

Abstract
Pancreatic ductal adenocarcinoma (PDAC) patients do not benefit from immune checkpoint blockade (ICB) along the PD-1/PD-L1 axis. Variable PD-L1 expression in PDAC indicates a potential access issue of PD-L1-targeted therapy. In order to monitor target engagement of PD-L1 targeted therapy, we generated a PD-L1 targeted PET tracer labeled with zirconium-89 (89Zr). As the MAPK signaling pathway (MEK and ERK) is known to modulate PD-L1 expression in other tumor types, we used [89Zr]Zr-DFO-anti-PD-L1 as a tool to non-invasively assess whether manipulation of the MAPK signaling cascade could be leveraged to modulate PD-L1 expression and thereby immunotherapeutic outcomes in PDAC. In this study, we observed that the inhibition of MEK or ERK is sufficient to increase PD-L1 expression, which we hypothesized could be leveraged for anti-PD-L1 immune checkpoint therapy. We found that the combination of ERK inhibition and anti-PD-L1 therapy indeed corresponded with a significant improvement of overall survival in a syngeneic mouse model of PDAC. Furthermore, immunohistochemical analysis indicates that the survival benefit may be CD8+ T-cell mediated. The therapeutic and molecular imaging tool kit developed could be exploited to better structure clinical trials and address the therapeutic gaps in challenging malignancies such as PDAC.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal diseases with limited treatment options and poor prognosis. Currently, surgery followed by adjuvant chemotherapy is the single curative option to combat PDAC, but only a small fraction of PDAC patients (~10%) of are likely to qualify and benefit. Additionally, approximately 80% of patients present with advanced and/or non-resectable disease (1). The clinical standard of care for non-resectable PDAC are chemotherapeutic agents gemcitabine or nab-paclitaxel, which largely fail to provide a long-term survival benefit. The last significant therapeutic stride for pancreatic cancer was the discovery that FOLFIRINOX provided a survival benefit compared to gemcitabine 10 years ago (2). More recent clinical trials have demonstrated a modest survival benefit in patients treated with combination chemotherapeutic drugs or radiotherapy (3–6).
One of the most difficult challenges in PDAC is that no treatments are curative, and tumors recur eventually. Immunotherapy, specifically PD-L1 checkpoint blockade, offers the opportunity to generate immune memory and thereby prevent or extend the period before recurrence. PDAC is currently unresponsive to PD-L1 inhibition as a monotherapy (7, 8) and this has been attributed to low PD-L1 expression in the TME. We purport increased expression of PD-L1 will enable targetability by anti-PD-L1 therapy and thereby improved efficacy.
In other tumor types, inhibition of the MAPK signaling cascade has primed the TME and worked synergistically with PD-L1 checkpoint blockade (9). Aberrant signaling of KRAS occurs in more than 95% of PDAC cases (10, 11), which is directly tied to MAPK and ERK signaling downstream. Since PDAC tumors are known to express low levels of PD-L1 (12), we hypothesized that pharmacological inhibition along the MAPK pathway can increase the targetability of PD-L1, thus enabling therapeutic response to anti-PD-L1 checkpoint blockade (13). The current diagnostic standard for asssesing PD-L1 status in many tumors, including PDAC, relies on biopsy and subsequent immunohistochemistry (IHC), which can be extremely invasive and often impossible to repeat. This method has many caveats due to the inter- and intratumoral heterogeneity of PD-L1, dynamic PD-L1 expression, and limited sampling via biopsy. With patient well-being in mind, we examined the effect of inhibition along the MAPK cascade on PD-L1 expression using a novel PD-L1-targeted non-invasive imaging tool.
We developed an anti-PD-L1 PET imaging agent by radiolabeling an anti-PD-L1 antibody with positron emitter zirconium-89 to assess PD-L1 expression in preclinical PDAC. Using this tool, we were able to show noninvasively that PD-L1 expression increases upon administration of an ERK inhibitor in orthotopic KPC models of PDAC, supporting the hypothesis for improved PD-L1 targetability upon tumor priming with ERK modulation. A schematic detailing our hypothesis is depicted in Fig. 1. We implemented a combination therapy strategy with ERK inhibition + anti-PD-L1 immune checkpoint blockade in orthotopic PDAC mice and were able to demonstrate significantly improved overall survival (OS) as well as decreased tumor burden. Further, the combination therapy arm demonstrated a significant increase in CD8+ T cells in a mechanism cohort relative to monotherapy arms, indicating a T-cell mediated mechanism.
Figure 1. Inhibitors that target MAPK signaling will increase PD-L1 and alter the tumor microenvironment in PDAC.
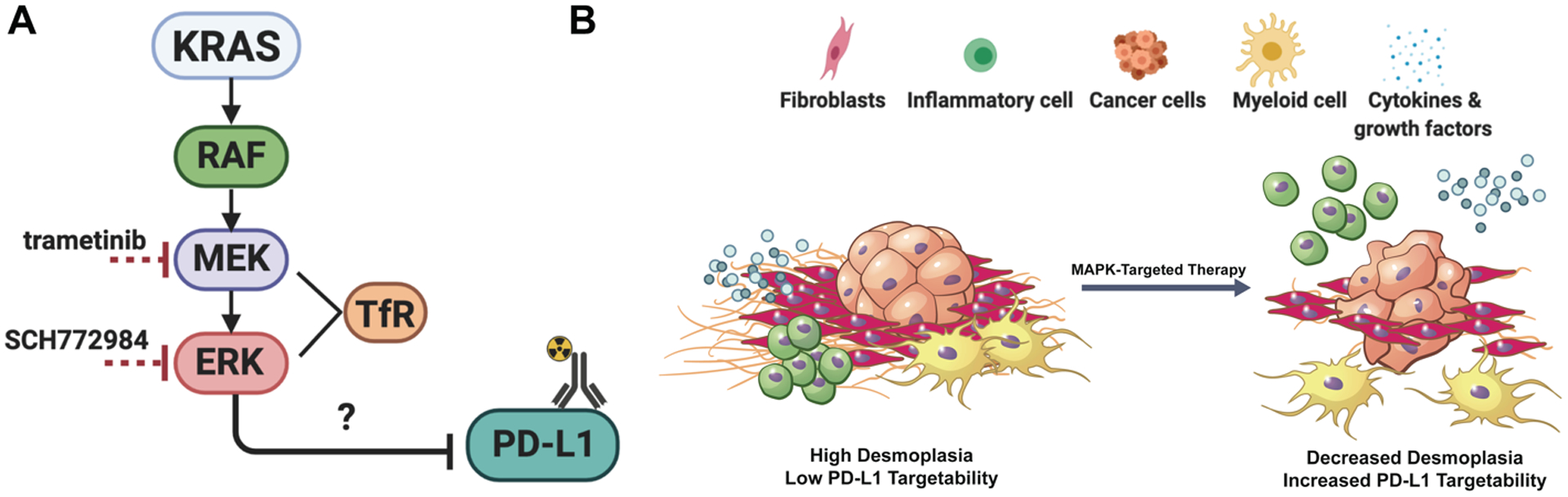
A. KRAS plays a central role in tumor development, regulating downstream proteins (RAF, MEK, and ERK) that are involved in proliferation, survival, metastasis, and angiogenesis. Oncogenic KRAS induces secretion of molecules that affect surrounding components of the stroma, such as fibroblasts, along with innate and adaptive immune cells. These cells within the tumor microenvironment promote cancer malignancy and provide opportunities for druggable targets in order to facilitate combination therapy strategies. Transferrin receptor (TfR) is a downstream readout for MEK and ERK inhibition. B. Proposed model of how MAPK-targeted therapy (ERK inhibitor SCH772984: abbreviated SCH, and MEK inhibitor trametinib) will alter the tumor microenvironment by activating the immune system and breaking down the stromal components, resulting in a release of cytokines and growth factors, along with a migration of inflammatory cells and macrophages. We also expect there to be an infiltration of immune cells when combined with ICB, and mechanistic studies will determine if and how they are functional.
