

Abstract
Nestin, a class IV intermediate filament protein, is generally considered as a putative marker of neural stem and progenitor cells in the central nervous system. Glioma is a common type of adult brain tumors, and glioblastoma (GBM) represents the most aggressive form of glioma. Here, we report that Nestin expression is significantly up-regulated in human GBM, compared with other types of glioma. Nestin knockdown or deletion in U251 cells and tumor cells from GBM patients derived xenografts (PDX), resulted in G2/M arrest, finally leading to apoptosis in tumor cells. Using proximity-dependent Biotin identification method, we identified βII-tubulin as an interacting protein of Nestin in U251 cells. Nestin stabilized βII-tubulin in U251 cells through physical interaction. Knockdown of Nestin or βII-tubulin disrupted spindle morphology in tumor cells. Our studies further revealed that Nestin deficiency in U251 cells and GBM PDX cells repressed tumor growth upon transplantation. Finally, we found that Nestin deficiency sensitized GBM cells to microtubule-destabilizing drugs such as vinblastine and vincristine. Our studies demonstrate the essential functions and underlying mechanisms of Nestin in the growth and drug response of GBM cells. Implications: Through interaction with βII-tubulin, Nestin facilitates cell cycle progression and spindle assembly of tumor cells in glioblastoma.
Keywords: Glioblastoma, Nestin, Cell cycle, Spindle, βII-tubulin
Introduction
Nestin, an intermediate filament protein, was originally identified in neural stem cells, which has been used as a putative marker for neural stem cells. Nestin expression is recently found in many tumor tissues including brain tumors, as well as pancreatic cancers and gastrointestinal tumors1,2. Nestin-expressing progenitors are found to serve as tumor initiating cells in medulloblastoma and glioblastoma (GBM)3,4. Nestin itself also plays important functions during tumor progression. We previously showed that Nestin mediates medulloblastoma tumorigenesis by augmenting the hedgehog signaling5. Nestin expression is required for the cellular plasticity and the expansion of cancer stem cell population in hepatocellular carcinomas or cholangiocarcinomas6. In addition, Nestin can enter cell nucleus and protect tumor cells from cellular senescence7.
Glioma is the most common brain tumors, which are classified as Grade I to IV based on histology and clinical criteria8. Grade I gliomas are benign and curable with complete resection, and represent an entity distinct from Grade II-IV tumors. Grade II gliomas consists of astrocytoma, oligodendroglioma and oligo-astrocytoma (or called mixed gliomas). All Grade II tumors eventually progress to high-grade gliomas. Grade IV tumors are also known as GBM, which is the most common primary malignant brain tumor. Despite all aggressive treatment, the median survival of GBM remains approximately 12–15 months8. In the last decades, no significant progress has been made in the prognosis of GBM due to the limited understanding of the tumorigenesis of GBM. Nestin expression is often detected in tumor tissue of GBM as well as cell lines derived from human or mouse GBM9–11. However, the functions of Nestin in GBM remain to be determined.
Chromosome segregation during cell division is strictly regulated in order to preserve genomic integrity. To facilitate chromosome segregation, cells assemble a special microtubule-based structure-the mitotic spindle, to capture and align chromosomes. Formation of a bipolar spindle is critical for segregation of chromosomes between dividing cells. Failure of this process often results in genomic instability and aneuploidy, characteristic of most cancers12,13. Microtubules are highly dynamic polymers consisting of a head-to tail arrangement of α/β tubulin heterodimers14. The intrinsic polarity of microtubules is formed with β-tubulin subunits facing the plus end and the α-tubulin subunit facing the minus end. Microtubule ends undergo stochastic changes from a polymerizing to a depolymerizing state, a phenomenon known as dynamic instability. Dynamic instability of microtubules is essential for a bipolar spindle assembly and chromosome segregation during cell division15,16.
Here, we found that Nestin expression is significantly upregulated in GBM tissues compared with other low-grade gliomas including astrocytoma, oligodendrocytoma and mixed gliomas. Knockdown Nestin by ShRNA or Nestin deletion by CRISPR resulted in G2/M arrest in U251 cells, a GBM cell line, as well as GBM PDX cells, leading to apoptosis of tumor cells. Our studies further revealed that Nestin deficiency caused abnormal spindle formation in GBM cells. By using proximity BioID analyses, we found that Nestin physically interacted with βII-tubulin in U251 cells. Nestin knockdown significantly decreased the half-life of βII-tubulin in GBM cells, suggesting that Nestin stabilizes β-tubulin through physical interaction in GBM cells. Finally, we demonstrate that Nestin knockdown significantly suppressed the growth of U251 cells and PDX cells after transplantation. In addition, Nestin deficiency sensitizes GBM cells to microtubule-destabilizing drugs. Our studies reveal the critical role of Nestin in mitotic progression and drug responses of GBM cells to anti-microtubule agents.
Materials and Methods
Cell culture
U251, U87, 293T, A172, DBTRG, M059J, U118MG were purchased from the American Type Culture Collection (ATCC) and cultured according to the guidelines recommended by the ATCC. All above cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/high glucose (Basal media) containing 10% fetal bovine serum (FBS, GIBCO). All cell lines were regularly tested for mycoplasma infection (MycoAlert Mycoplasma Detection Kit, #LT07-118; Lonza).
Human GBM PDX lines were provided by Dr. Minghua Wu in Central South University, which were previous published17–19. The basic information for PDX cells used in our studies, includes PDX1: GBM glioblastoma multiforme, characterized by Olig2+, GFAP+, MGMT methylation+, IDH1 wild, 1p/19q deleted; PDX2: GBM glioblastoma multiforme, characterized by Olig2+, GFAP+, MGMT methylation+, IDH1 wild, 1p/19q deleted; PDX3: grade II Diffuse astrocytic glioma, characterized by Olig2+, GFAP+, MGMT methylation+, IDH1 wild, 1p/19q deleted.
Animals and xenograft model in nude mice
Nude mice and SCID mice (at 5 weeks of age) were obtained from the Beijing Vital River Laboratory Animal Technology Co., Ltd and all experiments were performed in accordance with procedures approved by the laboratory animal committee of Soochow university. For mouse xenograft models, animals were randomly allocated into groups receiving cell line injections. U251 cells were infected with a lentivirus carrying Nestin ShRNA or scrambled ShRNA. 48hrs following the infection, cells were harvested after removing dead cells/debris by centrifugation (300 g for 5 mins). U251 cells (4×106/100 μl) were suspended in 100 μl PBS and injected subcutaneously into flanks of nude mice (n =4 mice/group). Tumor size was measured every 3 days and tumor volume was calculated as described previously20. G*power software was used to calculate required mouse number in each group for the transplantation (effect size=1.5–2, power=0.8, α error probability=0.05).
For intracranial transplantation of GBM PDX cells, SCID mice were purchased from the Beijing Vital River Laboratory Animal Technology Co., Ltd. PDX-1 cells were virally infected and harvested as mentioned above. PDX cells (3×105) were then suspended in 5μl PBS and intracranially injected into forebrains of SCID mice (6 mice/group) as previously described21.
Western blotting, Immunostaining and immunoprecipitation
Whole-cell extracts were obtained by lysing cells with RIPA buffer (Solarbio; R0010) containing protease inhibitors (5 μg/ml PMSF and cocktails) and then centrifuged at 15,000 g for 15 minutes at 4°C. Equal amounts of protein were separated by SDS PAGE and subsequently transferred onto nylon membrane. The protein samples were analyzed using antibodies including anti-Nestin (1:500, Santa Cruz), anti-GAPDH (1:1000; Proteintech), anti-βIII-tubulin (1:1000; Novus), anti-α-tubulin (1:1000; Proteintech), anti-βII-tubulin (1:500; HuaBio), anti-βI-tubulin (1:1000; Abcam).
For immunofluorescence, cells were fixed for 15 mins with 4% paraformaldehyde (PFA), permeabilized in 0.1% Triton X-100 for 10 min. Cells were then blocked for 1 hr with PBS containing 0.1% Triton X-100 and 1% BSA, and incubated with primary antibodies overnight at 4°C, and incubated with secondary antibodies for 2 hrs at room temperature. Cells were counterstained with DAPI and mounted with Fluoromount-G before being visualized using a Nikon Eclipse Ti. Primary antibodies used in this study include: anti-Nestin (1:200; R&D Systems), anti-α-tubulin (1:500; proteintech), anti-βII-tubulin (1:100; HuaBio), anti-Ki67 (1:200; Abcam) and anti-cleaved caspase-3 (1:200; Abcam).
Human glioma TMA was purchased from US Biomax, Inc. (GL803c), containing a total of 80 cores from 80 cases (containing 15 cases of astrocytoma, 40 cases of GBM, 4 mixed glioma, 9 oligodendroglioma, plus 5 adjacent normal brain tissue, single core per case) were used for analyzing Nestin expression in different types of glioma. After immunostaining with antibodies against Nestin, the slides were independently viewed and scored by two experienced pathologists. H-score was generated for Nestin expression on the slide. The H score consists of the product of the intensity of immunostaining (0–3+) and the percentage of cells with nuclear staining. The range of the H score was 0 to 300 (0% cells positive to 100% cells 3+positive).
For immunoprecipitation experiments, total cell lysate was mixed with GFP traps (Chromotek) and incubated 2hrs at 4°C. The traps were washed with PBS and resuspended in 80 μL 2×SDS loading buffer and subjected to Western blot analysis.
Genetic manipulation in GBM cells
To delete Nestin gene in U251 cells by CRISPR, the exon 1 of Nestin was selected for gRNA design. To select the candidate sgRNAs (single-guide RNA) for Nestin genome editing, the integral genomic sequence from the exon 1 of Nestin was submitted individually to the online CRISPR Design Tool (http://crispr.mit.edu/). The candidate sgRNAs were transformed into pLKO-puro. To construct the Nestin-overexpression (OE) plasmid, the whole coding sequence of Nestin was generated by PCR amplification and Homologous reorganization into HindIII the and KPnI sites of the PCS2-vector. All ligation products were transformed into DH5α competent cells and spread on solid Amp+ or kana+ Luria broth (LB) medium. After single colonies were inoculated into liquid LB medium and extracted plasmid, validated and sequenced plasmids were stored at −20 °C. Two days after U251 cells were co-infected with the lentivirus carrying sgRNA and the lentivirus with Cas-9, monoclones were sorted by flow cytometry and cultured on 96-well plates. Two weeks later, surviving cells were harvested to detect the Nestin deletion by sequencing.
ShRNAs used to knock down the expression of Nestin or βII-tubuin in U251 cells or GBM PDX cells. The sequences for shRNAs are below:
ShCtrl: GTCGCTTACCGATTCAGAATGGCTCGAGCCATTCTGAATCGGTAAGCGACTTTTT
ShNES#1: GGCTAGTCCCTGCCTGAATAACTCGAGTTATTCAGGCAGGGACTAGCCTTTTT
ShNES#2: GCAGACATCATTGGTGTTAATCTCGAGATTAACACCAATGATGTCTGCTTTTT
ShβII# 1: GGTGGAAAACACAGATGAACTCGAGTTCATCTGTGTTTTCCACC
ShβII# 2: GAGCAGATGTTCGACTCCAACTCGAGTTGGAGTCGAACATCTGCTC
BioID method and construction of bait plasmids
The BioID method was used to detect the binding partners of Nestin in vivo through proximity-dependent biotinylation22. The bait protein plasmid encodes Nestin fused to the promiscuous biotinylase BirA. U251 cells were transfected with the bait protein plasmid, and the culture was incubated with biotin-containing media for 24hrs. Cells were lysed and biotinylated proteins were captured on streptavidin beads and subjected to identification by mass spectrometry.
