

Abstract
Metastatic pancreatic neuroendocrine tumors (PNETs) remain an unmet clinical problem. Chronological treatment in PNETs includes observation (watchful protocol), surgery, targeted therapy, and chemotherapy. However, increasing evidence illustrates that the outcomes of targeted therapeutic options for the treatment of advanced PNETs show minimal response. The FDA approved mTOR inhibitor everolimus does not shrink patient’s tumors. It only delays disease progression that too in only a subset of patients while a significant fraction acquire resistance and show disease progression. Thus, there is a need for more effective targeted approaches to sensitize PNETs to everolimus for better treatment outcomes. Previously, we showed that mTOR regulator p21 activated kinase 4 (PAK4) and nicotinamide adenine dinucleotide biosynthesis enzyme nicotinamide phosphoribosyl transferase (NAMPT) were aberrantly expressed in PNET tissue and promoted everolimus resistance. In this report, we demonstrate that PAK4-NAMPT dual inhibitor KPT-9274 can synergize with everolimus (growth inhibition, colony suppression and glucose uptake assays). KPT-9274-everolimus disrupted spheroid formation in multiple PNET models. Molecular analysis showed alteration of mTORC2 through downregulation of RICTOR as a mechanism supporting synergy with everolimus in vitro. KPT-9274 suppressed β-catenin activity via inhibition of PAK4 highlighting the crosstalk between Rho GTPases and Wnt signaling in PNETs. KPT-9274, given at (150 mg/kg) in combination with sub-MTD everolimus (2.5 mg/kg) significantly suppressed two PNET derived xenograft. These studies bring forward a well-grounded strategy for advanced PNETs that fail to respond to single-agent everolimus.
Introduction
Pancreatic Neuroendocrine Tumors (PNETs) are rare and make about 1–2% of all pancreatic neoplasms, which represents about 1000 new cases every year in the United States [1],[https://www.cancer.gov/types/pancreatic/hp/pnet-treatment-pdq]. However, the incidence of PNETs has significantly increased over the years due to advanced early-stage disease detection [2]. In 2020, the American Cancer Society estimates that about 4032 people will be diagnosed with PNETs this current year; suggesting an incidence of 7% of all significant pancreatic neoplasm [https://www.cancer.org/cancer/pancreatic-neuroendocrine-tumor/about/key-statistics.html].
The management of PNETs is significantly difficult given that they have an unpredicted behavior from reasonably indolent to extremely aggressive [3]. The unpredicted course of this disease makes PNET one of the most heterogeneous and intractable subtypes of cancers. Although PNETs have a relatively low somatic mutation frequency compared to other tumor types, hyperactivation of the PI3K/AKT/mTOR signaling alongside chromatin alterations and errors in the DNA damage response pathway are considered the major drivers of this subtype of pancreatic cancer [4]. It has been well established that the tumor suppressors PTEN and TSC2 are two major regulators of the PI3K/AKT/mTOR cascade. Low expression levels of PTEN and TSC2 have been associated with PNETs progression, migration, and poor survival; thus, emphasizing the critical role of PI3K/AKT/mTOR signaling in these lesions [5]. The mTOR inhibitors are therefore used in a significant fraction of patients with PNETs. FDA-approved mTOR inhibitor in PNETs everolimus does not provide a clinically meaningful objective response rate (5% partial response) but it does stabilize the disease and hence prolong the progression-free survival. The RADIANT-3 study found a median progression-free survival of 11 months with everolimus compared to 4.6 months with the placebo (Hazard Ratio (HR) 0.35; 95% confidence interval (CI), 0.27 to 0.45; P<0.001). Additionally, 44% of patients participating in the latter trial were removed from the study due to disease progression [6]. It is important to note that mTOR interacts with several binding partners to form two distinct multiprotein complexes including mTORC1 and mTORC2. Everolimus inhibits the activity of mTORC1 by preventing this complex to interact with its intracellular receptor FKPB12 that results in the inhibition of the downstream effector p70S6K and 4EBP1 [7]. Resistance to everolimus and other mTOR inhibitors has been mainly attributed to mTORC2 in a feedback reaction to mTORC1 inhibition. Upon inhibition of the mTORC1, mTORC2 is activated to promote Akt signaling which inhibits TSC2; thus, promoting mTORC1 signaling [8]. Resistance to everolimus has also been attributed to the activation of the RAS/MEK/ERK signaling pathway [9–10]. This brings to the scope of this study that is to find alternative and more effective strategies to tame mTOR signaling in PNETs.
PAK4 is a member of group II P21-activated Kinase family initially identified as an effector of the Rho GTPases signaling pathway that controls cell morphology, adhesion, and migration [11]. PAK4 is known as a core molecule linking major signaling pathway including the Ras/Erk pathway (involved in everolimus resistance [9–10]), WNT/β-CATENIN pathway (an emerging target for PNETs [12]), AR/ER-depending pathway, and more importantly the PI3K/AKT/mTOR signaling pathway that regulates the pathogenesis and evolution of PNETs. Thus, PAK4 cross talks PNETs driver pathways making this kinase a significant target for resistant PNETs. On the other hand, NAMPT is the rate-limited enzyme responsible for the highest source of physiological NAD biosynthesis in eukaryotic cells. Tumor cells undergoing metabolic adaptation have highly active glycolysis, pentose, and fatty acid biosynthesis [13]. Since NAD is central to these metabolic changes, the cellular level of NAD must be balanced to modulate these processes. More importantly and related to Gastroenteropancreatic Neuroendocrine Tumors (GEP-NETs), NAMPT has been identified as a master regulator proteins for these subtypes of cancers. FK866 the specific NAMPT inhibitor was among the top six small-molecule compounds that invert the metastatic progression of GEP-NETs activity suggesting the critical role of NAMPT in the survival and the progression of these lesions [14].
In this report, we demonstrate the critical role of PAK4 and NAMPT in conferring resistance to mTOR inhibitors in PNET models. We evaluate the impact of 1st in class, oral PAK4-NAMPT dual inhibitor KPT-9274 (currently in Phase 1 clinical trials; NCT02702492) in combination with everolimus in vitro cellular models, 3D models, and in vivo PNETs xenograft models.
Materials and Methods
Cell lines and Reagents
Human PNET cells BON-1 (RRID: CVCL_3985) and QGP-1 (RRID: CVCL_3143) were used in this investigation. QGP-1 cells were purchased from the Japanese Cancer Research Resources Bank (JCRB) (Ibaraki, Osaka, Japan) on 04-05-2016. BON-1 cells were obtained under a material transfer agreement from Dr. Hellmich and Dr. Townsend (University of Texas Medical Branch, Galveston, TX, USA) on 11-09-2016. BON-1 and QGP-1 cells were authenticated using short tandem repeat (STR) profiling approach. BON-1 and QGP-1 cells are routinely treated for mycoplasma (recently done on 03-112021) using PCR technique and maintained at 37° C and 5% CO2. QGP-1 cells were maintained in RPMI (catalog no. 11875-093; Gibco, ThermoFisher Scientific, Waltham, MA, USA) culture medium and BON-1 cells were maintained in DMEM/F12 Ham (catalog no. 21041-025; Gibco, ThermoFisher Scientific, Waltham, MA, USA) culture medium. The medium was supplemented with 10% FBS (catalog no. S12550; Atlanta Biologicals, Minneapolis, MN, USA) and 1% penicillin/streptomycin (catalog no. SV30010; GE Healthcare, ThermoFisher Scientific, Waltham, MA, USA) except where otherwise specified. All the experiments were performed within 20 passages of the cell lines.