MATERIALS AND METHODS
Reagents and small molecule inhibitors.
All reagents were purchased from Sigma Aldrich unless otherwise indicated. ERK inhibitor SCH772984 (14) was purchased from Active Biochem and used at a concentration of 1000 nM for in vitro experiments. The MEK inhibitor trametinib was purchased from Selleckchem and used at a concentration of 100 nM for in vitro experiments. All drugs were reconstituted in sterile, dry DMSO, aliquoted, and stored at 80 °C until further use.
Cell culture.
The KPC cell line was a generous gift from the Steven Leach Lab at MSKCC and grown in DME:F12 + 1% Glutamax. KPC cells infected with luciferin green fluorescent protein were a kind gift from the Scott Lowe Lab at MSKCC (15) and grown in high-glucose DME. The cells were not authenticated, regularly tested for Mycoplasma (every 6–12 months), and used under passage ten.
Small animal models and fibroblast isolation.
All animal experiments were performed in accordance with the Institutional Animal Care and Use Committee at Memorial Sloan Kettering Cancer Center. KPC cells (10,000 in 20 μL 1:1 mixture of cells: Matrigel) were injected orthotopically into the pancreas of female C57/BL6J animals. Tumor growth was characterized by ultrasound and palpation. Tumors were excised 3 weeks post-implantation. Fibroblasts were isolated using a migratory stimulus base on previous published procedures (16) and expanded for 10 days in conditioned media.
In vivo therapy.
Mice bearing orthotopic PDAC xenografts were treated via intraperitoneal injection of ERK inhibitor SCH772984 (prepared in 5% DMSO vol/vol, 10% PEG-300 vol/vol, in 10% m/v 2-hydroxypropyl-β-cyclodextrin). A solution with the same concentration of DMSO and PEG-300 in 10% m/v 2-hydroxypropyl-β-cyclodextrin was prepared as the vehicle control for the SCH772984 therapy. For therapy studies, animals were administered ERK inhibitor 90 mg/kg once daily. For combination therapy studies, animals were administered ERK inhibitor 75 mg/kg once daily and 200 μg of anti-PD-L1 every other day for a total of four doses. Every other day, ERK inhibition and anti-PD-L1 were administered as a co-injection. Saline was used as an untreated control and was volume-matched to anti-PD-L1 monotherapy. IgG was used as a concentration-matched isotype control for anti-PD-L1 monotherapy. Vehicle was used as a concentration matched DMSO control for ERK inhibitor.
Flow cytometry.
For all experiments, flow cytometry was performed on BD LSRFortessa flow cytometer at Memorial Sloan Kettering. The following fluorescently labeled antibodies were used from BioLegend: APC-PD-L1 (B7-H1, Clone 10F.9G2), PE-anti CD71 (TfR, clone RI7217), APC-IgG2b (Rat isotype control), and PE-IgG2a (Rat isotype control) to quantify murine PD-L1 and TfR expression levels pre- and post-treatment with MAPK-targeted therapy. KPC cells were seeded in 6-well plates (200K/well) and adhered overnight. Cells were then incubated with SCH772984 (1000 nM), or trametinib (100 nM), or DMSO (concentration-matched vehicle control). After incubation, cells were washed with PBS and harvested with trypsin. Cells were treated with Fc blocking solution for 30 minutes prior to surface staining of PD-L1 and TfR. DAPI staining was used to access cell viability. Data was analyzed by FlowJo. For ex vivo flow cytometry, orthotopic KPC tumors were harvested from mice that were treated with SCH772984. Tumors were prepared for flow cytometry using the gentleMACS tumor dissociation kit (Miltenyi Biotech). Each tumor was cut into small pieces (2 – 4 mm) and tissue pieces were transferred into a gentleMACS C tube containing an enzyme mixture. A homogenous single cell suspension was obtained by the gentleMACS dissociator machine. The cells were then washed with PBS and treated with Fc blocking solution for 30 minutes prior to surface staining of PD-L1, TfR (see antibodies above), and anti-mouse CD3 (Alexa Fluor 700, clone 17A2). DAPI staining was used to access cell viability. Data was analyzed by FlowJo software.
Preparation and radiolabeling of [89Zr]Zr-DFO-anti-PD-L1.
Anti-PD-L1 (clone 6E11) was functionalized with p-isothiocyanatobenzyl desferrioxamine (DFO-Bn-NCS; Macrocyclics, Inc.) as described previously(17). 89Zr was produced through proton beam bombardment of yttrium foil and isolated in high purity as [89Zr]Zr-oxalate at Memorial Sloan Kettering Cancer Center (New York, NY) according to a previously published procedure(18).
In vivo Imaging.
D-Firefly Luciferin (80 μL, 15 mg/kg) was injected intraperitoneally into the mice under anesthesia. Bioluminescence images were obtained 10 minutes post-injection (IVIS, PerkinElmer). Images were normalized via region of interest (ROI) analysis using the Living Image software. For PET imaging studies, [89Zr]Zr-DFO-anti-PD-L1 (16–20 nmol, 14–15 MBq in PBS, 130–150 μL) was injected intravenously and 20 min static PET/CT images were acquired at 24, 48, and 72 h using a dual microPET/CT scanner (Inveon, Siemens). Blocking study was performed using a 30× stoichiometric (mass) excess of unlabeled DFO-PD-L1 co-injected with imaging doses (details aforementioned) of [89Zr]Zr-DFO-anti-PD-L1.
Biodistribution studies.
Blood was collected via cardiac puncture and all tissues were harvested and weighed wet. Radioactivity within each organ was counted using a Perkin Elmer γ-counter. Tracer uptake expressed as percentage injected dose per gram (% ID/g) was calculated as the radioactivity in each tissue divided by the organ mass and the decay-corrected injected dose at the time of counting using the standard formulation method.
Statistical Considerations.
GraphPad Prism (version 7; GraphPad Software) was used for statistical analyses of the data. Biodistribution and IHC data were analyzed by unpaired, two-tailed Student’s t-tests and differences at the 95% confidence level. The Log-rank Mantel-Cox text was used for survival curve analysis. P values of 0.05 or less were considered significant.
Immunofluorescence and immunohistochemistry.
Tumors were collected in 10% neutral buffered formalin and fixed for 48 h prior to transfer to 70% ethanol for further analysis. Tissues were embedded in Paraffin wax and cut at 5 μm sections prior to staining. The immunofluorescence detections of c-myc and PD-L1 were performed at Molecular Cytology Core Facility of Memorial Sloan Kettering Cancer Center using Discovery XT processor (Ventana Medical Systems, Roche-AZ). Detailed procedures can be found in the Supplementary Information. IF quantification was done in imageJ using mean fluorescence intensity on random selected tissue areas and normalized by total selected area. IHC was performed on a Leica Bond RX automated stainer using Bond reagents (Leica Biosystems, Buffalo Grove, IL), including a polymer detection system (DS9800, Novocastra Bond Polymer Refine Detection, Leica Biosystems). Further details can be found in the Supplementary Data, along with a table with specific antibodies and concentrations.