For construction of BioID bait proteins, human Nestin ORF (NM_006617) from OriGene Technologies was transferred using the OriGene Precision Shuttle cloning sites Sgf I and Mlu I. The BirA* N- and C-terminal fusions vectors developed by the Roux laboratory (Kim et al., 2014) were obtained from Addgene. The multi-cloning sites of these vectors, pcDNA3.1 BirA-HA (Plasmid #36047) and pcDNA3.1 myc-BioID (Plasmid #35700), were first reengineered to accept fragments from the OriGene Precision Shuttle system. Nestin ORF derived from the pCMV6-AN-mGFP N-terminal monomeric GFP fusion vector was fused in frame to create a C-terminal BirA fusion (C-BirA-HA-hNestin), and also fused in frame to create a N-terminal BirA fusion (N-BirA-myc-hNestin).
BioID mass spectrometry and data analysis
Nestin BirA* fusion plasmids were used to transfect U251 cells, and BioID was carried out as previously described23,24. At 24 hrs following the transfection, U251 cells were lysed and beads were resuspended in 50 μl of 8 M urea/50 mM Ammonium Bicarbonate, and proteins were reduced by adding 2 μl of 0.5 M Tris (2 carboxyethyl) phosphine (TCEP) to 50 μl of beads-proteins suspension mix. Proteins were reduced at 30°C for 60 mins, and the reaction was cooled to room temperature before alkylation by adding 4 μl of 0.5 M Iodoacetamide at room temperature in the dark for 30 min. Sample volume was adjusted by adding 350 μl of 50 mM ammonium bicarbonate to dilute the 8M urea to 1M before trypsin digestion with trypsin (mass spectrometry grade). 5μg trypsin per sample was added, and incubation was overnight at 30°C using Eppendorf Thermomixer at 700 rpm. Digested peptides were separated from beads by centrifugation and peptide digests were transferred to a new tube followed by an extra wash step with 50 μl of 50mM ammonium bicarbonate. Formic acid was added to the peptide solution (to 2%), followed by desalting with C18 TopTip (PolyLC) and finally drying in a SpeedVac. Tryptic peptides were re-suspended in 100 μl of 2% Acetonitrile in 0.1% formic acid, and 10 μl of total tryptic peptides were utilized for the LC-MS/MS analysis, consisting of an EASY-nLC 1000 HPLC Acclaim PepMap peptide trap, a 25 cm-2 μm Easy-Spray C18 column, Easy Spray Source, and a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific).
The LC-MS/MS analysis was performed by the proteomics facility at Sanford Burnham Preby Medical Discovery Institute in California. For protein Identification and data analysis, the LC-MS/MS raw data were submitted to Sorcerer Enterprise v.3.5 release (Sage-N Research Inc.) with SEQUEST algorithm as the search program for peptide/protein identification. SEQUEST was set up to search the target-decoy UniProt Human Reviewed protein fasta database containing protein sequences using trypsin for enzyme with the allowance of up to 2 missed cleavages, Semi Tryptic search, fixed modification of 57 Da for cysteine to account for carboxyamidomethylation, and precursor mass tolerance of 50 ppm. Differential search includes 16 Da for methionine oxidation, and 226 Da on lysine for biotinylation. The search results were viewed, sorted, filtered, and statically analyzed by using comprehensive proteomics data analysis software, Peptide/Protein prophet v.4.02 (ISB). The minimum trans-proteomic pipeline (TPP) probability score for proteins was set to 0.9, to assure very low error (much less than FDR 2%) with reasonably good sensitivity. The differential spectral count analysis was done by QTools, an open source in-house developed tool for automated differential peptide/protein spectral count analysis.
For interaction networks analysis, we used STRING v10.0 (www.string-db.org), keeping default parameters.
Q-PCR
Total RNA was isolated from cells using the Trizol reagent (ThermoFisher), cDNA was synthesized using oligo (dT) and Superscript II reverse transcriptase (Takara). Quantitative PCR reactions were performed in triplicate using SYBR qPCR Master Mix and ABI 7500 Real-Time PCR Detection System. Primers specific to GAPDH, Nestin, TUBB2A and TUBB2B were available upon request.
Flow cytometric analyses of cell cycling and apoptosis
For flow cytometry-based cell cycle analysis, U251 or PDX cells were labelled with propidium iodide (PI; MultiSciences). Cellular DNA contents were measured using a FACS Calibur flow cytometer (BD). The cell cycle results were analyzed by the ModFit software. At least 20000 cells were collected for each sample. For apoptosis analysis, cells were labelled with Annexin V and PI (MultiScience), and analyzed by flow cytometry. Apoptotic cells were positive for Annexin V.
CCK-8 assay and colony formation assay
Cell Counting kit-8 (CCK-8, APExBIO) was used to measure the cell viability/proliferation. Cells were seeded in 96-well plates in triplicate, CCK-8 reagent was added to each well and incubated at 37°C for 1 hr. Absorbance at 450 nm was measured by a microplate reader. Three biological replicates were performed for each experiment. GBM cells were treated with chemotherapeutic agents including vinblastine, vincristine or etoposide (APExBIO) for 48hrs before being harvested for viability measurement. Note that the cell viability of Nestin-deficient GBM cells after each drug treatment, was normalized with that of naïve Nestin-deficient cells (without drug treatment), to remove the effect of Nestin knockdown on cell survival.
U251 cells (1×103) were plated in triplicate into 6-well plates and cultured at 37°C. After 11 days, colonies were fixed with 4% paraformaldehyde for 15 mins and stained with crystal violet solution (C0121, Beyotime Biotechnology) for 20 mins. The plates were then washed, photographed, and visible colonies were counted using Image-Pro Plus.
Statistical analysis
All Q-PCR analyses, colony formation assays, flow cytometry analyses and immunochemical assays were repeated for over 3 times. All data are presented as the mean ± SD from at least three independent experiments. p-values were analyzed using the Student’s t-test unless stated otherwise. p values less than 0.05 were considered statistically significant, and the level of significance was indicated as *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
1. Nestin expression is upregulated in GBM
To examine the correlation between Nestin expression and GBM, we assessed the abundance of Nestin protein by immunohistochemical staining of tissue microarray (TMA) consisting of 15 astrocytoma, 40 GBM, 9 oligodendroglioma, 4 mixed glioma and 5 adjacent normal brain tissue. Strong, moderate or negative expression of Nestin was detected in GBM tissues (figure 1a). The abundance of Nestin protein in tissues was determined by H score for each TMA core (Supplementary table 1). Nestin protein was barely detected in tumor cells from astrocytoma, oligodendroglioma or mixed glioma (figure 1b). No Nestin protein was found in adjacent normal brain tissues (figure 1c). As shown in figure 1d, the levels of Nestin immunostaining were significantly higher in GBM than astrocytoma, oligodendroglioma or mixed glioma. These data suggest that Nestin protein is abundantly present in tumor cells from GBM. We also generated a mouse GBM model by forced expression of Ras and Akt in neural stem cells through viral infection, as previously described25. As shown in figure 1e–1f, Nestin protein was readily detected in tumor tissue, but not in adjacent normal brain tissue. Increased Nestin expression in tumor tissue was further confirmed by western blotting (figure 1g). The combined clinical and experimental data suggest that increased expression of Nestin is associated with GBM.
Figure 1. Nestin is highly expressed in GBM cells.
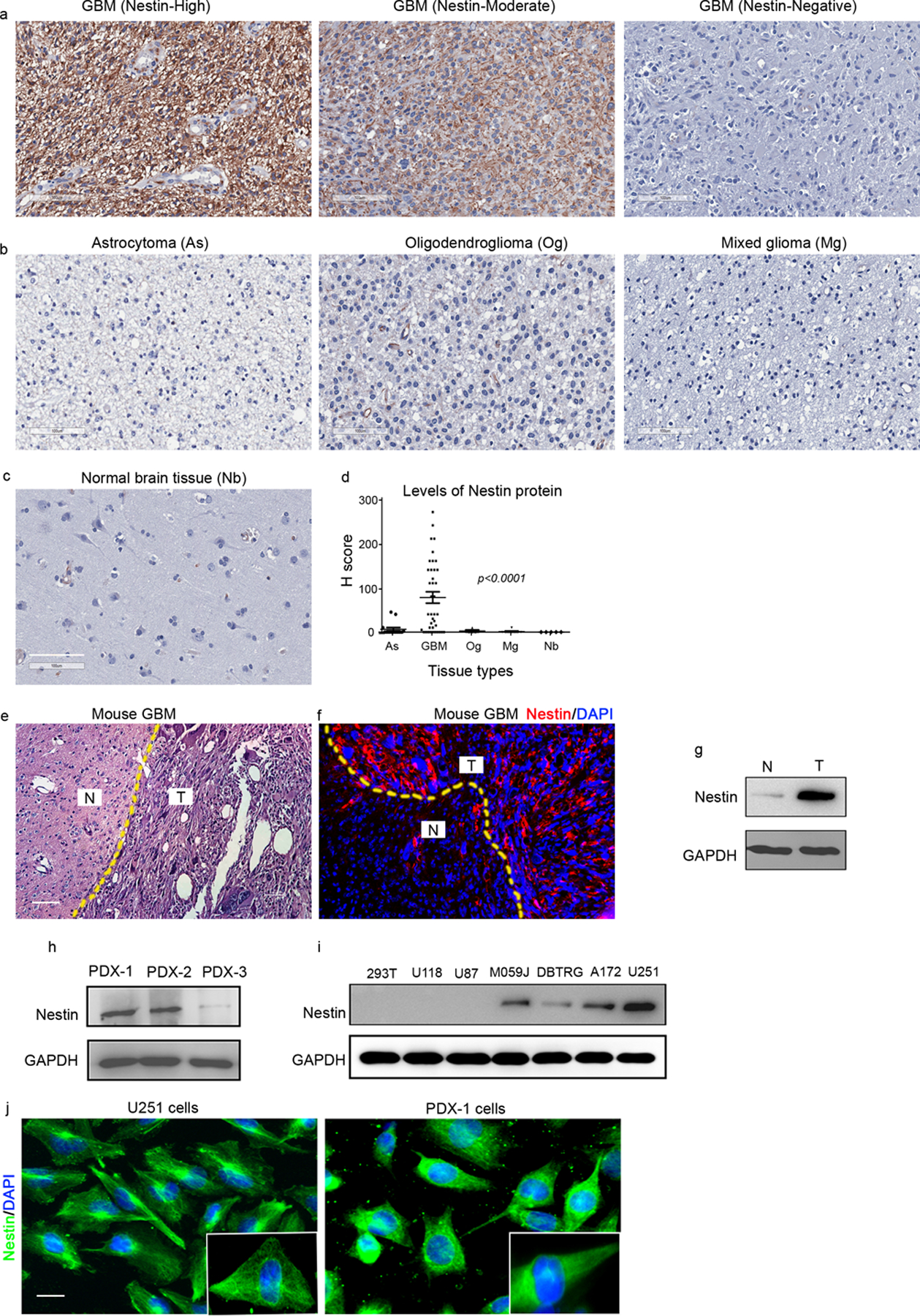
a-d, Representative images of immunohistochemical staining of Nestin from TMA cores of GBM specimens with high, moderate or negative expression of Nestin (a); from astrocytoma (As), oligodendroglioma (Og) or mixed glioma (Mg) (b), as well as from adjacent normal brain tissue (Nb) (c). The levels of Nestin proteins in TMA cores were compared by H score (d), p<0.0001, one way ANOVA. scale bar: 100 μm
e-g, HE staining (e) and immunostaining of Nestin (f) from a brain from a GBM mouse model. DAPI was used to counterstain cell nuclei. The abundance of Nestin protein in tumor tissue and normal brain tissue was examined by western blotting. N, adjacent normal brain tissue; T, GBM tumor tissue. scale bar: 50 μm
h-i, Nestin protein in tumor cells from 3 GBM PDX lines (h) and available GBM cell lines (i) was examined by western blotting. GAPDH was used as a loading control.
j, Nestin protein in U251 cells and PDX-1 cells was examined by immunocytochemistry. Insets show the magnified images of intracellular distribution of Nestin. scale bar: 20 μm.
