

Abstract
Expression of programmed death-ligand 1 (PD-L1) on cancer cells is a critical mechanism contributing to immunosuppression and immune escape. PD-L1 expression may also impact therapeutic outcomes of epidermal growth factor receptor (EGFR)-targeted therapy (e.g., with osimertinib/AZD9291) against EGFR mutant non-small cell lung cancers (NSCLCs) and can even be altered during the treatment albeit with largely undefined mechanisms. This study primarily focuses on elucidating the mechanism by which osimertinib induces PD-L1 degradation in addition to validating osimertinib’s effect on decreasing PD-L1 expression in EGFR mutant NSCLC cells and tumors. Osimertinib and other EGFR inhibitors effectively decreased PD-L1 levels primarily in EGFR mutant NSCLCs and xenografted tumors. Osimertinib not only decreased PD-L1 mRNA expression, but also prompted proteasomal degradation of PD-L1 protein, indicating both transcriptional and posttranslational mechanisms accounting for osimertinib-induced reduction of PD-L1. Knockdown of β-TrCP or inhibition of GSK3 failed to prevent PD-L1 reduction induced by osimertinib. Rather, knockdown of membrane-associated RING-CH 8 (MARCH8) that encodes a membrane-bound E3 ubiquitin ligase rescued osimertinib-induced PD-L1 reduction. Furthermore, manipulation of MARCH8 expression accordingly altered PD-L1 degradation rate. Critically, MARCH8 interacted with PD-L1 through its N-terminal region and also ubiquitinated PD-L1 in cells. Collectively, these results strongly suggest that MARCH8 is a previously undiscovered E3 ubiquitin ligase responsible for PD-L1 degradation including osimertinib-induced PD-L1 degradation, establishing a novel connection between MARCH8 and PD-L1 regulation.
Keywords: EGFR, osimertinib/AZD9291, PD-L1, MARCH8, degradation
Introduction
Epidermal growth factor receptor (EGFR)-targeted therapy against EGFR mutant (EGFRm) non-small cell lung cancers (NSCLCs), particularly with the third generation EGFR inhibitor, osimertinib (AZD9291), as the first line treatment, has achieved great success in the clinic with an overall survival of > 38 months (1,2). Unfortunately, all patients eventually relapsed and developed resistance, limiting the long-term benefit of the treatment (3,4). Hence, effective strategies that can overcome the acquired resistance are urgently needed in the clinic. To this end, it is essential to fully understand the biology of osimertinib and other EGFR-TKIs against EGFRm NSCLC cells including mechanisms of acquired resistance.
It is now recognized that programmed death-ligand 1 (PD-L1) expression on cancer cells is a critical mechanism contributing to immunosuppression and immune escape through inactivation of T cells via interacting with program death-1 (PD-1) on T cells. Accordingly, immunotherapy that targets the PD-L1/PD1 checkpoint represents a promising therapeutic option for several types of cancers including NSCLC, although only a small portion of NSCLC patients benefit from this immunotherapy (5–8). It is generally thought that EGFRm NSCLCs do not respond well to anti-PD1 or anti-PD-L1 immunotherapy (5,9). However, EGFR signaling positively regulates PD-L1 levels (10–12). Moreover, PD-L1 expression impacts therapeutic outcomes of EGFR-TKI treatment of EGFRm NSCLCs (13–19), albeit with conflicting reports (20,21) and is even linked to acquired resistance to EGFR-TKIs (10,22).
Interestingly, PD-L1 expression in EGFRm NSCLC can be downregulated by EGFR-TKIs including osimertinib as reported previously (11,23,24) and demonstrated in this study. Although protein degradation is suggested to be involved in mediating PD-L1 degradation by osimertinib beyond transcriptional regulation, the underlying mechanism, particularly the E3 ubiquitin ligase, accounting for PD-L1 degradation induced by osimertinib, possibly other EGFR-TKIs, is largely unknown. Thus, this study focused on understanding the molecular mechanism by which EGFR-TKIs, particularly osimertinib, induces PD-L1 degradation. Membrane-associated RING-CH 8 (MARCH8) is known to be a membrane-bound E3 ubiquitin ligase that is responsible for degradation of several host transmembrane and immune-associated proteins, including major histocompatibility complex (MHC)-II, CD44, CD81, CD86, CD98, interleukin-1 receptor accessory protein, TNF-related apoptosis-inducing ligand (TRAIL) receptor 1/death receptor 4 (DR4) and the transferrin receptor (25,26). Whiling working on MARCH8-mediated regulation of DR4 degradation induced by osimertinib (27), we were curiously and logically wondering whether MARCH8 is also involved in regulation of the degradation of PD-L1, another transmembrane and immune-related protein. As a result, we have identified MARCH8 as a novel E3 ubiquitin ligase to ubiquitinate and facilitate PD-L1 degradation including osimertinib-induced PD-L1 degradation, thus revealing a previously undiscovered novel connection between MARCH8 and PD-L1 degradation.
Materials and Methods
Reagents.
Osimertinib was purchased from Active Biochemicals (Maplewood, NJ). Erlotinib was purchased from LC Laboratories (Woburn, MA). CO1686 was purchased from MedChemExpress USA (Monmouth Junction, NJ). Actinomycin D (Act D), cycloheximide (CHX), and MG132 were purchased from Sigma Chemical Co. (St. Louis, MO). These reagents were dissolved in DMSO at the concentration of 10 mM as stock, which were aliquoted and stored at −80°C before use. Recombinant human EGF and INF-γ protein were purchased from R&D Systems (Minneapolis, MN) and prepared according to product instructions. Rabbit anti-human (E1L3N) and mouse anti-human PD-L1 (405.9A11) antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA). MARCH8 and GAPDH antibodies were purchased from Proteintech (Rosemont, IL). Mouse monoclonal anti-β-actin and anti-tubulin antibodies were purchased from Sigma Chemical Co.
Plasmids.
Expression plasmids including HA-vector (pC-HA), N-terminal HA-tagged human MARCH8 (pC-HA-MARCH8), its RING-CH domain mutant pC-HA-MARCH8-CS, and another RING-CH mutant pC-HA-MARCH8-W114A, in which tryptophan at position 144 was mutated to alanine (26) were generously provided by Dr. Kenzo Tokunaga (National Institutes of Infectious Disease, Tokyo, Japan). His-ubiquitin (His-Ub), pcDNA3-PD-L1 and pcDNA3-HA-PD-L1 expression plasmids were described previously (28). The N-terminal and C-terminal Flag-tagged human PD-L1 expression constructs: pCMV3-CF-PD-L1 (HG10084-CF) and pCMV3-NF-PD-L1 (HG10084-NF), were purchased from Sino Biological Inc (North Wales, PA). HA-ubiquitin (HA-Ub) expression plasmid was described previously (29). Flag-tagged full length human MARCH8 (hMARCH8-full), MARCH8-RING and MARCH8-ΔC-tail expression constructs were made with pCMV-Flag vector and according PCR products generated using the following primers: hMARCH8-Xho1-full, forward 5’-GCATCTCGAGGCCACCatgagcatgccactgcatcag-3’ and hMARCH8-BamH1-full, reverse 5’-GCATGGATCCgacgtgaatgatttctgctcc-3’ for hMARCH8-full; hMARCH8-Xho1-full, forward 5’-GCATCTCGAGGCCACCatgagcatgccactgcatcag-3’ and hMARCH8-BamH1-ΔC, reverse 5’-GCATGGATCCcactttacactgaacatac-3’ for MARCH8-ΔC-tail; and hMARCH8-Xho1-Ring, forward 5’-GCATCTCGAGGCCACCcttctatcacgccatccagc-3’ and hMARCH8-BamH1-Ring, reverse 5’-CATGGATCCcatgatcttcctgcgctcgc-3’ for MARCH8-RING.
Cell lines and cell culture.
The AZD9291-resistant (AR) cell lines, PC-9/AR and PC-9/GR/AR and HCC827/AR, were established in our laboratory as described previously (30,31). SH416, SH416/AR, SH450 and SH450/AR cell lines were kindly provided by Dr. Christine M. Lovly (Vanderbilt University School of Medicine, Nashville, TN). The gefitinib-resistant (GR) cell line, PC-9/GR, was provided by Dr. P. A. Jänne (Dana Farber Cancer Institute, Boston, MA). Other NSCLC cell lines were described in our previous studies (32,33). These cell lines except for H157 were not genetically authenticated. Mycoplasma test was performed regularly or as needed using MycoAlert@ Mycoplasma Detection Kit (Lonza; Rockland, ME). These cell lines were cultured in RPMI 1640 medium with 5% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Western blotting analysis.
Preparation of whole cell lysates from cell lines and procedure for detection of tested protein with Western blotting were described previously (30,31). Band intensities were scanned and quantified by NIH image J software.
Detection of cell surface PD-L1 with flow cytometry.
The tested cells were collected after treatments using trypsin into 15 ml centrifuge tube, pelleted at 250g for 5 min to remove medium. The cells were washed once with 2 ml cold PBS before incubation with PE-conjugated mouse anti-human PD-L1 (CD274/B7-H1; BioLegend, San Diego, CA) or PE mouse IgG1 isotype control (MOPC-21/P3; ThermoFisher Scientific/eBioscience, San Diego, CA) in 100 μl cold PBS containing 2% BSA according to the manufacturer’s instructions. The mean fluorescent index (MFI) that represents antigenic density on a per cell basis was used to assess cell surface PD-L1 levels.
CHX chase assay.
After treatment or transfection with different plasmids, CHX (10 μg/ml) was added to the cells, which were then collected at different time points after CHX addition. The tested proteins in the whole-cell protein lysates were detected with Western blotting. The band intensity was quantified by NIH image J software and compared to 0 time point post CHX treatment to calculate the relative percentage of proteins retained after suppressing de novo protein synthesis. The data were plotted to show the protein degradation rates.