RESULTS
The PDAC TME consists primarily of fibroblasts within the stroma and tumor cells. To determine whether inhibition along the MAPK cascade induces PD-L1 expression in cell types within the TME, we targeted ERK and MEK, two key players in the MAPK cascade with the leading inhibitors SCH772984 (SCH) and trametinib respectively. To do so, fibroblasts were isolated using a migratory stimulus26 and expanded for 10 days in conditioned media (Supplementary Fig. 1A). Fibroblasts were characterized for alpha smooth muscle actin to confirm the phenotype their morphology suggested (Supplementary Fig. 2B). These cells were plated and treated with ERK inhibitor SCH (1000 nM) or MEK inhibitor trametinib (100 nM) for 48 h. Our previous work showed that target engagement of ERK inhibitor SCH and MEK inhibitor trametinib can be monitored through the downregulation of transferrin receptor (TfR) (17). Flow cytometry showed ERK or MEK inhibition resulted in a significant decrease (P < 0.001) in TfR expression (as expected based on previous studies)20 which correlated to a significant increase for PD-L1 expression (Fig. 2A). This same trend was observed in plated KPC cancer cells, a mouse tumor cell line derived from the KPC transgenic mouse model of PDAC (Fig. 2B). The effect of ERK inhibition on PD-L1 expression was more pronounced than MEK. As a result, we sought to determine whether ERK inhibition induces PD-L1 expression in vivo. To do so in a clinically relevant manner, we developed a PD-L1 targeted non-invasive imaging tool.
Figure 2. MAPK-targeted therapy increases PD-L1 expression in cells comprising the PDAC tumor microenvironment.
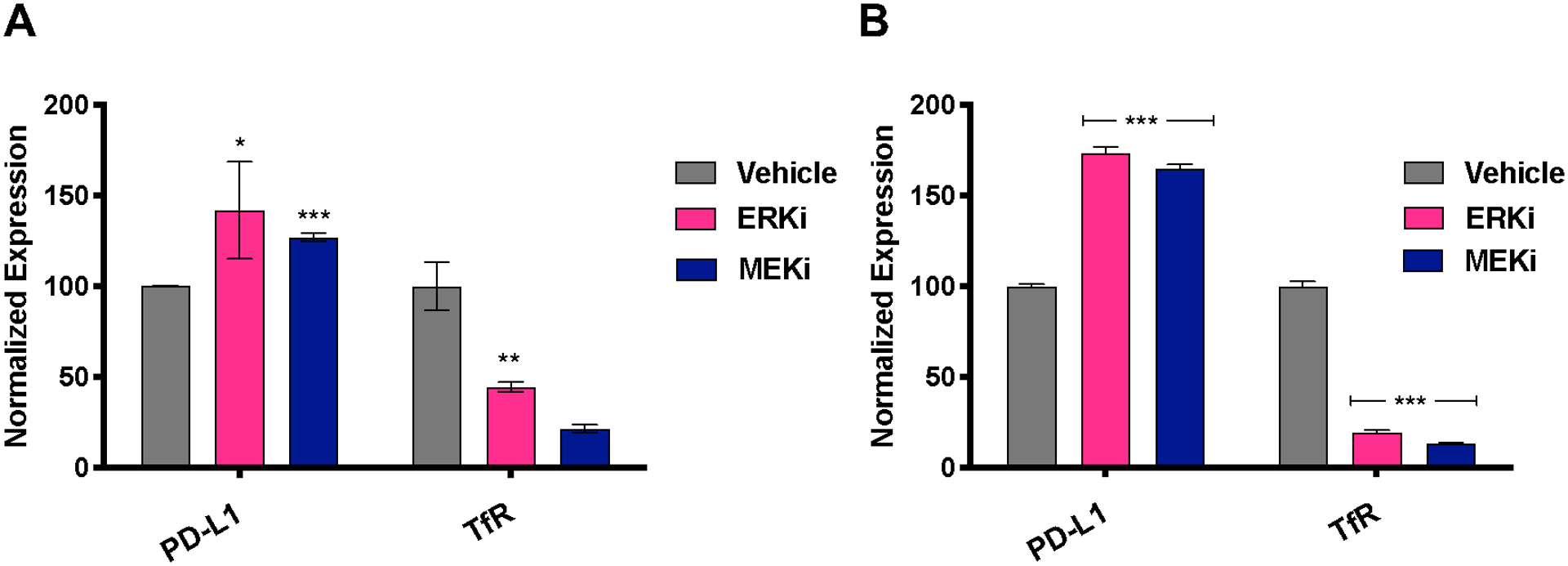
A. Transferrin receptor (TfR) expression decreases upon inhibition of KPC CAFs with ERK inhibitor SCH, and MEK inhibitor trametinib. MAPK inhibition (drugs against ERK and MEK) show a significant increase in PD-L1 expression on CAFs. B. TfR expression decreases upon inhibition of plated KPC cells with ERK inhibitor SCH, and MEK inhibitor trametinib. MAPK inhibition (drugs against ERK and MEK) show a significant increase in PD-L1 expression in KPC cells.
PET Imaging of PD-L1 expression in orthotopic PDAC models.
In order to detect PD-L1 status in a noninvasively in vivo, we generated a PD-L1 targeted positron emission tomography (PET) imaging agent. Radiolabeled anti-PD-L1 has been used as an immuno-PET agent in human (20) and several preclinical mouse models, laying the foundation for a PD-L1-targeted imaging tool in PDAC. To generate a PD-L1 targeted PET tracer, we synthesized a chelator-antibody conjugate for radiolabeling with 89Zr using the Genentech anti-PD-L1 clone 6E11 (Fig. 3A). We were able show positive (~60%) immunoreactivity for the PD-L1 target and that the anti-PD-L1 tracer is blockable (Fig. 3B), indicating specific binding of our radiolabeled construct. A PET imaging study with [89Zr]Zr-DFO-anti-PD-L1 showed PD-L1 expression in orthotopic KPC tumors over the course of 72 h (Fig. 3C). In vivo blocking with an excess of unlabeled DFO-anti-PD-L1 demonstrated significant decrease in tumor uptake and thereby specificity of the tracer in the tumor (Supplementary Fig. 3). Signal was also blocked peripherally in PD-L1 expressing tissues including the spleen and bone, along with a significant increase in [89Zr]Zr-DFO-anti-PD-L1 uptake observed in the blood, further demonstrating that the “antigen sink” is being blocked by the excess dose.
Figure 3. Synthesis, characterization, and PET imaging of [89Zr]Zr-DFO-anti-PD-L1 in KPC mice.