The PAK4-NAMPT dual inhibitor (KPT-9274) was obtained from Karyopharm Therapeutics (Newton, MA, USA). Structure of KPT-9274 has been described earlier [15]. KPT-9274 is a CRISPRres validated NAMPT inhibitor [16] and a SILAC validated PAK4 inhibitor [17]. Everolimus [(RAD001) mTOR inhibitor (catalog no. S1120), FK866 (APO866/Daporinad specific inhibitor of NAMPT; catalog no. S2799), and PF3758309 (specific inhibitor of PAK4; catalog no. S7094) were purchase from SelleckChem (Houston, TX, USA). The latter drugs were dissolved in dimethyl sulfoxide (DMSO, D-1391; Fisher Scientific, Waltham, MA, USA). Primary and secondary antibodies were purchased from multiple vendors including Cell Signaling Technology (Danvers, MA, USA), Proteintech (Rosemont, IL, USA), and/or Santa Cruz Biotechnology (Dallas, TX, USA). Glucose uptake cell-based assay (catalog no. 600470) and caspase-3/7 fluorescence assay (catalog no. 100009135) kit were purchased from Cayman Chemical (Ann Arbor, MI, USA). NAD/NADH-Glo™ assay (catalog no. G907) kit was obtained from Promega (Madison, WI, USA). The assays were done in triplicate and according to the manufacturer protocol.
Colonogenic Assay
50,000 BON-1 cells and 100,000 QGP-1 cells were seeded in triplicate in 15 X 60 mm petri dishes with appropriate medium and incubated overnight so that the cells could be 80% confluent the next day. Next, the cells were exposed to 600 nM of KPT-9274, 6μM of everolimus, and the combination of both drugs for 72 hours. After the treatment interval, the PNET cells were washed with warm PBS and 1000 cells were collected from each treatment condition then re-seeded in 15 X 60 mm petri dish and allowed to grow for 4 weeks (BON-1) and 6 weeks (QGP-1) at 37° C in 5% CO2 incubator. After the incubation interval, the supernatant was removed from each dish, and cells were exposed to 2 ml of methanol for 5 min. Next, colonies were stained with 2% crystal violet, allowed to dry, photographed, and quantified. Only colonies with greater or equal to 50 cells were quantified. This experiment was done in triplicate and repeated by a different person to remove individual bias.
Western Blot Analysis
50,000 BON-1 cells or 100,000 QGP-1 were grown in 100 mm petri dishes overnight. The following day, each cell line was treated with specified concentrations of KPT-9274, PF-3758308, Everolimus, FK866, and combinations for 72 hours. A total of 50 μg protein lysates from treated and untreated cells were separated in a 10–12% SDS-PAGE and transferred into a nitrocellulose membrane (GE Healthcare, ThermoFisher Scientific, Waltham, MA, USA). Mouse monoclonal antibodies anti-PAK4 (catalog no. sc-81532), anti-NAMPT (PEBF, catalog no. sc-393510), anti-NAPRT (catalog no. sc-398404), anti-β-TUBULIN (catalog no. sc-5274), anti-β-ACTIN (catalog no. sc-8432) from Santa Cruz Biotechnology, and anti-pmTOR (catalog no. 5536S), anti-mTOR (catalog no. 2972S), anti-RICTOR (catalog no. 9476S), anti-RAPTOR (catalog no. 2280S), anti-pP70S6K (catalog no. 9204S), anti-P70S6K (catalog no. 2708S) from cell signaling technology and were used at a 1:1000 dilution in (3% non-fat milk or 5% BSA) PBS with 0.1% Tween-20 (catalog no. P7949, Sigma-Aldrich, St. Louis, MO, USA).
Annexin V FITC
PNET cellular models BON-1 and QGP-1 were seeded in 60 mm petri dishes. The next day and except for the control samples, BON-1 and QGP-1 cell lines were treated with 9 μM of Octreotide and 9 μM of lanreotide for 72 hours. After the treatment period, the cells were washed with warm PBS, exposed to a reasonable amount of trypsin, and incubated for 5 minutes to allow the cells to detach from the plates. Next, treated and control cells were stained with Annexin V and Propidium Iodide (Annexin V FITC) [18]. Apoptotic cells were sorted using Annexin V FITC kit (Catalog no. K101-400; Biovision, Danvers, MA, USA) according to the manufacturers’ protocol. Stained cells were sorted using the Becton Dickinson flow cytometer at the Karmanos Cancer Institute Flow Cytometry Core. This experiment was done in triplicate as shown in supplemental figures file.
Quantitative Real-Time PCR
After each described treatment, RNA was isolated from treated cells, RT-qPCR was done using SYBR Green PCR master mix (Applied Biosystems, Waltham, MA, USA) on a StepOnePlus Real-Time PCR System according to the manufacturer’s instructions. Primers used in this study have shown in the Supplementary Table S1. Each sample was run in triplicates and repeated by a different person to remove individual bias. The protocol for this PCR included a denaturation (95° C for 10 min), then 40 cycles of amplification and quantification (95° C for 15 sec, 60° C for 1 min).
PNETs Spheroids formation
To further examine the efficacy of KPT-9274 single agent and/or the combination with everolimus, BON-1, and QGP-1 cells were seeded and grown in sphere formation culture medium (DMEM/F12 medium supplemented with insulin (5μg/ml; I9278), basic fibroblast growth factor (10 ng/mL, F0291), B27 (0.1x; Catalog no. 17504044) and human recombinant EGF (20 ng/mL, E5036; Sigma-Aldrich, St. Louis, MO, USA)) in flat bottom ultra-low attachment surface polystyrene plates (Corning, catalog no. 3261; Tewksbury, MA, USA) and then exposed to indicated drug concentration of either KPT-9274 alone, everolimus alone, or their combination. After five days treatment, PNET spheroids were counted under an inverted microscope and photographed. This experiment was repeated in a blinded fashion to remove individual bias.
Animal Studies
All animal studies have been conducted in accordance with, and with the approval of, an Institutional Animal Care and Use Committee (IACUC Protocol # 18-12-0887). Post adaptation in our animal housing facility, 4–5 weeks old female ICR-SCID mice (Taconic, Rensselaer, NY, USA) were subcutaneously injected with BON-1 or QGP-1 cell lines. Each cell line was suspended PBS (1×106 in 200 μL) and loaded in BD 26Gx 5/8 1ml Sub-Q syringe and carefully injected into the flanks of donor mice. Once tumors reached about 5–10% of the donor mice body weight (using a caliper and calculation to confirm the seize (LxW2/2)), the donor mice were euthanized, tumor harvested and fragments implanted into recipient mice. Three days post the implant, the recipient mice were randomly divided in 4 groups and treated by oral gavage with vehicle or diluent, KPT-9274, everolimus, or their combination (one time a day, three times a week for 4 weeks). Tumor size and body weight were recorded 5 times weekly. After 6 weeks the tumors were harvested, weighed, and photographed. The same treatment scheme was done using two different cell xenografts to remove cell specific response to the treatment regimen.
Statistical Analysis
Statistical assessments were done using GraphPad Prism 4 software (RRID: SCR_002798). As needed, the data were subjected to an unpaired two-tailed Student’s t-test and two-way ANOVA and presented as mean +/− standard error of the mean of at least three replicate experiments. We considered a P-value <0.05 as statistically significant.