Quantitative reverse transcription-PCR (qRT-PCR).
Cells, after treatment, were collected in Trizol (Sigma Chemical Co.) for extraction of total RNA, which was then reverse transcribed to cDNA template using the OneScript® cDNA Synthesis Kit (Applied Biological Materials Inc.; Richmond, BC, Canada) according to the manufacturer’s instructions. qPCR analysis was performed using the SYBR Green reagent from Bio-Rad (Hercules, CA) according to manufacturer’s instructions. The PD-L1 primers used were forward 5’-GGCATTTGCTGAACGCAT-3’; and reverse 5’-CAATTAGTGCAGCCAGGT-3’ (34). The MARCH8 primers were forward 5′-CTCTCGCACTTCTATCACGCCA-3′ and reverse 5′-AAGTGGAGGCTTCCTGTGCAGT-3′ (26). GAPDH was used as internal reference as previously reported (35).
RNA sequencing (RNA-seq) analysis.
Cell pellets from cell lines exposed to DMSO or osimertinib were shipped to MedGenome Inc., (Foster City, CA) for RNA-seq analysis with NovaSeq sequencer. Differential expression analysis was performed using DESeq2. The expression values for each gene were presented in FPKM (fragments per kilobase per million) units. Significant differential expression cutoffs were P value < 0.05 & fold change >=2 or <=−2.
Short-hairpin RNA (shRNA).
Lentiviral MARCH8 shRNAs in pLKO.1 plasmid (TRCN0000073235 and TRCN0000073236) were purchased from Sigma Chemical Co. The preparation of lentiviruses and cell infection were performed according to the manufacturer’s instructions. MARCH8 stable knockdown cell lines were obtained after selection with puromycin for 5 days. MARCH8 transient knockdown was also performed by infecting cells with shMARCH8 lentiviruses without selection for 48 h. MARCH8 knockdown was confirmed with Western blotting.
Protein immunoprecipitation (IP).
HEK293T cells were transfected with given plasmids using PolyJet transfection reagent (SignaGen Laboratories; Rockville, MD) following manufacturer’s guides. At 24 to 36 h after transfection, MG132 (10 μM) was added for additional 6 h before harvest using RIPA buffer containing protease and phosphatase inhibitor. The lysates were then incubated with Flag M2 beads overnight at 4°C with rotation. The beads were washed 5 times with RIPA buffer and once with 50 mM HEPES buffer. Then the pulldown proteins were then denatured and subject to SDS-PAGE for detection of given proteins.
Cellular ubiquitination assay.
HEK293T cells were co-transfected with plasmids carrying His-Ub, HA-PD-L1 with/without FLAG-MARCH8/CA. After 24 h, cells were exposed to MG132 (10 μM) for additional 12 h. The cells were then lysed in buffer A containing 6 M guanidine-HCl, 0.1 M Na2HPO4/NaH2PO4, and 10 mM imidazole [pH 8.0]) with sonication. The lysates were incubated with nickel-nitrilotriacetic acid (Ni-NTA) matrices (QIAGEN) for 3 h at room temperature. The His pull-down products were washed twice with buffer A, once with buffer A/TI (1 volume buffer A and 3 volumes buffer TI), and twice with buffer TI (25 mM Tris-HCl and 20 mM imidazole [pH 6.8]). At last, the pull-down proteins were resolved by SDS-PAGE for immunoblotting with HA antibody. Alternatively, HEK293T cells were co-transfected with Flag-PD-L1 (Sino Biological, Inc.), HA-Ub, and/or HA-March8 (WT and W114A) and processed similarly as above mentioned.
In vivo xenograft experiment.
The experiment was approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University. PC-9 cells (2×106) were injected subcutaneously into the flank of nude mice to establish xenograft tumors. Mice were orally given vehicle control or osimertinib (10 mg/kg/day) once tumor size reached approximately 300 mm3. Three mice of each group were sacrificed on the days of 1, 3, and 9 post treatment, respectively, for collecting tumor tissues and preparing protein lysates. Tumor volume is calculated using the formula V=π(length × width2)/6.
Human NSCLC tissues and PD-L1 detection.
The paired tissue samples from EGFR mutant NSCLC patients before treatment (i.e., baseline) and after relapse to treatment with first generation EGFR-TKIs including gefitinib, erlotinib or icotinib were collected at the Henan Cancer hospital (Zhengzhou, Henan, China) under the Ethics Review Committee (IRB)-approved protocol (2019–274). PD-L1 was detected with IHC with the same conditions described in our previous study (36).
Statistical analysis.
The statistical comparisons of differences between two treatments were analyzed using two-sided unpaired Student’s t tests by the Graphpad InStat 8.0 software. A P-value less than 0.05 is considered statistically significant.
Results
Osimertinib and other EGFR-TKIs decrease PD-L1 levels in EGFRm NSCLC cells and tumors.
We first determined the effects of osimertinib on modulating PD-L1 levels in EGFRm PC-9 cells (19del) and found that, at concentrations ranging from 5 to 100 nM, it decreased PD-L1 levels in a concentration-dependent manner (Fig. 1A). This reducing effect was apparent at 4 h treatment with osimertinib and sustained up to 16 h (Fig. 1B). In other EGFRm NSCLC cell lines including HCC827 (19del), H1975 (L858R/T790M) and PC-9/GR (19del/T790M), osimertinib also decreased PD-L1 levels (Fig. 1C). However, this effect was not or minimally observed in NSCLC cell lines (i.e., H157 and H460) with wild-type (WT) EGFR (Fig. 1C). Beyond osimertinib, the other EGFR-TKIs including erlotinib (first generation), afatinib (second generation), CO1686 (third generation), and EGF816 (third generation) reduced PD-L1 levels as well (Fig. 1D). In addition to cellular total levels of PD-L1 reduction, osimertinib significantly decreased cell surface levels of PD-L1 in both PC-9 and HCC827 cells (Fig. 1E). Notably, treatment of PC-9 xenografts with osimertinib caused robust and significant reduction of PD-L1 across the tested times from day 1 to day 9 post treatment (Fig. 1F). These results collectively demonstrate that osimertinib as well as other EGFR-TKIs decrease PD-L1 primarily in EGFRm NSCLC cells both in vitro and in vivo.
Fig. 1. Osimertinib and other EGFR-TKIs decrease PD-L1 levels in EGFR mutant NSCLC cell lines (A-E) and in xenografts (F).
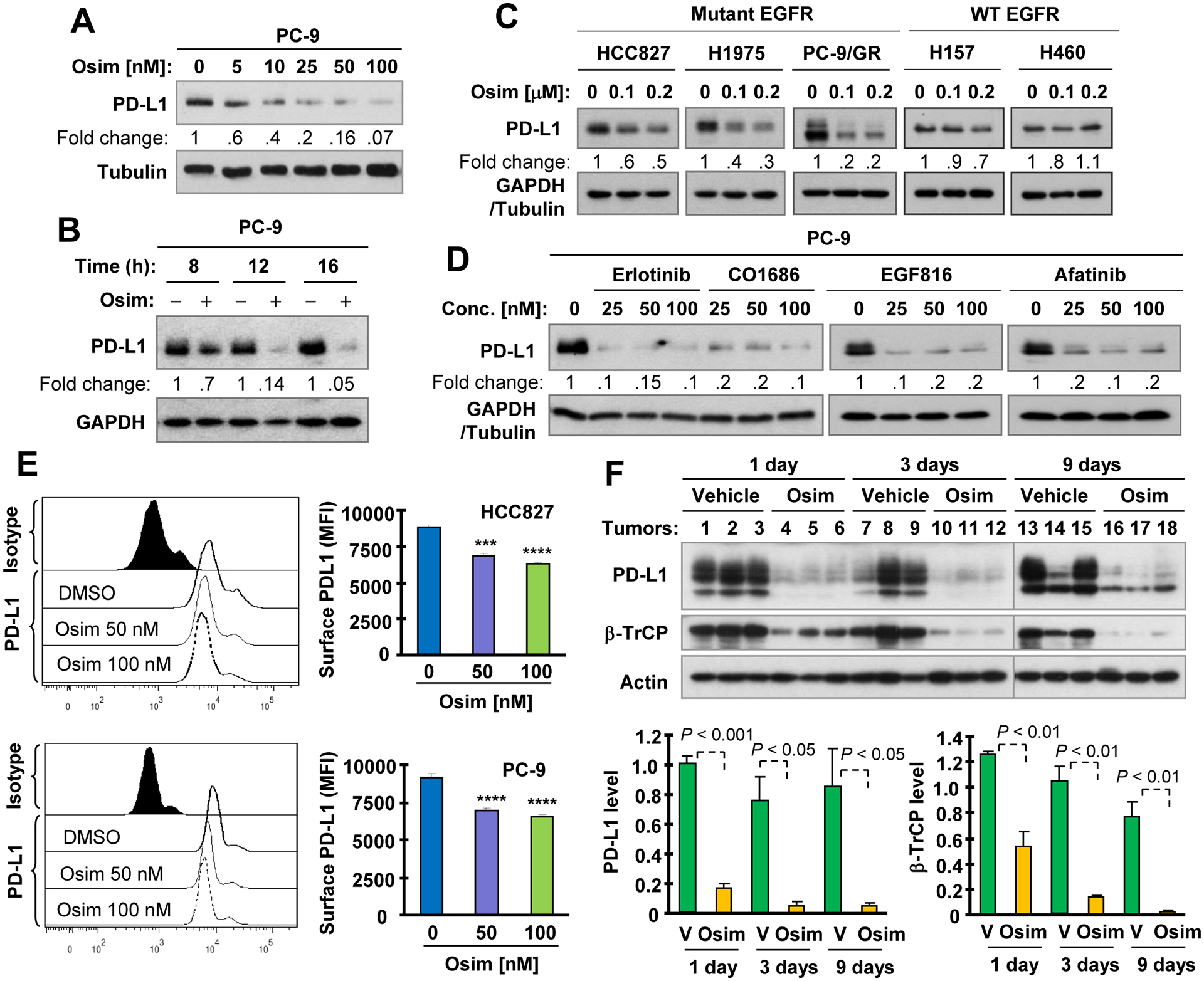
A-D, The indicated cell lines were exposed to varied concentrations of osimertinib (Osim) for 16 h (A and C), to 100 nM osimertinib for different time as indicated (B) or to different concentrations of EGFR-TKIs as indicated for 16 h (D). Whole cell lysates were then made from these cells and used for detection of PD-L1 and other proteins with Western blotting. E, Both PC-9 and HCC827 cell lines were treated with different concentrations of osimertinib for 36 h. Cell surface PD-L1 was then detected with flow cytometry. ***, P < 0.001 and ****, P < 0.0001 compared with DMSO. F, Whole cell lysates were prepared from PC-9 xenografts treated with osimertinib at 10 mg/kg body weight (og, once/daily) for the indicated times and then subject to Western blotting. Each column is the mean ± SD of triplicate determinations. V, vehicle.