We next examined GBM patient-derived xenografts and found that 2 of the 3 PDXs (PDX-1 and PDX-2, figure 1h) expressed substantial amounts of Nestin protein. However, very weak expression of Nestin was detected in tumor cells from PDX-3 (figure 1h). In addition, we examined the abundance of Nestin protein in established human glioma cell lines including U118, U87, M059J, DBTRG, A172 and U251, by western blotting (figure 1i). Human embryonic kidney 293T cells were used a negative control. As shown in figure 1i, Nestin protein was barely detected in 293T cells as expected. Nestin expression varied amongst the glioma cell lines, with U251 cells expressing the highest levels of Nestin, whereas almost no Nestin expression was detected in U87 and U118 cells. These data suggest that Nestin expression levels differ among established GBM PDX lines or cell lines. To investigate the function of Nestin in GBM cells, we focused on U251 cells and PDX-1/PDX-2 cells with the highest Nestin expression.
In addition, we examined Nestin protein in U251 cells and PDX-1 cells by immunocytochemistry. As shown in figure 1j, Nestin protein was detected in all tumor cells, as opposed to discrete subpopulations of tumor cells. Nestin was predominately localized in the cytoplasm of cells, forming a dense meshwork of intermediate filaments. Consistent with previous studies10,26, Nestin was also detected in the vicinity of the nucleus in GBM cells.
2. Nestin is required for cell cycle progression in GBM cells
We previously reported that Nestin promoted medulloblastoma formation by augmenting hedgehog (Hh) signaling in tumor cells5. To test whether Nestin exerts similar functions in GBM, we examined Hh pathway activation in U251 cells. U251 cells were treated with 3 μg/ml recombinant sonic hedgehog (Shh) or vehicle control for 48hrs. No increased expression of Hh pathway target genes including Gli1 and Ptch2 was observed in Shh-treated U251 cells compared with the control (supplementary figure 1a). Comparable levels of Gli1 protein were detected in U251 cells after treatment with Shh or vehicle (supplementary figure 1b). In addition, we treated U251 cells with vismodegib, a potent antagonist of Smo, or DMSO. 48hrs following the treatment, no alterations in Gli1 protein levels were found in U251 cells (supplementary figure 1b). These data suggest that Hh pathway is not activated in U251 cells.
To investigate possible functions of Nestin in GBM, we infected U251 cells with a lentivirus carrying shRNAs specific for Nestin, or a scrambled shRNA as a control. As shown in figure 2a, viral infection with Nestin shRNA effectively repressed Nestin expression in U251 cells. We next compared the colony forming capacity of Nestin-deficient cells and control cells (infected with scrambled shRNA) (figure 2b). The number of colonies derived from Nestin-deficient cells was significantly reduced compared with the control cells (figure 2b and 2c), suggesting that Nestin knockdown compromised the expansion of U251 cells. We also collected U251 cells transduced with Nestin shRNA or scrambled shRNA (passage 0), or after passaging them for 4 times (passage 4). Declined levels of Nestin protein in Nestin-shRNA transduced U251 cells were confirmed by western blotting (supplementary figure 2a). The number of colonies from Nestin-deficient U251 cells of passage 4 was increased compared with those of passage 0, but was still significantly less than that from control U251 cells transduced with scrambled shRNA (supplementary figure 2b and 2c).
Figure 2. Nestin deficiency induces G2/M phase arrest of GBM cells.
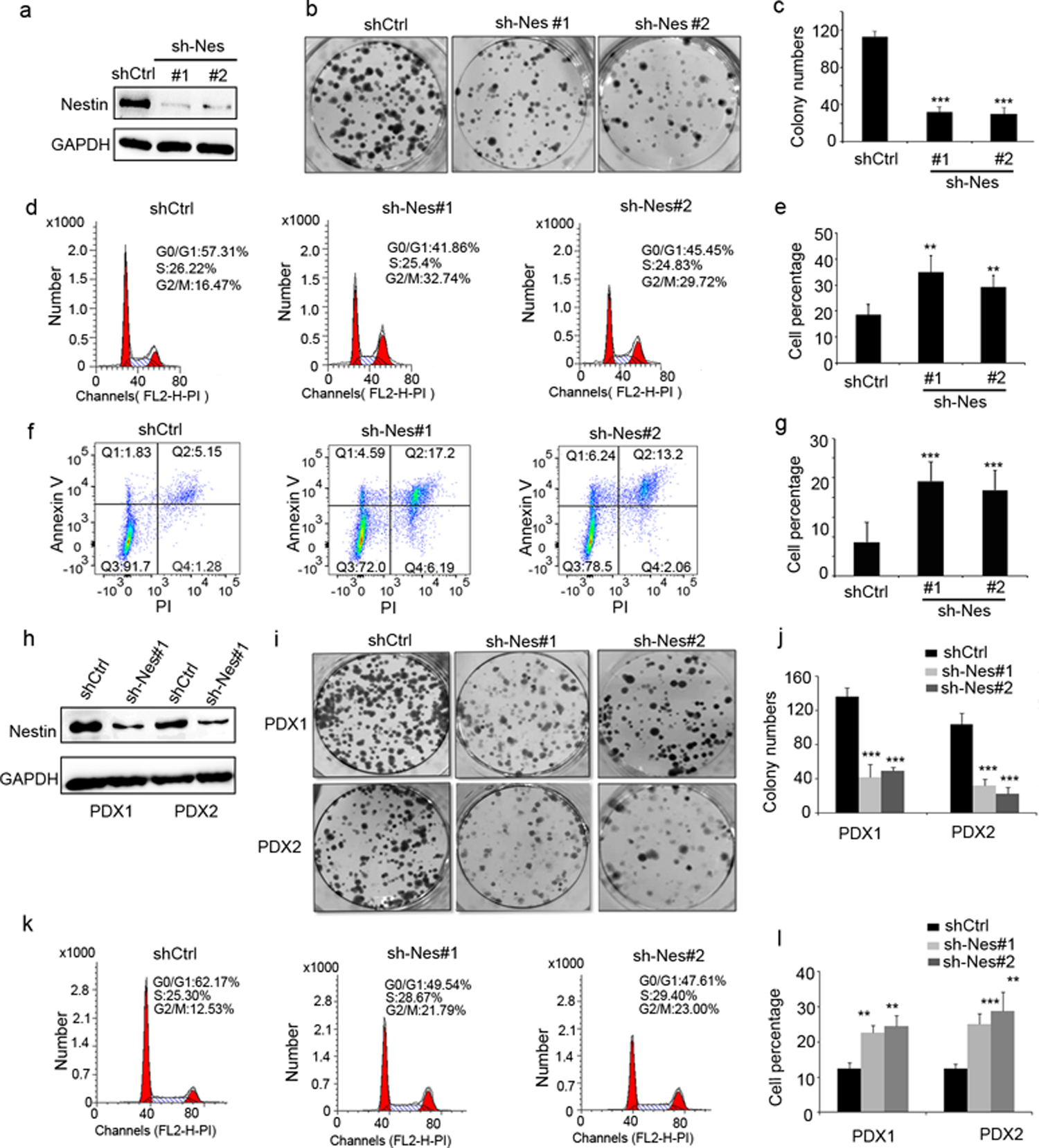
a, Nestin protein levels in U251 cells transduced with a lentivirus carrying shRNAs specific for Nestin (sh-Nes#1 and sh-Nes#2) or scrambled shRNA (shCtrl), examined by western blotting.
b–c, Images of colony formation of Nestin-deficient U251 cells (sh-Nes#1, sh-Nes#2) and control cells (transduced with scrambled shRNA) (b). The number of colonies from Nestin-deficient U251 cells and control cells was quantified (c).
d–e, Cell cycle distribution of U251 cells infected with Nestin shRNAs or scrambled shRNA, examined by flow cytometry (d). Percentages of cells in M phase among Nestin-deficient and control U251 cells was quantified (e).
f–g, Flow cytometry of U251 cells after labelling with Annexin V and PI (f). The percentage of apoptotic cells (Annexin V+), including cells at an early stage of apoptosis (Annexin V+/PI−) as well as cells at a late stage of apoptosis (Annexin V+/PI+), was quantified (g).
h–l, Nestin protein levels in tumor cells from GBM PDX1 and PDX2 lines, after viral infection with Nestin shRNA (sh-Nes#1) or scrambled shRNA, examined by western blotting. Colony formation of tumor cells from PDX-1 pr PDX-2, after infection with Nestin shRNA (sh-Nes#1 or sh-Nes#2) or scrambled shRNA (i). The number of colonies from Nestin deficient tumor cells and control tumor cells was quantified (j). Cell cycle distribution of Nestin-deficient or control tumor cells from PDX-1 (k). The percentage of PDX1 cells and PDX2 cells in G2/M phase, after infection with Nestin shRNAs or scramble shRNA, was quantified (l).
We then analyzed the cell cycle distribution of U251 cells after infection with Nestin shRNAs or scrambled shRNA. As shown in figure 2d and 2e, the percentage of cells in G2/M phase was markedly increased among Nestin-deficient cells compared with the control cells, suggesting that Nestin knockdown may result in G2/M phase arrest in U251 cells. Annexin V staining further revealed that over 20% of Nestin-deficient cells were undergoing apoptosis (Annexin V+), whereas 8.6±2.7% of control cells were apoptotic (figure 2f and 2g), suggesting that Nestin deficiency induces the apoptosis in U251 cells. The above data indicate that Nestin knockdown leads to G2/M arrest and apoptosis in U251 cells.
We next examined the effect of Nestin knockdown in human GBM cells isolated from PDX lines (PDX-1 and PDX-2) that highly expressed Nestin (figure 1h). As observed in U251 cells, Nestin shRNA significantly suppressed Nestin expression in PDX cells (figure 2h). Consistent with our findings in U251 cells, Nestin knockdown inhibited colony formation of tumor cells from both PDX-1 and PDX-2 (figure 2i and 2j). Moreover, increased number of Nestin-deficient cells was arrested in G2/M phase, compared with the control cells (infected with scrambled shRNA (figure 2k and 2l). Enhanced apoptosis was also observed among PDX tumor cells after Nestin knockdown (supplementary figure 3).
We also generated Nestin-null GBM cells (#1 and #19 lines) by deleting Nestin gene in U251 cells using CRISPR technology (supplementary figure 4a–4c). An accumulation of G2/M arrest and increased apoptosis were detected in Nestin-null U251 cells (supplementary figure 4d–4g), but to a lesser extent, compared with U251 cells after Nestin knockdown by shRNA. This may be due to some genetic compensation in U251 cells after Nestin deletion, as previously reported in CRISPR-mediated knockout cell lines27.
