

Abstract
p53 transcription factor is activated upon exposure to various cellular stresses, leading to growth suppression. However, aberrant activation of p53 can lead to defects in embryonic development and other abnormalities. Here, we identified zinc finger protein Zfp871 as a p53 target gene. We showed that as an RNA-binding protein, Zfp871 binds to Mdm2 3’UTR and stabilizes Mdm2 mRNA, which in turn suppresses p53 expression through increased expression of Mdm2 E3 ubiquitin ligase. Consistently, Zfp871 deficiency increases p53 expression, leading to growth suppression in a p53-dependent manner. To determine the role of Zfp871 in the p53 pathway, we utilized Zfp871-deficient mouse model and found that Zfp871-null mice were prone to embryonic/pre-weaning lethality, which can be partially rescued by simultaneous deletion of Trp53. We also found that mice heterozygous for Zfp871 had a short lifespan and were susceptible to steatohepatitis but not to spontaneous tumors. To determine the underlying mechanism, RNA-seq analysis was performed and showed that an array of genes involved in development, lipid metabolism and inflammation are regulated by Zfp871 in conjunction with p53. Taken together, we conclude that the Zfp871-Mdm2-p53 pathway plays a critical role in tumor-free survival and development.
Keywords: Zfp871, p53, Mdm2, mRNA stability, steatohepatitis
Introduction
Tumor suppressor p53 is a potent transcription factor and can be activated upon exposure to numerous cellular stresses. p53 manifests its tumor suppressor effects by inducing senescence, cell-cycle arrest and/or apoptosis (1,2). Due to its cogent transcriptional activity towards pro-apoptotic and cell-cycle arrest pathways, several layers for negative regulation of p53 exist that are required to maintain a fine equilibrium of p53 levels (3). This equilibrium is vital for tumor suppression, embryonic development as well as for maintaining tissue homeostasis (4). Abnormal activation of p53 during embryonic development has been linked to lethality as well as several other abnormalities that manifest through increased cell-cycle arrest and apoptosis (5,6). Even after years of extensive research on p53, most of the pathways regulating the p53 network are yet to be elucidated (1,2).
Zinc finger proteins are one of the most abundant and diverse groups of proteins with a wide range of cellular functions (7). They exhibit their key functions by binding to DNA, RNA and proteins, thereby regulating transcription, mRNA stability and protein ubiquitination, respectively (8,9). There are several families of proteins containing zinc fingers, of which the C2H2 zinc finger family is the most abundant and primarily consists of proteins with multiple repeats of zinc fingers fused with a functional domain such as the KRAB domain (8,10–12). Despite their abundance, the physiological functions for a majority of KRAB-containing zinc finger proteins remain to be characterized. Interestingly, several regulators of p53, such as Mdm2, Pirh2, and Apak (ZNF420), contain zinc fingers (13–15).
Previously, we showed that Zfp871, a KRAB-containing C2H2 zinc finger protein, interacts with Mdm2 and regulates p53 expression (16). Here, we established Zfp871 as a p53 target gene that in turn negatively regulates p53 via increased mRNA stability of Mdm2, a p53 E3 ubiquitin ligase (17). We showed the significance of the Zfp871-Mdm2-p53 pathway in growth suppression and regulation of an array of genes involved in development, lipid metabolism and inflammation. We also showed that simultaneous deletion of Trp53 in Zfp871-null mice could partially rescue the embryonic/pre-weaning lethality. Notably, mice heterozygous for Zfp871 had a shorter lifespan and were susceptible to steatohepatitis but not to spontaneous tumors. Our findings suggest that Zfp871 is an important regulator of p53 in development and disease.
Materials and Methods
Cell culture, cell line generation and reagents
SCp2 mouse mammary epithelial cells were a generous gift from Pierre-Yves Desprez (California Pacific Medical Center Research Institute) and were maintained as previously described (18). The mouse myoblast cell line C2C12, the mouse normal liver FL83B cells, human cancer cell lines HCT116, MCF-7 and RKO were obtained from ATCC between 2007 and 2018 and used below passage 20 or within 2 months of thawing. As ATCC authenticated and tested these cell lines, no further authentication and Mycoplasma testing were performed, especially considering that these cell lines were used at low passages. Mouse C2C12, mouse FL83B cells, human HCT116, MCF-7 and RKO cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT). Generation of Zfp871-KO C2C12 and SCp2 and Trp53-KO SCp2 cell lines was achieved by pSpCas9(BB)-2A-Puro vector expressing guide RNAs (Zfp871–1: 5′- GCA AAA TTT GCA GTA TAA GG −3′; Zfp871–2: 5′- TTT CAA ACT CAA CTA CCC TC −3′; p53: 5’- GAA GTC ACA GCA CAT GAC GG - 3’). The cells were selected with puromycin, and individual clones picked, genotyped, sequenced, and confirmed by western blot analysis. The primers used for genotyping were: (1) For Zfp871, forward primer, 5’- CCC TTT AAT CGG TGA AAG AAA GCA −3’, and reverse primer, 5’- GGC ACA ATG ATG CTC ACG TTA T −3’; (2) For mouse p53, forward primer, 5’- CAG CTG GCG AAG ACG TGC −3’, and reverse primer, 5’- CGG GAT ACA AAT TTC CTT CCA CC −3’.
Western blot analysis
Western blotting was performed as previously described (19). Briefly, cell lysates were collected as indicated, resolved on 8–11% SDS-polyacrylamide gels and transferred to nitrocellulose membrane. After the transfer, the membranes were blocked with PBST containing 2.5% milk at room temperature (RT) for 1 hour, followed by overnight 4°C incubation with primary antibodies prepared in 2.5% milk containing PBST. The following day, after 3X washings with PBST, the blots were incubated at RT for 1 hour in secondary antibody prepared in PBST containing 2.5% milk. After 3X washings with PBST, the blots were soaked in enhanced chemiluminescence reagents (ThermoFisher Scientific™), and then visualized with the BioSpectrum 810 Imaging System (UVP LLC, Upland, CA). Antibodies used were: Mdm2 (sc-965), p21 (sc-53870), actin (sc-133155), GFP (sc-9996), PML (sc-5621), and p130 (sc-374521) purchased from Santa Cruz Biotechnology (Santa Cruz, CA); HA (W15093A) from BioLegend (San Diego, CA); p53 (#1C12) from Cell Signaling (Beverly, MA); Mdm2 (OP115) from Millipore-Sigma (Burlington, MA). Zfp871 antibody was custom-made in rabbit using amino acids 41–189 of mouse Zfp871 by Cocalico biologicals, Inc.