Results
KPT-9274-everolimus combination inhibits the growth of PNETs cell lines
To examine whether the combination of KPT-9274 and everolimus has long-term growth inhibition in PNET cells in vitro, we performed a clonogenic assay. PNETs cells were treated with 600 nM of KPT-9274, 6 μM of everolimus, and then their combination for 72 hours. Treated cells were then collected and seeded in 15 X 60 mm Petri dishes and colonies were allowed to grow for four weeks at 37° C in a 5% CO2 incubator. We observed a statistically significant reduction in colony formation post-KPT-9274 single agent and KPT-9274-everolimus treatments in the PNETs cell line (Fig. 1A–1C). The latter suggests that the efficacy of KPT-9274-everolimus was sustained 4 weeks post-treatment in vitro. Supportive evidence of the efficacy of KPT-9274-everolimus came from caspase3/7 activity analysis. KPT-9274 and everolimus demonstrated minimal activity up to 600 nM and 6 μM, respectively. However, the combination of KPT-9274-everolimus showed a statistically significant enhancement in caspase3/7 activity (p<0.001) (Fig. 1D). 3D models such as spheroids are excellent and more significant tumor models to test small molecule inhibitors for anticancer therapy. Therefore, to further assess the efficacy of KPT-9274-everolimus, we developed PNETs spheroids and exposed them to KPT-9274 single agent and/or in combination with everolimus. We found that the combination therapy significantly disrupts and inhibits PNET’s sphere formation in vitro (Fig. 1E; 1F). We also examine the effect of everolimus treatment on NAD pool levels in PNET cells. As anticipated, we found that mTOR inhibition minimally decreases the NAD pool level; and such decrease in the combination treatment mainly came from the inhibition of NAMPT by KPT-9274 (Fig. 1G).
Figure 1: KPT-9274 in combination with everolimus dramatically inhibits the growth of PNET cell lines in vitro.
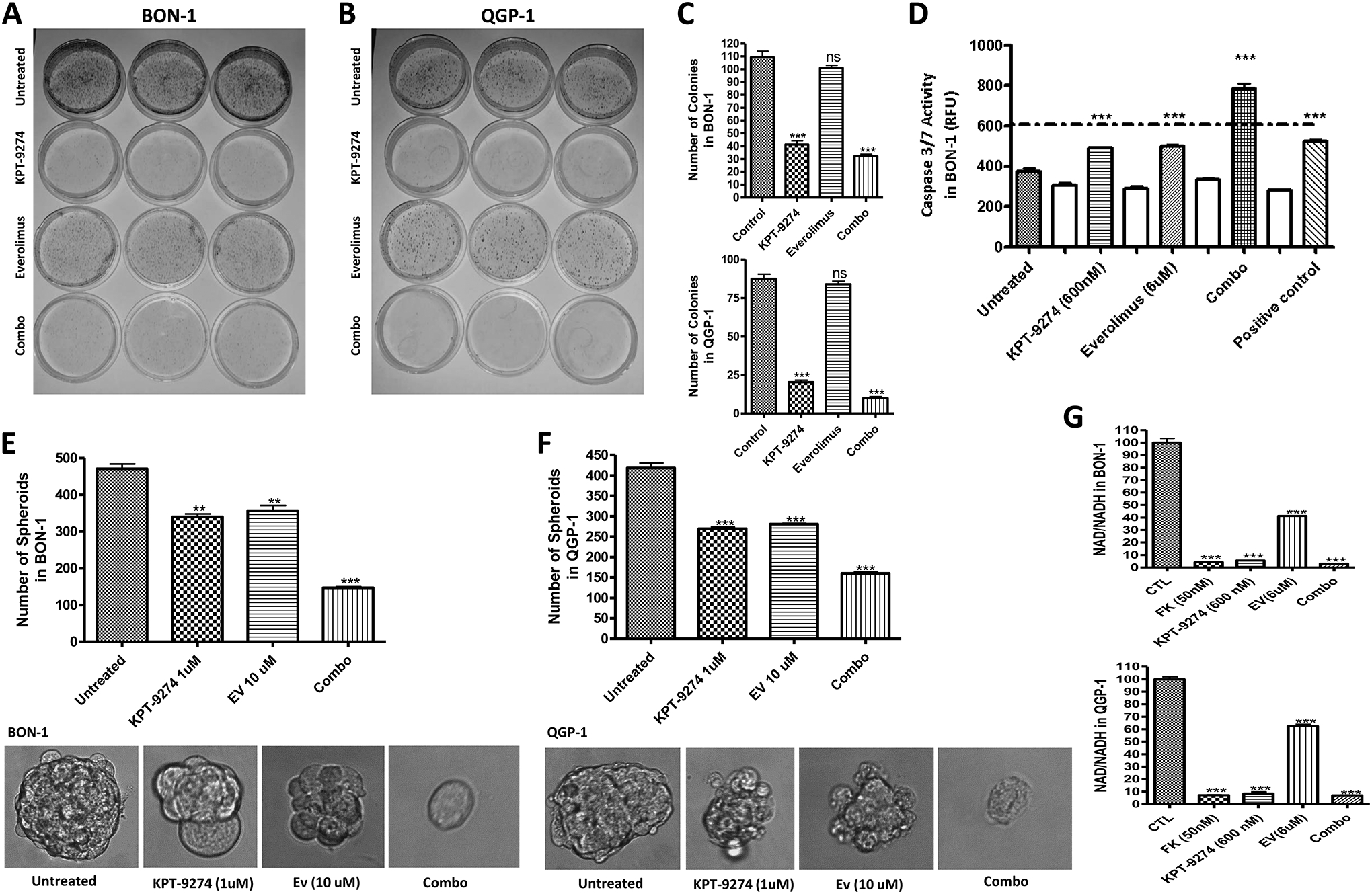
[A-C] Colony formation assay showing that KPT-9274 plus everolimus promotes a long-term inhibition of PNET cell line in vitro. [D] Caspase3/7 activity assay performed according to the manufacturers’ protocol. The assay shows activation of the mechanism of apoptosis 72 hours post-treatment with the indicated drugs. Caspase activity was enhanced in the combination treatment. The result is representative of three independent experiments. [E, F] BON-1 and QGP-1 spheroids, KPT-9274 in combination with everolimus significantly inhibit the growth and the number PNET spheroids in vitro. [G] NAD Cell Titer-Glo assay was performed according to the manufacturer’s protocol. The graphs show a statically significant reduction of the NAD pool after treatment with the indicated drugs. FK866 was used as a positive control. The reduction of NAD in the control treatment was mainly due to the effect of KPT-9274 rather than everolimus. RFU, relative fluorescence unit; *P<0.05, **P<0.01, and *** P<0.005). Each graph represents 3 independent experiments.
KPT-9274 demonstrates engagement with the targets in PNETs cell lines
Here, we wanted to examine whether KPT-9274 interacts with its respective targets in PNETs cellular models using Western blot and RT-qPCR. Thus, we treated PNET cell lines with increasing concentrations of KPT-9274 (150 nM, 300 nM, and 600 nM) in vitro. KPT-9274 demonstrated superior inhibition of PAK4 and NAMPT at 600 nM (Fig. 2A; 2C; 2E). Next, we compared the efficacy of target engagement of KPT-9274 to the combination of PF03798309 (the specific inhibitor of PAK4) and FK866 (the specific inhibitor of NAMPT) at 100 nM and 50 nM respectively. KPT-9274 given at 600 nM demonstrated similar efficacy to the combination of 100 nM PF-03798309 and 50 nM FK866 (Fig. 2B; 2D). The concentration of KPT-9274 used here is higher compared to PF-03798309 and FK866 because PNETs cell lines BON-1 and QGP-1 are not viable at >100 nM and >50 nM of PF-03798309 and FK866 respectively. We also found that exposure of QGP-1 cells to FK866 resulted in hyperactivation of NAPRT-1 to compensate for inhibition of NAMPT as can be seen in figure 2 (Fig. 2B; 2F). This suggests that inhibition of NAMPT by FK866 leads to activation of the Preiss-Handler pathway in this specific cell line. However, activation of NAPRT-1 in QGP-1 post-treatment with FK866 does not lead to increase NAD synthesis because the medium used to culture these cells do not contain niacin (essential for the Preiss-Handler pathway). Activation of NAPRT-1 was not seen in BON-1 cells post-treatment with FK866 (Fig. 2D) suggesting that NAPRT-1 could be dysfunctional in this specific cell line. We can conclude here that PNET cells rely preferentially on the salvage and the Preiss-Handler pathways for NAD biosynthesis.