To further examine whether decreased colony formation in GBM cells after infection with Nestin shRNAs was indeed due to Nestin deficiency, we virally infected U87 cells (that do not express Nestin in figure 1i) with Nestin shRNA or scrambled shRNA. As shown in supplementary figure 5a, no Nestin protein was detected in U87 cells after infection, as expected. No difference in the colony formation from U87 cells infected with Nestin shRNA was observed, compared with the control (supplementary figure 5b–5c). These data suggest that compromised colony formation from GBM cells after infection with Nestin shRNA, is not likely due to off-target effects of the shRNA.
Collectively, above data demonstrate the important role of Nestin in the cell cycle progression and survival of GBM cells.
3. Nestin deficiency impairs spindle organization in GBM cells
Given the G2/M arrest of U251 cells after Nestin knockdown, we analyzed Nestin-deficient U251 cells in mitosis by immunocytochemistry. U251 cells were virally infected with Nestin shRNA or scrambled shRNA. 96hrs following the infection, we examined the spindle morphology of mitotic U251 cells by immunocytochemistry using an antibody against α-tubulin. A majority of mitotic U251 cells infected with scrambled shRNA, had assembled a bipolar mitotic spindle (figure 3a). In contrast, a significant number of Nestin-deficient U251 cells exhibited abnormal spindle morphology including distorted spindles, multipolar as well as monopolar spindles (figure 3a). The percentage of cells with aberrant spindles was significantly increased among Nestin-deficient U251 cells compared with U251 cells infected with scrambled shRNA (figure 3b). As expected, the fraction of U251 cells with bipolar spindles dramatically decreased after Nestin knockdown (figure 3b). These data suggest that Nestin deficiency compromises the spindle formation in U251 cells, likely leading to a delay or arrest in mitosis. Similarly, the proportion of mitotic cells with typical bipolar spindles was markedly reduced in PDX-1 cells after Nestin knockdown, whereas the percentage of cells with disorganized spindles increased among Nestin-deficient PDX cells (figure 3c). The above data demonstrate that Nestin is critical for spindle assembly during mitosis of GBM cells.
Figure 3. Nestin deficiency impairs spindle organization in GBM cells.
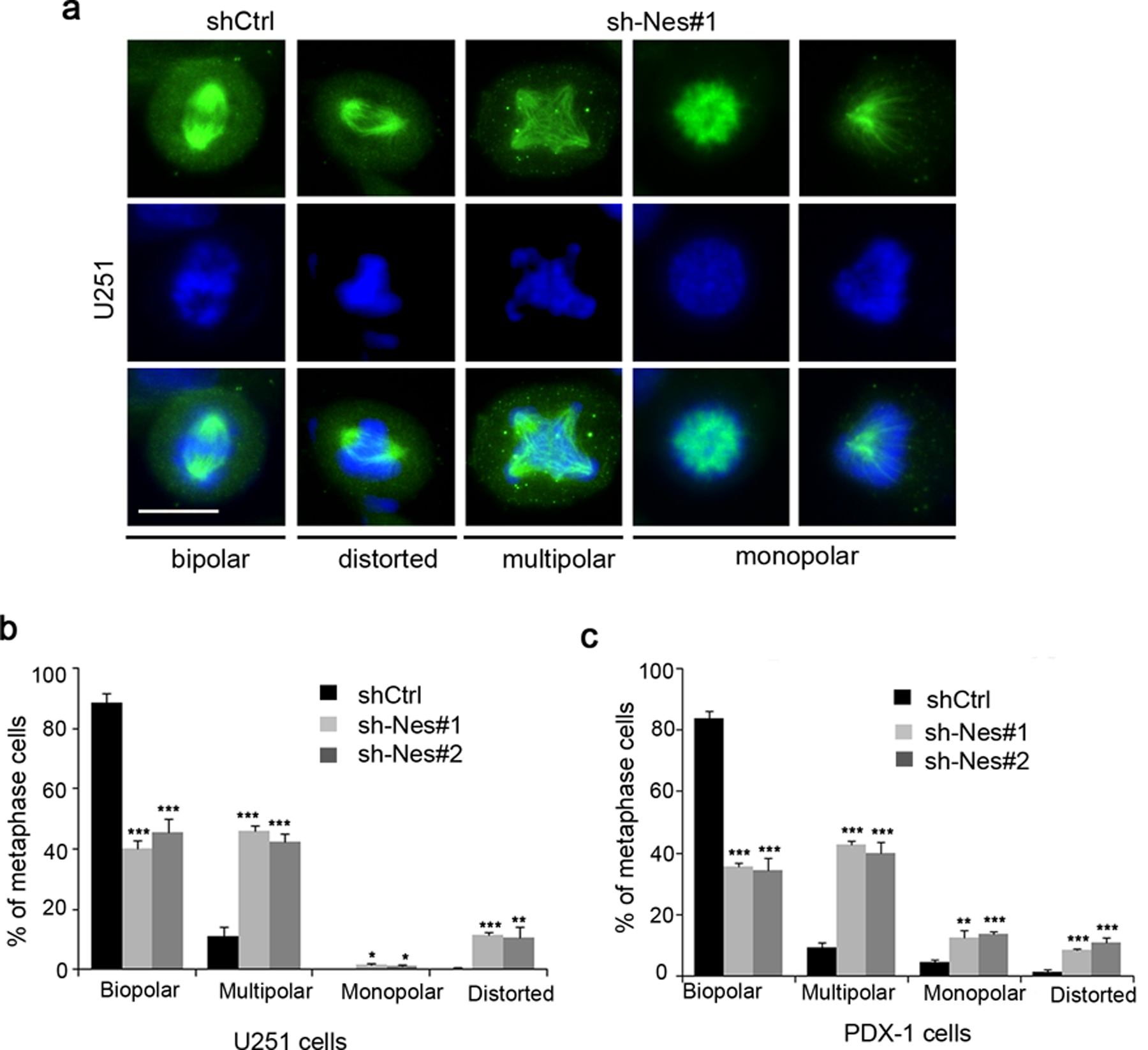
a-b, U251 cells infected with scrambled shRNA (shCtrl) or Nestin shRNA (sh-Nes#1) for 96 hrs, and harvested to examine the spindle morphology by immunofluorescence with an antibody specific to α-tubulin (a). DAPI was used to counterstain cell nuclei (a). The percentage of cells (after infection with scrambled shRNA, sh-Nes#1 or sh-Nes#2) with normal (bipolar) or abnormal spindles including distorted, multipolar or monopolar spindles among metaphase cells was quantified (b). scale bar: 20 μm.
c, GBM PDX-1 cells were infected with scrambled shRNA or Nestin shRNAs (sh-Nes#1, sh-Nes#2) for 96 hrs, and collected to examine the spindle morphology by immunofluorescence of α-tubulin. The percentage of cells with normal or abnormal spindles among PDX cells in metaphase was quantified.
4. Nestin physically interacts with tubulins in U251 cells
To further our understanding of the essential role of Nestin in the viability of U251 cells and PDX cells, we screened for binding partners of Nestin in GBM cells by the proximity-dependent biotinylation assay (BioID), a method based on biotinylation of proteins that are near neighbors of a fusion protein directly within cells28. As shown in figure 4a, we generated two constructs with human Nestin encoding sequence (C-BirA-HA-hNestin and N-BirA-myc-hNestin) fused to biotin protein ligase derived from Escherichia coli called BirA28 (details in Materials and Methods). U251 cells were transfected with either C-BirA-HA-hNestin or N-BirA-myc-hNestin. Untransfected U251 cells were used as a negative control. After addition of biotin, U251 cells were lysed and biotinylated proteins were subsequently captured with streptavidin-coupled beads. Bound proteins were then analyzed by mass-spectrometry (figure 4a). The strategy for filtering candidate Nestin BioID protein hits is outlined in figure 4b.
Figure 4. Nestin stabilizes βII-tubulin in GBM cells by physical interaction.
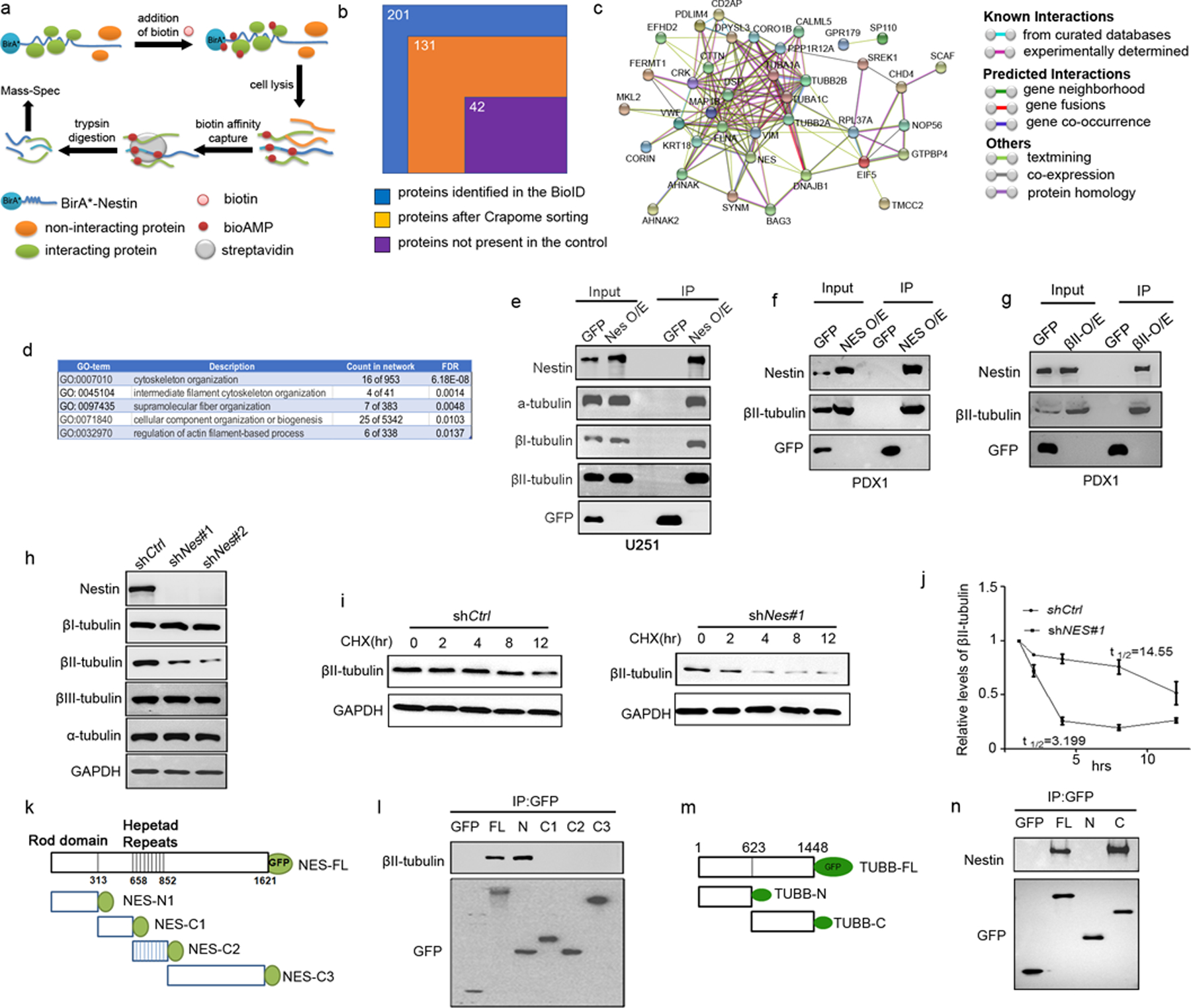
a, A schematic representation of the BioID approach to identify Nestin-interacting proteins.