RNA isolation and RT-PCR
Total RNA was harvested using TRIzol reagent (Invitrogen) and isolated according to the manufacturer’s instructions. RevertAid First Strand cDNA Synthesis kit (ThermoFisher Scientific™) was used to synthesize cDNA from 2 μg total RNA as per manufacturer’s protocol. The primers used to amplify mouse actin were forward primer, 5’-TCC ATC ATG AAG TGT GAC GT-3’, and reverse primer, 5’-TGA TCC ACA TCT GCT GGA AG-3’. The primers used to amplify Zfp871 were the same as previously described (16). The primers used to amplify mouse p21 were forward primer, 5’- GTA CTT CCT CTG CCC TGC TG −3’, and reverse primer, 5’- TCT GCG CTT GGA GTG ATA GA −3’. The primers used to amplify mouse Mdm2 were forward primer, 5’- ATG AGG TCT ATC GGG TCA CAG T −3’, and reverse primer, 5’- CAC ATC CAA GCC TTC TTC TGC −3’. The primers for mouse Mdm2 pre-mRNA were forward primer, 5′- CCT CAT GTT CTT TGC CTA TTT ATG −3′, and reverse primer, 5′- AGC CAG TTC TCA CGA AGG GT −3. The following program was used for PCR amplification: (i) 95 °C for 3 min, (ii) 95 °C for 30 s, (iii) 60 °C for 30 s, (iv) 72 °C for 45 s and (v) 72 °C for 5 min. From steps 2–4, the cycle was repeated 23 times for Actin and Gapdh, 25 times for p21 and Mdm2, or 26 times for Zfp871.
sgRNAs to generate knockout cell lines and siRNAs for knockdown
To generate Zfp871-KO cells, two single-guide RNA (sgRNA) expression vectors pSpCas9(BB)-2A-Puro-sgZfp871–1 and pSpCas9(BB)-2A-Puro-sgZfp871–2 were used to remove exon 3 and create frame shift deletions. The generation of sgRNA expression vector was performed as described previously (20). The oligonucleotides for sgZfp871–1 are sense, 5’- CAC CGG CAA AAT TTG CAG TAT AAG G −3’, and antisense, 5’- AAA CCC TTA TAC TGC AAA TTT TGC C −3’; for sgZfp871–2 are sense, 5’- CAC CGT TTC AAA CTC AAC TAC CCT C −3’, and antisense, 5’- AAA CGA GGG TAG TTG AGT TTG AAA C −3’. To generate Trp53-KO cells, one sgRNA expression vector pSpCas9(BB)-2A-Puro-sgp53Ex5 was used to target coding region in exon 5 to create frame shift deletions. The oligonucleotides for sgp53Ex5 are sense, 5’- CAC CGG AAG TCA CAG CAC ATG ACG G −3’, and antisense, 5’- AAA CCC GTC ATG TGC TGT GAC TTC C −3’.
All small interfering RNAs (siRNAs) were purchased from Dharmacon RNA Technologies (Chicago, IL). Zfp871 specific siRNAs have been described previously (16). The scrambled siRNA, 5’-GCA GUG UCU CCA CGU ACU A dTdT-3’, was used as a control.
RNA-IP assay
RNA immunoprecipitation was carried out as described previously (21). Briefly, SCp2 cells and MEFs (WT and Zfp871−/−) were grown to 80–90% confluence in a 100 mm dish (~2 × 106 cells) and cell extracts were prepared in immunoprecipitation buffer (10mM HEPES, pH 7.0, 100mM KCl, 5mM MgCl2, 0.5% Nonidet P-40, and 1mM DTT), pre-cleared with protein A/G beads, and then incubated with 2 μg of anti-Zfp871 or an isotype control IgG overnight at 4 °C. The RNA–protein immunocomplexes were brought down using magnetic protein A/G beads (MedChemExpress). Following washing and RNA-purification, RT-PCR analysis was carried out to determine the levels of Mdm2 and Gapdh transcripts.
mRNA half-life assay
To measure the mouse Mdm2 mRNA stability, wild type and Zfp871−/− MEF cells were treated for various times with 5, 6-dichloro-1-beta-Dribofuranosylbenzimidazole (DRB), an inhibitor of transcription. The relative level of Mdm2 mRNA was quantified using ImageJ software and normalized using the level of Actin mRNA. Values were plotted versus time and the half-life of Mdm2 mRNA was calculated.
Mice
The use of animals and the study protocols were approved by the University of California at Davis Institutional Animal Care and Use Committee. Zfp871+/− mice were generated by the Mouse Biology Program at the University of California, Davis (Davis, CA, USA). Trp53+/− mice were obtained from the Jackson Laboratory (Sacramento, CA, USA) as described previously (22). Zfp871+/−; p53+/− mice were generated by intercrossing Zfp871+/− mice with Trp53+/− mice. To genotype the Zfp871-WT allele, we used the forward primer, 5’- TAG TCT GGC CTC GAA CTC AGA ACC −3’, and the reverse primer, 5’- TCT CAG AGG CTG CTG TGC AAG G −3’. To genotype the Zfp871-KO allele, we used the forward primer, 5’- GGG ATC TCA TGC TGG AGT TCT TCG −3’ and reverse primer same as for WT allele. The primers to genotype Trp53-WT and KO alleles have been described previously (23).
Mouse embryonic fibroblast (MEF) isolation
MEFs were isolated from 12.5–13.5 days post-coitum (pc) mouse embryos, as described previously (22). To generate WT, Trp53−/− and Zfp871−/−; Trp53−/− MEFs, Zfp871+/−; Trp53+/− mice were inter-crossed. To generate WT, Zfp871+/− and Zfp871−/− MEFs, Zfp871+/− mice were interbred. The MEFs were cultured in DMEM supplemented with 10% FBS (Hyclone Laboratories, Erie, PA, USA), 55 μM β-mercaptoethanol and 1x non-essential amino acids solution (Cellgro, Manassas, VA, USA).
Histological analysis
Mouse tissues were fixed in 10% neutral buffered formalin, processed, and embedded in paraffin blocks. Tissue blocks were sectioned (6 μm) and stained with Hematoxylin and Eosin.
Statistical Analysis
Mice survival analysis was performed by Kaplan–Meier survival analysis using the log rank test. Data are presented as means ± standard error of the mean (SEM) or means ± standard deviation (SD). The p values for qRT-PCRs and colony formation assays was calculated using the two-tailed Student’s t-tests, and p < 0.05 was considered statistically significant. For each experimental data point, n = 3. Excel (Microsoft, Redmond, WA) was used for statistical analyses.
Additional Materials and Methods
The detailed methods and information for RNA-seq, chromatin immunoprecipitation (ChIP), colony formation, proliferation assays and EGFP reporter assays are provided in Supplementary Materials and Methods.
Results
Zfp871 is induced by DNA damage in a p53-dependent manner
As Zfp871 is implicated in PCBP4-dependent p53 regulation (16), we examined whether Zfp871 is induced following cytotoxic stress. For this, mouse SCp2 mammary epithelial cells, in which endogenous p53 is wild-type (WT), were treated with either camptothecin (CPT) or doxorubicin (DOX) for 24 hours and examined for proteins and mRNAs of interest. We observed that the levels of Zfp871 protein and mRNA were increased with CPT or DOX treatment (Figure 1A, B and E). As positive controls, we showed that p53 and its target p21 were also increased (Figure 1A, B and E). To further test this, mouse C2C12 and FL83B cells, both of which carry an endogenous WT p53, were treated with DOX for various times. We observed that the levels of Zfp871 protein were increased 24-hour post-treatment, which sustained till 48 hours (Figures 1C and D). Similarly, we observed that the levels of Zfp871 mRNA were increased 12 hours after DNA damage and remained elevated 48 hours post DNA damage (Figures 1F and G).