Osimertinib decreases PD-L1 mRNA levels in EGFRm NSCLC cells.
From our RNA-seq data generated with EGFRm NSCLC cell lines exposed to DMSO and osimertinib, respectively, we noticed that PD-L1 mRNA expression was significantly decreased in osimertinib-treated HCC827 and PC-9 cell lines (Fig. S1A–B). With qRT-PCR, we also detected significant decrease of PD-L1 mRNA levels in the tested EGFRm NSCLC cell lines including PC-9, HCC827, and H1975 at both 4 h and 8 h post osimertinib treatment (Figs. S2A and B), thus validating the RNA-seq results. This effect was also confirmed with RT-PCR (Fig. S2C). Hence, it is clear that osimertinib suppresses PD-L1 transcription.
Osimertinib promotes MARCH8-mediated proteasomal degradation of PD-L1 independent of GSK3 and β-TrCP.
EGF and INFγ are known to upregulate PD-L1 expression albeit with different mechanisms: the former elevates PD-L1 levels primarily through enhancing protein stability, whereas the latter induces PD-L1 expression primarily at the transcriptional level (37). When checking the effects of osimertinib on induced PD-L1 expression by EGF and INFγ, respectively, we noticed that osimertinib abolished PD-L1 elevation induced by EGF (Fig. S2D), but not INFγ, although it did attenuate INFγ-induced PD-L1 elevation (Fig. S2E). The presence of Act D, a general transcription inhibitor, abolished PD-L1 elevation induced by INFγ, but not by EGF in both PC-9 and HCC827 cells (Fig. S2F). These results support the notion that these two proteins apparently induce PD-L1 expression through distinct mechanisms as previously demonstrated (37). On the other hand, these findings also suggest that osimertinib may affect PD-L1 degradation since it abrogated EGF-induced PD-L1 elevation.
We then determined whether osimertinib indeed affects posttranslational process of PD-L1. Using the CHX chase assay, we found that PD-L1 was degraded much faster in osimertinib-treated cells than in their corresponding control cell lines exposed to DMSO (Fig. 2A). Moreover, the presence of the proteasome inhibitor, MG132, largely rescued the reduction of PD-L1 induced by osimertinib (Fig. 2B) and other EGFR-TKIs (Fig. 2C). These results together demonstrate that osimertinib and other EGFR-TKIs destabilize PD-L1 by prompting its proteasomal degradation.
Fig. 2. Osimertinib facilitates PD-L1 protein degradation (A), which can be rescued by proteasome inhibition (B and C) and by knockdown of MARCH8 (E-H), but not by knockdown of β-TrCP (D).
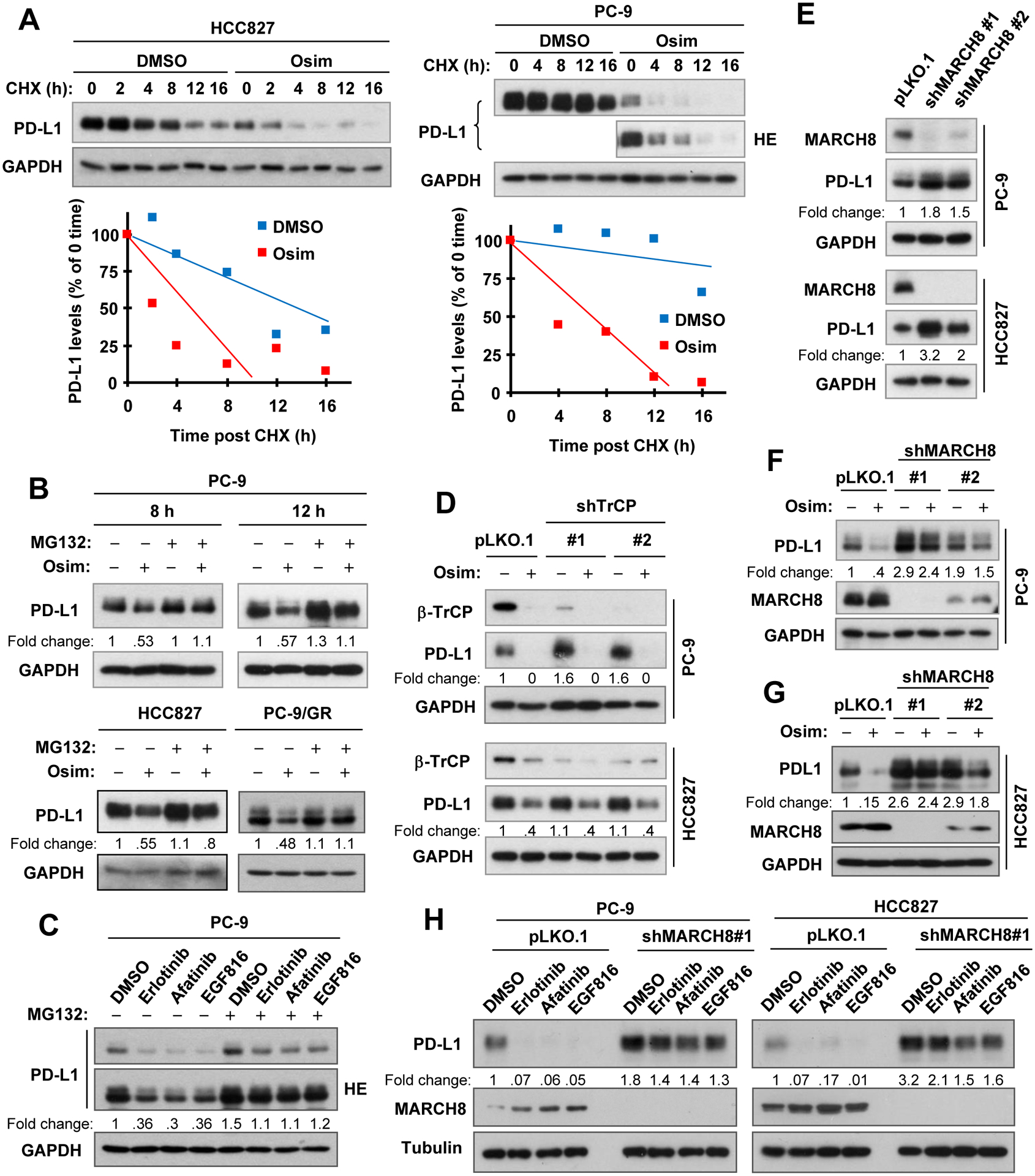
A, PC-9 and HCC827 cells were exposed to DMSO or 100 nM osimertinib (Osim) for 8 h, followed by the addition of 10 μg/ml CHX. Cells were then harvested at the indicated times post CHX for Western blotting. PD-L1 levels were plotted relative to those at time 0 of CHX treatment after being quantified by NIH Image J software and normalized to GAPDH. B and C, The indicated cell lines were pre-treated with 10 μM MG132 for 30 min and then co-treated with 100 nM osimertinib for an additional 8 h (HCC827) or 8 h and 12 h (PC-9) or with 100 nM EGFR-TKIs as indicated for 8 h (C). D-F, Whole cell lysates were prepared from the indicated cell lines expressing different β-TrCP shRNAs (D) or MARCH8 shRNAs (F) treated with DMSO or 100 nM osimertinib for 8 h or from the indicated cell lines expressing different MARCH shRNAs (E). G and H, The indicated cell lines were infected with shMARCH8 lentiviruses for approximately 48 h followed with 100 nM osimertinib (G) or the given EGFR-TKIs (H) for 16 h. The specified proteins were detected with Western blotting. HE, high exposure.
Since PD-L1 was suggested to undergo GSK3-dependent and β-TrCP-mediated proteasomal degradation (37), we then determined whether this mechanism is involved in osimertinib-induced PD-L1 degradation. We first knocked down β-TrCP expression in both PC-9 and HCC827 cell lines and then checked its impact on PD-L1 reduction induced by osimertinib. In fact, knockdown of β-TrCP gene did not affect the ability of osimertinib to decrease PD-L1 levels at all (Fig. 2D). Interestingly, we found that osimertinib itself reduced β-TrCP levels while decreasing PD-L1 levels both in vitro (Figs. 2D and S3A) and in vivo (Fig. 1F). In agreement, the presence of the GSK3 inhibitor, either CHIR99021 or SB216763, alone decreased PD-L1 levels and did not prevented PD-L1 reduction induced by osimertinib (Fig. S3B). Similarly, GSK3 knockdown failed to rescue osimertinib-induced PD-L1 reduction either (Fig. S3C). In cell lines we examined, we noted that both CHIR99021 and SB216763 actually decreased PD-L1 levels (Figs. S3B and D). Collectively, these results strongly suggest that osimertinib-induced PD-L1 reduction is likely to be independent of GSK3 and β-TrCP.
