hepatic steatosis, inflammation, and fibrosis. 25 178 179 However, clinical studies did not show improvement of NASH by Colesevelam. 180
As an alternative to BA sequestrants, pharmacological inhibition of ASBT to prevent intestinal BA reuptake is being increasingly tested in models of NASH. 24 In contrast to resin-bound BAs which activate TGR5 therefore increasing GLP-1 levels, free intestinal BAs increased by ASBT inhibition activate FXR signaling, which in turn downregulates GLP-1 secretion. 47 181 Interestingly, intestinal ASBT inhibition led to increased fecal BA excretion and a shift in BA composition toward more FXR agonistic BAs in line with normalization of glucose tolerance and improvement of hepatic steatosis and NAS in HFD-fed mice. 182 However, a recent 24-week interim analysis of a phase II trial involving ASBT inhibitor Volixibat showed that despite target affection seen by dose-dependent increase of C4 serum levels and decrease of cholesterol levels, no liver-specific therapeutic benefit could be noted. 183 Currently, a wide range of ASBT inhibitors are explored for pediatric cholestasis, but their potential benefit for NASH remains to be determined (reviewed in Karpen et al 184 ).
Finally, norUDCA, recently renamed as norucholic acid, is a side chain shortened, conjugation-resistant UDCA derivative that undergoes cholehepatic shunting therefore allowing for liver-targeted therapy in multiple liver diseases. 185 norUDCA has been shown to elicit anti-inflammatory, antiapoptotic, and antifibrotic effects in multiple experimental NASH models. 186 187 Cholehepatic shunting is particularly beneficial in targeting ductular reaction, which is a histopathological feature typically seen in cholestatic liver injury but also in NAFLD. 188 Ductular reaction in NASH has been associated with fibrosis development as well as the extent of hepatocytes in replicative arrest, therefore reflecting hepatocyte senescence. 189 Additionally, cholangiocytes have been shown to be susceptible to lipoapoptosis primarily through saturated free FAs, suggesting their engagement in the pathogenesis of NAFLD. 190 Recently, a multicenter phase IIa trial investigated the effect of norUDCA in NASH revealing a dose-dependent reduction of serum ALT 191 inciting a currently ongoing phase IIb study whose primary endpoint will involve histological improvement or resolution of NASH (EudraCT-Nr.:2018–003443–31). Due to the underappreciated role of microcholestasis and ductular changes in NASH, future combination therapies may need to focus on anticholestatic agents in NASH.
Conclusion and Outlook
Beyond their traditional role as detergents facilitating lipid uptake, BAs have recently emerged as important signaling molecules of controlling cellular metabolism throughout the enterohepatic system by FXR-dependent and FXR-independent pathways. The effect of FXR signaling on hepatic BA, lipid, and glucose metabolism in addition to its immunomodulatory properties holds promise for impeding development or progression of NASH. Moreover, recent findings have established the concept of microcholestasis as a key feature of NAFLD, further underlining the therapeutic potential of FXR modulation. While most preclinical and experimental NASH models have shown markedly positive results, clinical data on FXR-related and other BA-targeted treatment strategies remain promising, but rather modest. Since the mechanism of action is rather novel, particular attention has to be directed to occurrence of side effects such as dyslipidemia and pruritus and long-term safety observations. Future studies will have to show whether combination strategies with other metabolic, anti-inflammatory, and/or antifibrotic strategies result in synergistic therapeutic benefit and future studies are warranted to investigate the effect of targeting BA-related and anticholestatic pathways in NAFLD by FXR and beyond.
Main Concepts and Learning Points
In addition to their detergent actions, bile acids (BAs) have important signaling function via the nuclear receptor FXR, which regulates their own homeostasis as well as lipid/glucose metabolism and inflammation.
Alterations of BA homeostasis are a key pathogenetic and prognostic factor in nonalcoholic fatty liver disease (NAFLD) and its progressive variant with steatohepatitis (NASH).
Microcholestasis is an underappreciated feature of the pathophysiology of NAFLD/NASH, which can be targeted by FXR and other BA-based therapies.
Results from preclinical and clinical studies indicate that targeting FXR and other BA-related pathways is a promising treatment strategy for NASH.
Footnotes
Conflicts of Interest Michael Trauner reports personal fees from Albireo; grants, personal fees, and other from Gilead; grants, personal fees, and other from Falk Foundation; personal fees from Novartis; grants, personal fees, and other from Intercept; grants, personal fees, and other from MSD; personal fees from Phenex; grants from Takeda; personal fees from BMS; personal fees from Boehringer Ingelheim; personal fees from BioMX; personal fees from Genfit; personal fees from Regulus; other from Abbvie; grants from Cymabay; personal fees from Shire; personal fees from Pliant outside the submitted work. In addition, he has a patent Medical use of nor-UDCA (WO2006119803 and WO20099013334 licensed to Medical University of Graz.