Figure 1-. Zfp871 is induced by DNA damage in a p53-dependent manner.

(A, B) The levels of p53, p21, Zfp871 and Actin proteins were measured by western blotting in SCp2 cells mock-treated or treated with 250 nM CPT (A) or 350 μg/ml Dox (B) for 24 h. (C, D) The levels of p53, Zfp871 and Actin were measured in C2C12 (C) and FL83B (D) cells mock-treated or treated with 250 μg/ml Dox for 12–48 h. (E) The levels of Zfp871 and p21 mRNAs were measured by qRT-PCR in SCp2 cells mock-treated or treated with 250 nM CPT or 350 μg/ml Dox for 24 h. (F, G) The levels of p21, Zfp871 and Actin mRNA were measured by RT-PCR in C2C12 (F) and FL83B (G) cells mock-treated or treated with 250 μg/ml Dox for 12–48 h. (H, I) The levels of p53, p21, Zfp871, and Actin proteins (H) and mRNAs (I) were measured in isogenic control and Trp53−/− SCp2 (clone #25 or #35) cells, which were mock-treated or treated with 350 μg/ml Dox for 24 h. (J, K) The levels of p53, p21, Zfp871, and Actin proteins (J) and mRNAs (K) were measured in Trp53−/− SCp2 (clone #25) cells that were transiently transfected with either empty vector or mouse p53 for 24 h. (L) Schematic diagrams of the Zfp871 and p21 promoters along with putative p53-RE sites. Also shown are the locations of primers on Zfp871 and p21 promoters for chromatin immunoprecipitation (ChIP) assays. (M) Cell lysates purified from SCp2 cells mock-treated or treated with 200 nM CPT for 16 h were immunoprecipitated with control IgG or anti-p53 antibody, followed by RT-PCR to determine the binding of p53 to the p53 REs in the Zfp871 and p21 promoters.
To determine whether Zfp871 is induced by DNA damage in a p53-dependent manner, multiple Trp53−/− SCp2 cell lines were generated and used. As expected, the levels of basal and DNA-damage induced p21 protein and mRNA were much lower in two separate Trp53-KO clones as compared to that in isogenic control cells (Figure 1H and I). Similarly, the levels of Zfp871 protein were much lower in Trp53-KO cells as compared to isogenic control. Moreover, the level of Zfp871 was not further induced by DNA damage in Trp53-KO cells as compared to isogenic control (Figure1H, I; Supplementary Figure S1A, B). Furthermore, the levels of Zfp871 mRNA and protein were increased by ectopic expression of p53 in Trp53−/− SCp2 cells (Figure 1J and K). These data suggest that Zfp871 is induced by DNA damage in a p53-dependent manner.
To determine whether Zfp871 is a target of p53, we searched for potential p53-responsive element(s) (p53-RE) in the genomic locus of the Zfp871 gene. We found three putative REs – one in the promoter region and two in intron 1 of the Zfp871 gene (Figure 1L). To determine whether these putative p53-REs are recognized by p53, chromatin immunoprecipitation (ChIP) was performed and showed that p53-RE1 and p53-RE2 but, not p53-RE3, were recognized by p53 following DNA damage (Figure 1M). The binding of p53 to the p21 promoter was also detected and used as a positive control (Figure 1L-M). Thus, p53 directly regulates Zfp871 expression upon cellular stress.
Zfp871 negatively regulates mouse and human p53 expression
Several p53 target genes, such as Mdm2 and Rbm38, in turn regulate p53 expression forming a feedback loop (24,25). Thus, mouse Zfp871 was transiently expressed in multiple mouse and human cancer cells to corroborate our previous findings (16). We showed that the levels of p53 protein were decreased by over-expression of Zfp871 in mouse SCp2 and C2C12 cells as well as in human RKO, MCF7, and HCT116 cells (Figure 2A-E). These data suggest that the Zfp871-p53 loop is conserved across species, which is likely carried out by some close relatives of murine Zfp871 in humans. Conversely, we showed that upon knockdown of Zfp871, the level of p53 protein was markedly increased in mouse SCp2, C2C12, and FL83B cells (Figures 2F-H). To confirm this, we generated a cohort of WT, Zfp871+/− and Zfp871−/− MEFs. We observed that the levels of p53 protein along with its target p21 were increased in Zfp871+/− and Zfp871−/− MEFs as compared to WT MEFs from two separate MEF isolates (Figure 2I). These data indicate that Zfp871 negatively regulates mouse and human p53 expression.
Figure 2 -. Zfp871 negatively regulates mouse and human p53 expression.

(A-E) The levels of Zfp871, p53 and Actin proteins were measured by western blotting in SCp2 (A), C2C12 (B), RKO (C), MCF7 (D) and HCT116 (E) cells that were transiently transfected with either empty vector or HA-Zfp871 for 24 h. (F-H) The levels of Zfp871, p53 and Actin proteins were measured in SCp2 (F), C2C12 (G) and FL83B (H) cells that were transfected with 50 nM control or Zfp871 siRNA for 72 h. (I) The levels of Zfp871, p53, p21, and Actin were measured in two sets of WT, Zfp871+/−, and Zfp871−/− MEFs. Low and high exposures of the same p53 blot are shown in the right panel.
Zfp871 binds and positively regulates Mdm2 mRNA stability
Mdm2 is a major regulator of p53 that controls p53 protein stability through its E3 ubiquitin ligase activity (17). Based on our earlier observations, we found that Zfp871 can regulate p53 protein stability as a part of the Mdm2 E3 ligase complex (16). However, it is also possible that Zpf871 may directly regulate Mdm2 expression to modulate p53 protein stability. Indeed, we found that the level of Mdm2 protein was decreased, whereas the level of p53 protein was increased, in Zfp871-KO SCp2 cells as compared to that in isogenic control (Figure 3A). On the contrary, overexpression of Zfp871 led to increased Mdm2 expression along with decreased expression of p53 (Figure 3B). These data suggest that Zfp871 regulates Mdm2 expression independent of p53. Next, to determine whether an increased expression of Mdm2 protein was due to increased expression of Mdm2 transcript by Zfp871, we measured Mdm2 mRNA in SCp2 cells transfected with Zfp871. We found that the level of Mdm2 mRNA was increased by ectopic expression of Zfp871 (Figure 3C). To confirm this, the levels of Mdm2 protein and mRNA were examined in three different sets of MEFs. We observed that the levels of Mdm2 protein and mRNA were markedly decreased in Zfp871−/− MEFs as compared to their littermate control WT MEFs (Figure 3D-F; Supplementary Figure S2A-B). Of note, we did not observe a significant decrease of Mdm2 protein and only a slight decrease in Mdm2 mRNA in Zfp871+/− MEFs compared to WT MEFs (Supplementary Figure S2C, S2D and S2E), suggesting that Zfp871 may be haplo-insufficient in regulating Mdm2 expression. To corroborate that Zfp871 regulates Mdm2 independent of p53, the levels of Mdm2 protein were determined in WT, Zfp871−/−, Trp53−/− and Zfp871−/−; Trp53−/− MEFs. Consistent with the above observations, the level of Mdm2 protein was decreased in Zfp871−/− MEFs as compared to WT MEF (Figure 3G). As a target of p53, Mdm2 expression was decreased in Trp53−/− MEFs as compared to WT MEFs, as expected (Figure 3G). Notably, we observed that the level of Mdm2 protein was further decreased in Zfp871−/−; Trp53−/− MEFs as compared to that in Zfp871−/− or Trp53−/− MEFs (Figure 3G). Moreover, Mdm2 expression was increased by ectopic expression of Zfp871 in Zfp871−/−; Trp53−/− double knockout MEFs (Figure 3H). These data provide strong evidence that Zfp871 regulates Mdm2 independent of p53.