In an effort to identify the real upstream E3 ubiquitin ligase that mediates PD-L1 degradation induced by osimertinib, MARCH8, a membrane-specific E3 ubiquitin ligase, captured our attention because it is responsible for the degradation of multiple membrane and immune-related proteins such as MHC-II and DR4 (25,26). We curiously and logically wanted to know whether there is a connection between MARCH8 and PD-L1 degradation induced by osimertinib and other EGFR-TKIs. Indeed, we found that MARCH8 knockdown elevated basal levels of PD-L1 in both PC-9 and HCC827 cell lines (Fig. 2E). Moreover, osimertinib’s ability in decreasing PD-L1 levels was compromised in both PC-9 and HCC827 cells in which MARCH8 was silenced (Figs. 2F and 2G). Similarly, PD-L1 reduction induced by other different EGFR-TKIs was also largely rescued by MARCH8 knockdown (Fig. 2H). These results strongly suggest that MARCH8 is likely involved in mediating PD-L1 reduction or degradation induced by osimertinib and other EGFR-TKIs.
Enforced MARCH8 expression decreases PD-L1 levels and enhances its degradation.
Following the above finding, we then focused on demonstrating whether MARCH8 functions as a novel E3 ubiquitin ligase for PD-L1 degradation. When different amounts of construct harboring ectopic MARCH8 gene were transfected into HEK293T cells, we observed dose-dependent expression of ectopic MARCH8 accompanied with gradually decreased levels of endogenous PD-L1 (Fig. 3A). Co-transfection of the expression plasmids carrying MARCH8 and PD-L1 (non-tagged and tagged) genes, respectively, into the HEK293T cells also showed that the ectopic PD-L1 levels gradually decreased when the expression amounts of MARCH8 were increased (Figs. 3B and C). Hence, it is clear that enforced expression of MARCH8 decreases the levels of both endogenous and ectopic PD-L1. The CHX chase assay further showed that enforced expression of ectopic WT MARCH8, but not the ligase-deficient mutant, MARCH8-W114A, facilitated the degradation rates of both endogenous (Fig. 3D) and ectopic (Fig. 3E) PD-L1, indicating that MARCH8 indeed enhances PD-L1 degradation.
Fig. 3. Enforced expression of ectopic MARCH8 decreases PD-L1 levels (A-C) and enhances PD-L1 degradation (D and E).
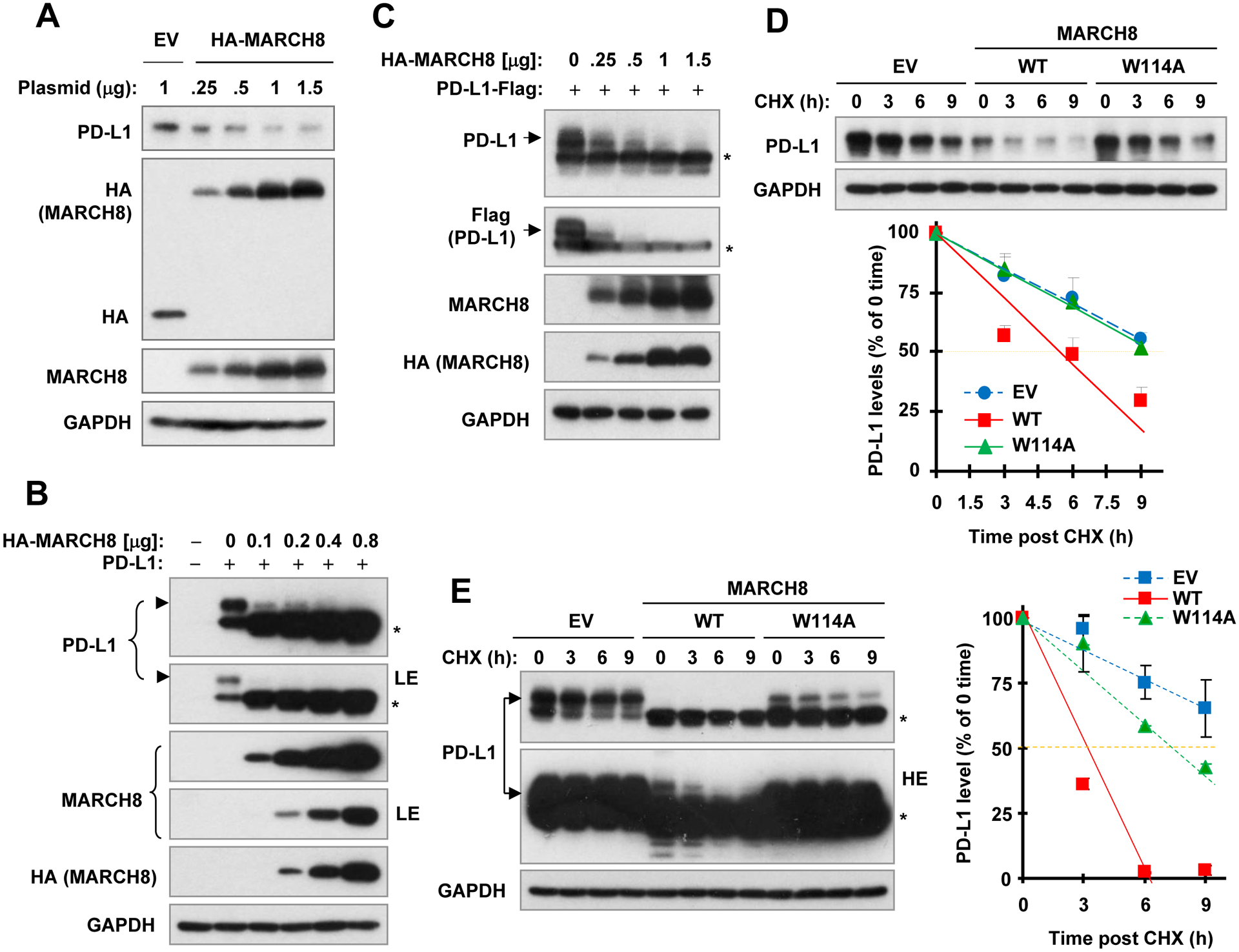
A-C, HEK293T cells were transfected with vector control (V) or wild-type (WT) MARCH8 expression plasmid at different amounts as indicated (A), or co-transfected with MARCH8 plus a plasmid expressing non-tagged PD-L1 (B) or flag-tagged PD-L1 (C) for 48 h. D and E, HEK293T cells were transfected with vector, WT or mutated MARCH8 (W114A) for 40 h or co-transfected with MARCH8 and PD-L1 (non-tagged) expression plasmids for 24 h, followed by addition of 10 μg/ml CHX for varied time as indicated. Whole cell lysates were then prepared from the above treatments for Western blot analysis to detect the indicated proteins. PD-L1 levels were plotted relative to those at time 0 of CHX treatment after being quantified by NIH Image J software and normalized to GAPDH (D and E). LE, low exposure; HE, high exposure; *, uncharacterized band generated by ectopic expression of PD-L1.
MARCH8 E3 ubiquitinase activity is required for decreasing PD-L1 levels.
The W114A mutant did not facilitate PD-L1 degradation as demonstrated above, suggesting the possible involvement of MARCH8 ubiquitinase activity in mediating PD-L1 degradation. Hence, we compared the effect of WT MARCH8 on PD-L1 reduction with that caused by the ubiquitinase-inactive MARCH8 mutant, W114A. Enforced expression of WT, but not W114A, decreased the levels of endogenous PD-L1 (Fig. 4A). Similar results were also generated with co-expression of these MARCH8 with untagged PD-L1 (Fig. 4B), C-terminal tagged PD-L1 (Fig. 4C) or N-terminal tagged PD-L1 (Fig. 4D). In some experiments, we further included an additional MARCH8 mutant named MARCH8-CS, in which all cysteines in the RING-CH domain were mutated, for its effect on PD-L1. Like W114A, MARCH8-CS did not decrease PD-L1 either (Figs. 4C and D). We also checked cell surface PD-L1 alterations in cells expressing these genes and found that WT MARCH8, but not the MARCH8 mutants, significantly reduced cell surface levels of endogenous PD-L1 (Fig. 4E). These results clearly indicate that the ubiquitinase activity of MARCH8 is likely required for MARCH8 to decrease PD-L1 levels or enhance PD-L1 degradation.
Fig. 4. Enforced expression of WT, but not ubiquitinase-dead, MARCH8 decreases both endogenous (A and E) and exogenous PD-L1 levels (B-D) including cell surface PD-L1 levels (E).