References
- 1.European Association for the Study of the Liver (EASL) ; European Association for the Study of Diabetes (EASD) ; European Association for the Study of Obesity (EASO) . EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64(06):1388–1402. doi: 10.1016/j.jhep.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Chalasani N, Younossi Z, Lavine J E. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67(01):328–357. doi: 10.1002/hep.29367. [DOI] [PubMed] [Google Scholar]
- 3.Wong V W-S, Chan W-K, Chitturi S. Asia-Pacific Working Party on Non-alcoholic Fatty Liver Disease guidelines 2017-part 1: definition, risk factors and assessment. J Gastroenterol Hepatol. 2018;33(01):70–85. doi: 10.1111/jgh.13857. [DOI] [PubMed] [Google Scholar]
- 4.Younossi Z M. Non-alcoholic fatty liver disease - a global public health perspective. J Hepatol. 2019;70(03):531–544. doi: 10.1016/j.jhep.2018.10.033. [DOI] [PubMed] [Google Scholar]
- 5.Younossi Z, Tacke F, Arrese M. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2019;69(06):2672–2682. doi: 10.1002/hep.30251. [DOI] [PubMed] [Google Scholar]
- 6.Brunt E M, Wong V W-S, Nobili V. Nonalcoholic fatty liver disease. Nat Rev Dis Primers. 2015;1:15080. doi: 10.1038/nrdp.2015.80. [DOI] [PubMed] [Google Scholar]
- 7.Ekstedt M, Nasr P, Kechagias S. Natural history of NAFLD/NASH. Curr Hepatol Rep. 2017;16(04):391–397. doi: 10.1007/s11901-017-0378-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Satapathy S K, Sanyal A J. Epidemiology and natural history of nonalcoholic fatty liver disease. Semin Liver Dis. 2015;35(03):221–235. doi: 10.1055/s-0035-1562943. [DOI] [PubMed] [Google Scholar]
- 9.Vanni E, Marengo A, Mezzabotta L, Bugianesi E. Systemic complications of nonalcoholic fatty liver disease: when the liver is not an innocent bystander. Semin Liver Dis. 2015;35(03):236–249. doi: 10.1055/s-0035-1562944. [DOI] [PubMed] [Google Scholar]
- 10.Eslam M, Newsome P N, Sarin S K. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73(01):202–209. doi: 10.1016/j.jhep.2020.03.039. [DOI] [PubMed] [Google Scholar]
- 11.International Consensus Panel . Eslam M, Sanyal A J, George J. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158(07):19990–20140. doi: 10.1053/j.gastro.2019.11.312. [DOI] [PubMed] [Google Scholar]
- 12.Adams L A, Anstee Q M, Tilg H, Targher G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut. 2017;66(06):1138–1153. doi: 10.1136/gutjnl-2017-313884. [DOI] [PubMed] [Google Scholar]
- 13.Mantovani A, Petracca G, Beatrice G, Tilg H, Byrne C D, Targher G. Non-alcoholic fatty liver disease and risk of incident diabetes mellitus: an updated meta-analysis of 501 022 adult individuals. Gut. 2021;70(05):962–969. doi: 10.1136/gutjnl-2020-322572. [DOI] [PubMed] [Google Scholar]
- 14.NASH CRN Research Group . David K, Kowdley K V, Unalp A, Kanwal F, Brunt E M, Schwimmer J B. Quality of life in adults with nonalcoholic fatty liver disease: baseline data from the nonalcoholic steatohepatitis clinical research network. Hepatology. 2009;49(06):1904–1912. doi: 10.1002/hep.22868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newton J L, Jones D EJ, Henderson E. Fatigue in non-alcoholic fatty liver disease (NAFLD) is significant and associates with inactivity and excessive daytime sleepiness but not with liver disease severity or insulin resistance. Gut. 2008;57(06):807–813. doi: 10.1136/gut.2007.139303. [DOI] [PubMed] [Google Scholar]
- 16.Younossi Z M, Wong V W-S, Anstee Q M. Fatigue and pruritus in patients with advanced fibrosis due to nonalcoholic steatohepatitis: the impact on patient-reported outcomes. Hepatol Commun. 2020;4(11):1637–1650. doi: 10.1002/hep4.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong R J, Aguilar M, Cheung R. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology. 2015;148(03):547–555. doi: 10.1053/j.gastro.2014.11.039. [DOI] [PubMed] [Google Scholar]
- 18.Burra P, Becchetti C, Germani G. NAFLD and liver transplantation: disease burden, current management and future challenges. JHEP Rep. 2020;2(06):100192. doi: 10.1016/j.jhepr.2020.100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldberg D, Ditah I C, Saeian K. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology. 2017;152(05):10900–10990. doi: 10.1053/j.gastro.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Younossi Z M, Stepanova M, Ong J. Nonalcoholic steatohepatitis is the most rapidly increasing indication for liver transplantation in the United States. Clin Gastroenterol Hepatol. 2021;19(03):5.8E7–5.89E7. doi: 10.1016/j.cgh.2020.05.064. [DOI] [PubMed] [Google Scholar]
- 21.Holmer M, Melum E, Isoniemi H. Nonalcoholic fatty liver disease is an increasing indication for liver transplantation in the Nordic countries. Liver Int. 2018;38(11):2082–2090. doi: 10.1111/liv.13751. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Jadhav K, Zhang Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochem Pharmacol. 2013;86(11):1517–1524. doi: 10.1016/j.bcp.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan L, Bambha K. Bile acid receptors and nonalcoholic fatty liver disease. World J Hepatol. 2015;7(28):2811–2818. doi: 10.4254/wjh.v7.i28.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arab J P, Karpen S J, Dawson P A, Arrese M, Trauner M. Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology. 2017;65(01):350–362. doi: 10.1002/hep.28709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaap F G, Trauner M, Jansen P LM. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11(01):55–67. doi: 10.1038/nrgastro.2013.151. [DOI] [PubMed] [Google Scholar]
- 26.Arab J P, Arrese M, Trauner M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu Rev Pathol. 2018;13:321–350. doi: 10.1146/annurev-pathol-020117-043617. [DOI] [PubMed] [Google Scholar]
- 27.Trauner M, Fuchs C D, Halilbasic E, Paumgartner G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology. 2017;65(04):1393–1404. doi: 10.1002/hep.28991. [DOI] [PubMed] [Google Scholar]