b, Sorting process of the proteins identified by the Nestin BioID assay. 201 proteins were identified by MS, 70 proteins were excluded based on CRAPome analysis and 89 proteins were present in the control U251 cells, resulting in 42 proteins identified by the BioID assay as Nestin-interacting proteins in U251 cells.
c, PPI network of Nestin interacting proteins (revealed by the BioID approach) based on STRING-based protein interacting networks.
d, Top 5 terms based on GO enrichment analyses of Nestin interacting proteins.
e, Whole cell extracts from U251 cells virally infected with a Nestin-GFP vector or an empty GFP vector, were immunoprecipitated using an antibody against GFP. GFP, α-tubulin, βI and βII-tubulins, Nestin proteins were detected in the input (whole cell extracts) and immunoprecipitates by western blotting.
f–g, PDX-1 cells were infected with a lentivirus carrying a GFP-tagged vector encoding Nestin (f) or βII-tubulin (g), or an empty vector as a control. Whole cell lysates from PDX-1 cells were immunoprecipitated using an antibody against GFP (f and g). Nestin, βII-tubulin and GFP were detected in the input and immunoprecipitates by western blotting.
h, U251 cells were virally infected with Nestin shRNA (shNes#1, shNes#2) or scrambled shRNA (shCtrl). 96hrs following the infection, U251 cells were harvested for examination of Nestin, α-, βI-, βII- and βIII-tubulin proteins by western blotting. GAPDH was used as a loading control.
i–j, Nestin-deficient U251 cells and control U251 cells were treated with CHX for 12hrs and collected at designated timepoints to examine the levels of βII-tubulin by western blotting (i). The relative levels of βII-tubulin in U251 cells (normalized to the level at 0hr) were quantified based on the densitometry of the western blot band. The half-life of βII-tubulin in U251 cells was calculated by Prism 7.
k–l, Schematic representation of the GFP-tagged Nestin fragments (k). U251 cells were trans fected with full-length or fragments of Nestin. The cell lysate was immunoprecipitated with an antibody against GFP, and examined for βII-tubulin and GFP by western blotting (l).
m–n, Schematic representation of the GFP-tagged βII-tubulin fragments (m). The cell lysate of U251 cells transfected with the full-length or fragments of βII-tubulin, was immunoprecipitated with an antibody against GFP, and examined for Nestin and GFP by western blotting (n).
201 proteins were detected by BioID with a spectral count of 1 or greater in either C-BirA-HA-hNestin- or N-BirA-myc-hNestin-transfected U251 cells. We then removed 70 proteins that were contained in the Contaminant Repository for Affinity Purification (www.crapome.org), as previously described29. Cytoskeletal proteins were not removed at this stage, in that Nestin is known to interact with cytoskeletal proteins30. Among the remaining 131 proteins, 89 proteins were eliminated because they were also detected from the untransfected U251 cells. This resulted in a final list of 42 candidate interactors of Nestin in U251 cells (Supplementary table 2). We mapped the 42 proteins on the STRING-based protein interaction networks31. 40 proteins composed a single large network (p value of protein interaction enrichment <1.0e-13) (figure 4c), suggesting that our BioID experiments captured biologically meaningful and proximal proteins to Nestin. By Gene Ontology (GO) enrichment analyses, we found that GO terms related to “cytoskeletal organization”, were significantly enriched in the Nestin-interacting proteins (figure 4d), consistent with the established role of Nestin as a major component of the cytoskeleton. Importantly, tubulin α chains (TUBA1A and TUBA1C) and tubulin β chains (TUBB2A and TUBB2B) were identified among cytoskeletal proteins associated with Nestin, suggesting that Nestin may physically interact with α- and β-tubulins.
We next examined the physical interaction between Nestin and tubulins in U251 cells by immunoprecipitation (figure 4e). U251 cells were infected with a lentivirus encoding GFP-tagged Nestin or an empty GFP vector. α-, βI- and βII-tubulins were detected in U251 cells infected with either Nestin-GFP or GFP alone. Moreover, α-, βI- and βII-tubulins were readily precipitated in Nestin-GFP-infected U251 cells using an antibody against GFP, whereas no tubulins were precipitated in U251 cells infected with the empty GFP vector. These data indicate that Nestin interacts with α- and β-tubulins in U251 cells. We next tested the interaction between Nestin and βII-tubulin in PDX cells. PDX-1 cells were infected with a lentivirus encoding GFP-tagged Nestin or an empty GFP vector. As shown in figure 4f, βII-tubulin was precipitated by the GFP antibody in Nestin-GFP infected cells, but not in control cells (infected with GFP alone). In addition, we introduced GFP-tagged βII-tubulin or an empty GFP vector in PDX-1 cells by viral infection (figure 4g). The GFP antibody precipitated endogenous Nestin protein in PDX-1 cells after infection with the βII-tubulin-GFP vector, but not in cells after infection with the empty GFP vector. These results further confirm the physical interaction between Nestin and βII-tubulin in GBM cells.
5. Nestin stabilizes β-II tubulin in GBM cells
We next examined whether the levels of tubulin proteins in U251 cells are affected by Nestin knockdown. As expected, Nestin expression was effectively repressed in U251 cells virally infected with Nestin shRNAs (shNes#1, shNes#2) (figure 4h). The protein levels of α-, βI- and βIII-tubulins were comparable in Nestin-deficient U251 cells and control U251 cells infected with scrambled shRNA (shCtrl) (figure 4h). However, the levels of βII-tubulin were significantly reduced in U251 cells after Nestin knockdown. No significant alterations in the mRNA expression of TUBB2A and TUBB2B (encoding βII-tubulin) were detected in Nestin-deficient U251 cells compared with the control cells (supplementary figure 6a). In addition, we overexpressed Nestin or GFP alone in U251 cells by lentivirus infection. As shown in supplementary figure 6b–6c, forced expression of Nestin markedly enhanced βII-tubulin protein level in U251 cells, but caused no changes in the mRNA expression of βII-tubulin. These data suggest that Nestin may post-transcriptionally regulate the stability of βII-tubulin in U251 cells.
To further confirm the regulatory effects of Nestin on the stability of βII-tubulin, we examined the half-life of βII-tubulin in Nestin-deficient U251 cells and control U251 cells (with scrambled shRNA) following cycloheximide (CHX) treatment. As shown in figure 4i, in the presence of CHX, the level of βII-tubulin remained unchanged until 8 hrs in the control U251 cells. However, in Nestin-deficient cells, the levels of βII-tubulin dramatically declined within 4 hrs following CHX treatment. Almost no βII-tubulin was detected in Nestin-deficient U251 cells at 8 hrs and 12 hrs after CHX treatment. The half-life of βII-tubulin was significantly reduced in Nestin-deficient cells compared with that in the control cells (14.6 hrs vs 3.2 hrs, p<0.001) (figure 4j), demonstrating that Nestin deficiency compromises the stability of βII-tubulin in U251 cells.
Nestin is comprised of an N-terminal rod domain and 41 heptad repeats in an extensive C-terminal region30. To identify the domain of Nestin responsible for interaction with βII-tubulin, we generated the following GFP-tagged Nestin fragments (figure 4k): Nes-FL (containing full length of Nestin), NES-N1 (containing N-terminal domain), Nes-C2 (containing all heptad repeats), Nes-C1 (containing the linker region between Nes-N1 and Nes-C2), and Nes-C3 (the C-terminal region), as we previously described5. U251 cells were transfected with Nestin fragments or an empty GFP vector for immunoprecipitation assay. As shown in figure 4l, endogenous βII-tubulin was precipitated with Nes-FL and Nes-N1, but not with the C-terminal fragments or GFP alone. In addition, we prepared GFP-tagged constructs containing βII-tubulin fragments: TUBB-FL (containing full length of βII-tubulin), TUBB-N (consisting of N terminal of βII-tubulin) and TUBB-C (containing C terminal of βII-tubulin) (figure 4m). U251 cells were transfected with the above βII-tubulin fragments or an empty GFP vector as a control. The interaction between βII-tubulin fragments with endogenous Nestin was examined by immunoprecipitation. Both TUBB-FL and TUBB-C precipitated with endogenous Nestin in U251 cells. No physical interaction between Nestin and TUBB-N or GFP alone was detected (figure 4n) These data indicate that the interaction between Nestin and βII-tubulin in U251 cells is mediated by N-terminal of Nestin and C-terminal of βII-tubulin.
6. βII-tubulin knockdown recapitulates the phenotype of Nestin-deficient GBM cells
To investigate the functions of βII-tubulin in GBM cells, we knocked down βII-tubulin in U251 cells by viral infection of shRNAs specific for βII-tubulin or scrambled shRNA as a control. 48 hrs following the infection, the protein levels of βII-tubulin were significantly declined in U251 cells infected with βII-tubulin shRNAs (shβII#1, shβII#2), compared with scrambled shRNA (shCtrl) (figure 5a). However, no changes in the levels of βI-tubulin and βIII-tubulin were observed in U251 cells after βII-tubulin knockdown (figure 5a). In addition, expression of Nestin mRNA and protein was not altered in U251 cells after βII-tubulin knockdown (supplementary figure 7a–7b). These data indicate that shβII#1 or shβII#2 specifically repress the expression of βII-tubulin in U251 cells.
Figure 5. βII-tubulin knockdown phenocopies Nestin deficiency in U251 cells.