Figure 3 -. Loss of Zfp871 leads to decreased expression of Mdm2 protein and transcript.

(A) The levels of Zfp871, p53, Mdm2 and Actin proteins were measured by western blotting in isogenic control and Zfp871−/− clone #33. (B, C) The levels of Zfp871, p53, Mdm2 and Actin proteins (B) and mRNAs (C) were measured in SCp2 cells transiently transfected with either empty vector or HA-Zfp871 for 24 h. The Zfp871, p53 and Actin blots depicted in Figure 3B are the same as the ones shown in Figure 2A as the same lysates were used to probe Mdm2 and Actin. (D, E) The levels of Zfp871, Mdm2, and Actin proteins (D) and mRNAs (E) were measured in two sets of WT and Zfp871−/− MEFs. (F) The levels of Mdm2 mRNA were measured by qRT-PCR in WT and Zfp871−/− MEFs. (G) The levels of Zfp871, Mdm2, and Actin proteins were measured in WT, Zfp871−/−, Trp53−/−, and Zfp871−/−; Trp53−/− MEFs. Low and high exposures of the same Mdm2 blot are shown. (H) The levels of Zfp871, p53, Mdm2 and Actin proteins were measured in Zfp871−/−; Trp53−/− MEFs transiently transfected with empty vector or HA-Zfp871 for 24 h. (I, J) The levels of Mdm2 pre-mRNA were measured by RT-PCR in WT, Zfp871−/−, Trp53−/−, and Zfp871−/−; Trp53−/− MEFs as indicated. Low and high exposures of the Mdm2 pre-mRNA from the same gel are shown in Figure 3J.
To determine whether the level of Mdm2 mRNA increased by Zfp871 is due to increased transcription or mRNA stability, we measured Mdm2 pre-mRNA by RT-PCR with one primer corresponding to the unspliced intron 11 and the other corresponding to exon 12. We showed that the levels of Mdm2 pre-mRNA were increased in Zfp871−/− and Zfp871+/− MEFs as compared to WT MEFs (Figure 3I; Supplementary Figure S2F). We hypothesized that loss of Zfp871 leads to increased expression of p53 (Figure 3A), which in turn promotes transcription of Mdm2, resulting in an increased level of Mdm2 pre-mRNA. To test this, Mdm2 pre-mRNA was measured in WT, Zfp871−/−, Trp53−/− and Zfp871−/−; Trp53−/− MEFs. We observed that the increase in Mdm2 pre-mRNA seen in Zfp871−/− MEFs disappeared in Zfp871−/−; Trp53−/− MEFs (Figure 3J), suggesting that the increased pre-mRNA observed upon Zfp871-KO is indeed a consequence of increased p53.
Based on above observations that Zfp871 increased Mdm2 pre-mRNA but decreased mature Mdm2 mRNA, we theorized that Zfp871 might bind Mdm2 mRNA and positively regulate its stability. To test this, RNA immunoprecipitation (RNA-IP) assay was performed and showed that Mdm2 mRNA was detected in anti-Zfp871 immunoprecipitates from SCp2 cells (Figure 4A) and WT MEFs but, not Zfp871−/− MEFs (Figure 4B). The binding of Zpf871 to Gapdh mRNA was measured as a negative control and found to be negligible (Figure 4A-B). Furthermore, to test whether Zfp871 regulates Mdm2 mRNA stability, the half-life of Mdm2 mRNA was measured in WT and Zfp871−/− MEFs treated with DRB (5,6-Dichlorobenzimidazole-β-D-ribofuranoside) for various times. DRB inhibits RNA polymerase II-mediated transcription elongation step and thus, impedes de novo mRNA synthesis (26). We showed that DRB treatment led to rapid decrease of Mdm2 transcript in Zfp871−/− MEFs as compared to that in WT MEFs (Figure 4C). The half-life was decreased from 3.75 hours in WT MEFs to 1.65 hours in Zfp871−/− MEFs (Figure 4D).
Figure 4 -. Zfp871 binds to Mdm2 transcript and positively regulates Mdm2 mRNA stability.

(A) Lysates from SCp2 cells were subjected to RNA-IP using anti-Zfp871 antibody to determine the binding of Zfp871 to Mdm2 mRNA. The binding of Zfp871 to Gapdh mRNA was used as a negative control. Control IgG was used as a RNA-IP control. (B) RNA-IP experiment was performed as in (A) except lysates from WT and Zfp871−/− MEFs were used. (C) The levels of Zfp871, Mdm2 and Actin transcripts were measured in WT and Zfp871−/− MEFs mock-treated or treated with DRB for indicated times. Low and high exposures of the Mdm2 transcript from the same gel are shown. (D) The relative levels of Mdm2 transcript in (C) were normalized to actin and the half-life of Mdm2 transcript was calculated by plotting the remaining mRNA over time. (E) Schematic representation of the mouse Mdm2 transcript and the locations of three 3’UTR fragments in EGFP reporter vectors. (F-I) Various amounts of HA-Zfp871 expression vector were transfected into Zfp871−/−; Trp53−/− MEFs together with a fixed amount of EGFP only (F) or EGFP plus Full-length (FL) Mdm2 3’UTR (G), EGFP plus fragment-A (H), or EGFP plus fragment-B (I). The levels of HA-Zfp871, EGFP and actin proteins were examined by western blotting.