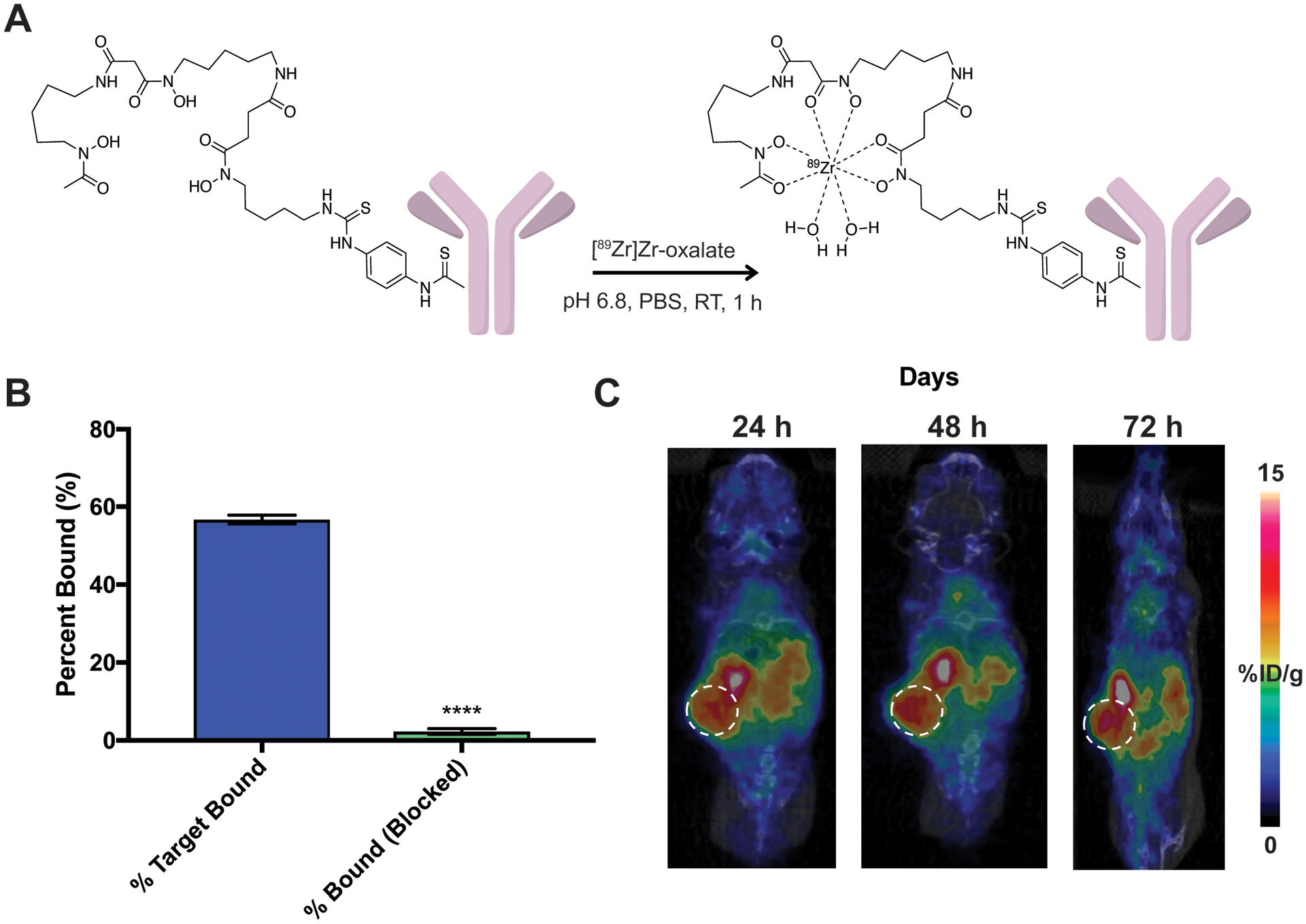
A. Radiolabeling schema of anti-PD-L1 antibody-DFO with zirconium-89. B. [89Zr]Zr-DFO-anti-PD-L1 specifically binds PD-L1 and is blockable via an unlabeled excess of anti-PD-L1. C. Representative PET images of [89Zr]Zr-DFO anti-PD-L1 out to 72 h shows delineated tumor uptake in orthotopic KPC mice.
[89Zr]Zr-DFO-anti-PD-L1 uptake increases with ERK inhibition in multiple KPC mouse models.
To determine whether ERK inhibition increases PD-L1 in vivo, we implemented a therapy study in KPC orthotopic models. KPC cells that were stably infected with luciferin-GFP and injected 10,000 cells/mouse into the pancreas of C57BL6 mice and three weeks after implantation, were able to visualize tumor burden by bioluminescent imaging (Fig. 4A). At week three, all mice were stratified based on tumor size (determined by ROI analysis) and treated tumor-bearing mice with either an ERK inhibitor (90 mg/kg, once daily) or vehicle control. Four days after therapy began, mice were injected with [89Zr]Zr-DFO-anti-PD-L1 (timeline represented in Fig. 4B) and PET imaging was performed 72 h post-injection (Fig. 4C, n = 4/group). The tissues were subsequently resected for biodistribution (Fig. 4D) via ex vivo gamma counting. There was a significant increase of [89Zr]Zr-DFO-PD-L1 in the tumor (P < 0.01), with no significant differences in other tissues, indicating increased PD-L1 expression following ERK inhibition. A duplicate cohort was used for ex vivo flow cytometry (Fig. 4E, n = 3–4/group) to determine ERK inhibitor target engagement in vivo. Tumor-bearing mice treated with the ERK inhibitor demonstrated a decrease in TfR expression (indicating ERK target engagement) and an increase of PD-L1 expression from therapy (P < 0.01). These data suggest ERK inhibition primes the TME for PD-L1-targeted immune checkpoint blockade. To determine whether PD-L1 expression visualized by PET in vivo corresponded to PD-L1 expression ex vivo, we excised tumors and stained for PD-L1. Excised tumors showed a significant increase in PD-L1 expression following ERK inhibition. In addition, excised tumors demonstrated a decreased expression of MYC, a downstream target of ERK activation, corroborating ERK inhibitor target engagement (Fig. 2F). Expression of PD-L1 and MYC was quantified using mean fluorescence intensity per unit area (Fig. 2G) on serial tissue sections of mouse KPC tumors from different treatment groups (n = 3/group, P < 0.001). Immunofluorescence experiments were optimized using controls to mitigate channel bleeding and non-specific secondary interactions (Supplementary Fig. 3).
Figure 4. ERK inhibition increased uptake of [89Zr]Zr-DFO-anti-PD-L1 in orthotopic KPC-Luc-GFP mice.