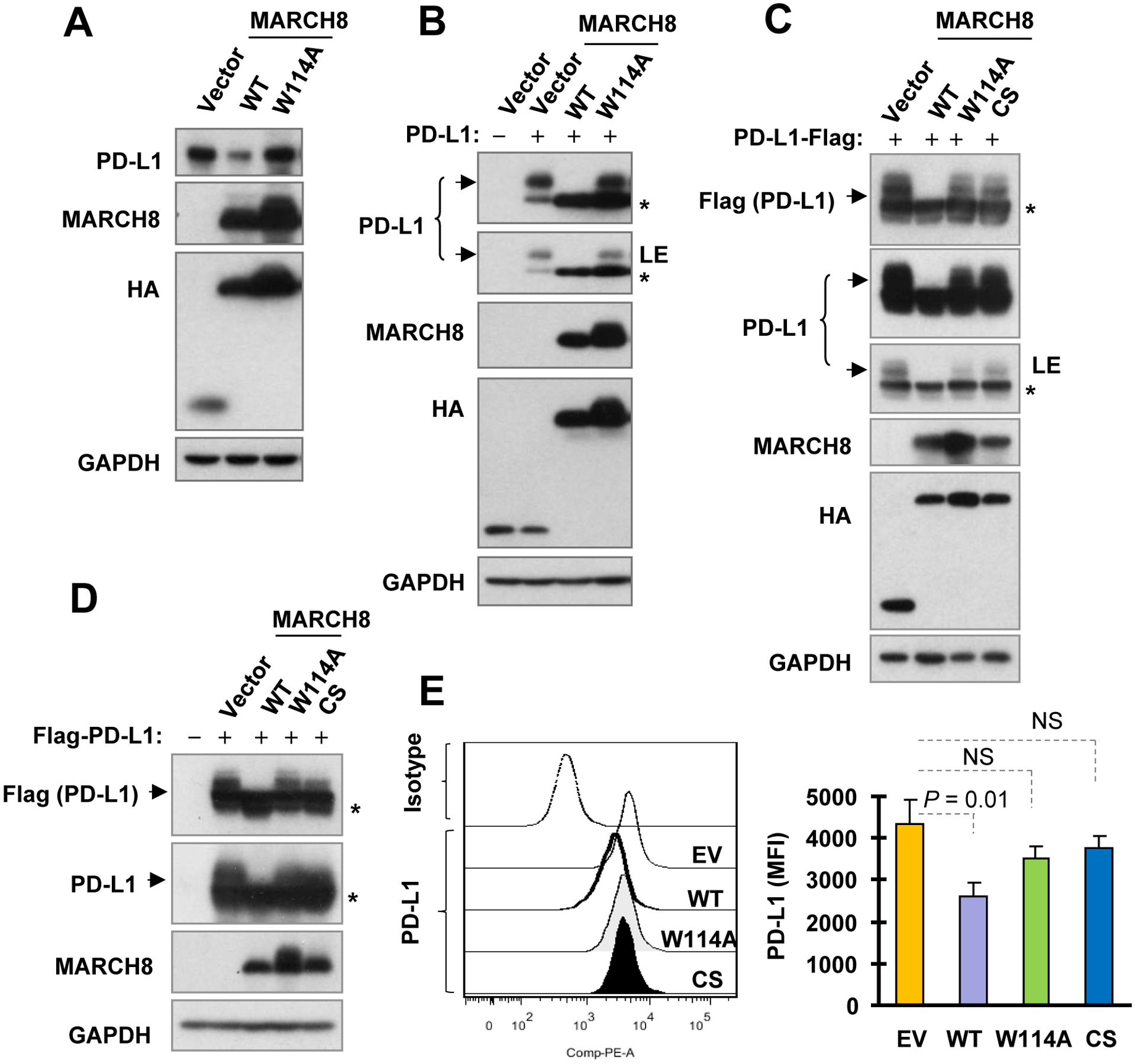
A and E, HEK293T cells were transfected with the plasmids harboring different MARCH8 genes as indicated for 36 h. B-D, HEK293T cells were co-transfected with MARCH8 plus a plasmid expressing non-tagged PD-L1 (B) or tagged PD-L1 (C and D) for 36 or 48 h. Total cellular and cell surface PD-L1 were detected with Western blotting (A-D) and flow cytometry (E), respectively. LE, low exposure; EV, empty vector; NS, not significant; *, uncharacterized band generated by ectopic expression of PD-L1.
MARCH8 interacts with PD-L1 in cells.
To further characterize the relationship between MARCH8 and PD-L1, we then determined whether MARCH8 and PD-L1 have an interaction. In HEK293T cells co-transfected with Flag-tagged PD-L1 and individual HA-tagged MARCH8 gene, we could detect both WT and mutant MARCH8 with either HA or MARCH8 antibody in the complexes pulled down with the Flag antibody (Fig. 5A), suggesting an interaction between MARCH8 and PD-L1, and that mutations in the RING-CH domain do not interfere MARCH8 interaction with PD-L1. To further demonstrate which domain of MARCH8 is essential for PD-L1 interaction, we generated expression constructs harboring different deletions of MARCH8 gene, which encodes MARCH8 protein with two transmembrane domains and a RING-CH domain (25,38), as depicted in Fig. 5B. By performing IP with either Flag antibody or HA antibody from the cell lysates derived from HEK293T cells co-transfected with HA-PD-L1 and variant Flag-MARCH8 encoding constructs, we could detect the interaction between PD-L1 and full-length MARCH8 or ΔC-tail MARCH8, but not between PD-L1 and RING MARCH8 (Fig. 5C). These results suggest that MARCH8 binds to PD-L1 with its N-terminal region. Following these findings, we lastly examined whether the MARCH8 and PD-L1 interaction is phosphorylation-dependent. To address this question, we prepared the cell lysates from HEK293T cells co-transfected with HA-PD-L1 and Flag-MARCH8 constructs with lysis buffer without phosphatase inhibitors followed by treatment with or without λ-phosphatase under the condition that was conventionally used in our previous studies to inhibit phosphatase activity (39,40) before the IP assay. We could detect PD-L1 in the complexes pulled down with the Flag (MARCH8) antibody from the lysates treated both with and without λ-phosphatase (Fig. 5D), suggesting that de-phosphorylation does not affect the interaction between MARCH8 and PD-L1. In other words, this preliminary result suggest that MARCH8 may interact with PD-L1 in a phosphorylation-independent manner. These results collectively suggest that MARCH8 interacts with PD-L1 in cells through its N-terminal region, likely independent of phosphorylation.
Fig 5. MARCH8 interacts with PD-L1 (A) with its N-terminal region (B and C) in a phosphorylation-independent manner (D) and ubiquitinates PD-L1 in cells (E and F).
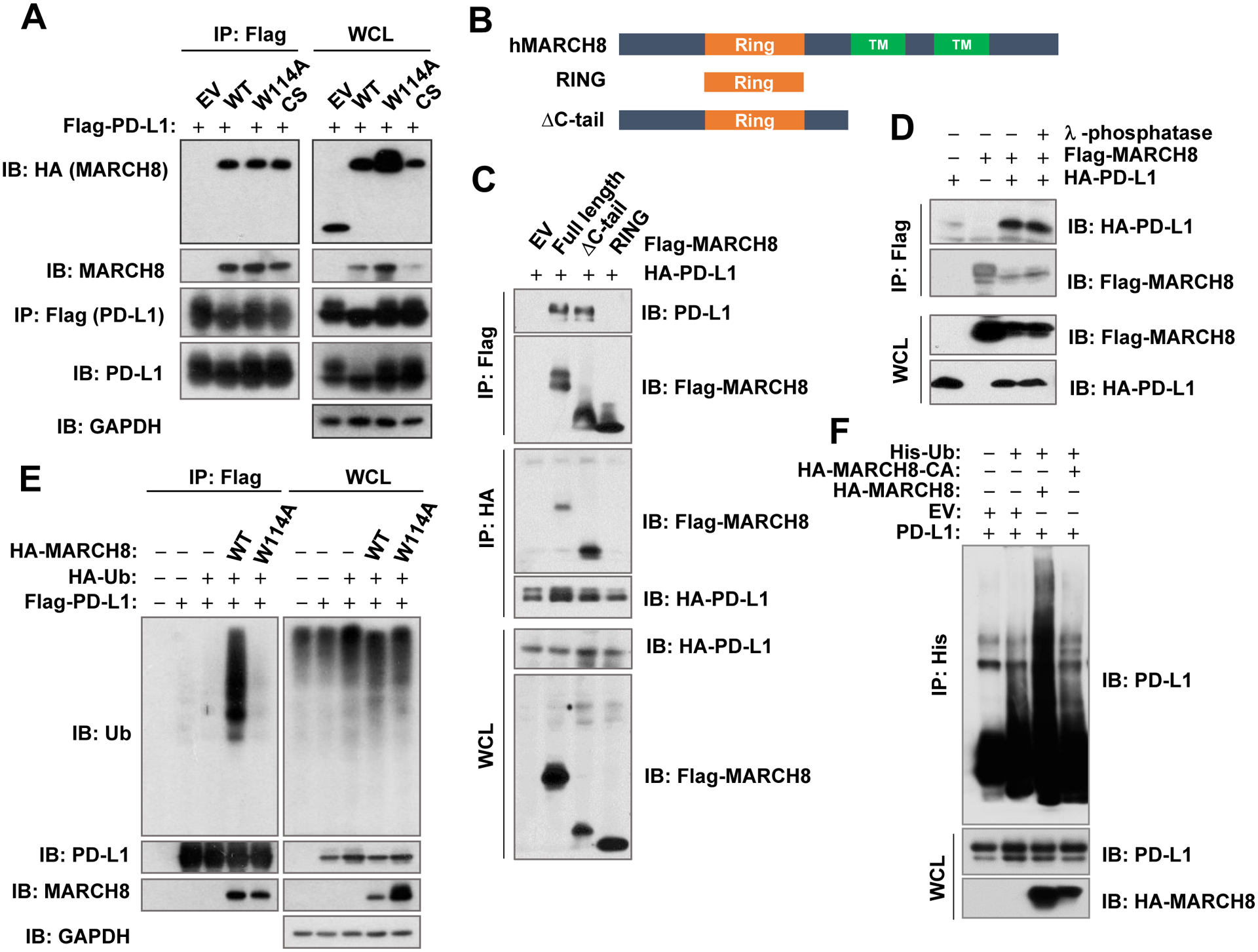
A and E, HEK293T cells were co-transfected with the indicated plasmids for 36 h followed by addition of MG132 (10 μM) for 9 h (A) or 2 h (E) and then harvested for preparation of whole cell lysates (WCL) for subsequent IP/Immunoblotting (IB) to detect different proteins as indicated. B, Schematic illustration of the MARCH8 domains, in which the RING domain was separated and indicated as RING and ΔC-tail. C, HEK293T cells were co-transfected with HA-PD-L1 construct and an indicated plasmid carrying a given MARCH8 gene as illustrated in B for 48 h. Cells were treated with 10 μM MG132 for 12 h before harvested for preparation of whole cell lysates and subsequent IP/IB (C). D, HEK293T cells were co-transfected with HA-PD-L1 and MARCH8 constructs for 48 h including treatment with 10 μM MG132 for 12 h before harvesting. The cells were harvested and lysed with non-phosphatase inhibitors-containing EBC buffer and treated with or without γ-phosphatase for 30 min at 37°C before being subject to IP/IB assay. F, HEK293T cells were co-transfected with different plasmids as indicated for 36 h followed by treatment with 10 μM MG132 for additional 12 h. Cells were then harvested for preparation of whole cell lysates and subsequent IP/IB.