Figure 2: KPT-9274 downregulates the expression of PAK4 and NAMPT in BON-1 and QGP-1.
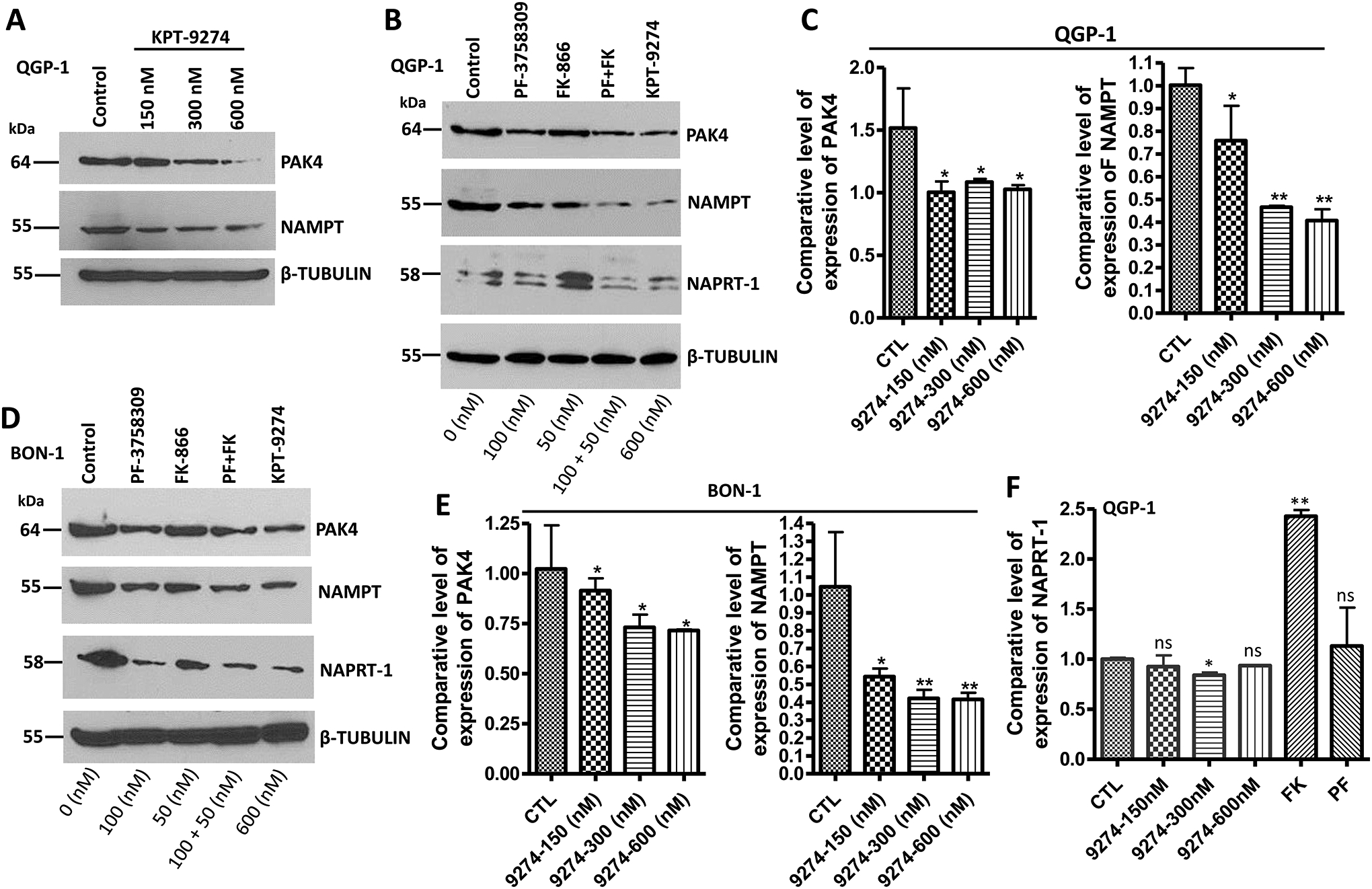
[A, B, and D] 50000 BON-1 and 100000 QGP-1 cells were grown in 100 mm Petri dishes and exposed for 72 hours to the shown concentrations of each small molecule inhibitor as described in the methods section. 50 μg of protein lysates were resolved on a 12% SDS-PAGE following by western blot comparing the expression level of PAK4, NAMPT, NAPRT-1. β-TUBULIN was used as an internal control. [C, E, and F] RT-qPCR comparing the expression level of PAK4, NAMPT, and NAPRT-1 in PNET cell line BON-1 and QGP-1. Here, we used mRNA samples with RIN# greater than 2. Each expression level was normalized with actin mRNA. Each graph represents 3 independent experiments.
KPT-9274 inhibit glucose uptake in PNETs cells
Recently, WNT/β-CATENIN signaling has been revealed to play a role in neuroendocrine tumor cell growth and survival [8, 12]. It has also been demonstrated that PAK4 activates β-CATENIN and carries this transcription factor from the cytoplasm to the nucleus where it binds to the TCF to promote the transcription of several WNT signaling dependent genes [19]. Thus, PAK4 is a major regulator of WNT/β-CATENIN signaling in cells and more importantly an attractive target to modulate wnt signaling in PNETs. Elghazi and colleagues have also shown that deletion of β-CATENIN in pancreatic progenitor leads to a decrease in neuroendocrine cell mass and impaired glucose tolerance [20]. To confirm this hypothesis, we first evaluated the expression of WNT and β-CATENIN post-treatment with KPT-9274 by RT-PCR. We exposed PNET cell lines BON-1 and QGP-1 to escalating concentrations of KPT-9274 (150 nM, 300 nM, and 600 nM) for 24 hours. As anticipated, inhibition of PAK4 led to a downregulation of β-CATENIN mRNA level in PNET cell lines (Fig. 3A; 3B). Alteration of β-CATENIN activity and subsequently induced negative feedback resulting in high mRNA levels of WNT in BON-1 and QGP-1 (Fig. 3A; 3B). The latter suggests that WNT signaling is a genetic dependency (addiction) in PNET cell lines BON-1 and QGP-1. The latter further demonstrates that KPT-9274 modulates the activity of PAK4 in PNET cell lines. We further confirm the hypothesis that deletion of β-CATENIN impair glucose tolerance in neuroendocrine cells, we evaluated glucose uptake in PNET cells post-treatment with KPT-9274. In this assay Apigenin (GLUT-1 inhibitor) was used as a positive control for reduced glucose uptake. We found that PNETs cells exposed to KPT-9274 resulted in a statistically significant reduction of glucose uptake (Fig. 3C; 3D). In conclusion, PNET cells treated with KPT-9274 results in a reduction of cellular level of NAD and glucose uptake which justifies the reduction of cellular ATP as shown in our previous publication [18].
Figure 3: WNT/β-CATENIN regulates glucose uptake in PNETs cells.