- 28.Hofmann A F. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159(22):2647–2658. doi: 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- 29.Hofmann A F. The enterohepatic circulation of bile acids in mammals: form and functions. Front Biosci. 2009;14:2584–2598. doi: 10.2741/3399. [DOI] [PubMed] [Google Scholar]
- 30.Jansen P LM, Ghallab A, Vartak N. The ascending pathophysiology of cholestatic liver disease. Hepatology. 2017;65(02):722–738. doi: 10.1002/hep.28965. [DOI] [PubMed] [Google Scholar]
- 31.Allen K, Jaeschke H, Copple B L. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178(01):175–186. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66(06):1300–1312. doi: 10.1016/j.jhep.2017.02.026. [DOI] [PubMed] [Google Scholar]
- 33.Li T, Chiang J YL. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66(04):948–983. doi: 10.1124/pr.113.008201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiang J YL. Bile acid metabolism and signaling. Compr Physiol. 2013;3(03):1191–1212. doi: 10.1002/cphy.c120023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu C, Fuchs C D, Halilbasic E, Trauner M.Bile acids in regulation of inflammation and immunity: friend or foe? Clin Exp Rheumatol 201634(04, Suppl 98):25–31. [PubMed] [Google Scholar]
- 36.Makishima M, Okamoto A Y, Repa J J.Identification of a nuclear receptor for bile acids Science 1999284(5418):1362–1365. [DOI] [PubMed] [Google Scholar]
- 37.Parks D J, Blanchard S G, Bledsoe R K.Bile acids: natural ligands for an orphan nuclear receptor Science 1999284(5418):1365–1368. [DOI] [PubMed] [Google Scholar]
- 38.Wang H, Chen J, Hollister K, Sowers L C, Forman B M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3(05):543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 39.Studer E, Zhou X, Zhao R. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology. 2012;55(01):267–276. doi: 10.1002/hep.24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodwin B, Jones S A, Price R R. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6(03):517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 41.Huang L, Zhao A, Lew J-L. Farnesoid X receptor activates transcription of the phospholipid pump MDR3. J Biol Chem. 2003;278(51):51085–51090. doi: 10.1074/jbc.M308321200. [DOI] [PubMed] [Google Scholar]
- 42.Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf D J, Suchy F J. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276(31):28857–28865. doi: 10.1074/jbc.M011610200. [DOI] [PubMed] [Google Scholar]
- 43.Denson L A, Sturm E, Echevarria W. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology. 2001;121(01):140–147. doi: 10.1053/gast.2001.25503. [DOI] [PubMed] [Google Scholar]
- 44.Lu T T, Makishima M, Repa J J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6(03):507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 45.Neimark E, Chen F, Li X, Shneider B L. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology. 2004;40(01):149–156. doi: 10.1002/hep.20295. [DOI] [PubMed] [Google Scholar]
- 46.Song K-H, Li T, Owsley E, Strom S, Chiang J YL. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology. 2009;49(01):297–305. doi: 10.1002/hep.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chávez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. 2017;152(07):1.679E6–1.694E6. doi: 10.1053/j.gastro.2017.01.055. [DOI] [PubMed] [Google Scholar]
- 48.Sinal C J, Tohkin M, Miyata M, Ward J M, Lambert G, Gonzalez F J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(06):731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Lee F Y, Barrera G. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A. 2006;103(04):1006–1011. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watanabe M, Houten S M, Wang L. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113(10):1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuchs C D, Traussnigg S A, Trauner M. Nuclear receptor modulation for the treatment of nonalcoholic fatty liver disease. Semin Liver Dis. 2016;36(01):69–86. doi: 10.1055/s-0036-1571296. [DOI] [PubMed] [Google Scholar]
- 52.Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart J-C, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. 2003;17(02):259–272. doi: 10.1210/me.2002-0120. [DOI] [PubMed] [Google Scholar]
- 53.Potthoff M J, Boney-Montoya J, Choi M. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab. 2011;13(06):729–738. doi: 10.1016/j.cmet.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carulli N, Ponz de Leon M, Podda M. Chenodeoxycholic acid and ursodeoxycholic acid effects in endogenous hypertriglyceridemias. A controlled double-blind trial. J Clin Pharmacol. 1981;21(10):436–442. doi: 10.1002/j.1552-4604.1981.tb01746.x. [DOI] [PubMed] [Google Scholar]
- 55.Hashim S A, Vanitallie T B. Cholestyramine resin therapy for hypercholesteremia: clinical and metabolic studies. JAMA. 1965;192:289–293. doi: 10.1001/jama.1965.03080170017004. [DOI] [PubMed] [Google Scholar]
- 56.Garg A, Grundy S M. Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann Intern Med. 1994;121(06):416–422. doi: 10.7326/0003-4819-121-6-199409150-00004. [DOI] [PubMed] [Google Scholar]
- 57.Crouse J R., III Hypertriglyceridemia: a contraindication to the use of bile acid binding resins. Am J Med. 1987;83(02):243–248. doi: 10.1016/0002-9343(87)90692-9. [DOI] [PubMed] [Google Scholar]