To further corroborate the regulation of Mdm2 mRNA stability by Zfp871 and to determine the Zfp871-binding region in Mdm2 3’UTR, 4 reporter constructs were generated (Figure 4E), which were then transfected into Zfp871−/−; Trp53−/− MEFs together with varying amounts of HA-Zfp871 expression vector. We found that ectopic expression of Zfp871 had little if any effect on expression of GFP reporter from the construct that contained only the GFP coding sequence (Figure 4F). Interestingly, Zfp871 was able to increase GFP expression from the construct in which the GFP ORF was either fused to the full-length mouse Mdm2 3’UTR or fragment A of Mdm2 3’UTR but, not fragment B (Figure 4G-I). These results suggest that Zfp871 binds the A-fragment of mouse Mdm2 3’UTR and stabilizes it.
Cells deficient in Zfp871 are prone to cellular senescence and growth suppression in a p53-dependent manner
To assess the effect of the Zfp871-Mdm2-p53 axis on growth suppression, we created Zfp871-KO C2C12 and SCp2 mouse cell lines using CRISPR-Cas9. We showed that the levels of p53 protein were increased in Zfp871-KO and Zfp871-het cells (Figure 5A, D and Supplementary Figures S3A, S3D), consistent with the above observations that siRNA knockdown of Zfp871 led to increased expression of p53 in SCp2 and C2C12 cells (Figure 2F-G). To examine whether Zfp871 deficiency has an effect on cell proliferation, clonogenic assay was performed and showed that the ability of Zfp871-KO and Zfp871-het cells to form colonies was significantly decreased as compared to isogenic controls (Figures 5B, C, E and F; Supplementary Figures S3B, S3C, S3E and S3F).
Figure 5 -. Cells deficient in Zfp871 are prone to cellular senescence and growth suppression in a p53-dependent manner.
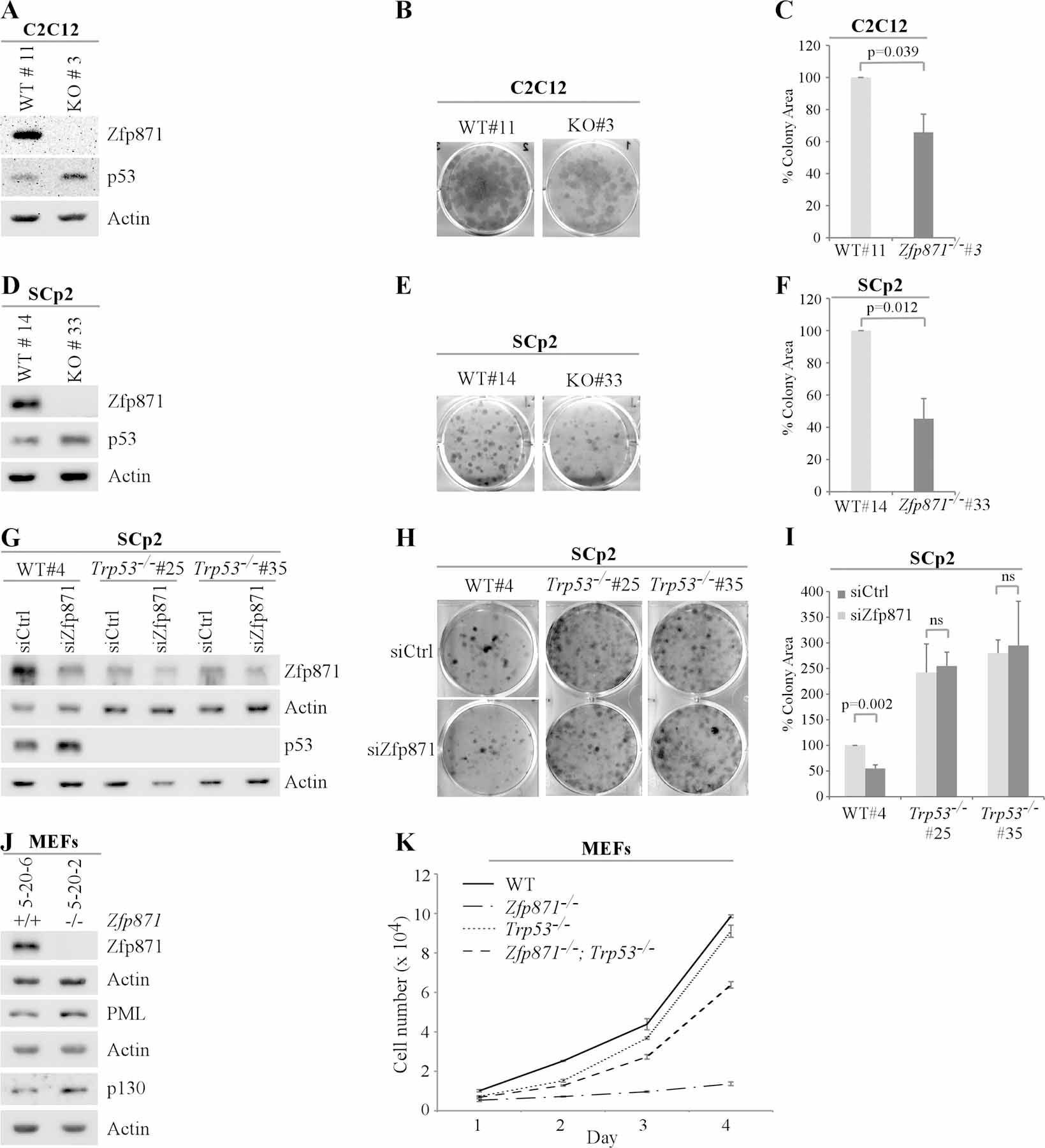
(A, D) The levels of Zfp871, p53 and Actin proteins were measured in isogenic control and Zfp871−/− C2C12 (clone #3) cells (A) or isogenic control and Zfp871−/− SCp2 (clone #33) cells (D). (B, E) Isogenic control and Zfp871−/− C2C12 (clone #3) cells (B) or isogenic control and Zfp871−/− SCp2 (clone #33) cells (E) were plated for colony formation assays and grown for one week followed by fixation with methanol: acetic acid (7:1) and staining with crystal violet. (C, F) The colonies obtained from the experiments in B and E were quantified using ColonyArea in ImageJ software and plotted as percentage of colony area. The density of colonies from isogenic control cells was set at 100%. (G) The levels of Zfp871, p53, and Actin proteins (G) were measured in isogenic control, Trp53−/− #25, and Trp53−/− #35 SCp2 cells transfected with control or Zfp871 siRNA for 72 h. (H) The cells treated as in (G) were plated for colony formation assays 24 h after transfection and grown for one week followed by fixing with fixation with methanol: acetic acid (7:1) and staining with crystal violet. (I) The colonies obtained from the experiment in (H) were quantified using ColonyArea in ImageJ software and plotted as percentage colony area. The density of colonies from isogenic control cells transfected with control siRNA was set at 100%. (J) The levels of Zfp871, PML, p130, and Actin proteins were measured in WT and Zfp871−/− MEFs. (K) The rate of cell proliferation was measured for WT, Zfp871−/−, Trp53−/−, and Zfp871−/−; Trp53−/− MEFs plated in a 6-well plate over a 4-day period.