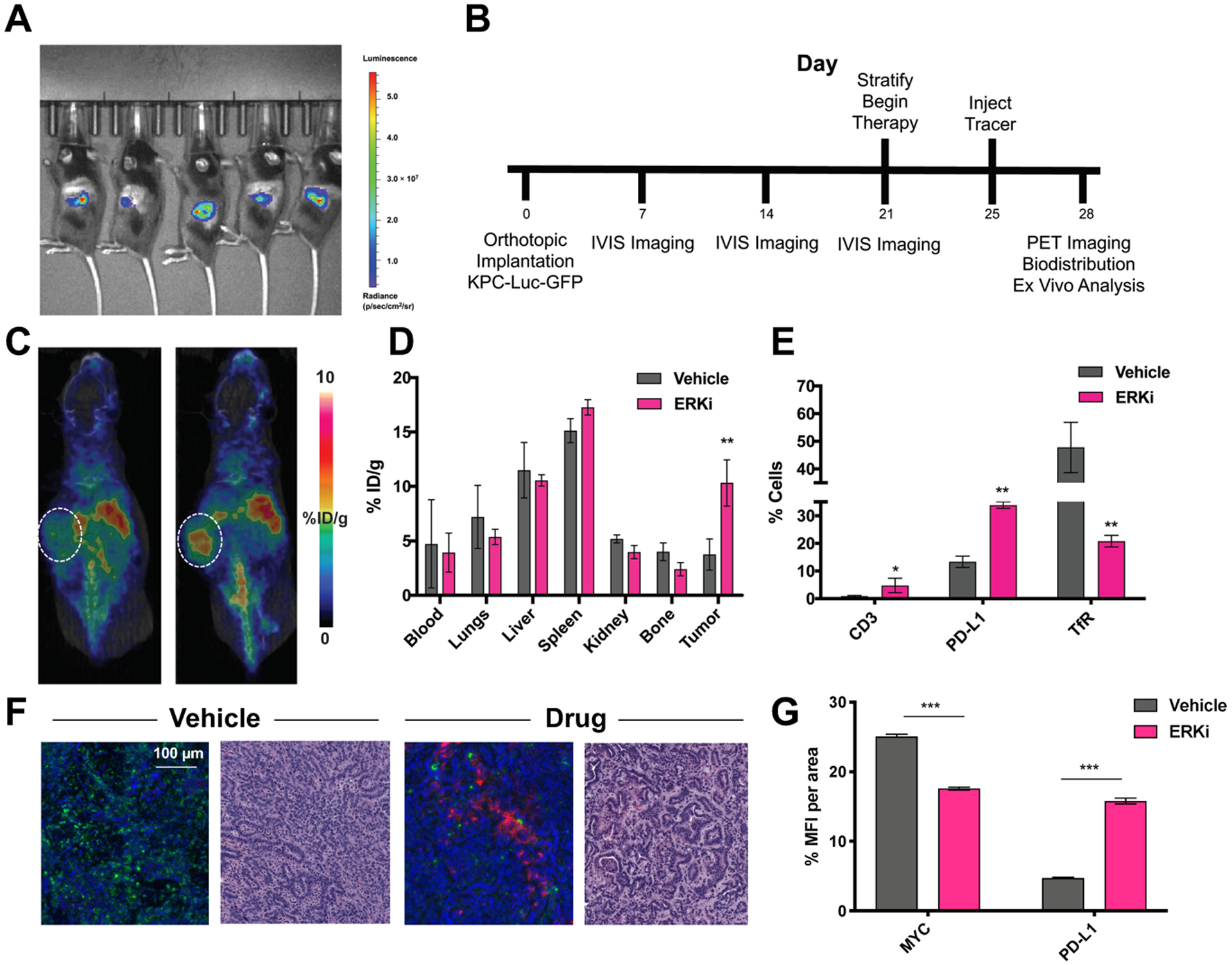
A. Bioluminescent imaging of KPC-Luc-GFP tumors 2 weeks post-orthotopic injection. B. Timeline of implantation and drugging. Animals received ERK inhibitor SCH772984 once daily at 90 mg/kg for 7 days. C. Uptake of [89Zr]Zr-DFO-anti-PD-L1 increase upon ERK inhibition in vivo and is quantified via ex vivo biodistribution (D). E. Ex vivo flow cytometry of KPC-Luc-GFO tumors shows an increase in PD-L1 expression an CD3+ T cells correlated to a decrease in TfR expression. **P < 0.01. F. Immunofluorescent analysis of MYC (green) and PD-L1 (pink) expression ex vivo in KPC tumors and H&E stains of serial sections. G. Quantification of immunofluorescent analysis of MYC+ and PD-L1+ cells in KPC PDAC tumors. Abbreviation: MFI: mean fluorescent intensity.
Combination ERK inhibition and immune checkpoint blockade decreases tumor growth and improves overall survival in orthotopic KPC-Luc-GFP mice.
To determine whether ERK induced PD-L1 expression is sufficient to reduce tumor size and produce a survival benefit to PD-L1 checkpoint blockade, we performed a therapy study in subcutaneous flank models of the KPC-Luc-GFP. Tumor bearing mice were treated with the ERK inhibitor alone or in combination with anti-PD-L1 therapy. Tumor size was quantified by bioluminescence imaging. Treatment with the ERK inhibitor as a single agent or in combination with anti PD-L1 therapy resulted in the same marginal benefit in median survival when compared to saline, vehicle or anti-PD-L1 monotherapy (Supplementary Fig. 4). However, tumor volume changes had no correlation to the marginal survival benefits. As subcutaneous models lack components of the PDAC TME, we investigated whether these results were recapitulated in an orthotopic PDAC model. As such, we sought to determine whether combination ERK and PD-L1 inhibition provides a survival benefit in an orthotopic model of PDC (n = 5–6/group). We observed a significant improvement of survival upon combination therapy with ERK inhibitor + PD-L1 checkpoint blockade, suggesting tumor location is central to the study of this signaling axis (Fig. 5A, P < 0.01 based on Log-rank Mantel Cox statistical test). Since we had observed slight toxicities at the 90 mg/kg dose in our previous experiments, we elected to drug mice once daily with ERK inhibitor (75 mg/kg) and once every other day with anti-PD-L1 (200 mg) for a total of 4 doses for 1 week total for each of the combination therapy studies. Implantation, imaging, therapy, and resection timeline is described in Fig. 5B. Each therapy arm, with the exception of PD-L1 vs. IgG or vehicle, was significant compared to its respective control (P < 0.05 for ERKi vs. vehicle). The combination therapy arm showed significant increased survival compared to each monotherapy (P < 0.01 when compared to PD-L1 monotherapy, and P < 0.05 compared to ERKi monotherapy). Treatment with ERKi also significantly reduced tumor growth compared to its vehicle control (Fig. 5C, P < 0.05). Despite significant regression in tumor growth (Fig. 5C, P < 0.01 for combination arm vs. ERKi, P < 0.05 for combination vs. PD-L1), tumors grew back after cessation of therapy, resulting in only 30 days total survival.
Figure 5. Combination ERK inhibition and anti PD-L1 improves survival and tumor growth in KPC-Luc-GFP mice.