- 58.Ma K, Saha P K, Chan L, Moore D D. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116(04):1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2010;51(04):771–784. doi: 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mazuy C, Helleboid A, Staels B, Lefebvre P. Nuclear bile acid signaling through the farnesoid X receptor. Cell Mol Life Sci. 2015;72(09):1631–1650. doi: 10.1007/s00018-014-1805-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Staels B, Handelsman Y, Fonseca V. Bile acid sequestrants for lipid and glucose control. Curr Diab Rep. 2010;10(01):70–77. doi: 10.1007/s11892-009-0087-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kir S, Beddow S A, Samuel V T.FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis Science 2011331(6024):1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y-D, Chen W-D, Wang M, Yu D, Forman B M, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008;48(05):1632–1643. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McMahan R H, Wang X X, Cheng L L. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem. 2013;288(17):11761–11770. doi: 10.1074/jbc.M112.446575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fickert P, Fuchsbichler A, Moustafa T. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol. 2009;175(06):2392–2405. doi: 10.2353/ajpath.2009.090114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Verbeke L, Farre R, Trebicka J. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology. 2014;59(06):2286–2298. doi: 10.1002/hep.26939. [DOI] [PubMed] [Google Scholar]
- 67.Verbeke L, Mannaerts I, Schierwagen R. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep. 2016;6:33453. doi: 10.1038/srep33453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Laleman W, Van Landeghem L, Van der Elst I, Zeegers M, Fevery J, Nevens F. Nitroflurbiprofen, a nitric oxide-releasing cyclooxygenase inhibitor, improves cirrhotic portal hypertension in rats. Gastroenterology. 2007;132(02):709–719. doi: 10.1053/j.gastro.2006.12.041. [DOI] [PubMed] [Google Scholar]
- 69.Schwabl P, Hambruch E, Seeland B A. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol. 2017;66(04):724–733. doi: 10.1016/j.jhep.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 70.Zhou J, Cui S, He Q. SUMOylation inhibitors synergize with FXR agonists in combating liver fibrosis. Nat Commun. 2020;11(01):240. doi: 10.1038/s41467-019-14138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Begley M, Gahan C GM, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29(04):625–651. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 72.Ridlon J M, Kang D J, Hylemon P B, Bajaj J S. Bile acids and the gut microbiome. Curr Opin Gastroenterol. 2014;30(03):332–338. doi: 10.1097/MOG.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Inagaki T, Moschetta A, Lee Y-K. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103(10):3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lorenzo-Zúñiga V, Bartolí R, Planas R. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology. 2003;37(03):551–557. doi: 10.1053/jhep.2003.50116. [DOI] [PubMed] [Google Scholar]
- 75.Verbeke L, Farre R, Verbinnen B. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol. 2015;185(02):409–419. doi: 10.1016/j.ajpath.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 76.Gadaleta R M, van Erpecum K J, Oldenburg B. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60(04):463–472. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 77.Campbell C, McKenney P T, Konstantinovsky D.Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells Nature 2020581(7809):475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gonzalez F J, Jiang C, Patterson A D. An intestinal microbiota-farnesoid X receptor axis modulates metabolic disease. Gastroenterology. 2016;151(05):845–859. doi: 10.1053/j.gastro.2016.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jia W, Xie G, Jia W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol. 2018;15(02):111–128. doi: 10.1038/nrgastro.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sayin S I, Wahlström A, Felin J. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17(02):225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 81.Li F, Jiang C, Krausz K W. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun. 2013;4:2384. doi: 10.1038/ncomms3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pagadala M, Kasumov T, McCullough A J, Zein N N, Kirwan J P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol Metab. 2012;23(08):365–371. doi: 10.1016/j.tem.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jiang C, Xie C, Li F. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest. 2015;125(01):386–402. doi: 10.1172/JCI76738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Parséus A, Sommer N, Sommer F. Microbiota-induced obesity requires farnesoid X receptor. Gut. 2017;66(03):429–437. doi: 10.1136/gutjnl-2015-310283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ridaura V K, Faith J J, Rey F E.Gut microbiota from twins discordant for obesity modulate metabolism in mice Science 2013341(6150):1.241214E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jiao N, Baker S S, Chapa-Rodriguez A. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut. 2018;67(10):1881–1891. doi: 10.1136/gutjnl-2017-314307. [DOI] [PubMed] [Google Scholar]
- 87.Puri P, Daita K, Joyce A. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology. 2018;67(02):534–548. doi: 10.1002/hep.29359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bechmann L P, Kocabayoglu P, Sowa J-P. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology. 2013;57(04):1394–1406. doi: 10.1002/hep.26225. [DOI] [PubMed] [Google Scholar]
- 89.Ferslew B C, Xie G, Johnston C K. Altered bile acid metabolome in patients with nonalcoholic steatohepatitis. Dig Dis Sci. 2015;60(11):3318–3328. doi: 10.1007/s10620-015-3776-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dasarathy S, Yang Y, McCullough A J, Marczewski S, Bennett C, Kalhan S C. Elevated hepatic fatty acid oxidation, high plasma fibroblast growth factor 21, and fasting bile acids in nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol. 2011;23(05):382–388. doi: 10.1097/MEG.0b013e328345c8c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mouzaki M, Wang A Y, Bandsma R. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS One. 2016;11(05):e0151829. doi: 10.1371/journal.pone.0151829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kalhan S C, Guo L, Edmison J. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism. 2011;60(03):404–413. doi: 10.1016/j.metabol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aranha M M, Cortez-Pinto H, Costa A. Bile acid levels are increased in the liver of patients with steatohepatitis. Eur J Gastroenterol Hepatol. 2008;20(06):519–525. doi: 10.1097/MEG.0b013e3282f4710a. [DOI] [PubMed] [Google Scholar]