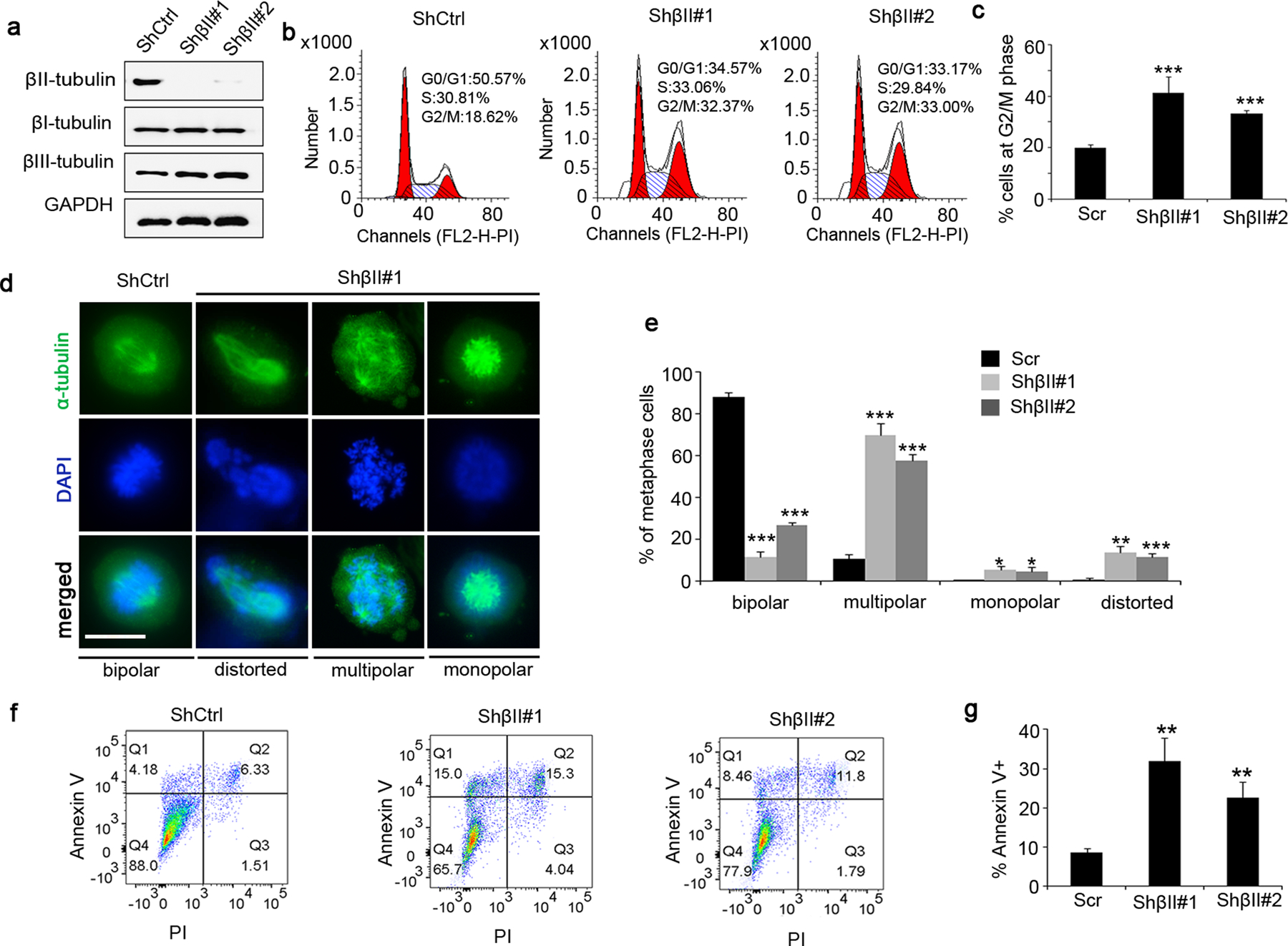
a, U251 cells were infected with a lentivirus carrying shRNA specific for βII-tubulin (shβII#1, shβII#2) or scrambled shRNA (shCtrl). 48hrs following the infection, U251 cells were harvested to examine protein levels of βI-, βII- and βIII-tubulin by western blotting. GAPDH was used as a loading control.
b–c, Cell cycle distribution of βII-tubulin deficient U251 cells and control U251 cells (with shCtrl) was analyzed by flow cytometry. The percentage of cells at G2/M phase among cells at metaphase was quantified (c).
d–e, U251 cells (βII-tubulin deficient cells and control cells) at metaphase of mitosis were immunostained for α-tubulin to examine spindle morphology (d). DAPI was used to counterstain cell nuclei. The percentage of cells with normal spindles or abnormal spindles (including multipolar, monopolar or distorted spindles) was quantified (e). scale bar: 15 μm.
f–g, Apoptosis in U251 cells (βII-tubulin deficient cells and control cells) was examined by flow cytometry after labelling with Annexin V and PI (f). The percentage of apoptotic cells (Annexin V+) was quantified (g).
We next examined the cell cycle distribution of U251 cells after βII-tubulin knockdown. As shown in figure 5b–5c, the ratio of cells at G2/M phase was significantly increased among U251 cells after βII-tubulin knockdown, compared with that in control U251 cells infected with scrambled shRNA. These data suggest that βII-tubulin deficiency induces G2/M arrest in U251 cells. Moreover, abnormal spindle assembly (distorted, multipolar or monopolar spindles) was frequently observed in U251 cells after βII-tubulin knockdown, during metaphase of mitosis (figure 5d). As expected, a majority of U251 cells infected with scrambled shRNA exhibited typical bipolar spindles (figure 5d). Increased percentage of cells with abnormal spindle morphology after βII-tubulin knockdown (figure 5e), suggesting that βII-tubulin deficiency interferes spindle assembly in U251 cells. Finally, the ratio of annexin V+ cells was much greater in βII-tubulin deficient cells than that in the control cells (figure 5f–5g), indicating that βII-tubulin deficiency leads to apoptosis in U251 cells. Collectively, the above data suggest that βII-tubulin knockdown results in G2/M arrest, disrupted spindle assembly and apoptosis in U251 cells, consistent with the phenotypes observed in Nestin-deficient cells. These findings support the notion that Nestin facilitates cell cycle progression of GBM cells through stabilizing βII-tubulin.
We attempted to overexpress βII-tubulin in Nestin-deficient cells, to rescue the apoptosis and G2/M arrest of U251 cells after Nestin knockdown. However, forced expression of βII-tubulin alone caused extensive apoptosis in U251 cells (data not shown), suggesting that the precise regulation of βII-tubulin dynamics may be critical for the survival of U251 cells.
7. Nestin is required for in vivo growth of GBM cells
To further examine the role of Nestin in the growth of GBM, we knocked down Nestin expression in U251 cells by viral infection of Nestin shRNAs or scrambled shRNA as a control. Nestin-deficient cells or control cells were subcutaneously transplanted into the flanks of nude mice. Following the transplantation, in vivo growth of injected cells was monitored by measuring the volume of flank mass. As shown in figure 6a, the control U251 cells grew rapidly in vivo, after 10 days following the transplantation. Until 35 days following the transplantation, the volumes of tumors from control U251 cells reached 1500mm3 (the size limit for subcutaneous tumors). In contrast, the growth rate of Nestin-deficient U251 cells was significantly declined compared with the control cells. By the time when the experiment was terminated (35 days after the transplantation), the size of tumors from Nestin-deficient U251 cells was markedly reduced than that from control U251 cells (figure 6b). These data suggest that Nestin deficiency represses the in vivo growth of U251 cells.
Figure 6. Nestin knockdown inhibits in vivo growth of GBM.
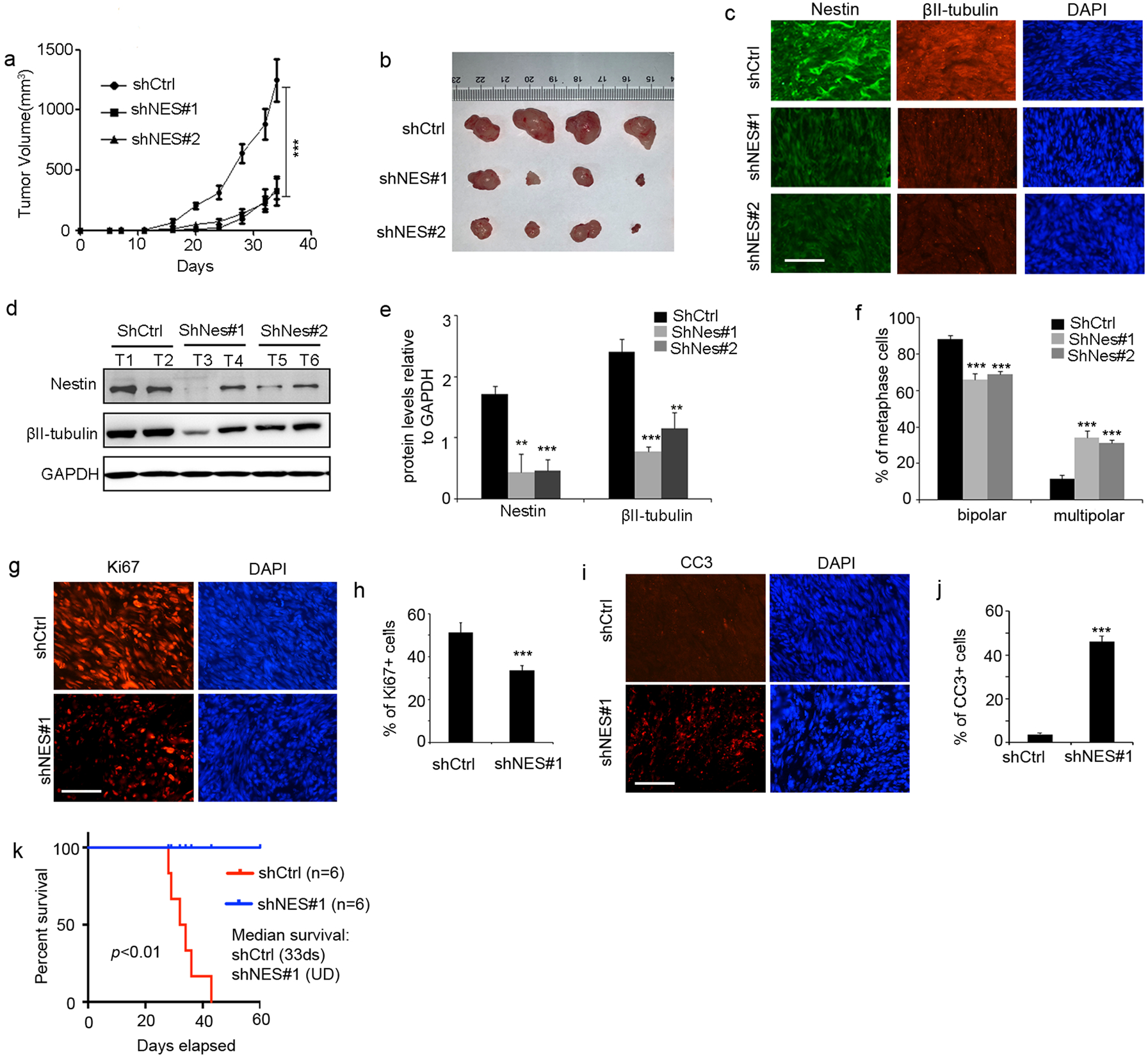
a–f, U251 cells virally infected with scrambled shRNA (shCtrl) or Nestin shRNA (shNes#1 or shNes#2), were subcutaneously injected into flanks of nude mice. Tumor volume was measured every 3 days (a). Tumors from Nestin-deficient cells or control cells were harvested (b) and examined for Nestin and βII-tubulin proteins by immunohistochemistry (c) western blotting (d). T1–T6, tumor tissues from U251 cells transduced with shCtrl, shNes#1 or shNes#2.
The levels of Nestin and βII-tubulin proteins were quantified in tumor tissues (e). The percentage of cells with bipolar or multipolar spindles among metaphase cells in tumor tissues (f). scale bar: 50 μm.
g–j, Tumors from Nestin-deficient U251 cells or control cells were collected to examine tumor cell proliferation (Ki67+) in tumor tissues by immunohistochemistry (g). DAPI was used to counterstain cell nuclei. The percentage of proliferative cells in tumor tissue was quantified (h). Apoptosis (CC3+) in tumor tissues was also examined by immunohistochemistry (i) and quantified in j. scale bar: 50 μm.
k. Survival curves of SCID mice after transplantation of GBM PDX-1 cells infected with Nestin shRNA (shNes#1) or scrambled shRNA (shCtrl), which were intracranially transplanted into the forebrains of SCID mice. The median survival for mice transplanted with control cells was 33 ds, and for mice with Nestin-deficient cells was undefined (UD). Log-rank test, p<0.01.