To determine whether the effect of Zfp871 deficiency on cell growth is p53-dependent, Zfp871 was transiently knocked down in two separate Trp53-KO SCp2 clones as well as in an isogenic control clone (Figure 5G). These cells were then used for colony formation assays. When transfected with control siRNA, Trp53-KO cells were highly competent to form colonies as compared to isogenic control cells (Figure 5H, top panel). However, when Zfp871 was knocked down by siRNA, the ability of Trp53-KO cells to form colonies was not changed whereas the ability of isogenic control cells to form colonies was markedly inhibited (Figures 5H, bottom panel and Figure 5I). These data suggest that the decreased cell proliferation in Zfp871-deficient cells is p53-dependent.
Since increased p53 expression often leads to cellular senescence, we examined whether Zfp871 deficiency has any effect on cellular senescence in Zfp871−/− MEFs. As the Zfp871−/− MEFs contained a LacZ cassette (Supplementary Figure S4A), senescence-associated β-gal staining would not be feasible to measure the extent of cells undergoing cellular senescence. Instead, various senescence markers, such as promyelocytic leukemia protein (PML) and p130, were measured. As shown in Figure 5J, PML and p130 were highly expressed in Zfp871−/− MEFs as compared to wild-type MEFs (Figure 5J). To further test this, a cohort of MEFs with various genotypes (WT, Trp53−/−, Zfp871−/− and Trp53−/−; Zfp871−/−) was used for cell proliferation assay. WT and Trp53−/− MEFs showed similar growth rates (Figure 5K). Interestingly, the rate of cell proliferation was suppressed by knockout of Zfp871, which was restored by simultaneously knockout of Trp53 (Figure 5K). Together, these findings indicate that increased p53 is indeed responsible for decreased cell proliferation observed in Zfp871−/− MEFs and Zfp871-KD SCp2 cells.
Zfp871−/− mice exhibit embryonic/pre-weaning lethality in part through accumulation of p53
To determine the biological function of Zfp871, we obtained Zfp871 heterozygous mice, which were generated using the knock-out first approach at Mutant Mouse Regional Resource Center (MMRRC) (Supplementary Figure S4A and S4B). We found that Zfp871+/− mice developed normally and were fertile. However, out of 52 pups and 10 litters from Zfp871-het mouse interbreed, one Zfp871−/− mouse was born alive with less than half the size of Zfp871+/− littermate and survived for 17 days prior to weaning (Supplementary Table S1 and Figure S4C). These results suggest that Zfp871 is required for embryonic development and survival.
Deletion of the Mdm2 gene in mice leads to embryonic lethality due to aberrant expression of p53, which can be rescued by simultaneous deletion of Trp53 (27,28). Since Zfp871 deletion increased p53 expression, we postulated that deletion of Trp53 may render Zfp871−/− mice viable. Therefore, Zfp871+/−; Trp53+/− mice were interbred, resulting in 7 litters and 33 pups. Three Zfp871−/−; Trp53+/− pups were born alive, although four Zfp871−/−; Trp53+/− and two Zfp871−/− pups were expected from the interbreeds based on Mendelian ratio (Supplementary Table S2). Additionally, Zfp871−/−; Trp53+/− pups were only half the size of Zfp871+/−; Trp53+/− littermate. Two died a day after birth and one survived for 24 days (Supplementary Figure S4C). These data suggest that embryonic lethality and pre-weaning death by deletion of Zfp871 can be partially rescued by simultaneous deletion of Trp53.
Zfp871+/− mice have a short lifespan and are prone to steatohepatitis
As Zfp871+/− mice are viable, a cohort of Zfp871+/− mice were generated and monitored for their long-term survival and predisposition to tumors and other abnormalities. To minimize the number of animals used, the data for some of the WT mice were adapted from two previous studies (23,29). All the mice were derived from the same C57BL/6 background and maintained in the same facility. Since some Zfp871+/− and WT littermates were kept in the same cage, we observed that Zfp871+/− mice were smaller in size compared to age-matched WT littermates (Supplementary Figure 4D). Notably, Kaplan-Meier analysis showed that the median survival for Zfp871+/− mice (n=27; 89.3 weeks) was significantly shorter than that for WT mice (n=56; 117 weeks) (Figure 6A). Interestingly, like WT mice, Zfp871+/− mice were not prone to spontaneous tumors (Figure 6B, Supplementary Tables S3 and S4). The tumor spectrum in Zfp871+/− mice appeared very similar to WT mice with a few exceptions (Figure 6B). Zfp871+/− mice had one incidence of lung adenocarcinoma and hemangiopericytoma, a slightly elevated incidence of B-cell lymphoma (16.7% compared to 11.8% in WT), and no incidence of T-cell lymphoma (compared to the 3.9% in WT) (Figure 6B, Supplementary Tables S3 and S4).
Figure 6 -. Zfp871+/− mice have a short lifespan and are prone to steatohepatitis.

(A) Kaplan-Meier survival curve for WT and Zfp871+/− mice. (B) Tumor spectra and penetrance in WT and Zfp871-heterozygous mice. (C, D, E) The number and percentage of WT and Zfp871+/− mice with splenic and/or thymic hyperplasia (C), with inflammation in two or more organs (D), or with steatohepatitis (E). (F) Representative images of hematoxylin and eosin (H&E)-stained livers from a WT mouse with normal liver and three Zfp871+/− mice with steatohepatitis.
As Zfp871+/− mice were not prone to spontaneous tumor but had a short lifespan, histological analysis was performed to examine the major organs for other potential abnormalities. We found that 96% (23 out of 24) of Zfp871+/− mice, whereas only 4% (2 out of 51) of WT mice, had splenic/thymic hyperplasia (Figure 6C). We also found that 42% of Zfp871+/− but none of WT mice were prone to inflammation in 2 or more organs (Figure 6D). Notably, 50% (12 out of 24) of Zfp871+/− mice, whereas just 6% (3 out of 51) of WT mice, developed steatohepatitis (Figure 6E-F). As steatohepatitis often leads to liver cirrhosis and eventually liver failure (30), we conclude that the high incidence of steatohepatitis and chronic inflammation are likely responsible for the shorter lifespan observed in Zfp871+/− mice.