- 94.Legry V, Francque S, Haas J T. Bile acid alterations are associated with insulin resistance, but not with NASH, in obese subjects. J Clin Endocrinol Metab. 2017;102(10):3783–3794. doi: 10.1210/jc.2017-01397. [DOI] [PubMed] [Google Scholar]
- 95.Haeusler R A, Astiarraga B, Camastra S, Accili D, Ferrannini E. Human insulin resistance is associated with increased plasma levels of 12α-hydroxylated bile acids. Diabetes. 2013;62(12):4184–4191. doi: 10.2337/db13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Watkins J B, III, Sanders R A. Diabetes mellitus-induced alterations of hepatobiliary function. Pharmacol Rev. 1995;47(01):1–23. [PubMed] [Google Scholar]
- 97.Garcia-Marin J J, Villanueva G R, Esteller A. Diabetes-induced cholestasis in the rat: possible role of hyperglycemia and hypoinsulinemia. Hepatology. 1988;8(02):332–340. doi: 10.1002/hep.1840080224. [DOI] [PubMed] [Google Scholar]
- 98.Carnovale C E, Roma M G, Monti J A, Rodriguez Garay E A. Studies on the mechanism of bile salt-independent bile flow impairment in streptozotocin-induced hepatotoxicity. Toxicology. 1991;68(03):207–215. doi: 10.1016/0300-483x(91)90070-h. [DOI] [PubMed] [Google Scholar]
- 99.González J, Fevery J. Spontaneously diabetic biobreeding rats and impairment of bile acid-independent bile flow and increased biliary bilirubin, calcium and lipid secretion. Hepatology. 1992;16(02):426–432. doi: 10.1002/hep.1840160222. [DOI] [PubMed] [Google Scholar]
- 100.Cazanave S, Podtelezhnikov A, Jensen K. The transcriptomic signature of disease development and progression of nonalcoholic fatty liver disease. Sci Rep. 2017;7(01):17193. doi: 10.1038/s41598-017-17370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Min H-K, Kapoor A, Fuchs M. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15(05):665–674. doi: 10.1016/j.cmet.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nobili V, Alisi A, Mosca A. Hepatic farnesoid X receptor protein level and circulating fibroblast growth factor 19 concentration in children with NAFLD. Liver Int. 2018;38(02):342–349. doi: 10.1111/liv.13531. [DOI] [PubMed] [Google Scholar]
- 103.Eren F, Kurt R, Ermis F, Atug O, Imeryuz N, Yilmaz Y. Preliminary evidence of a reduced serum level of fibroblast growth factor 19 in patients with biopsy-proven nonalcoholic fatty liver disease. Clin Biochem. 2012;45(09):655–658. doi: 10.1016/j.clinbiochem.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 104.Schreuder T CMA, Marsman H A, Lenicek M. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2010;298(03):G440–G445. doi: 10.1152/ajpgi.00322.2009. [DOI] [PubMed] [Google Scholar]
- 105.Alisi A, Ceccarelli S, Panera N. Association between serum atypical fibroblast growth factors 21 and 19 and pediatric nonalcoholic fatty liver disease. PLoS One. 2013;8(06):e67160. doi: 10.1371/journal.pone.0067160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sydor S, Best J, Messerschmidt I. Altered microbiota diversity and bile acid signaling in cirrhotic and noncirrhotic NASH-HCC. Clin Transl Gastroenterol. 2020;11(03):e00131. doi: 10.14309/ctg.0000000000000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Appleby R N, Moghul I, Khan S. Non-alcoholic fatty liver disease is associated with dysregulated bile acid synthesis and diarrhea: a prospective observational study. PLoS One. 2019;14(01):e0211348. doi: 10.1371/journal.pone.0211348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yoshimoto S, Loo T M, Atarashi K.Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome Nature 2013499(7456):97–101. [DOI] [PubMed] [Google Scholar]
- 109.Lelouvier B, Servant F, Païssé S. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: a pilot analysis. Hepatology. 2016;64(06):2015–2027. doi: 10.1002/hep.28829. [DOI] [PubMed] [Google Scholar]
- 110.Fickert P, Zollner G, Fuchsbichler A. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology. 2002;123(04):1238–1251. doi: 10.1053/gast.2002.35948. [DOI] [PubMed] [Google Scholar]
- 111.Masyuk A I, Gradilone S A, Banales J M. Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors. Am J Physiol Gastrointest Liver Physiol. 2008;295(04):G725–G734. doi: 10.1152/ajpgi.90265.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Segovia-Miranda F, Morales-Navarrete H, Kücken M. Three-dimensional spatially resolved geometrical and functional models of human liver tissue reveal new aspects of NAFLD progression. Nat Med. 2019;25(12):1885–1893. doi: 10.1038/s41591-019-0660-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pizarro M, Balasubramaniyan N, Solís N. Bile secretory function in the obese Zucker rat: evidence of cholestasis and altered canalicular transport function. Gut. 2004;53(12):1837–1843. doi: 10.1136/gut.2003.037689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Geier A, Dietrich C G, Grote T. Characterization of organic anion transporter regulation, glutathione metabolism and bile formation in the obese Zucker rat. J Hepatol. 2005;43(06):1021–1030. doi: 10.1016/j.jhep.2005.05.031. [DOI] [PubMed] [Google Scholar]