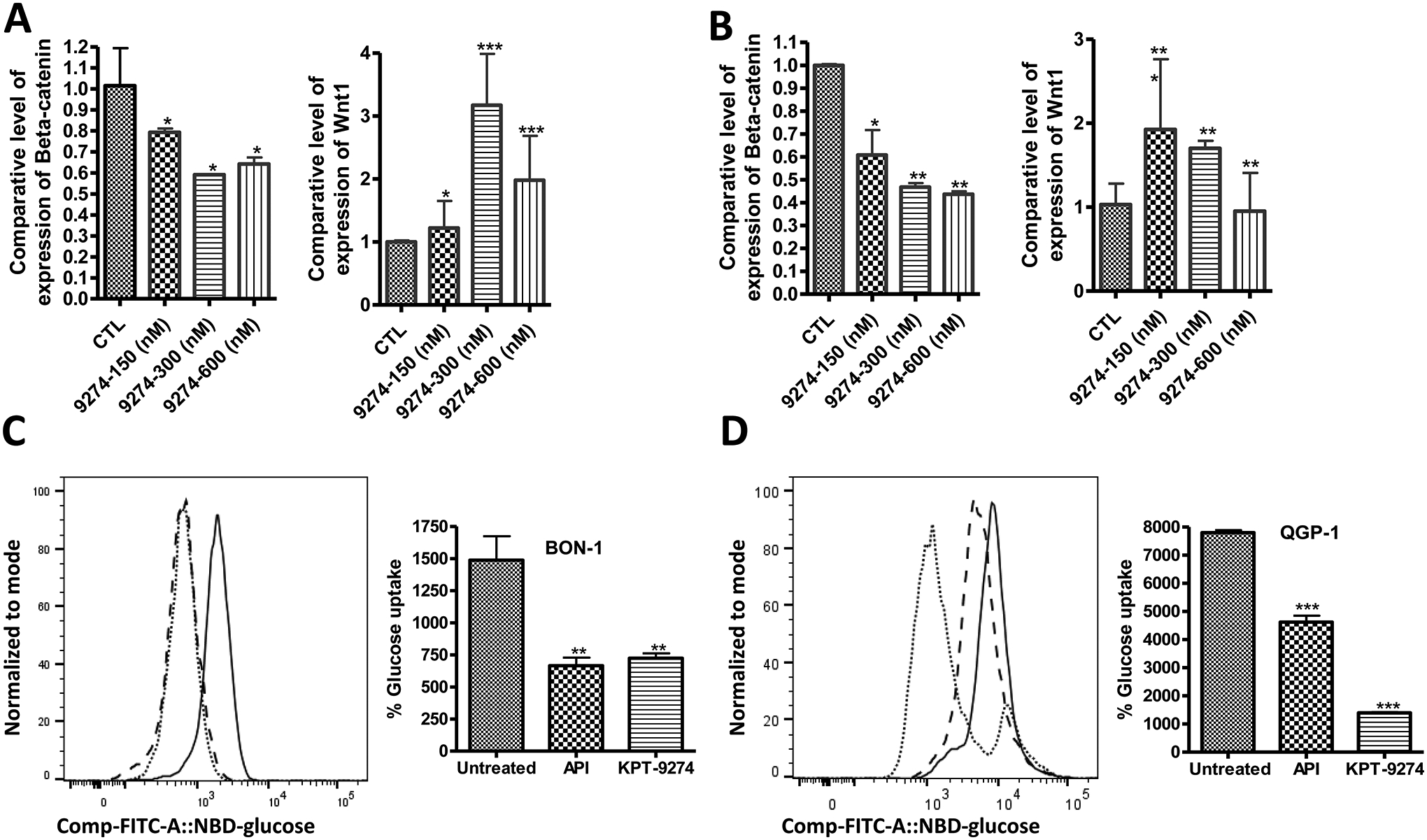
50000 BON-1 and 100000 QGP-1 cells were grown in 100 mm Petri dishes and exposed for 24 hours to the shown concentrations of the small molecule inhibitor KPT-9274 as described in the methods section. [4A, and 4B] represent RT-qPCR comparing the expression level of β-CATENIN, WNT in PNET cell lines post-treatment with increasing concentration of KPT-9274. Each expression level was normalized with actin mRNA. We used samples with RNA integrity numbers greater than 2. Each graph represents 3 independent experiments. [4C, and 4D] Glucose uptake in PNET cells 24 hours post-treatment with KPT-9274 (solid, dashed and dottle lines represent untreated, apigenin treated and KPT-9274 treated cells respectively), each condition was done in triplicate.
KPT-9274 synergizes with everolimus through the downregulation of mTORC2.
In our previous study, we showed that KPT-9274 synergizes with everolimus and Sunitinib in vitro [18]. Here, we first wanted to investigate whether KPT-9274 also synergizes with other FDA approved targeted drugs in PNETs such as the somatostatin analogs octreotide and lanreotide. In the MTT assay, no synergy was observed with octreotide and lanreotide in either BON-1 or QGP-1 (Supplementary Fig. S1). Supportive evidence for this lack of synergy comes from Annexin V FITC analysis. Octreotide (9 μM) and lanreotide (9 μM) demonstrated minimal apoptosis compared to the control samples in BON-1 and QGP-1 (Supplementary Fig. S2). It has been shown that these somatostatin analogs do not inhibit the growth of BON-1 and QGP-1 because these cell lines have a very low expression of somatostatin receptors [21]. This finding suggests that KPT-9274 can only synergize with drugs that can alter PI3K/AKT/mTOR signaling in cells. To confirm this scenario, we look at the expression level of PI3K, AKT, and mTOR post-treatment with KPT-9274 by western blot and RT-PCR. As anticipated, treatment with KPT-9274 downregulated the expression level of PI3K, AKT, and mTOR in PNET cells (Fig. 4A–4F). It was unclear whether KPT-9274 inhibits the mTORC1 or the mTORC2; therefore, we evaluated the expression level of RAPTOR and RICTOR two major components of the mTORC1 and the mTORC2, respectively. KPT-9274 dramatically inhibits the expression of RICTOR but minimally alters the expression of RAPTOR at the mRNA level (Fig. 4D; 4E). Nevertheless, western blot shows that KPT-9274 does not alter the expression of RAPTOR at the protein level. It has been shown that RICTOR is aberrantly activated when PAK4 is overexpressed [22]. We have already shown that PAK4 is overexpressed in PNET cell lines and patients tissue [18]. Thus, resistance to everolimus that is mainly due to activation of mTORC2 can be overcome by KPT-9274. In conclusion, inhibition of the mTORC2 explains how KPT-9274 synergizes with everolimus in PNETs cell lines BON-1 and QGP-1.
Figure 4: Inhibition of mTORC2 by KPT-9274 promote synergy with everolimus in PNET cell lines.