MARCH8 ubiquitinates PD-L1 in cells.
Since MARCH8 is an E3 ubiquitin ligase, we determined whether MARCH8 is able to ubiquitinate PD-L1 in cells. As presented in Fig. 5E, we could detect clear PD-L1 ubiquitination in HEK293T cells co-transfected with constructs harboring PD-L1 and WT MARCH8, respectively, but not in the cells expressing PD-L1 and MARCH8-W114A, when the cell lysates were pulled down with the Flag (PD-L1) antibody followed by detection of ubiquitination with a ubiquitin antibody. Similarly, when the cell lysates were pulled down with a His (ubiquitin) antibody followed with PD-L1 detection with a PD-L1 antibody, increased PD-L1 ubiquitination was detected only in cells expressing WT MARCH8, but not in those expressing the mutant MARCH8, MARCH8-CA, in which the RING-CH domain was deleted (Fig. 5F). These results together clearly indicate that MARCH8 ubiquitinates PD-L1 in cells.
Osimertinib increases MARCH8 expression in EGFRm NSCLC cells.
We noted that osimertinib increased MARCH8 levels while decreasing PD-L1 levels (Fig. 2E). We, therefore, further examined the effects of osimertinib on MARCH8 expression in PC-9 and HCC827 cells in a more detailed way. Indeed, treatment with osimertinib at the tested concentration ranges of 25–250 nM elevated MARCH8 levels while decreasing PD-L1 levels (Fig. 6A). Similar results could also be achieved by erlotinib and CO1686 treatment (Fig. 6B). Osimertinib did not affect the degradation of MARCH8 as demonstrated in a CHX chase assay (Fig. 6C). In the presence of the transcriptional inhibitor, Act D, osimertinib failed to increase MARCH8 levels (Fig. 6D). Moreover, osimertinib significantly increased MARCH8 mRNA levels (Fig. 6E). Hence it appears that osimertinib increases MARCH8 expression primarily through enhancing its transcription levels.
Fig. 6. Osimertinib and other EGFR-TKIs (A and B) increase MARCH8 levels primarily via enhancing gene transcription (C-E).

A and B, Indicated cell lines were exposed to different concentrations of osimertinib (Osim) for 12 h (A) or 100 nM of the indicated EGFR-TKIs for 8 h (B). C, PC-9 cells were treated with DMSO or 100 nM osimertinib for 10 h followed by addition of 10 μg/ml CHX. Cells were then harvested at the indicated times post CHX. D, Indicated cell lines were pre-treated with 2.5 μg/ml Act D for 30 min followed by co-treatment with 100 nM osimertinib for an additional 8 h. The indicated proteins in A-D were detected with Western blotting. MARCH8 levels in C were plotted relative to those at time 0 of CHX treatment after being quantified by NIH Image J software and normalized to tubulin. E, PC-9 and HCC827 cells were treated with the indicated concentrations of osimertinib for 4 or 8 h. MARCH8 mRNA levels were detected with qRT-PCR. The data are the means ± SDs of triplicate determinations.
PD-L1 levels are elevated in osimertinib-resistant cells and in EGFRm NSCLC tissues relapsed to EGFR-TKI therapy and resistant to modulation by osimertinib in osimertinib-resistant cells.
To know whether there is an alteration of PD-L1 expression in EGFRm NSCLC cells when acquiring resistance to EGFR-TKIs, we compared baseline PD-L1 expression between PC-9 and its derived osimertinib-resistant PC-9/AR cell lines by checking RNA-seq data and found that PD-L1 mRNA expression was significantly increased in PC-9/AR cells comparing to PC-9 cells (Fig. 7A). Hence, we further detected PD-L1 protein levels in a few more EGFRm NSCLC cell lines with acquired resistance to osimertinib in comparison with their corresponding parental cell lines. As presented in Fig. 7B, baseline levels of PD-L1 in PC-9/AR, PC-9/GR/AR, HCC827/AR and SH416/AR were clearly higher than those in their corresponding parental cell lines. Under the conditions of osimertinib treatment, PD-L1 levels were reduced in PC-9 and HCC827 cells, but not or minimally in PC-9/AR and HCC827/AR cells (Fig. 7C), indicating the lost ability of osimertinib to downregulate PD-L1 expression in EGFRm NSCLC cells with acquired osimertinib resistance. Moreover, we detected PD-L1 expression using IHC in 19 paired tissue samples collected from EGFRm NSCLC patients before the treatment with first generation EGFR-TKIs (baseline) and after relapse to the treatments. We found that PD-L1 expression was elevated in 11 cases of relapsed samples (58%), while it remained reduced in 3 cases of relapsed samples (16%) or unchanged in 5 cases (26%), indicating that PD-L1 expression is increased in the majority of EGFRm NSCLC after acquiring resistance to EGFR-TKI treatment (Figs. 7D and 7E).
Fig. 7. Osimertinib-resistant cells possess elevated basal levels of PD-L1 (A and B), which are minimally modulated by osimertinib (C) and PD-L1 elevation occurs in EGFRm NSCLC tissues after relapse to EGFR-TKI treatment (D and E).

A, RNA-seq analyses show that PD-L1 mRNA expression in PC-9AR was significantly elevated in comparison with PC-9 cells (N = 3 for each cell lines). B and C, Whole-cell protein lysates were prepared from cell lines as indicated as comparable cell densities (B) or the cell lines treated with different concentrations of osimertinib for 16 h (C). Western blotting was used to detect PD-L1 in parental and osimertinib-resistant NSCLC cell lines. GAPDH was used as a loading control. D and E, PD-L1 expression was detected using immunohistochemical staining in 19 paired human NSCLC tissues (D). The representative images were shown (E).
Discussion
Downregulation of PD-L1 by EGFR-TKIs including osimertinib in EGFRm NSCLC cells were reported previously (11,23,24). The current study has further validated the downregulation of PD-L1 in EGFRm NSCLC cells with a variety of EGFR-TKIs including the first, the second, and the third generation EGFR-TKIs and in EGFRm tumors in vivo exposed to osimertinib. Beyond suppression of PD-L1 transcription possibly involving inhibition of NF-κB (23) and IL-6/JAK/STAT3 pathway (11), posttranslational regulation of PD-L1 levels by EGFR-TKIs involving enhancement of protein degradation was also suggested (24). In agreement, our findings also suggest that both transcriptional and posttranslational mechanisms are involved in mediating osimertinib-induced PD-L1 reduction in EGFRm NSCLC cells. However, it is unclear how osimertinib as well as other EGFR-TKIs enhances PD-L1 degradation.
PD-L1 has been suggested to undergo GSK3-dependent and β-TrCP-mediated proteasome degradation (37). Based on suppression of osimertinib-induced PD-L1 reduction by LiCl, a previous study suggested that osimertinib induces GSK3-dependent PD-L1 degradation (24). However, we failed to demonstrate the GSK3 dependency of osimertinib-induced PD-L1 degradation based on the following findings: 1) inhibition of GSK3 with both small molecule GSK3 inhibitors (SB216763 and CHIR99021) and GSK3 siRNA did not rescue PD-L1 reduction by osimertinib and even enhanced osimertinib-induced PD-L1 reduction in some cell lines (e.g. PC-9); and 2) Both SB216763 and CHIR99021 in fact effectively decreased PD-L1 levels. Thus, our data suggest that osimertinib induces GSK3-independent PD-L1 degradation. Moreover, we found that β-TrCP knockdown did not rescue PD-L1 reduction induced by osimertinib and osimertinib induced PD-L1 reduction accompanied with decrease in β-TrCP levels as well. Hence, it is clear that osimertinib is unlikely to induce β-TrCP-mediated PD-L1 degradation. Collectively, our findings in this study strongly suggest that osimertinib induces GSK3/β-TrCP-independent PD-L1 degradation.
The intriguing finding in this study is MARCH8 as a previously undiscovered novel E3 ubiquitin ligase to mediate PD-L1 degradation including osimertinib-induced PD-L1 degradation. In this study, knockdown of MARCH8 substantially increased the basal levels of PD-L1 and rescued PD-L1 reduction induced by osimertinib as well as other EGFR-TKIs. Complementarily, enforced expression of ectopic MARCH8 decreased PD-L1 levels and enhanced PD-L1 degradation; these could be achieved only by expressing WT MARCH8, but not E3 ligase-dead MARCH8 mutants although these mutants remained the ability to interact with PD-L1. Critically, MARCH8 could interact with PD-L1 through its N terminal region and effectively ubiquitinated PD-L1 in cells. Taking these findings together, we strongly believe that MARCH8 is a novel E3 ubiquitin ligase that mediates PD-L1 degradation induced by osimertinib and other EGFR-TKIs. We noted that MARCH8 knockdown showed partial protective effects on PD-L1 reduction induced by osimertinib and other EGFR-TKIs in the tested cell lines. This should be reasonable since transcriptional regulation mechanism also plays a role in regulation of PD-L1 levels induced by EGFR-TKIs
The human MARCH family comprises eleven members (termed MARCH-1 to 11), of which nine are transmembrane proteins that often share over-lapping substrate specificity (25,38). Among these transmembrane MARCH proteins, MARCH8 and MARCH1 share high sequence homology (25). Therefore, whether other MARCH proteins, particularly MARCH1, exerts similar function as MARCH8 does in mediating PD-L1 degradation including osimertinib-induced PD-L1 degradation needs to be investigated. Different from MARCH1 that is expressed at very low levels in most tissues, except for lymph nodes and spleen, MARCH8 is often expressed in many human tissues, particularly lung (highest expression) (38). Interestingly, most of transmembrane proteins such as MHC-II, whose degradation is mediated by MARCH8 (25,26), are immunomodulatory receptors, strongly suggesting a significant role of MARCH8 in immune regulation (41). The current finding on MARCH8 negative regulation of PD-L1 levels through its E3 ubiquitin ligase function further support the important role of MARCH8 in immune regulation.