- 115.Canet M J, Merrell M D, Hardwick R N. Altered regulation of hepatic efflux transporters disrupts acetaminophen disposition in pediatric nonalcoholic steatohepatitis. Drug Metab Dispos. 2015;43(06):829–835. doi: 10.1124/dmd.114.062703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dzierlenga A L, Cherrington N J. Misregulation of membrane trafficking processes in human nonalcoholic steatohepatitis. J Biochem Mol Toxicol. 2018;32(03):e22035. doi: 10.1002/jbt.22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schaap F G, van der Gaag N A, Gouma D J, Jansen P LM. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology. 2009;49(04):1228–1235. doi: 10.1002/hep.22771. [DOI] [PubMed] [Google Scholar]
- 118.Zollner G, Fickert P, Zenz R. Hepatobiliary transporter expression in percutaneous liver biopsies of patients with cholestatic liver diseases. Hepatology. 2001;33(03):633–646. doi: 10.1053/jhep.2001.22646. [DOI] [PubMed] [Google Scholar]
- 119.Zhang Y, Li F, Patterson A D. Abcb11 deficiency induces cholestasis coupled to impaired β-fatty acid oxidation in mice. J Biol Chem. 2012;287(29):24784–24794. doi: 10.1074/jbc.M111.329318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fuchs C D, Krivanec S, Steinacher D. Absence of Bsep/Abcb11 attenuates MCD diet-induced hepatic steatosis but aggravates inflammation in mice. Liver Int. 2020;40(06):1366–1377. doi: 10.1111/liv.14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Okushin K, Tsutsumi T, Ikeuchi K. Heterozygous knockout of Bile salt export pump ameliorates liver steatosis in mice fed a high-fat diet. PLoS One. 2020;15(08):e0234750. doi: 10.1371/journal.pone.0234750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Krawczyk M, Rusticeanu M, Grünhage F, Lammert F. The common bile salt export pump (ABCB11) p.A444V variant is associated with BMI levels in the general population. Z Gastroenterol. 2009;47(09):s-0029–s-1241387. [Google Scholar]
- 123.Acalovschi M, Tirziu S, Chiorean E, Krawczyk M, Grünhage F, Lammert F. Common variants of ABCB4 and ABCB11 and plasma lipid levels: a study in sib pairs with gallstones, and controls. Lipids. 2009;44(06):521–526. doi: 10.1007/s11745-009-3300-z. [DOI] [PubMed] [Google Scholar]
- 124.Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. 2018;68(02):280–295. doi: 10.1016/j.jhep.2017.11.014. [DOI] [PubMed] [Google Scholar]
- 125.Carr R M, Reid A E. FXR agonists as therapeutic agents for non-alcoholic fatty liver disease. Curr Atheroscler Rep. 2015;17(04):500. doi: 10.1007/s11883-015-0500-2. [DOI] [PubMed] [Google Scholar]
- 126.Goto T, Itoh M, Suganami T. Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci Rep. 2018;8(01):8157. doi: 10.1038/s41598-018-26383-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang S, Wang J, Liu Q, Harnish D C. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51(02):380–388. doi: 10.1016/j.jhep.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 128.Mencarelli A, Renga B, Distrutti E, Fiorucci S. Antiatherosclerotic effect of farnesoid X receptor. Am J Physiol Heart Circ Physiol. 2009;296(02):H272–H281. doi: 10.1152/ajpheart.01075.2008. [DOI] [PubMed] [Google Scholar]
- 129.Hartman H B, Gardell S J, Petucci C J, Wang S, Krueger J A, Evans M J. Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR-/- and apoE-/- mice. J Lipid Res. 2009;50(06):1090–1100. doi: 10.1194/jlr.M800619-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hambruch E, Miyazaki-Anzai S, Hahn U. Synthetic farnesoid X receptor agonists induce high-density lipoprotein-mediated transhepatic cholesterol efflux in mice and monkeys and prevent atherosclerosis in cholesteryl ester transfer protein transgenic low-density lipoprotein receptor (-/-) mice. J Pharmacol Exp Ther. 2012;343(03):556–567. doi: 10.1124/jpet.112.196519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li Y TY, Swales K E, Thomas G J, Warner T D, Bishop-Bailey D. Farnesoid x receptor ligands inhibit vascular smooth muscle cell inflammation and migration. Arterioscler Thromb Vasc Biol. 2007;27(12):2606–2611. doi: 10.1161/ATVBAHA.107.152694. [DOI] [PubMed] [Google Scholar]
- 132.Gai Z, Gui T, Hiller C, Kullak-Ublick G A. Farnesoid X receptor protects against kidney injury in uninephrectomized obese mice. J Biol Chem. 2016;291(05):2397–2411. doi: 10.1074/jbc.M115.694323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Han C Y. Update on FXR biology: promising therapeutic target? Int J Mol Sci. 2018;19(07):E2069. doi: 10.3390/ijms19072069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Đanić M, Stanimirov B, Pavlović N. Pharmacological applications of bile acids and their derivatives in the treatment of metabolic syndrome. Front Pharmacol. 2018;9:1382. doi: 10.3389/fphar.2018.01382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mudaliar S, Henry R R, Sanyal A J. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145(03):5740–820. doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 136.NASH Clinical Research Network Neuschwander-Tetri B A, Loomba R, Sanyal A J.Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial Lancet 2015385(9972):956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.REGENERATE Study Investigators Younossi Z M, Ratziu V, Loomba R.Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial Lancet 2019394(10215):2184–2196. [DOI] [PubMed] [Google Scholar]
- 138.Gege C, Hambruch E, Hambruch N, Kinzel O, Kremoser C. Nonsteroidal FXR ligands: current status and clinical applications. Handb Exp Pharmacol. 2019;256:167–205. doi: 10.1007/164_2019_232. [DOI] [PubMed] [Google Scholar]
- 139.Patel K, Harrison S A, Elkhashab M. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. 2020;72(01):58–71. doi: 10.1002/hep.31205. [DOI] [PubMed] [Google Scholar]
- 140.ATLAS Investigators . Loomba R, Noureddin M, Kowdley K V. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology. 2021;73(02):625–643. doi: 10.1002/hep.31622. [DOI] [PubMed] [Google Scholar]
- 141.Tully D C, Rucker P V, Chianelli D. Discovery of tropifexor (LJN452), a highly potent non-bile acid FXR agonist for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH) J Med Chem. 2017;60(24):9960–9973. doi: 10.1021/acs.jmedchem.7b00907. [DOI] [PubMed] [Google Scholar]