In addition, we harvested tumors from Nestin-deficient cells or control cells to examine protein levels of Nestin and βII-tubulin by immunohistochemistry (figure 6c) or western blotting (figure 6d). High levels of Nestin and βII-tubulin proteins were detected in tumor tissues from control cells as expected (figure 6d). The levels of Nestin proteins were significantly declined in tumor tissue derived from Nestin-deficient cells (figure 6e). Accordingly, the amount of βII-tubulin proteins was decreased dramatically in tumor tissues from Nestin-deficient cells (figure 6e). These data further confirm that the stability of βII-tubulin is regulated by Nestin in GBM cells. We next examined spindle morphology of cells in tumor tissue, and found that the percentage of cells with multipolar spindles was significantly increased in tumor tissues from Nestin-deficient cells (figure 6f). As shown in figure 6g, extensive proliferation (Ki67+) was detected in tumor tissue from control U251 cells, whereas the proliferation of Nestin-deficient tumor cells was significantly inhibited. Suppressed proliferation of Nestin-deficient cells compared with the control cells (figure 6h), reinforces the critical role of Nestin in the proliferation of GBM cells. Consistent with in vitro apoptosis in U251 cells induced by Nestin knockdown, extensive apoptosis (positive for cleaved caspase-3, CC3) was detected in tumor tissue from Nestin-deficient cells (figure 6i–6j). The above data further demonstrate that Nestin deficiency suppresses tumor growth by inhibiting tumor cells proliferation and promoting the apoptosis of tumor cells.
Finally, we examined the growth of GBM PDX-1 cells after Nestin knockdown. For this purpose, we infected GBM PDX-1 cells with a lentivirus carrying Nestin shRNA (shNes#1) or scrambled shRNA (shCtrl). Nestin deficient PDX cells and control cells were intracranially transplanted into forebrains of SCID mice as previously described32. PDX cells infected with the control shRNA, developed tumors with 100% penetrance (median survival, 33 ds), whereas no tumors were formed from Nestin-deficient PDX cells (figure 6k). These data indicate that the orthotopic growth of GBM cells also relies on Nestin expression.
8. Nestin deletion sensitizes GBM cells to microtubule-targeting agents
Having observed the function of Nestin in stabilizing βII-tubulin in GBM cells, we sought to investigate whether Nestin knockdown could affect the response of GBM cells to Vinca alkaloids including vinblastine and vincristine. Vinca alkaloids are widely used in the treatment of human cancers, predominately by disrupting the stability of β-tubulin33. Nestin-deficient cells and control cells were prepared by infecting U251 cells with a lentivirus carrying Nestin shRNA or scrambled shRNA. Infected U251 cells were treated with vinblastine or vincristine at designated concentrations. Etoposide, a chemotherapeutic drug inhibiting DNA synthesis by forming a complex with topoisomerase34, was also used to treat U251 cells as a control. At 48hrs following the treatment, U251 cells were harvested to examine cell viability by CCK8 assay. As shown in figure 7a, no significant difference in the viability between Nestin-deficient U251 cells and control cells was observed after etoposide treatment. Comparable IC50 values of etoposide were observed in Nestin-deficient U251 cells and control cells. These data suggest that Nestin deficiency did not alter the sensitivity of U251 cells to etoposide. However, the viability of Nestin-deficient U251 cells was overall reduced compared with the control cells, in the presence of vinblastine or vincristine. The IC50 of vinblastine or vincristine in Nestin-deficient U251 was significantly decreased than that in control U251 cells (vinblastine: 2.0 nM vs 3.6 nM, p<0.01; vincristine: 14.7 nM vs 4.3 nM, p<0.001), suggesting that Nestin deficiency sensitizes U251 cells to vinblastine or vincristine.
Figure 7. Nestin deficiency sensitizes GBM cells to vinblastine.
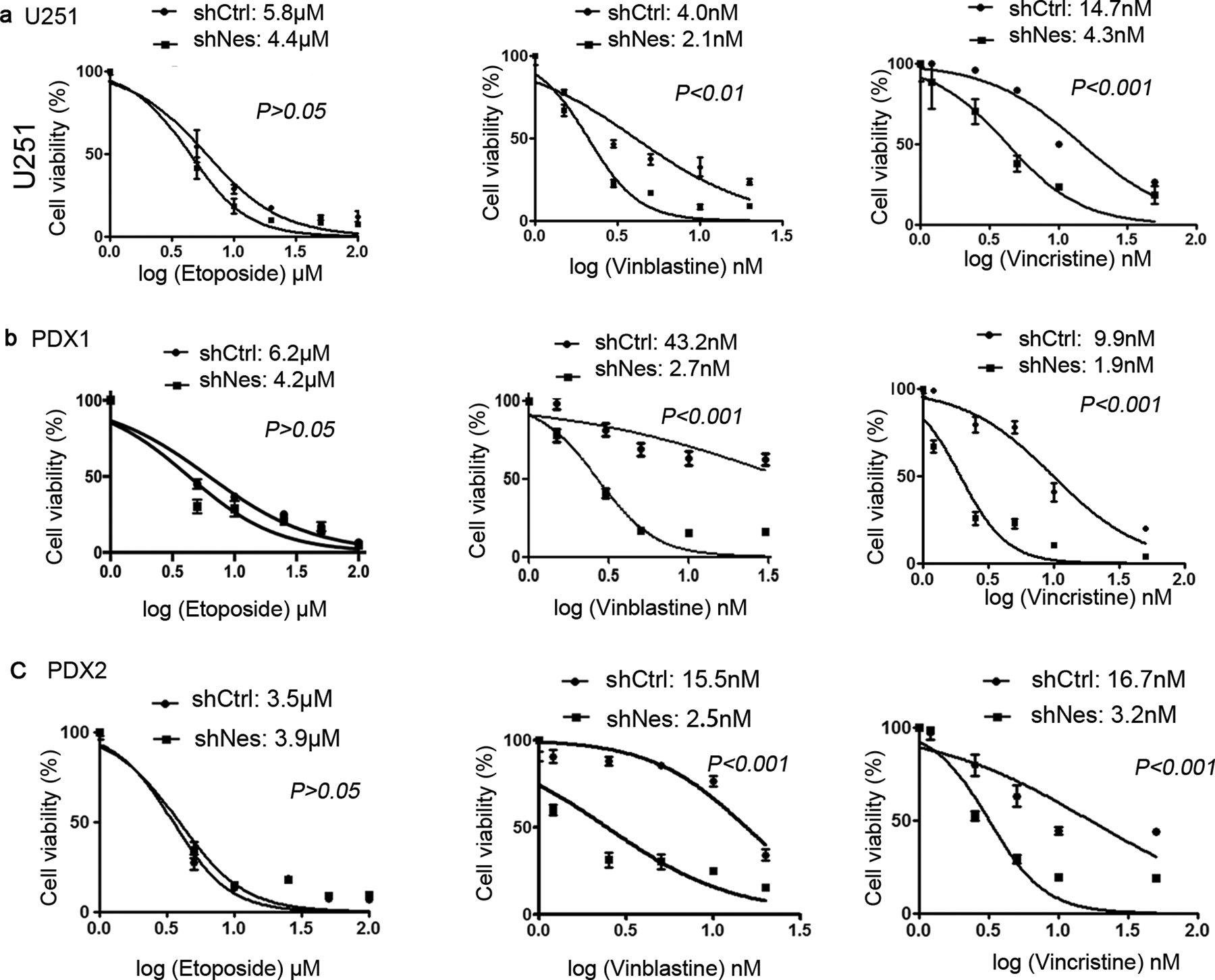
U251 cells (a), GBM PDX1 cells (b) and PDX2 cells (c) were virally infected with Nestin shRNA (shNes) or scrambled shRNA (shCtrl). At 96 hrs following the infection, cells were treated with etoposide (1 μM, 5 μM, 10 μM, 25 μM, 50 μM, 100 μM), vinblastine (1.5 nM, 3 nM, 5 nM, 10 nM, 20 nM) or vincristine (0.2 nM, 1.2 nM, 2.5 nM, 5 nM, 10 nM, 50 nM) for 48 hrs. The cell viability was measured by CCK8 assay, and plotted against the concentrations. IC50 of vinblastine and etoposide in GBM cells was quantified. Drug responses of Nestin deficient cells and control cells were compared by Log-rank test.
In addition, GBM PDX-1 cells and PDX-2 cells were utilized to test their responses to vinblastine, vincristine or etoposide. GBM PDX cells infected with Nestin shRNA or scrambled shRNA, were treated with vinblastine, vincristine or etoposide for 48 hrs. Similar with our observations in U251 cells, the IC50 of etoposide was comparable in Nestin-deficient PDX cells and control cells (figure 7b and 7c). However, PDX-1 cells and PDX-2 cells became more sensitive to vinblastine or vincristine after Nestin knockdown. These data further confirm that Nestin deficiency enhances the effectiveness of microtubule-destabilizing agents in GBM cells.
Discussion
Instead of being merely a structural protein, Nestin participates in the control of important cellular processes, especially in human malignancies. Nestin is known to contribute to the migration and invasion of pancreatic cancer cells35. In addition, Nestin was found preventing the senescence of non-small cell lung carcinoma cells, through inactivation of the proapoptotic cyclin-dependent kinase 57. Our previous studies revealed that Nestin enhances the hedgehog signaling, thereby promoting the progression of medulloblastoma. Here, we demonstrate an indispensable role of Nestin in cell cycle progression in GBM cells. Knockdown of Nestin by shRNA induced the G2/M arrest and apoptosis in U251 cells and GBM PDX cells. Nestin participated in spindle formation in mitotic tumor cells through stabilizing βII-tubulin, and Nestin deficiency caused spindle abnormalities in GBM cells. Nestin knockdown significantly repressed the in vivo growth of U251 cells and GBM PDX cells. Consistent with our findings, a recent study reported that Nestin is required for efficient progression though the G2/M phase of the cell cycle in human hepatocellular carcinoma cells6. Finally, Nestin deficiency sensitizes GBM cells to vinblastine as well as vincristine, two tubulin-destabilizing drugs, in agreement with the important role of Nestin in stabilizing tubulin proteins in tumor cells. In summary, our studies reveal the essential functions of Nestin in spindle formation during mitosis of GBM cells, and demonstrate the important role of Nestin in GBM progression.
Matsuda et. al. previously reported that Nestin knockdown repressed the phosphorylation of heat shock cognate 71 KDa protein (HSC71) in A172 cells, a glioblastoma cell line, through physical interaction36. GBM cells that initially express low levels of Nestin, were found to initiate Nestin expression during tumor progression in vivo37. These studies highlight the importance of Nestin expression in GBM growth. In our studies, Nestin knockdown interfered the bipolar spindle formation in GBM cells, leading to the G2/M arrest as well as apoptosis of tumor cells. These data demonstrate the important functions of Nestin in cell cycle progression and survival of GBM cells. Nestin knockdown compromised the stability of βII-tubulin in GBM cells, and suggests the interaction with Nestin promotes the stability of βII-tubulin. The mitotic defects observed after Nestin depletion can be attributed to the loss of βII-tubulin as knockdown of βII-tubulin also disrupted the spindle formation, delayed G2/M and increased apoptosis in GBM cells. These findings are fully consistent with the known functions of tubulins in the mitosis and apoptosis in tumor cells38,39. The physical interaction between Nestin and βII-tubulin is supported by the findings that these two proteins are co-expressed in neural stem cells during human fetal brain development40. The critical role of Nestin and βII-tubulin in G2/M progression of GBM cells, implies that the expression of Nestin and βII-tubulin may vary across phases of cell cycle in these cells. Future studies are warranted to investigate the possible association between the expression of Nestin and βII-tubulin genes/proteins and cell cycle phases in GBM cells.