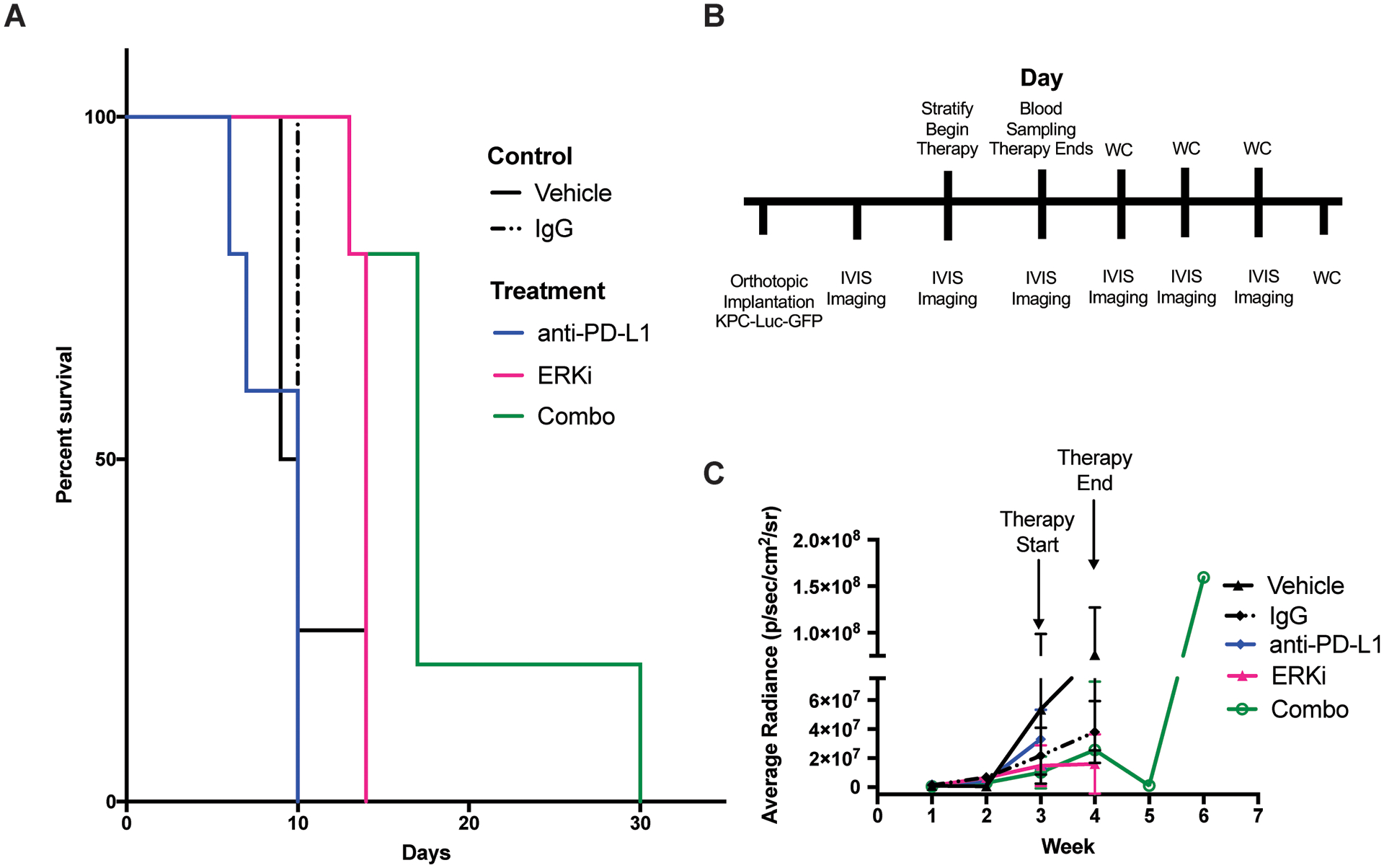
A. Survival curve of treatment arms over 30-day time period, drugging period was during the first week. Significant survival was observed in ERK inhibitor treatment groups (P < 0.05) vs. vehicle, saline and IgG. Significant survival was observed in combination treatment groups (P < 0.005) vs. all control groups (P < 0.005 for vehicle, saline and IgG) and when compared to each monotherapy (P < 0.05 for anti-PD-L1 and ERKi alone). B. Timeline of implantation, therapy, and BLI. C. ROI analysis of IVIS images taken throughout therapy until study endpoint.
Immunohistochemical analysis of ex vivo endpoint tissues indicates potential immunosuppressive mechanisms.
We found a significant decrease in CD4+ T cells (P < 0.05 when comparing combination therapy arm (0.32 % ± 0.1), represented in % positive tumors cells to each respective monotherapy: ERKi (0.91 ± 0.2), PD-L1 (1.67 ± 0.6) (Supplementary Fig. 5). Upon further inspection, we observe a trending decrease in FoxP3+ cells, albeit not significant when comparing different therapy arms. Quantitative analysis of CD3, CD8, MYC, and PD-L1 in endpoint tissues can also be found in Supplementary Fig. 5. Supplementary Fig. 6 shows representative images of CD4 and FoxP3 analysis in all treatment arms. A therapy cohort was repeated to investigate T cell engagement through tissues collected at a single endpoint for each therapy arm. Quantitation from cytonuclear and area analysis for CD3, CD8, CD4, MYC, and PD-L1 are represented in Fig. 6A. Of note is a significant increase in CD8+ T cells in the combination therapy arm when compared to vehicle and ERK inhibition alone (P < 0.01, Fig. 6B). Additionally, increase in CD4+ T cells trend similarly with CD8+ T cells (Fig. 6C). Furthermore, the expected increase in PD-L1 expression and decrease in MYC expression (in the ERK inhibitor and combination therapy arms) is also represented in this cohort. Representative images highlight these trends. Supplementary Fig. 7 highlights representative IHC for MYC, PD-L1, and CD3 in different therapy arms at 1× magnification.
Figure 6. Immunohistochemical analysis of ex vivo tissues collected at single endpoint indicate a potential T-cell mediated mechanism.
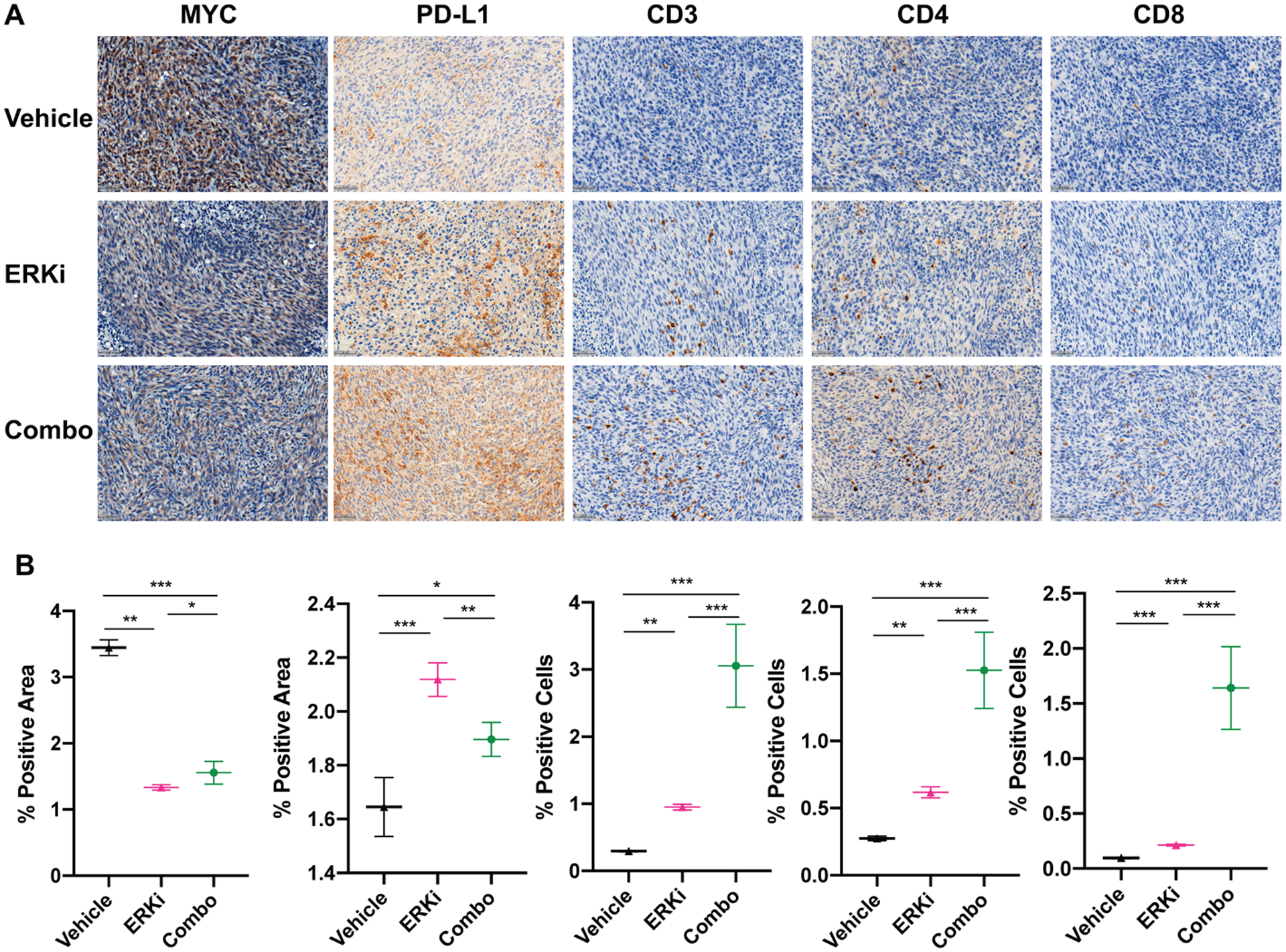
A. Representative images of serial sections from the same tumors stained for MYC, PD-L1, CD3, CD4, and CD8 with trends quantified in (B). Significant trends are observed along with decreases in MYC staining and increase in PD-L1 staining (along with CD3, CD4 and CD8) in each treatment arm compared to respective controls. *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
According to our cell viability and pathway activity data on these model cell lines only cells harboring the rare G12C KRAS mutation and low EGFR expression are sensitive to single MEK inhibitor (trametinib) treatment. The common G12D KRAS mutation leads to elevated baseline Akt activity, thus treatment with single MEK inhibitors fails. However, combination of MEK and Akt inhibitors are synergistic in this case. In case of wild-type KRAS and high EGFR expression MEK inhibitor induced Akt phosphorylation leads to trametinib resistance which necessitates for MEK and EGFR or Akt inhibitor combination treatment. In all we provide strong preclinical rational and possible molecular mechanism to revisit MEK inhibitor therapy in pancreatic cancer in both monotherapy and combination, based on molecular profile analysis of pancreatic cancer samples and cell lines. According to our most remarkable finding, a small subgroup of patients with G12C KRAS mutation may still benefit from MEK inhibitor monotherapy.Monotherapy is rarely effective in treating PDAC patients. Clinical trials across tumor types for KRAS, MEK, and ERK inhibitors have failed or had mixed results across patient cohorts trending towards no response. These failures can be attributed to intrapatient heterogeneity and complicated feedback mechanisms that are still not well understood (21, 22). However, MEK inhibitors in the context of combination therapy have shown some biological response in PDAC (23, 24). In other tumors types, ERK inhibition has shown to be safe in PDAC (25) and the ERK pathway shown to modulate PD-L1 expression in other tumors types (13, 26, 27).