- 142.Hernandez E D, Zheng L, Kim Y. Tropifexor-mediated abrogation of steatohepatitis and fibrosis is associated with the antioxidative gene expression profile in rodents. Hepatol Commun. 2019;3(08):1085–1097. doi: 10.1002/hep4.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Badman M K, Chen J, Desai S. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel non-bile acid FXR agonist tropifexor (LJN452) in healthy volunteers. Clin Pharmacol Drug Dev. 2020;9(03):395–410. doi: 10.1002/cpdd.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lucas K J, Lopez P, Lawitz E J. Tropifexor, a highly potent FXR agonist, produces robust and dose-dependent reductions in hepatic fat and serum alanine aminotransferase in patients with fibrotic NASH after 12 weeks of therapy: FLIGHT-FXR Part C interim results. Dig Liv Dis. 2020;52(01):E38. [Google Scholar]
- 145.Papazyan R, Liu X, Liu J. FXR activation by obeticholic acid or nonsteroidal agonists induces a human-like lipoprotein cholesterol change in mice with humanized chimeric liver. J Lipid Res. 2018;59(06):982–993. doi: 10.1194/jlr.M081935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu ; European Association for the Study of the Liver . EASL Clinical Practice Guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67(01):145–172. doi: 10.1016/j.jhep.2017.03.022. [DOI] [PubMed] [Google Scholar]
- 147.Zhou M, Wang X, Phung V. Separating tumorigenicity from bile acid regulatory activity for endocrine hormone FGF19. Cancer Res. 2014;74(12):3306–3316. doi: 10.1158/0008-5472.CAN-14-0208. [DOI] [PubMed] [Google Scholar]
- 148.Gadaleta R M, Scialpi N, Peres C. Suppression of hepatic bile acid synthesis by a non-tumorigenic FGF19 analogue protects mice from fibrosis and hepatocarcinogenesis. Sci Rep. 2018;8(01):17210. doi: 10.1038/s41598-018-35496-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Loomba R, Ling L, Dinh D M. The commensal microbe veillonella as a marker for response to an FGF19 analog in NASH. Hepatology. 2021;73(01):126–143. doi: 10.1002/hep.31523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Scheiman J, Luber J M, Chavkin T A. Meta-omics analysis of elite athletes identifies a performance-enhancing microbe that functions via lactate metabolism. Nat Med. 2019;25(07):1104–1109. doi: 10.1038/s41591-019-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Harrison S A, Rossi S J, Paredes A H. NGM282 improves liver fibrosis and histology in 12 weeks in patients with nonalcoholic steatohepatitis. Hepatology. 2020;71(04):1198–1212. doi: 10.1002/hep.30590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Harrison S A, Neff G, Guy C D. Efficacy and safety of aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology. 2021;160(01):2190–2310. doi: 10.1053/j.gastro.2020.08.004. [DOI] [PubMed] [Google Scholar]
- 153.Hirschfield G M, Chazouillères O, Drenth J P. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: a multicenter, randomized, double-blind, placebo-controlled phase II trial. J Hepatol. 2019;70(03):483–493. doi: 10.1016/j.jhep.2018.10.035. [DOI] [PubMed] [Google Scholar]
- 154.Harrison S A, Rinella M E, Abdelmalek M F.NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial Lancet 2018391(10126):1174–1185. [DOI] [PubMed] [Google Scholar]
- 155.Rinella M E, Trotter J F, Abdelmalek M F. Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis. J Hepatol. 2019;70(04):735–744. doi: 10.1016/j.jhep.2018.11.032. [DOI] [PubMed] [Google Scholar]
- 156.Fang S, Suh J M, Reilly S M. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med. 2015;21(02):159–165. doi: 10.1038/nm.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Xie C, Jiang C, Shi J. An intestinal farnesoid X receptor-ceramide signaling axis modulates hepatic gluconeogenesis in mice. Diabetes. 2017;66(03):613–626. doi: 10.2337/db16-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mueller M, Thorell A, Claudel T. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J Hepatol. 2015;62(06):1398–1404. doi: 10.1016/j.jhep.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Buko V U, Kuzmitskaya-Nikolaeva I A, Naruta E E, Lukivskaya O Y, Kirko S N, Tauschel H-D. Ursodeoxycholic acid dose-dependently improves liver injury in rats fed a methionine- and choline-deficient diet. Hepatol Res. 2011;41(07):647–659. doi: 10.1111/j.1872-034X.2011.00820.x. [DOI] [PubMed] [Google Scholar]
- 160.Wang W, Zhao J, Gui W. Tauroursodeoxycholic acid inhibits intestinal inflammation and barrier disruption in mice with non-alcoholic fatty liver disease. Br J Pharmacol. 2018;175(03):469–484. doi: 10.1111/bph.14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ozcan U, Yilmaz E, Ozcan L.Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes Science 2006313(5790):1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Laurin J, Lindor K D, Crippin J S. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: a pilot study. Hepatology. 1996;23(06):1464–1467. doi: 10.1002/hep.510230624. [DOI] [PubMed] [Google Scholar]
- 163.NASH Study Group . Leuschner U FH, Lindenthal B, Herrmann G. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52(02):472–479. doi: 10.1002/hep.23727. [DOI] [PubMed] [Google Scholar]
- 164.Lindor K D, Kowdley K V, Heathcote E J. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology. 2004;39(03):770–778. doi: 10.1002/hep.20092. [DOI] [PubMed] [Google Scholar]
- 165.Swiss Association for the Study of the Liver . Dufour J-F, Oneta C M, Gonvers J-J. Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin e in nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2006;4(12):1537–1543. doi: 10.1016/j.cgh.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 166.FRESGUN . Ratziu V, de Ledinghen V, Oberti F. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol. 2011;54(05):1011–1019. doi: 10.1016/j.jhep.2010.08.030. [DOI] [PubMed] [Google Scholar]