To identify the interacting partners of Nestin, we utilized the recently established proximity-dependent Biotin identification method41. BioID allows to screen for proximate proteins in a relatively natural cellular environment. In our studies, we identified 42 interacting candidates of Nestin in U251 cells by using BioID. Based on gene ontology enrichment analysis, 40 of 42 proteins formed a single interactome, with significant GO terms including cytoskeletal network etc. Among them, α and βII-tubulin were among the highest ranked candidates of Nestin interacting proteins in BioID assays. The interaction between βII-tubulin and Nestin was further confirmed by immunoprecipitation. It should be noted that α-tubulin was found to interact with Nestin in U251 cells by BioID assay, and precipitate with Nestin in our immunoprecipitation assay. These data suggest that Nestin may be physically associated with α-tubulin. However, we cannot rule out the possibility that α-tubulin indirectly interact with Nestin through βII-tubulin, because of the heterodimer of α-tubulin and β-tubulin. Nevertheless, Nestin knockdown significantly compromised the stability of βII-tubulin in U251, without any alterations in the stability of α-tubulin. The members of the tubulin family share a high degree of structural homology and are distinguished from one another by highly divergent sequences at their carboxy-terminal tail42. Indeed, the interaction between Nestin and βII-tubulin was mediated by the C-terminal of βII-tubulin, providing a possible explanation that the stabilizing effects of Nestin is specific to βII-tubulin. Future studies are warranted to further investigate whether interaction with Nestin affects the posttranscriptional modification of βII-tubulin, a critical mechanism regulating protein-protein interaction with the microtubule cytoskeleton43.
It is well-known that tubulin expression is highly auto-regulated in mammalian cells44. The reduction of the initial portion of the tubulin chain often serves as a signal to the cells to enhance the stability or production of remaining tubulin mRNAs. For example, mice with deletion of Tubb2a or Tubb2b appear to have relatively subtle phenotypes in cortical development, which is believed to be due to the compensation from other tubulins45. However, no increased amounts of βI, or βIII-tubulin were observed in Nestin-deficient U251 cells with declined levels of βII-tubulin. The remaining question is why the declined levels of βII-tubulin was not compensated in U251 cells after knockdown of Nestin or βII-tubulin. One possible explanation is that the functions of βII-tubulin may be particularly required for the cell cycle progression in GBM cells. Indeed, recent studies reveal that expression levels and functions of tubulin isotypes are tumor type-specific38. For example, βIII-tubulin regulates the tumorigenesis and metastasis of lung cancer cells and ovarian cancer cells46–48, but appears not required for tumor growth of breast cancer49.
Suppression of microtubule dynamics by anti-microtubule drugs disrupt spindle formation, which leads to checkpoint arrest, eventually leading to apoptosis33. Vinka alkaloids such as vinblastine and vincristine, are widely utilized as cancer chemotherapeutic agents by disrupting the stability of tubulins, in particular, β-tubulin50. In our studies, Nestin knockdown sensitized U251 cells and GBM PDX cells to vinblastine and vincristine. In contrast, Nestin knockdown had no significant effects on the sensitivity to etoposide, a chemotherapeutic drug that does not target microtubules. These findings demonstrate the role of Nestin in the sensitivity of tumor cells to microtubule-destabilizing agents. Consistent with our findings, many other tubulin associated proteins such as Tau and MAP2 influence the sensitivity of tubule-targeting agents in tumor cells51. Most tubulin-targeting agents depolymerize microtubules by interacting with β-tubulin, Nestin deficiency may reinforce the tubulin-disrupting effects of vinblastine or and vincristine through compromising the stability of β-tubulin. These data suggest that agents that suppress Nestin expression could be combined with microtubule-targeting drugs in treating tumors including GBM. In addition, compounds that alter Nestin-tubulin interactions can be explored by high-throughput screening of small molecule libraries. Such compounds alone or in combination with microtubule-targeting drugs could represent an effective approach to treat GBM.
Supplementary Material
Acknowledgement
We would like to thank Dr. Richard Katz at Fox Chase Cancer Center for constructive discussion of this project. This research was supported by American Cancer Society (RSG1605301NEC to Z. Yang), PA CURE Health Research Fund (CURE 4100068716 to Z. Yang) and National Natural Science Foundation of China (82073873 and BK20170348 to Y. Wang).
Footnotes
Conflict of Interest: All authors declare no potential conflicts of interest.
References
- 1.Krupkova O Jr., Loja T, Zambo I & Veselska R Nestin expression in human tumors and tumor cell lines. Neoplasma 57, 291–298 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Schiffer D, Manazza A & Tamagno I Nestin expression in neuroepithelial tumors. Neuroscience letters 400, 80–85 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Chen J, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li P, et al. A population of Nestin-expressing progenitors in the cerebellum exhibits increased tumorigenicity. Nat Neurosci 16, 1737–1744 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li P, et al. Nestin Mediates Hedgehog Pathway Tumorigenesis. Cancer research 76, 5573–5583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tschaharganeh DF, et al. p53-Dependent Nestin Regulation Links Tumor Suppression to Cellular Plasticity in Liver Cancer. Cell 158, 579–592 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, et al. Nuclear Nestin deficiency drives tumor senescence via lamin A/C-dependent nuclear deformation. Nat Commun 9, 3613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louis DN, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta neuropathologica 131, 803–820 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Tohyama T, et al. Nestin expression in embryonic human neuroepithelium and in human neuroepithelial tumor cells. Lab Invest 66, 303–313 (1992). [PubMed] [Google Scholar]
- 10.Veselska R, et al. Nestin expression in the cell lines derived from glioblastoma multiforme. BMC Cancer 6, 32 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mangiola A, et al. Stem cell marker nestin and c-Jun NH2-terminal kinases in tumor and peritumor areas of glioblastoma multiforme: possible prognostic implications. Clinical cancer research : an official journal of the American Association for Cancer Research 13, 6970–6977 (2007). [DOI] [PubMed] [Google Scholar]
- 12.McIntosh JR, Grishchuk EL & West RR Chromosome-microtubule interactions during mitosis. Annu Rev Cell Dev Biol 18, 193–219 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Holland AJ & Cleveland DW Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep 13, 501–514 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desai A & Mitchison TJ Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13, 83–117 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Wittmann T, Hyman A & Desai A The spindle: a dynamic assembly of microtubules and motors. Nat Cell Biol 3, E28–34 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Goldstein LS & Philp AV The road less traveled: emerging principles of kinesin motor utilization. Annu Rev Cell Dev Biol 15, 141–183 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Li P, et al. Novel Therapy for Glioblastoma Multiforme by Restoring LRRC4 in Tumor Cells: LRRC4 Inhibits Tumor-Infitrating Regulatory T Cells by Cytokine and Programmed Cell Death 1-Containing Exosomes. Front Immunol 8, 1748 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng J, et al. Hypermethylated gene ANKDD1A is a candidate tumor suppressor that interacts with FIH1 and decreases HIF1alpha stability to inhibit cell autophagy in the glioblastoma multiforme hypoxia microenvironment. Oncogene 38, 103–119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Z, et al. The EGFR-ZNF263 signaling axis silences SIX3 in glioblastoma epigenetically. Oncogene 39, 3163–3178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du F, et al. Leukotriene Synthesis Is Critical for Medulloblastoma Progression. Clinical cancer research : an official journal of the American Association for Cancer Research 25, 6475–6486 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng Y, et al. Sustained hedgehog signaling in medulloblastoma tumoroids is attributed to stromal astrocytes and astrocyte-derived extracellular matrix. Lab Invest (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roux KJ, Kim DI, Burke B & May DG BioID: A Screen for Protein-Protein Interactions. Curr Protoc Protein Sci 91, 19 23 11–19 23 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim DI, et al. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proceedings of the National Academy of Sciences of the United States of America 111, E2453–2461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang P, et al. The Protein Encoded by the CCDC170 Breast Cancer Gene Functions to Organize the Golgi-Microtubule Network. EBioMedicine 22, 28–43 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holland EC, et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 25, 55–57 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Wang J, et al. Nestin regulates cellular redox homeostasis in lung cancer through the Keap1-Nrf2 feedback loop. Nat Commun 10, 5043 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Brolosy MA & Stainier DY R. Genetic compensation: A phenomenon in search of mechanisms. PLoS genetics 13, e1006780 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cronan JE Targeted and proximity-dependent promiscuous protein biotinylation by a mutant Escherichia coli biotin protein ligase. J Nutr Biochem 16, 416–418 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Mellacheruvu D, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10, 730–736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michalczyk K & Ziman M Nestin structure and predicted function in cellular cytoskeletal organisation. Histol Histopathol 20, 665–671 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Szklarczyk D, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordon RE, et al. Statins Synergize with Hedgehog Pathway Inhibitors for Treatment of Medulloblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 1375–1388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jordan MA & Wilson L Microtubules as a target for anticancer drugs. Nature reviews. Cancer 4, 253–265 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Wu CC, et al. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333, 459–462 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Matsuda Y, et al. Nestin is a novel target for suppressing pancreatic cancer cell migration, invasion and metastasis. Cancer Biol Ther 11, 512–523 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuda Y, Ishiwata T, Yoshimura H, Hagio M & Arai T Inhibition of nestin suppresses stem cell phenotype of glioblastomas through the alteration of post-translational modification of heat shock protein HSPA8/HSC71. Cancer letters 357, 602–611 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Lu WJ, et al. Inducible expression of stem cell associated intermediate filament nestin reveals an important role in glioblastoma carcinogenesis. International journal of cancer. Journal international du cancer 128, 343–351 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Parker AL, Kavallaris M & McCarroll JA Microtubules and their role in cellular stress in cancer. Front Oncol 4, 153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitchison TJ Microtubule dynamics and kinetochore function in mitosis. Annu Rev Cell Biol 4, 527–549 (1988). [DOI] [PubMed] [Google Scholar]
- 40.Nakamura Y, et al. Expression of tubulin beta II in neural stem/progenitor cells and radial fibers during human fetal brain development. Lab Invest 83, 479–489 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Roux KJ, Kim DI, Raida M & Burke B A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. The Journal of cell biology 196, 801–810 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sullivan KF & Cleveland DW Identification of conserved isotype-defining variable region sequences for four vertebrate beta tubulin polypeptide classes. Proceedings of the National Academy of Sciences of the United States of America 83, 4327–4331 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janke C & Bulinski JC Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 12, 773–786 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Cleveland DW Autoregulated control of tubulin synthesis in animal cells. Current opinion in cell biology 1, 10–14 (1989). [DOI] [PubMed] [Google Scholar]
- 45.Bittermann E, et al. Differential requirements of tubulin genes in mammalian forebrain development. PLoS genetics 15, e1008243 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrandina G, et al. Class III beta-tubulin overexpression is a marker of poor clinical outcome in advanced ovarian cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research 12, 2774–2779 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Mozzetti S, et al. Class III beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research 11, 298–305 (2005). [PubMed] [Google Scholar]
- 48.McCarroll JA, Gan PP, Liu M & Kavallaris M betaIII-tubulin is a multifunctional protein involved in drug sensitivity and tumorigenesis in non-small cell lung cancer. Cancer research 70, 4995–5003 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Kanojia D, et al. betaIII-Tubulin Regulates Breast Cancer Metastases to the Brain. Mol Cancer Ther 14, 1152–1161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calinescu AA & Castro MG Microtubule targeting agents in glioma. Transl Cancer Res 5, S54–S60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhat KM & Setaluri V Microtubule-associated proteins as targets in cancer chemotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research 13, 2849–2854 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.