PD-L1 checkpoint blockade has similarly failed as a monotherapy, and the majority of PDAC patients do not respond to PD-L1 inhibition (28, 29). Increasing the expression of PD-L1 to enable anti-PD-L1 target engagement may seem paradoxical. However, the complex role of PD-L1 expression in the TME has left much open to question. Our data shows that ERK inhibition increases the expression of PD-L1 and that is sufficient to improve anti-PD-L1 targetability and efficacy. In keeping with this notion, the combination of kinase inhibition to increase PD-L1 targetability has demonstrated potential survival benefits and immune memory in other tumor types (26).
A number of strategies to prime the TME for immune checkpoint blockade have been successful in mouse models of PDAC. These strategies include stromal remodeling (30, 31), immune priming (32, 33), and stereotactic radiation (34). Tumors with highly immunosuppressive tumor microenvironments, such as in PDAC, inhibit T cell infiltration/activation which causes low response rates to ICB. Recent work by Christmas et al demonstrated the sensitization of pancreatic tumors to ICB by epigenetic modulation of myeloid suppressor cells in the TME.(35) Others have also shown that degrading stromal hyaluronan in a metastatic murine PDAC model leads to decreased immunosuppressive signaling in myeloid cells and increases memory T cell infiltration (36). Both stromal modification and immune cell recruitment are both extremely important for the PDAC TME but need to be pursued with vigilance and careful dosing. Drug regimens in these realms have failed due to unanticipated toxicities as well as lack of significant improvement (37, 38).
In preclinical studies, scientists often resect tissues at multiple time points to monitor target engagement and biological response throughout treatment. While the mouse models offer continuous monitoring via sacrificial studies, it is impossible to obtain multiple biopsies in patients to assess biological response. Clinical trials for PDAC immunotherapy largely fail and it is unclear why due to the aforementioned limitation. Current criteria for assessing therapy response in clinical trials (including RECIST and iRECIST) often lack a specific imaging component that can noninvasively detect biologically relevant changes to the TME (39, 40). Targeted non-invasive imaging may enable understanding mechanistic underpinnings even when the clinical results do not reach end points. This allows the researchers to refine therapeutic approaches in a strategic manner, which can potentially improve both clinical outcomes long term and reduce the number of patients suffering unnecessary side effects in failed trials. We propose a therapeutic strategy in combination with a non-invasive imaging tool to enable clinicians to monitor anti-PD-L1 target engagement at iterative time points in patients, thereby expanding possible conclusions for both positive and negative clinical results.
Increased infiltration of T cells, specifically CD8+ T cells, has been a longstanding indicator of successful cancer immunotherapy (41). The role of CD4+ cells has been indicative of immunosuppression, but in reality has not been fully explored and largely remained in the background (42). There is increasing evidence that CD4+ T cells contribute to the anti-tumor immune response and may play a bigger role than historically thought. In addition to providing help to antitumor CD8+ T cells, CD4+ T cells are also involved in direct cytotoxicity against tumor (41). Quezada et al. showed that CD4+ T cells transferred to lymphopenic mice inhibit melanoma tumor growth in an MHC Class II dependent manner and this cytotoxic effect was enhanced with the combination of anti-CTLA4 therapy.(43) A more recent study shows that MHC-I− /MHC-II+ expressing tumors have a positive response to anti-PD-1 therapy associated with CD4+ T cell infiltration in a Classical Hodgkin Lymphoma (cHL) mouse model (44). These studies are encouraging for our own data and uncover a potential role of synergy between ERK inhibition and ICB that could be exploited further in challenging malignancies such as PDAC.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge the Small Animal Imaging, Flow Cytometry, Molecular Cytology, Pathology, and the Radiochemistry and Molecular Imaging Probe Core Facilities at MSK, all of which are supported in part by NIH grant P30 CA08748. We would also like to acknowledge financial support from NIH grant R35 CA232130 (J.S.L.) and the MSK Center for Molecular Imaging and Nanotechnology Tow Fellowship (K.E.H.). We thank Olivera Grbovic-Huezo from the Steven Leach Lab for the naïve KPC cell line and Marcus Ruscetti and John Morris IV from the Scott Lowe Lab for the KPC-Luc-GFP cell line. We acknowledge BioRender software and the MSK Graphics Department for aid in the preparation of Figure 1.