- 167.Kars M, Yang L, Gregor M F. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010;59(08):1899–1905. doi: 10.2337/db10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pols T WH, Noriega L G, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol. 2011;54(06):1263–1272. doi: 10.1016/j.jhep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Watanabe M, Houten S M, Mataki C.Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation Nature 2006439(7075):484–489. [DOI] [PubMed] [Google Scholar]
- 170.Roth J D, Feigh M, Veidal S S. INT-767 improves histopathological features in a diet-induced ob/ob mouse model of biopsy-confirmed non-alcoholic steatohepatitis . World J Gastroenterol. 2018;24(02):195–210. doi: 10.3748/wjg.v24.i2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Thomas C, Gioiello A, Noriega L. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10(03):167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.SB-756050 Project Team . Hodge R J, Lin J, Vasist Johnson L S, Gould E P, Bowers G D, Nunez D J. Safety, pharmacokinetics, and pharmacodynamic effects of a selective TGR5 agonist, SB-756050, in type 2 diabetes. Clin Pharmacol Drug Dev. 2013;2(03):213–222. doi: 10.1002/cpdd.34. [DOI] [PubMed] [Google Scholar]
- 173.Yasuda H, Hirata S, Inoue K, Mashima H, Ohnishi H, Yoshiba M. Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem Biophys Res Commun. 2007;354(01):154–159. doi: 10.1016/j.bbrc.2006.12.168. [DOI] [PubMed] [Google Scholar]
- 174.Casaburi I, Avena P, Lanzino M. Chenodeoxycholic acid through a TGR5-dependent CREB signaling activation enhances cyclin D1 expression and promotes human endometrial cancer cell proliferation. Cell Cycle. 2012;11(14):2699–2710. doi: 10.4161/cc.21029. [DOI] [PubMed] [Google Scholar]
- 175.Keitel V, Cupisti K, Ullmer C, Knoefel W T, Kubitz R, Häussinger D. The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology. 2009;50(03):861–870. doi: 10.1002/hep.23032. [DOI] [PubMed] [Google Scholar]
- 176.Li T, Holmstrom S R, Kir S. The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol Endocrinol. 2011;25(06):1066–1071. doi: 10.1210/me.2010-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Claudel T, Staels B, Kuipers F. The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol. 2005;25(10):2020–2030. doi: 10.1161/01.ATV.0000178994.21828.a7. [DOI] [PubMed] [Google Scholar]
- 178.Tsuji Y, Kaji K, Kitade M. Bile acid sequestrant, sevelamer ameliorates hepatic fibrosis with reduced overload of endogenous lipopolysaccharide in experimental nonalcoholic steatohepatitis. Microorganisms. 2020;8(06):E925. doi: 10.3390/microorganisms8060925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Takahashi S, Luo Y, Ranjit S. Bile acid sequestration reverses liver injury and prevents progression of nonalcoholic steatohepatitis in Western diet-fed mice. J Biol Chem. 2020;295(14):4733–4747. doi: 10.1074/jbc.RA119.011913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.San Diego Integrated NAFLD Research Consortium (SINC) . Le T-A, Chen J, Changchien C. Effect of colesevelam on liver fat quantified by magnetic resonance in nonalcoholic steatohepatitis: a randomized controlled trial. Hepatology. 2012;56(03):922–932. doi: 10.1002/hep.25731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Potthoff M J, Potts A, He T. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol. 2013;304(04):G371–G380. doi: 10.1152/ajpgi.00400.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rao A, Kosters A, Mells J E. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci Transl Med. 2016;8(357):357ra122. doi: 10.1126/scitranslmed.aaf4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Volixibat in Adults study group . Newsome P N, Palmer M, Freilich B. Volixibat in adults with non-alcoholic steatohepatitis: 24-week interim analysis from a randomized, phase II study. J Hepatol. 2020;73(02):231–240. doi: 10.1016/j.jhep.2020.03.024. [DOI] [PubMed] [Google Scholar]
- 184.Karpen S J, Kelly D, Mack C, Stein P. Ileal bile acid transporter inhibition as an anticholestatic therapeutic target in biliary atresia and other cholestatic disorders. Hepatol Int. 2020;14(05):677–689. doi: 10.1007/s12072-020-10070-w. [DOI] [PubMed] [Google Scholar]
- 185.Halilbasic E, Steinacher D, Trauner M. Nor-ursodeoxycholic acid as a novel therapeutic approach for cholestatic and metabolic liver diseases. Dig Dis. 2017;35(03):288–292. doi: 10.1159/000454904. [DOI] [PubMed] [Google Scholar]
- 186.Beraza N, Ofner-Ziegenfuss L, Ehedego H. Nor-ursodeoxycholic acid reverses hepatocyte-specific nemo-dependent steatohepatitis. Gut. 2011;60(03):387–396. doi: 10.1136/gut.2010.223834. [DOI] [PubMed] [Google Scholar]
- 187.Fickert P, Moustafa T, Fuchsbichler A. Differential effects of norUDCA and UDCA in the treatment of fatty liver and arteriosclerosis in western chow-fed ApoE knock out mice. J Hepatol. 2008;48:S42. [Google Scholar]
- 188.Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G. Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology. 2019;69(01):420–430. doi: 10.1002/hep.30150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Richardson M M, Jonsson J R, Powell E E. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology. 2007;133(01):80–90. doi: 10.1053/j.gastro.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 190.Natarajan S K, Ingham S A, Mohr A M. Saturated free fatty acids induce cholangiocyte lipoapoptosis. Hepatology. 2014;60(06):1942–1956. doi: 10.1002/hep.27175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Austrian/German NAFLD-norUDCA study group . Traussnigg S, Schattenberg J M, Demir M. Norursodeoxycholic acid versus placebo in the treatment of non-alcoholic fatty liver disease: a double-blind, randomised, placebo-controlled, phase 2 dose-finding trial. Lancet Gastroenterol Hepatol. 2019;4(10):781–793. doi: 10.1016/S2468-1253(19)30184-0. [DOI] [PubMed] [Google Scholar]