To date, studies of the MARCH8 protein has focused on its function in the immune system. The involvement of MARCH8 in cancer is largely unclear. MARCH8 was reported to be downregulated in human NSCLC in comparison with their adjacent normal tissue and high MARCH8 expression is associated with good prognosis (42), whereas MARCH8 expression was increased in gastric and esophageal cancers and associated with bad prognosis (43,44). Moreover, these studies also generated opposite results when examining the impact of MARCH8 knockdown or overexpression on alterations of cell growth, apoptosis and migration (42–44). Our findings on the involvement of MARCH8 in mediating PD-L1 degradation by osimertinib and other EGFR-TKIs in EGFRm NSCLC cells warrant further study on the role of MARCH8 in NSCLC and other types of cancer.
The impact of PD-L1 expression on EGFR-targeted therapy against ERGFRm NSCLC is unclear because the published data are quite conflicting. While some studies suggested that PD-L1 expression in EGFRm NSCLCs does not impact patients’ response to EGFR-TKIs including osimertinib (20,21), other studies reported that high PD-L1 expression correlates with primary resistance to EGFR-TKIs (13–15) or that increased PD-L1 is associated with acquired resistance to EGFR-TKIs (10,22). Interestingly, there are also studies showing that PD-L1 expression may serve as a favorable biomarker predicting better response to EGFR-TKIs (16–19). Osimertinib clearly has a direct effect on eliminating EGFRm NSCLC cells via induction of apoptosis as we previous demonstrated (30). The finding of downregulation of PD-L1 in EGFRm NSCLC cells by osimertinib and other EGFR-TKIs as demonstrated in this study and other studies (11,23,24) suggest that osimertinib may also have an indirect effect on eliminating EGFRm NSCLC cells in vivo through activating endogenous immune defense mechanism. This is also in agreement with our previous finding that osimertinib facilitates c-FLIP degradation and augments TRAIL-induced apoptosis, a critical mechanism for immune cells to eliminate cancer cells (45,46), in EGFRm NSCLCs (47). In a previous preclinical study with EGFRm NSCLCs, erlotinib initial treatment can trigger a transient immunostimulatory effect despite the immunosuppressive phenotype observed following longer-term treatment (48). A recent study with single-cell RNA sequencing of human NSCLC biopsies has also confirmed this early immunostimulatory phenotype in EGFR-targeted therapy (49).
Although EGFR-TKIs such as osimertinib decreased PD-L1 levels in the early phase of treatment when EGFRm cells were sensitive to the treatment, we observed a rebound elevation of PD-L1 in several osimertinib-resistant cell lines and in the majority of EGFRm tissues (58%) collected after relapse to EGFR-TKI treatment when compare with their corresponding baseline expression. Coincidently, other studies with preclinical models or clinical tissues demonstrated similar findings (50,51). Whether this is a potential mechanism for the late stage of immunosuppressive phenotype observed in previous studies (48,49) needs to be explored. It is also very likely that the rebound elevation of PD-L1 expression observed may contribute to the emergence of acquired resistance to EGFR-TKIs including osimertinib. The current study has not been able to address why PD-L1 expression was not increased in other patients relapsed from EGFR-TKIs, while it was increased in the majority of relapsed EGFRm NSCLC cases. Given the diversified mechanisms underlying EGFR-TKI acquired resistance (52) and different regulatory mechanisms accounting for downregulation of PD-L1 by EGFR-TKIs in EGFRm NSCLC cells as demonstrated in this study and others (11,23,24), the variable alterations of PD-L1 in EGFR-TKI relapsed tissues should be reasonable. Nonetheless, our findings warrant further investigation on the underlying mechanisms.
Another interesting question is whether EGFRm NSCLCs relapsed to osimertinib or other EGFR-TKIs with elevated PD-L1 expression will respond better to PD-1 and particularly PD-L1 immune checkpoint blockade therapy. Given that PD-L1 expression is a valid predictable biomarker for PD-1/PD-L1 checkpoint immunotherapy in advance NSCLC patients (53), it should be a valid question to see whether EGFR-TKI resistant NSCLC with elevated PD-L1 expression can be switched to receive PD-1, particularly PD-L1, checkpoint immunotherapy. Indeed, a clinical study by Isomoto et al. (51) showed that median progression-free survival (PFS) for subsequent treatment with antibodies to PD-1 after progression from EGFR-TKI treatment was significantly longer for patients with a high than those with a low PD-L1 expression (7.1 vs. 1.7 months), and two of five patients whose PD-L1 expression level changed from low to high after EGFR-TKI treatment achieved a PFS of > 6 months. Another recent clinical study has suggested that patients with short PFS to EGFR-TKIs predicted better response to subsequent PD-1/PD-L1 based immunotherapy in EGFRm NSCLC (54). It will be interesting to further investigate whether these patients have elevated PD-L1 expression, contributing to better response to PD-1/PD-L1 checkpoint immunotherapy.
In summary, the current study has demonstrated the effects of osimertinb and other EGFR-TKIs on downregulation of PD-L1 in EGFRm NSCLC cells and tumors through both transcriptional and post-translational mechanisms. The most important finding is the identification of MARCH8 as a previously undiscovered PD-L1 E3 ubiquitin ligase that mediates PD-L1 degradation induced by osimertinib and other EGFR-TKIs. The rebound elevation of PD-L1 in some NSCLCs with acquired resistance to EGFR-TKIs, which are resistant to further modulation by EGFR-TKIs, may suggest a late stage of immunosuppressive phenotype and may contribute to emergence of acquired resistance to EGFR-TKIs: this may also provide an opportunity for PD-1/PD-L1 immune checkpoint blockade therapy for those patients.
Supplementary Material
Implications.
This study has demonstrated a previously undiscovered function of MARCH8 in mediating PD-L1 degradation induced by EGFR inhibitors in EGFR mutant NSCLC cells, establishing a novel connection between MARCH8 and PD-L1 regulation.
Acknowledgement
We thank Dr. A. Hammond in our department for editing the manuscript.
Grant Support:
NIH/NCI R01 CA223220 (to SYS), R01 CA245386 (to SYS) and UG1 CA233259 (to SSR), Emory University Winship Cancer Institute lung cancer pilot fund (to SYS) and Lee Foundation Award to the Winship Lung Cancer Program (to SSR and SYS).
Abbreviations:
- NSCLC
non-small cell lung cancer
- EGFR
epidermal growth factor receptor
- EGFR-TKIs
EGFR-tyrosine kinase inhibitors
- CHX
cycloheximide
- shRNA
short-hairpin RNA
Footnotes
Conflicts of interest: SSR is on consulting/advisory board for AstraZeneca, BMS, Merck, Roche, Tesaro and Amgen. TKO is on consulting/advisory board for Novartis, Celgene, Lilly, Sandoz, Abbvie, Eisai, Takeda, Bristol-Myers Squibb, MedImmune, Amgen, AstraZeneca and Boehringer Ingelheim. Other authors declare that they have no conflict of interest.