Several key developmental pathways as well as genes involved in lipid metabolism and inflammation are regulated by Zfp871 in conjunction with p53
To explain the mouse phenotypes observed upon Zfp871 deficiency, RNA-seq was performed to determine the genes and pathways regulated by Zfp871. As Zfp871 deletion leads to increased p53 expression, we wanted to determine the pathways regulated by Zfp871 in conjunction with p53 and the ones regulated by Zfp871 independent of p53. For this, total RNAs were purified from a cohort of WT, Zfp871−/−, Trp53−/− and Zfp871−/−; Trp53−/− MEFs at passage 4 for RNA-seq. Venn diagram showed that both common and distinct groups of genes were expressed among these MEFs (Supplementary Figure S5A). Heat-map analysis revealed that several groups of genes were differentially expressed in Zfp871−/− MEFs in a p53-dependent manner (Supplementary Figure S5B). To determine the number of genes that were regulated by Zfp871 in conjunction with p53, we compared gene expression patterns between WT and Zfp871−/− MEFs (Supplementary Figure S5C). We found that Zfp871 and p53 coordinately up-regulated 643 genes and down-regulated 953 genes (Supplementary Figure S5C). Similarly, to determine genes regulated by Zfp871 independent of p53, we compared gene expression patterns between Trp53−/− and Zfp871−/−; Trp53−/− MEFs (Supplementary Figure S5D). Interestingly, we observed that the number of genes regulated by Zfp871 alone (493 genes) were less than a third of the number regulated by both Zfp871 and p53 (1496 genes) (Supplementary Figure S5D). This suggests that the repertoire of pathways regulated by Zfp871 is substantially increased by p53. Next, to look at various pathways regulated by Zfp871 alone or together with p53, we performed gene set enrichment analysis (Supplementary Figures S5E and S5F). Notably, several pathways vital for cell and embryonic development were found to be regulated by Zfp871 in combination with p53 (Supplementary Figure S5E). Nevertheless, Zfp871 alone was able to regulate pathways that are important for cell migration and for cellular response to interferon-beta (Supplementary Figure S5F). Furthermore, we looked for differentially expressed genes that are involved in the development of steatohepatitis. We found that several genes involved in fatty acid metabolism and inflammation were differentially expressed in Zfp871−/− MEFs as compared to WT MEFs (Supplementary Table S5), suggesting that altered lipid metabolism and inflammation by ZFP871 deficiency is likely responsible for steatohepatitis.
Discussion
The tumor suppressor p53 plays a vital role in cell growth and survival through its transcriptional activity (1,2). Interestingly, multiple p53-induced genes come back to negatively or positively regulate p53, forming a feedback loop. In this study, we found that p53 transcriptionally regulates Zfp871, which in turn negatively regulates p53. We also showed that Zfp871-KO leads to growth suppression and cellular senescence in a p53-dependent manner.
p53 is known to be a short-lived protein regulated through protein stability (31). Accordingly, several E3 ubiquitin ligases are known to target p53 for degradation, of which Mdm2 is a well-defined one (31). Previously, we showed that Zfp871 negatively regulates p53 protein stability as part of Mdm2 E3 ubiquitin ligase complex through association with Mdm2 (16). Additionally, through the KRAB domain present in its N-terminus, Zfp871 may interact with KRAB-associated protein-1 (KAP1, also called TRIM28), which in turn has been shown to regulate p53 protein stability as a part of Mdm2 E3 ubiquitin ligase complex (32). Here we found that as an RNA-binding protein, Zfp871 binds to Mdm2 3’UTR, positively regulating Mdm2 mRNA stability and consequently p53 protein stability. This is in accordance with the reported dual activities of Zfp871 to act as a transcription factor and as an RNA-binding protein (33). Interestingly, in contrast to the decrease in mature Mdm2 mRNA upon Zfp871 KO, we observed increased Mdm2 pre-mRNA. This contrast seems to be the result of increased levels of p53 protein upon depletion of Zfp871, which would then lead to increased transcription and subsequently, elevated levels of Mdm2 pre-mRNA. On the other hand, loss of Zfp871 leads to decreased stability of Mdm2 mature mRNA as most RNA-binding proteins are known to regulate the stability of processed mature mRNA and not the pre-mRNA (34). Thus, Zfp871 regulates p53 protein stability as a part of the Mdm2 complex but also as a regulator of Mdm2 expression.
KRAB-containing C2H2 zinc finger proteins form the largest family of mammalian transcription factors. However, the biological functions for most of the family proteins are not known (7). To determine the biological function of Zfp871, we employed a Zfp871-deficient mouse model. Interestingly, we were unable to obtain Zfp871 knock-out mice at Mendelian ratios at birth, which we theorized could be due to increased p53 levels, leading to embryonic lethality (6,27,28). Thus, we investigated if we could obtain Zfp871−/− mice with simultaneous deletion of Trp53. While we were able to obtain Zfp871−/−; Trp53+/− pups close to Mendelian ratio, these mice died soon after birth, indicating a partial rescue. Thus, RNA-seq was performed and showed that multiple key developmental pathways are regulated by Zfp871 in conjunction with p53, suggesting that alteration of these developmental pathways is likely responsible for the incomplete rescue observed in these mice. Nonetheless, our study indicated that p53 is a critical effector of Zfp871 function.
We found that Zfp871+/− mice have a short lifespan. Because Zfp871+/− mice are not prone to spontaneous tumors but to chronic inflammation in multiple organs, especially steatohepatitis, we postulate that steatohepatitis/NASH is likely responsible for the shortened lifespan. NASH is one of the most common forms of liver disease in the Western economies as well as a major cause of liver failure resulting in death (30). Although NASH is frequently associated with obesity and diabetes, it can be triggered by changes in gene expression (35). Accordingly, increased levels of p53 protein have been shown to correlate with increase in NASH severity in mice as well as in human patients (36–38). Additionally, loss of Zfp871 leads to altered expression of multiple genes necessary for lipid metabolism and inflammation, which would synergize with increased expression of p53 to promote steatohepatitis. It should be mentioned here that although we could not find a human zinc finger protein with more than 50% sequence homology with Zfp871 protein, there could be functional homologs of Zfp871 in humans that regulate p53 protein stability in a similar mechanism. Taken together, we conclude that the Zfp871-Mdm2-p53 pathway plays an indispensable role in development and long-term survival.
Supplementary Material
Implications:
A fine equilibrium of p53 is required for preventing damaging effects of aberrant p53 expression. We identify the Zfp871-Mdm2-p53 pathway that plays a critical role in development of mice and steatohepatitis.
Acknowledgements
We would like to thank members of the Chen-Zhang laboratory for suggestions and helpful discussions. This work is supported in part by National Institutes of Health grants [CA195828, CA224433, and CA250338 to XC].
Financial support:
NIH grants CA195828, CA250338, and CA224433
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Data Availability Statement
The RNA-sequencing datasets from this publication have been submitted to the GEO database and have been assigned the following Series record: GSE176582.