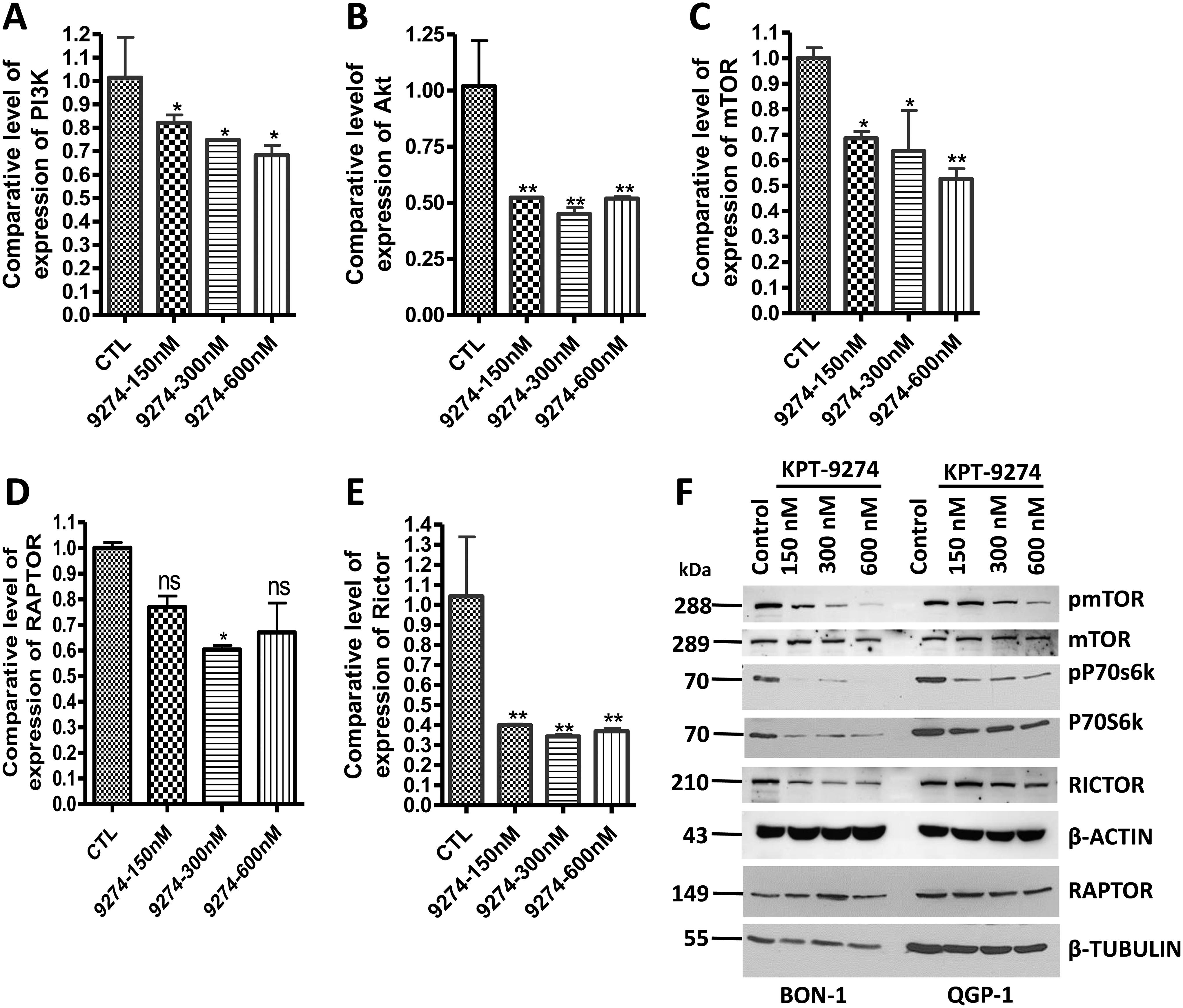
[A, B, C, D, and E] RT-qPCR comparing the expression level of PI3K, Akt, mTOR, RICTOR, and RAPTOR in PNET cell line QGP-1 post-treatment with increasing concentration of KPT-9274. Each expression level was normalized with actin mRNA. Each graph represents 3 independent experiments. [F] BON-1 and QGP-1 cells were grown in 100 mm Petri dishes and exposed to the shown concentrations of KPT-9274 as described in the methods section. 50 μg of proteins were resolved on a 10% PAGE following by western blot comparing the expression level of pmTOR, mTOR, pP70S6K, P70S6K, RICTOR, RAPTOR. β-TUBULIN was used as an internal control.
Combination of KPT-9274 and everolimus demonstrate tumors shrinkage in vivo
In our previous publication, we showed that KPT-9274 single-agent oral treatment at 150 mg/kg twice a day, five days a week (BID x 5) for four weeks resulted in a statistically significant reduction of tumor growth in BON-1 xenograft without apparent toxicity [18]. This led us to evaluate the anti-tumor activity of the combination KPT-9274-everolimus in BON-1 and QGP-1 xenograft. KPT-9274 dosed orally at 150 mg/kg in combination with everolimus at 2.5 mg/kg once a day three days a week for four weeks. As can be seen from the results, such treatment resulted in a statistically significant reduction of tumor growth of BON-1 and QGP-1 xenografts (Fig. 5A; 5D) [gross tumor images] and Fig. 5B, 5E [tumor weight]. More importantly, no overlapping toxicity (no bodyweight loss or any signs of discomfort) was observed in any of the groups undergoing treatment (Fig. 5C; 5F). Animals were randomly divided into 4 groups, however, there were slight differneces in body weights at the strating point of treatment as the animals age was between 4 and 5 weeks. These findings suggest that dual inhibition of PAK4 and NAMPT by KPT-9274 enhances the sensitivity of mTOR inhibition in PNET xenograft in vivo. A generalized schema of dual pathway inhibitions has shown in Fig. 6.
Figure 5: KPT-9274-everolimus shrink BON-1 and QGP-1 growth in vivo.
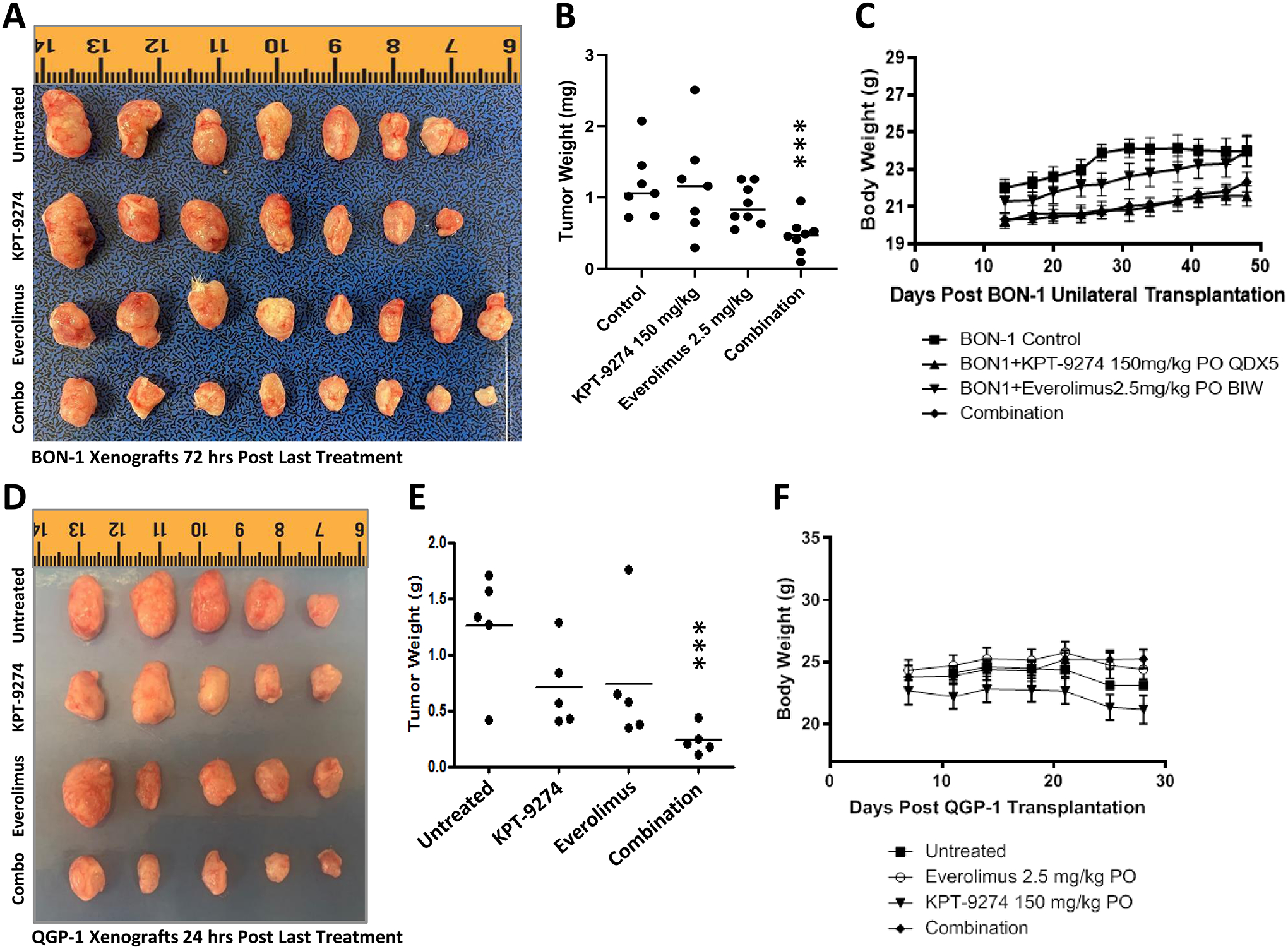
BON-1 and QGP-1 cells were grown as subcutaneous xenografts in ICR-SCID mice. [A, and D] Gross visualization of excised tumors. [B, and E] Graphical representation of tumor weight post-treatment. [C, and F] Animal body weight at the indicated days during the treatment. Five mice per group were used in the QGP-1 experiment and eight mice per group were used in the BON-1 experiment. Treatment in the BON-1 experiment was extended to 12 weeks to track eventual toxicity. [A] In the control group, one mouse developed malocclusion and was euthanized; in the KPT-9274 single-agent group, one mouse did not develop tumors (*** representing P<0.02).