References
- 1.Shah R, Lester JF. Tyrosine Kinase Inhibitors for the Treatment of EGFR Mutation-Positive Non-Small-Cell Lung Cancer: A Clash of the Generations. Clinical lung cancer 2020;21:e216–e28 [DOI] [PubMed] [Google Scholar]
- 2.Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N Engl J Med 2020;382:41–50 [DOI] [PubMed] [Google Scholar]
- 3.Carlisle JW, Ramalingam SS. Role of osimertinib in the treatment of EGFR-mutation positive non-small-cell lung cancer. Future oncology 2019;15:805–16 [DOI] [PubMed] [Google Scholar]
- 4.Piper-Vallillo AJ, Sequist LV, Piotrowska Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J Clin Oncol 2020:JCO1903123 [DOI] [PubMed] [Google Scholar]
- 5.Giroux Leprieur E, Dumenil C, Julie C, Giraud V, Dumoulin J, Labrune S, et al. Immunotherapy revolutionises non-small-cell lung cancer therapy: Results, perspectives and new challenges. Eur J Cancer 2017;78:16–23 [DOI] [PubMed] [Google Scholar]
- 6.Somasundaram A, Burns TF. The next generation of immunotherapy: keeping lung cancer in check. J Hematol Oncol 2017;10:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stambrook PJ, Maher J, Farzaneh F. Cancer Immunotherapy: Whence and Whither. Molecular cancer research : MCR 2017;15:635–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegde PS, Chen DS. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020;52:17–35 [DOI] [PubMed] [Google Scholar]
- 9.Remon J, Besse B, Soria JC. Successes and failures: what did we learn from recent first-line treatment immunotherapy trials in non-small cell lung cancer? BMC medicine 2017;15:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee BS, Park DI, Lee DH, Lee JE, Yeo MK, Park YH, et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem Biophys Res Commun 2017 [DOI] [PubMed] [Google Scholar]
- 11.Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu Z, et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol 2016;49:1360–8 [DOI] [PubMed] [Google Scholar]
- 12.Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2015;10:910–23 [DOI] [PubMed] [Google Scholar]
- 13.Hsu KH, Huang YH, Tseng JS, Chen KC, Ku WH, Su KY, et al. High PD-L1 expression correlates with primary resistance to EGFR-TKIs in treatment naive advanced EGFR-mutant lung adenocarcinoma patients. Lung Cancer 2019;127:37–43 [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto Y, Sawa K, Fukui M, Oyanagi J, Izumi M, Ogawa K, et al. Impact of tumor microenvironment on the efficacy of epidermal growth factor receptor-tyrosine kinase inhibitors in patients with EGFR-mutant non-small cell lung cancer. Cancer Sci 2019;110:3244–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su S, Dong ZY, Xie Z, Yan LX, Li YF, Su J, et al. Strong Programmed Death Ligand 1 Expression Predicts Poor Response and De Novo Resistance to EGFR Tyrosine Kinase Inhibitors Among NSCLC Patients With EGFR Mutation. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2018;13:1668–75 [DOI] [PubMed] [Google Scholar]
- 16.Lin C, Chen X, Li M, Liu J, Qi X, Yang W, et al. Programmed Death-Ligand 1 Expression Predicts Tyrosine Kinase Inhibitor Response and Better Prognosis in a Cohort of Patients With Epidermal Growth Factor Receptor Mutation-Positive Lung Adenocarcinoma. Clinical lung cancer 2015;16:e25–35 [DOI] [PubMed] [Google Scholar]
- 17.Tang Y, Fang W, Zhang Y, Hong S, Kang S, Yan Y, et al. The association between PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced non-small cell lung cancer patients treated with EGFR-TKIs. Oncotarget 2015;6:14209–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, Li H, Pang R, Huang J. Altered status of programmed death-ligand 1 after recurrence in resected lung adenocarcinoma patients. OncoTargets and therapy 2017;10:2003–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takashima Y, Sakakibara-Konishi J, Hatanaka Y, Hatanaka KC, Ohhara Y, Oizumi S, et al. Clinicopathologic Features and Immune Microenvironment of Non-Small-cell Lung Cancer With Primary Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Clinical lung cancer 2018;19:352–9 e1 [DOI] [PubMed] [Google Scholar]
- 20.Hsu PC, Wang CW, Kuo SC, Lin SM, Lo YL, Huang AC, et al. The Co-Expression of Programmed Death-Ligand 1 (PD-L1) in Untreated EGFR-Mutated Metastatic Lung Adenocarcinoma. Biomedicines 2020;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown H, Vansteenkiste J, Nakagawa K, Cobo M, John T, Barker C, et al. Programmed Cell Death Ligand 1 Expression in Untreated EGFR Mutated Advanced NSCLC and Response to Osimertinib Versus Comparator in FLAURA. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2020;15:138–43 [DOI] [PubMed] [Google Scholar]
- 22.Hsu PC, Jablons DM, Yang CT, You L. Epidermal Growth Factor Receptor (EGFR) Pathway, Yes-Associated Protein (YAP) and the Regulation of Programmed Death-Ligand 1 (PD-L1) in Non-Small Cell Lung Cancer (NSCLC). International journal of molecular sciences 2019;20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin K, Cheng J, Yang T, Li Y, Zhu B. EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-kappaB. Biochem Biophys Res Commun 2015;463:95–101 [DOI] [PubMed] [Google Scholar]
- 24.Jiang XM, Xu YL, Huang MY, Zhang LL, Su MX, Chen X, et al. Osimertinib (AZD9291) decreases programmed death ligand-1 in EGFR-mutated non-small cell lung cancer cells. Acta pharmacologica Sinica 2017;38:1512–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bauer J, Bakke O, Morth JP. Overview of the membrane-associated RING-CH (MARCH) E3 ligase family. N Biotechnol 2017;38:7–15 [DOI] [PubMed] [Google Scholar]
- 26.Tada T, Zhang Y, Koyama T, Tobiume M, Tsunetsugu-Yokota Y, Yamaoka S, et al. MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat Med 2015;21:1502–7 [DOI] [PubMed] [Google Scholar]
- 27.Zhang S, Qian G, Shi P, Zhang G, Fan S, He Y, et al. Downregulation of death receptor 4 is tightly associated with positive response of EGFR mutant lung cancer to EGFR-targeted therapy and improved prognosis. Theranostics 2021;11:3964–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018;553:91–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koo J, Wu X, Mao Z, Khuri FR, Sun SY. Rictor undergoes GSK3-dependent, FBXW7-mediated ubiquitination and proteasomal degradation. J Biol Chem 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi P, Oh YT, Deng L, Zhang G, Qian G, Zhang S, et al. Overcoming Acquired Resistance to AZD9291, A Third-Generation EGFR Inhibitor, through Modulation of MEK/ERK-Dependent Bim and Mcl-1 Degradation. Clin Cancer Res 2017;23:6567–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi P, Oh YT, Zhang G, Yao W, Yue P, Li Y, et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett 2016;380:494–504 [DOI] [PubMed] [Google Scholar]
- 32.Ren H, Chen M, Yue P, Tao H, Owonikoko TK, Ramalingam SS, et al. The combination of RAD001 and NVP-BKM120 synergistically inhibits the growth of lung cancer in vitro and in vivo. Cancer Lett 2012;325:139–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, et al. Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res 1997;57:4931–9 [PubMed] [Google Scholar]
- 34.Ou JN, Wiedeman AE, Stevens AM. TNF-alpha and TGF-beta counter-regulate PD-L1 expression on monocytes in systemic lupus erythematosus. Scientific reports 2012;2:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qian G, Wang D, Magliocca KR, Hu Z, Nannapaneni S, Kim S, et al. Human papillomavirus oncoprotein E6 upregulates c-Met through p53 downregulation. Eur J Cancer 2016;65:21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng L, Qian G, Zhang S, Zheng H, Fan S, Lesinski GB, et al. Inhibition of mTOR complex 1/p70 S6 kinase signaling elevates PD-L1 levels in human cancer cells through enhancing protein stabilization accompanied with enhanced beta-TrCP degradation. Oncogene 2019;38:6270–82 [DOI] [PubMed] [Google Scholar]
- 37.Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nature communications 2016;7:12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samji T, Hong S, Means RE. The Membrane Associated RING-CH Proteins: A Family of E3 Ligases with Diverse Roles through the Cell. Int Sch Res Notices 2014;2014:637295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inuzuka H, Tseng A, Gao D, Zhai B, Zhang Q, Shaik S, et al. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta-TRCP) ubiquitin ligase. Cancer Cell 2010;18:147–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukushima H, Ogura K, Wan L, Lu Y, Li V, Gao D, et al. SCF-mediated Cdh1 degradation defines a negative feedback system that coordinates cell-cycle progression. Cell Rep 2013;4:803–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng C The emerging roles of the MARCH ligases in antiviral innate immunity. Int J Biol Macromol 2021;171:423–7 [DOI] [PubMed] [Google Scholar]
- 42.Fan J, Tian L, Li M, Huang SH, Zhang J, Zhao B. MARCH8 is associated with poor prognosis in non-small cell lung cancers patients. Oncotarget 2017;8:108238–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin J, Ji Z, Hong Y, Song Z, Hu N, Zhuang M, et al. Sh-MARCH8 Inhibits Tumorigenesis via PI3K Pathway in Gastric Cancer. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 2018;49:306–21 [DOI] [PubMed] [Google Scholar]
- 44.Singh S, Saraya A, Das P, Sharma R. Increased expression of MARCH8, an E3 ubiquitin ligase, is associated with growth of esophageal tumor. Cancer cell international 2017;17:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anel A, Bosque A, Naval J, Pineiro A, Larrad L, Alava MA, et al. Apo2L/TRAIL and immune regulation. Front Biosci 2007;12:2074–84 [DOI] [PubMed] [Google Scholar]
- 46.Falschlehner C, Schaefer U, Walczak H. Following TRAIL’s path in the immune system. Immunology 2009;127:145–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi P, Zhang S, Zhu L, Qian G, Ren H, Ramalingam SS, et al. The Third-Generation EGFR Inhibitor, Osimertinib, Promotes c-FLIP Degradation, Enhancing Apoptosis Including TRAIL-Induced Apoptosis in NSCLC Cells with Activating EGFR Mutations. Transl Oncol 2019;12:705–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dominguez C, Tsang KY, Palena C. Short-term EGFR blockade enhances immune-mediated cytotoxicity of EGFR mutant lung cancer cells: rationale for combination therapies. Cell Death Dis 2016;7:e2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 2020;182:1232–51 e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang L, Guo F, Liu X, Li X, Qin Q, Shu P, et al. Continuous targeted kinase inhibitors treatment induces upregulation of PD-L1 in resistant NSCLC. Scientific reports 2019;9:3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Isomoto K, Haratani K, Hayashi H, Shimizu S, Tomida S, Niwa T, et al. Impact of EGFR-TKI Treatment on the Tumor Immune Microenvironment in EGFR Mutation-Positive Non-Small Cell Lung Cancer. Clin Cancer Res 2020;26:2037–46 [DOI] [PubMed] [Google Scholar]
- 52.Lim SM, Syn NL, Cho BC, Soo RA. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat Rev 2018;65:1–10 [DOI] [PubMed] [Google Scholar]
- 53.Wojas-Krawczyk K, Kubiatowski T. Imperfect Predictors for Lung Cancer Immunotherapy-A Field for Further Research. Frontiers in oncology 2020;10:568174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu S, Wu F, Li X, Zhao C, Jia Y, Jia K, et al. Patients With Short PFS to EGFR-TKIs Predicted Better Response to Subsequent Anti-PD-1/PD-L1 Based Immunotherapy in EGFR Common Mutation NSCLC. Frontiers in oncology 2021;11:639947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.