References
- 1.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009. page 413–31. [DOI] [PubMed]
- 2.Levine AJ, Oren M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009. page 749–58. [DOI] [PMC free article] [PubMed]
- 3.Choi J, Donehower LA. p53 in embryonic development: Maintaining a fine balance. Cell. Mol. Life Sci 1999. page 38–47. [DOI] [PMC free article] [PubMed]
- 4.Grier JD, Xiong S, Elizondo-Fraire AC, Parant JM, Lozano G. Tissue-Specific Differences of p53 Inhibition by Mdm2 and Mdm4. Mol Cell Biol 2006; [DOI] [PMC free article] [PubMed]
- 5.Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM, et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 2014;514:228–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang M, Zhang Y, Xu E, Mohibi S, De Anda DM, Jiang Y, et al. Rbm24, a target of p53, is necessary for proper expression of p53 and heart development. Cell Death Differ 2018;25. [DOI] [PMC free article] [PubMed]
- 7.Cassandri M, Smirnov A, Novelli F, Pitolli C, Agostini M, Malewicz M, et al. Zinc-finger proteins in health and disease. Cell Death Discov 2017. [DOI] [PMC free article] [PubMed]
- 8.Emerson RO, Thomas JH. Adaptive evolution in zinc finger transcription factors. PLoS Genet 2009;5. [DOI] [PMC free article] [PubMed]
- 9.Gamsjaeger R, Liew CK, Loughlin FE, Crossley M, Mackay JP. Sticky fingers: zinc-fingers as protein-recognition motifs. Trends Biochem. Sci 2007. page 63–70. [DOI] [PubMed]
- 10.Yang P, Wang Y, Macfarlan TS. The Role of KRAB-ZFPs in Transposable Element Repression and Mammalian Evolution. Trends Genet 2017. page 871–81. [DOI] [PMC free article] [PubMed]
- 11.Imbeault M, Helleboid PY, Trono D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017;543:550–4. [DOI] [PubMed] [Google Scholar]
- 12.Najafabadi HS, Mnaimneh S, Schmitges FW, Garton M, Lam KN, Yang A, et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat Biotechnol 2015;33:555–62. [DOI] [PubMed] [Google Scholar]
- 13.Lee JT, Gu W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ 2010. page 86–92. [DOI] [PMC free article] [PubMed]
- 14.Shloush J, Vlassov JE, Engson I, Duan S, Saridakis V, Dhe-paganon S, et al. Structural and functional comparison of the RING domains of two p53 E3 ligases, Mdm2 and Pirh2. J Biol Chem 2011;286:4796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian C, Xing G, Xie P, Lu K, Nie J, Wang J, et al. KRAB-type zinc-finger protein Apak specifically regulates p53-dependent apoptosis. Nat Cell Biol 2009;11:580–91. [DOI] [PubMed] [Google Scholar]
- 16.Yan W, Scoumanne A, Jung YS, Xu E, Zhang J, Zhang Y, et al. Mice deficient in poly(C)-binding protein 4 are susceptible to spontaneous tumors through increased expression of ZFP871 that targets p53 for degradation. Genes Dev [Internet]. Comparative Oncology Laboratory, School of Veterinary Medicine, School of Medicine, University of California at Davis, Davis, California 95616, USA.; 2016;30:522–34. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26915821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997;387:296–9. [DOI] [PubMed] [Google Scholar]
- 18.Desprez PY, Hara E, Bissell MJ, Campisi J. Suppression of mammary epithelial cell differentiation by the helix-loop-helix protein Id-1. Mol Cell Biol 1995;15:3398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohibi S, Zhang J, Chen X. PABPN1, a Target of p63, Modulates Keratinocyte Differentiation through Regulation of p63α mRNA Translation. J Invest Dermatol [Internet]. 2020;140:2166–2177.e6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022202X20312562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc [Internet]. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.; 2013;8:2281. Available from: 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peritz T, Zeng F, Kannanayakal TJ, Kilk K, Eiríksdóttir E, Langel U, et al. Immunoprecipitation of mRNA-protein complexes. Nat Protoc 2006;1:577–80. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Cho SJ, Shu L, Yan W, Guerrero T, Kent M, et al. Translational repression of p53 by RNPC1,a p53 target over expressed in lymphomas. Genes Dev [Internet]. 2011/07/19. Comparative Cancer Center, Schools of Medicine and Veterinary Medicine, University of California at Davis, USA.; 2011;25:1528–43. Available from: https://www.ncbi.nlm.nih.gov/pubmed/21764855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Qian Y, Zhang J, Yan W, Jung YS, Chen M, et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev 2017;31:1243–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shu L, Yan W, Chen X. RNPC1, an RNA-binding protein and a target of the p53 family, is required for maintaining the stability of the basal and stress-induced p21 transcript. Genes Dev [Internet]. 2006/10/20. Department of Cell Biology, The University of Alabama at Birmingham, Birmingham, AL 35294, USA.; 2006;20:2961–72. Available from: https://www.ncbi.nlm.nih.gov/pubmed/17050675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barak Y, Juven T, Haffner R, Oren M. Mdm2 Expression Is Induced By Wild Type P53 Activity. EMBO J [Internet]. 1993;12:461–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8440237%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC413229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zandomeni R, Carrera Zandomeni M, Shugar D, Weinmann R. Casein kinase type II is involved in the inhibition by 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole of specific RNA polymerase II transcription. J Biol Chem 1986;261:3414–9. [PubMed] [Google Scholar]
- 27.Luna RMDO, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; [DOI] [PubMed]
- 28.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995;378:206–8. [DOI] [PubMed] [Google Scholar]
- 29.Yang HJ, Zhang J, Yan W, Cho SJ, Lucchesi C, Chen M, et al. Ninjurin 1 has two opposing functions in tumorigenesis in a p53-dependent manner. Proc Natl Acad Sci U S A 2017;114:11500–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanches SCL, Ramalho LNZ, Augusto MJ, Da Silva DM, Ramalho FS. Nonalcoholic Steatohepatitis: A Search for Factual Animal Models. Biomed Res. Int 2015. [DOI] [PMC free article] [PubMed]
- 31.Pant V, Lozano G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev 2014. page 1739–51. [DOI] [PMC free article] [PubMed]
- 32.Wang C, Ivanov A, Chen L, Fredericks WJ, Seto E, Rauscher FJ, et al. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J 2005;24:3279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han H, Braunschweig U, Gonatopoulos-Pournatzis T, Weatheritt RJ, Hirsch CL, Ha KCH, et al. Multilayered Control of Alternative Splicing Regulatory Networks by Transcription Factors. Mol Cell 2017;65:539–553.e7. [DOI] [PubMed] [Google Scholar]
- 34.Mohibi S, Chen X, Zhang J. Cancer the’RBP’eutics–RNA-binding proteins as therapeutic targets for cancer. Pharmacol Ther [Internet]. 2019;203:107390. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0163725819301275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dongiovanni P, Anstee Q, Valenti L. Genetic Predisposition in NAFLD and NASH: Impact on Severity of Liver Disease and Response to Treatment. Curr Pharm Des 2013;19:5219–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomita K, Teratani T, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K, et al. P53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J Hepatol 2012;57:837–43. [DOI] [PubMed] [Google Scholar]
- 37.Yahagi N, Shimano H, Matsuzaka T, Sekiya M, Najima Y, Okazaki S, et al. p53 Involvement in the pathogenesis of fatty liver disease. J Biol Chem 2004;279:20571–5. [DOI] [PubMed] [Google Scholar]
- 38.Farrell GC, Larter CZ, Hou JY, Zhang RH, Yeh MM, Williams J, et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J Gastroenterol Hepatol 2009;24:443–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing datasets from this publication have been submitted to the GEO database and have been assigned the following Series record: GSE176582.