Figure 6: Mechanism of action of KPT-9274 in PNET cells.
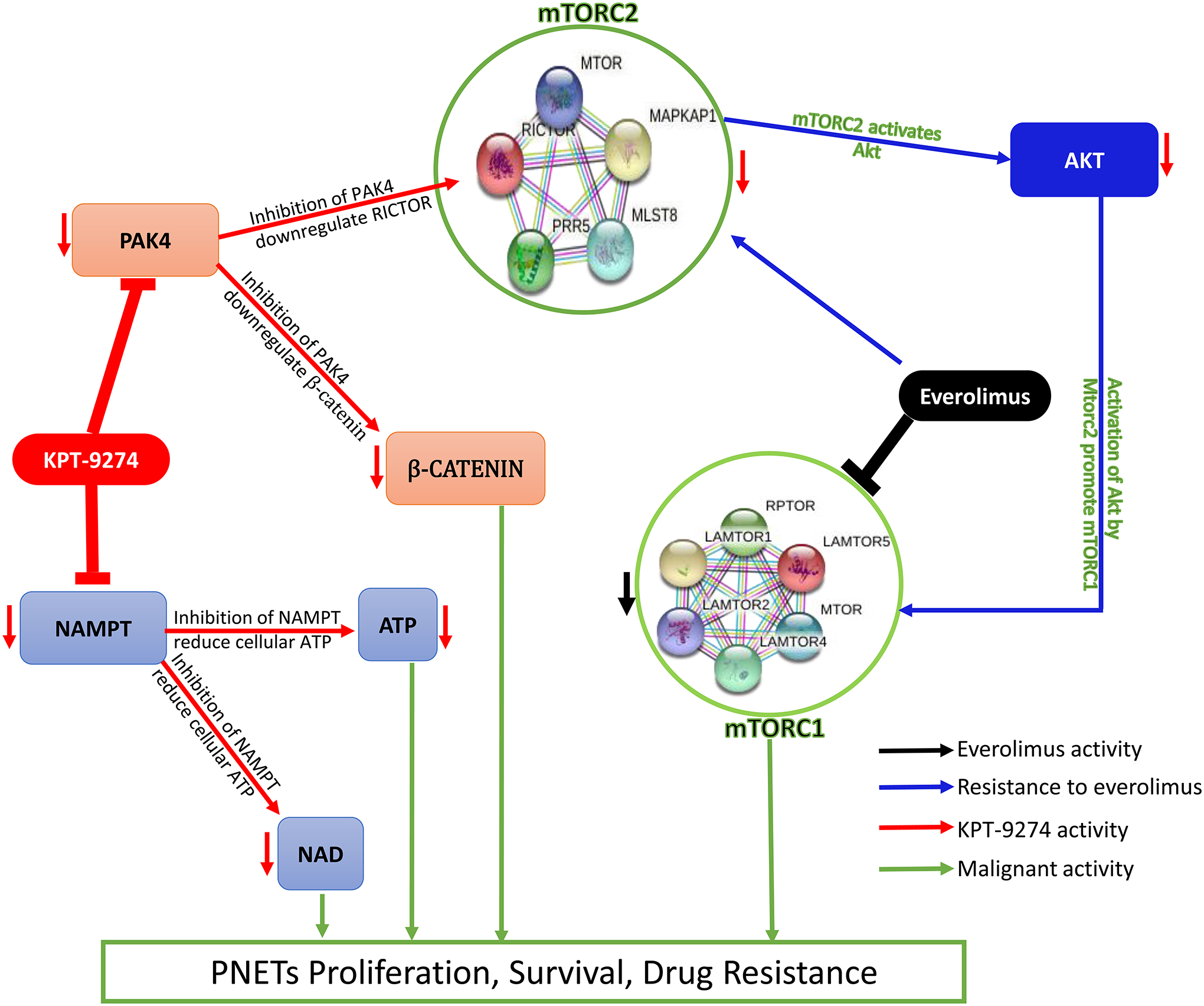
mTORC2 regulates resistance to everolimus in NET cells. Everolimus inhibits mTORC1 and its downstream effectors 4EBP-1 and P70S6K by preventing mTORC1 to interact with its intracellular receptor FKBP12 which induces mTORC2 to phosphorylate AKT thus promoting PI3K/AKT/mTOR signaling. KPT-9274 inhibits PAK4 which downregulates RICTOR then alters the activation of mTORC2. Inhibition of PAK4 also downregulates β-CATENIN. Inhibition of NAMPT reduces the cellular level of NAD and ATP. All the latter events promote PNETs shrinkage.
Discussion
In this study, we show that dual inhibition of mTOR regulator PAK and NAD biosynthesis rate-limiting enzyme NAMPT can synergize with everolimus in vitro against PNET tumor models. This combination could disrupt anchorage-independent cell growth i.e. spheroid inhibition and could efficiently suppress the growth of xenograft tumors in multiple pancreatic neuroendocrine tumor models.
Although the phosphatidylinositol-3-kinase (PI3K)/Protein Kinase B (PKB)/mammalian target of rapamycin (mTOR) is well established to play a central role in the pathogenesis and proliferation of PNETs, there is still a lack of effective approach to regulating these proteins cascade in the clinic. The mTOR inhibitor everolimus helps in delaying disease progression in a subset of patients with PNETs; however, this drug neither provides those patients with a clinically meaningful response rate nor improves their overall survival due to the development of drug resistance. Therefore, the current studies are focused on uncovering potential therapeutic targets to improve everolimus-sensitivity in PNETs. Among all genes associated with therapy resistance in cancers, we have identified PAK4 and NAMPT as two potential regulators of everolimus resistance in PNETs.
PAK4 is one of the mTOR pathway regulatory proteins and a member of the p21-activated kinases that act as regulatory switches to control important cellular processes including invasion and metastasis, survival, drug resistance, cell proliferation, and epithelial-to-mesenchymal transition [9,10,11]. Mutations leading to activation of PAK4 are rare; however, when activated by overexpression PAK4 has oncogenic signaling effects. NAMPT is the main protein that catalyzes the savage pathway of NAD synthesis in cells. This protein has been shown to be one of the mechanistic dependencies in neuroendocrine tumors in general. Earlier, we showed that PAK4 and NAMPT were aberrantly expressed in PNET cell lines and patients derived xenograft. Co-inhibition of PAK4 and NAMPT may lead to synergistic anti-tumor effects through energy depletion, inhibition of DNA repair, cell cycle arrest, inhibition of proliferation, and ultimately apoptosis. Hematologic and solid tumor cells that have become dependent on both PAK4 and NAMPT pathways may be susceptible to single-agent cytotoxicity by KPT-9274. As presented here oral KPT-9274, a PAK4-NAMPT dual inhibitor when combined with everolimus can produce a significant reduction in the size of the PNET tumors through downregulation of mTORC1 and mTORC2 signaling. Everolimus can synergize with various targeted agents. Vitale et al. showed a synergy between everolimus and demethylating agent 5-aza-2’-deoxycytidine (AZA) in chemo-radiotherapy resistant medullary thyroid cancer cells [23]. We did not observe any synergy with everolimus and somatostatin analogs which is in agreement with the findings of Krug et al. [24], however, a study hass demonstrated that lanreotide inhibited the cell growth syngergistically in everolimus resistant BON1 cells [25]. In contrast, Hue-Fontaine et al. did not observed synegy between everolimus and metformin in the diabetic patients [26] may be because of the targeting of same pathway.