FINANCIAL INFORMATION
This work was supported in part by NIH grant P30 CA08748, NIH grant R35 CA232130 (J.S.L.) and the MSK Center for Molecular Imaging and Nanotechnology Tow Fellowship (K.E.H.). Materials were provided by Genentech. No financial support was provided. The authors have nothing to disclose.
REFERENCES
- 1.Cameron JL, Riall TS, Coleman J, & Belcher KA (2006) One Thousand Consecutive Pancreaticoduodenectomies. Annals of Surgery 244(1):10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conroy T, et al. (2011) FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. New England Journal of Medicine 364(19):1817–1825. [DOI] [PubMed] [Google Scholar]
- 3.Picozzi VJ, et al. (2015) 90Y-clivatuzumab tetraxetan with or without low-dose gemcitabine: A phase Ib study in patients with metastatic pancreatic cancer after two or more prior therapies. European Journal of Cancer 51(14):1857–1864. [DOI] [PubMed] [Google Scholar]
- 4.Hingorani SR, et al. (2017) HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. Journal of Clinical Oncology:JCO.2017.2074.9564. [DOI] [PubMed] [Google Scholar]
- 5.Quan K, et al. (2017) Results of a prospective phase 2 clinical trial of induction gemcitabine/capecitabine followed by stereotactic ablative radiation therapy in borderline resectable or locally advanced pancreatic adenocarcinoma. Practical Radiation Oncology. [DOI] [PubMed] [Google Scholar]
- 6.Von Hoff DD, et al. (2013) Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. The New England journal of medicine 369(18):1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamath SD, et al. (2020) Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. The oncologist 25(5):e808–e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Reilly EM, et al. (2019) Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol 5(10):1431–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ebert PJR, et al. (2016) MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 44(3):609–621. [DOI] [PubMed] [Google Scholar]
- 10.Dimastromatteo J, Houghton JL, Lewis JS, & Kelly KA (2015) Challenges of Pancreatic Cancer. Cancer journal (Sudbury, Mass.) 21(3):188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tesfaye AA, Kamgar M, Azmi A, & Philip PA (2018) The evolution into personalized therapies in pancreatic ductal adenocarcinoma: challenges and opportunities. Expert Review of Anticancer Therapy 18(2):131–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng L (2017) PD-L1 Expression in Pancreatic Cancer. Journal of the National Cancer Institute 109(6):djw304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng HY, et al. (2018) Chemotherapeutic Treatments Increase PD-L1 Expression in Esophageal Squamous Cell Carcinoma through EGFR/ERK Activation. Translational oncology 11(6):1323–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaikuad A, et al. (2014) A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat Chem Biol 10(10):853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruscetti M, et al. (2020) Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 181(2):424–441.e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müerköster S, et al. (2004) Tumor Stroma Interactions Induce Chemoresistance in Pancreatic Ductal Carcinoma Cells Involving Increased Secretion and Paracrine Effects of Nitric Oxide and Interleukin-1β. Cancer Research 64(4):1331–1337. [DOI] [PubMed] [Google Scholar]
- 17.Henry KE, et al. (2018) A PET Imaging Strategy for Interrogating Target Engagement and Oncogene Status in Pancreatic Cancer. Clinical Cancer Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holland JP, Sheh Y, & Lewis JS (2009) Standardized methods for the production of high specific-activity zirconium-89. Nuclear medicine and biology 36(7):729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer AT & Gambhir SS (2018) The Immunoimaging Toolbox. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 59(8):1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bensch F, et al. (2018) 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nature Medicine 24(12):1852–1858. [DOI] [PubMed] [Google Scholar]
- 21.Ferrarelli LK (2020) Predicting resistance to KRAS inhibitors. Science Signaling 13(614):eaba8405. [Google Scholar]
- 22.Mullard A (2019) Cracking KRAS. Nat Rev Drug Discov 18(12):887–891. [DOI] [PubMed] [Google Scholar]
- 23.Ko AH, et al. (2016) A Multicenter, Open-Label Phase II Clinical Trial of Combined MEK plus EGFR Inhibition for Chemotherapy-Refractory Advanced Pancreatic Adenocarcinoma. Clinical Cancer Research 22(1):61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Infante JR, et al. (2014) A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. European Journal of Cancer 50(12):2072–2081. [DOI] [PubMed] [Google Scholar]
- 25.Janku F, et al. (2020) Phase I dose-finding study of oral ERK1/2 inhibitor LTT462 in patients (pts) with advanced solid tumors harboring MAPK pathway alterations. Journal of Clinical Oncology 38(15_suppl):3640–3640. [Google Scholar]
- 26.Brea EJ, et al. (2016) Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunology Research 4(11):936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stutvoet TS, et al. (2019) MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. The Journal of pathology 249(1):52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le DT, et al. (2017) Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (New York, N.Y.) 357(6349):409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu ZI, et al. (2018) Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clinical cancer research : an official journal of the American Association for Cancer Research 24(6):1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beatty GL, et al. (2011) CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (New York, N.Y.) 331(6024):1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Mackelenbergh MG, et al. (2019) Clinical Trials Targeting the Stroma in Pancreatic Cancer: A Systematic Review and Meta-Analysis. Cancers 11(5):588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson BA, Yarchoan M, Lee V, Laheru DA, & Jaffee EM (2017) Strategies for Increasing Pancreatic Tumor Immunogenicity. Clinical Cancer Research 23(7):1656–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vonderheide RH (2018) The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer cell 33(4):563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gajiwala S, Torgeson A, Garrido-Laguna I, Kinsey C, & Lloyd S (2018) Combination immunotherapy and radiation therapy strategies for pancreatic cancer-targeting multiple steps in the cancer immunity cycle. Journal of gastrointestinal oncology 9(6):1014–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christmas BJ, et al. (2018) Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer immunology research 6(12):1561–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blair AB, et al. (2019) Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer. Clinical Cancer Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hakim N, Patel R, Devoe C, & Saif MW (2019) Why HALO 301 Failed and Implications for Treatment of Pancreatic Cancer. Pancreas (Fairfax) 3(1):e1–e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rhim Andrew D, et al. (Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 25(6):735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beer L, et al. (2019) Comparison of RECIST, iRECIST, and PERCIST for the Evaluation of Response to PD-1/PD-L1 Blockade Therapy in Patients With Non–Small Cell Lung Cancer. Clinical Nuclear Medicine 44(7). [DOI] [PubMed] [Google Scholar]
- 40.Carter BW, Bhosale PR, & Yang WT (2018) Immunotherapy and the role of imaging. Cancer 124(14):2906–2922. [DOI] [PubMed] [Google Scholar]
- 41.Chen P-L, et al. (2016) Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discovery 6(8):827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tay RE, Richardson EK, & Toh HC (2021) Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther 28(1–2):5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quezada SA, et al. (2010) Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. Journal of Experimental Medicine 207(3):637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagasaki J, et al. (2020) The critical role of CD4+ T cells in PD-1 blockade against MHC-II–expressing tumors such as classic Hodgkin lymphoma. Blood Advances 4(17):4069–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.