Earlier attempts to develop PAK4 specific or NAMPT specific inhibitors were not that fruitful. The first PAK4 specific inhibitor PF3758309 (used in this study as control) was evaluated in phase I clinical trial (NCT00932126) but unfortunately, the trial was halted due to its poor pharmacokinetics (PK). The IV dosed FK866, the first specific inhibitor of NAMPT (also used in this study as control) was tested in phase I clinical trial on advanced/refractory solid tumors by Holen et al. [27] and it was noted that the dose escalation was limited by thrombocytopenia and there was no observed objective response to the drug.
KPT-9274 is a promising oral investigational medicine and, currently, its safety and efficacy are under vigorous investigation under a Phase 1 clinical trial (PANAMA Trial; NCT02702492) in patients with advanced solid malignancies and non-Hodgkin’s lymphoma. Early results of this phase I trial after treating 14 patients in 4 cohorts with an escalating dose of KPT-9274 starting from 10 mg and reaching up to 40 mg that grade-4 anemia was the only observed drug limiting toxicity seen at 40 mg dose but the maximally tolerated dose has not been reached yet. The most common adverse events seen in this trial were myalgia, arthralgia, and flu-like illness. The best response was stable disease seen in almost 30% of the patients (https://www.annalsofoncology.org/article/S0923-7534(20)37794-2/fulltext#%20). Preliminary data indicate that “treatment of KPT-9274 reduces NAD levels vs. baseline in circulating leukocytes and tumor biopsies. Analyses to correlate NAPRT-1 status with response is ongoing (2 of 4 SD pts are NAPRT-1)”.
In conclusion, our studies show that PAK4-NAMPT dual inhibition could be a feasible strategy either alone or in combination with everolimus in advanced PNETs that warrant further clinical investigations.
Supplementary Material
Acknowledgments
Work in the lab of Azmi AS is supported by R01CA24060701 and SKY Foundation Inc.
Conflict of Interest:
Yosef Landesman is an employee at Karyopharm Therapeutics Inc. Asfar Azmi received funding and speaker fees from Karyopharm Therapeutics Inc. for independent projects. Asfar Azmi received funding from Janssen, EISAI and Rhizen for independent projects. Asfar Azmi serves as a consultant for GLG and Guidepoint.
Authors’ Disclosures
A.S. Azmi received funding and speaker fees from Karyopharm Therapeutics Inc. for independent projects. Asfar Azmi received funding from Janssen, EISAI and Rhizen for independent projects. Asfar Azmi serves as a consultant for GLG and Guidepoint. A.S. Azmi reports grants from SKY Foundation outside the submitted work. No disclosures were reported by the other authors.
References:
- 1.Capelli P, et al. , Endocrine neoplasms of the pancreas: pathologic and genetic features. Arch Pathol Lab Med, 2009. 133(3): p. 350–64. [DOI] [PubMed] [Google Scholar]
- 2.Dasari A, et al. , Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol, 2017. 3(10): p. 1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ro C, et al. , Pancreatic neuroendocrine tumors: biology, diagnosis, and treatment. Chin J Cancer, 2013. 32(6): p. 312–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mafficini A and Scarpa A, Genomic landscape of pancreatic neuroendocrine tumours: the International Cancer Genome Consortium. J Endocrinol, 2018. 236(3): p. R161–R167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Missiaglia E, et al. , Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol, 2010. 28(2): p. 245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao JC, Shah MH, Ito T, et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N Engl J Med 2011;364:514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laplante M and Sabatini DM, mTOR signaling in growth control and disease. Cell, 2012. 149(2): p. 274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aristizabal Prada ET and Auernhammer CJ, Targeted therapy of gastroenteropancreatic neuroendocrine tumours: preclinical strategies and future targets. Endocr Connect, 2018. 7(1): p. R1–R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Svejda B, Kidd M, Kazberouk A, et al. Limitations in small intestinal neuroendocrine tumor therapy by mTor kinase inhibition reflect growth factor-mediated PI3K feedback loop activation via ERK1/2 and AKT. Cancer 2011;117:4141–4154. [DOI] [PubMed] [Google Scholar]
- 10.Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008;118:3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manser E, et al. , A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature, 1994. 367(6458): p. 40–6. [DOI] [PubMed] [Google Scholar]
- 12.Frost M, Lines K & Thakker R Current and emerging therapies for PNETs in patients with or without MEN1. Nat Rev Endocrinol 14, 216–227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Umesh PY, Tashvinder S, Pramit K, et al. Metabolic Adaptation in Cancer Stem Cells. Front. Oncol, 2020. [Google Scholar]
- 14.Alvarez MJ, et al. , A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nature Genetics, 2018. 50(7): p. 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abu Aboud O et al. (2016) Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer Growth. Mol Cancer Ther 15 (9), 2119–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neggers JE et al. (2018) Target identification of small molecules using large-scale CRISPR-Cas mutagenesis scanning of essential genes. Nat Commun 9 (1), 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aboukameel A et al. (2017) Novel p21-Activated Kinase 4 (PAK4) Allosteric Modulators Overcome Drug Resistance and Stemness in Pancreatic Ductal Adenocarcinoma. Mol Cancer Ther 16 (1), 76–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mpilla BG, et al. PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors. Cancers (Basel) in 2019. 11 (12): 1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, et al. , Nucleo-cytoplasmic shuttling of PAK4 modulates beta-catenin intracellular translocation and signaling. Biochim Biophys Acta, 2012. 1823(2): p. 465–75. [DOI] [PubMed] [Google Scholar]
- 20.Elghazi L, et al. , Importance of beta-Catenin in glucose and energy homeostasis. Sci Rep, 2012. 2: p. 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Exner S Prasad V Wiedenmann B Grötzinger C Octreotide Does Not Inhibit Proliferation in Five Neuroendocrine Tumor Cell Lines. Front Endocrinol (Lausanne). 2018;9:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cordover E, et al., KPT-9274, an Inhibitor of PAK4 and NAMPT, Leads to Downregulation of mTORC2 in Triple Negative Breast Cancer Cells. Chem Res Toxicol, 2020. 33(2): p. 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vitale G et al. , Synergistic activity of everolimus and 5-aza-2’-deoxycytidine in medullary thyroid carcinoma cell lines. Mol Oncol, 2017. 11 (8): p. 1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krug S et al. , Interaction between somatostatin analogues and targeted therapies in neuroendocrine tumor cells. PLoS One, 2019. 14 (6): p. e0218953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sciammarella C et al. (2020) Lanreotide Induces Cytokine Modulation in Intestinal Neuroendocrine Tumors and Overcomes Resistance to Everolimus. Front Oncol, 2020. 10, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hue-Fontaine L et al. (2020) Metformin and everolimus in neuroendocrine tumours: A synergic effect? Clin Res Hepatol Gastroenterol, 2020. 44 (6), 954–960. [DOI] [PubMed] [Google Scholar]
- 27.Holen K, et al. , The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest New Drugs, 2008. 26(1): p. 45–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.