

Abstract
Antibody-antigen co-crystal structures are a valuable resource for the fundamental understanding of antibody-mediated immunity. Determination of structures with antibodies in complex with their antigens, however, is a laborious task without guarantee of success. Therefore, homology modeling of antibodies and docking to their respective-antigens has become a very important technique to drive antibody and vaccine design. The quality of the antibody modeling process is critical for the success of these endeavors. Here, we compare different computational protocols for predicting antibody structure from sequence in the biomolecular modeling software Rosetta - all of which use multiple existing antibody structures to guide modeling. Specifically, we compare protocols developed solely to predict antibody structure (RosettaAntibody, AbPredict) with a universal homology modeling protocol (RosettaCM). Following recent advances in homology modeling with multiple templates simultaneously, we propose that the use of multiple templates over the same antibody regions may improve modeling performance. To evaluate whether multi-template comparative modeling with RosettaCM can improve the modeling accuracy of antibodies over existing methods, this study compares the performance of the three modeling algorithms when modeling human antibodies taken from antibody-antigen co-crystal structures. In these benchmarking experiments, RosettaCM outperformed other methods when modeling antibodies with long HCDR3s and few available templates.
Keywords: Antibody, homology modeling, loop prediction, Rosetta, benchmark, structure
Introduction
Antibody-antigen co-crystal structures are essential to the understanding of the molecular interactions of antibodies with their target epitopes. These structures are obtained through experimental methods such as X-ray crystallography or high-resolution electron microscopy.1–3 High-throughput sequencing has allowed for the identification of large numbers of antibody heavy and light chain variable region gene sequences to guide discovery of specific antibodies and drive vaccine development.4 However, with an estimated 1011 potential unique antibody variable gene sequences in a single individual, determining the structure of a large number of antibodies is not achievable with current experimental methods.5,6
Structural homologs identified through sequence similarity have been used to predict antibody variable region structure since the initial work of Kabat and Wu in 1972.7 While protein homology modeling mostly relies on templates with high sequence similarity, antibody modeling is more complex. The antibody structure generally contains regions of high conservation such as the framework, and regions of high diversity, specifically the complementarity-determining regions (CDRs). Antibodies are Y-shaped proteins, with two variable domains or fragments (Fv), a heavy chain and a light chain Fv, forming the tips of the arms that bind to the antigen. The variable domains are structured as two β-sandwiches each containing two β-sheets formed of antiparallel β-strands around a hydrophobic core. This highly conserved β-sheet structure is termed the immunoglobulin fold and provides a scaffold for the hypervariable loops at one end that form the antigen-binding site. These loops, known as complementarity-determining regions (CDRs), are encoded by genes produced through recombination of the V(D)J gene segments. V (variable), D (diversity), and J (joining) segments exist as multiple copy arrays on the chromosome, and their recombination is initiated by double-strand DNA breaks, followed by the deletion and sometimes inversion of segments and their subsequent ligation together.8 The variable domain of the heavy chain is encoded by genes formed through the recombination of VH, DH, and JH genes while the light chain variable domain is encoded by the VL and JL genes.8 This recombination process contributes to the massive immunoglobin diversity in vertebrate immune systems. Canonical structural classes of CDRs have been defined based on loop size and the presence of certain residues at key positions in the loop and framework regions from reported crystal structures.9,10 Five of the six CDRs fall into known canonical classes, approximately 85% of the time, but the structurally diverse HCDR3 has eluded classification. Current efforts focus on dividing the HCDR3 region in a torso and head region.10,11
The Rosetta software suite is an academically developed framework for protein structure prediction and design.12,13 RosettaAntibody is a popular antibody modeling application built on the Rosetta that utilizes conformational clusters from antibody canonical classes based on the classification by North et al. to predict antibody variable region structure.10,14 A significant limitation of RosettaAntibody is the unavailability of good structural templates for some CDR conformations, causing the grafting on templates with low sequence identity without the ability to refine this part of the structure. RosettaAntibody models the HCDR3 de novo instead of using data from existing antibody structures, as the inherent diverse nature of HCDR3 loops has yet escaped classification through clustering.15,16 A recent major improvement in RosettaAntibody has been achieved through the re-docking of the heavy-light chain Fv interface after modeling.16,17 RosettaAntibody continues to perform well in antibody modeling challenges. In an assessment of eleven antibodies, RosettaAntibody modeled the HCDR3 loop in five out of six cases with short HCDR3 loops (8 to 10 residues) at a backbone RMSD <2.0 Å, although it struggled more for long HCDR3 loops (11 to 14 residues), where backbone RMSDs averaged around 5 Å.16
AbPredict is another antibody modeling protocol built into the Rosetta modeling suite. AbPredict selects templates for each of four regions: the variable heavy chain domain (including HCDR1 and HCDR2), the variable light chain domain (including LCDR1 and LCDR2), HCDR3, and LCDR3, similar to the assembly of the antibody from during V(D)J recombination.18 Templates for the four regions are grafted onto a reference structure. Afterwards, a database is used to optimize the heavy chain-light chain orientation.18 AbPredict shares RosettaAntibody’s limitation of relying heavily on template quality without the ability to de novo model regions that lack good templates. However, because AbPredict operates independently of sequence homology, instead searching for combinations of backbone conformation observed in natural antibody structures, AbPredict can sample a greater conformational space, potentially improving accuracy in HCDR3 loops.18
RosettaCM is the generic comparative modeling method of the Rosetta modeling suite. Templates are selected manually based on the protein being modeled, and the sequence of interest is threaded onto its corresponding template structure. Rosetta de novo fragments are used to fill in any gaps not covered by the templates. The resulting model is optimized by all-atom refinement to generate full-chain models.19 The ability of RosettaCM to integrate multiple templates with de novo fragments to generate a single model allows it to sample a broader structural landscape and select sub-templates for different protein regions, with performance improving with higher sequence similarity. However, because Rosetta sampling is stochastic in nature, not every modeling trajectory will converge on to a local energy minimum on the score function. Because of time constraints, RosettaCM cannot exhaustively sample the complete structural space.20
In this study, we developed a RosettaCM-based protocol for homology modeling of antibodies that uses its power to use multiple templates for each antibody region. We compared our protocol to different Rosetta-based computational protocols for predicting the structure of an antibody from its sequence by using a benchmark set. We collected a template database of human antibody-antigen structures from the Protein Data Bank (PDB). We performed a basic local alignment search tool (BLAST)21 search for antibody sequences from next generation sequencing against the template database to evaluate the availability of high sequence similarity templates. 1,000 models of each antibody in the benchmark set were generated by the multi-template protocol, RosettaAntibody, and AbPredict. The ten best models selected based on Rosetta energy score were compared to the native crystal structure to determine modeling quality. Finally, we outlined our new protocol for modeling antibodies in Rosetta and compared it to existing protocols to provide a guideline for modeling antibodies with Rosetta.
Results
Database of human antibodies in complex with their antigen
Many benchmarks on antibody modeling are conducted on unbound antibody structures and on antibodies from multiple species.22 However, in many real-world tasks for antibody modeling, the final models are docked to respective antigens. Furthermore, often only antibodies from human origin are of interest, especially in the development of human therapeutic antibodies. For this reason, we constructed a template database of diverse, high-quality human antibodies that were co-crystalized with their antigen structures publicly available in the Protein Data Bank (PDB). The resolution was better than 4.5 Å and redundant antibodies were removed from the template database. This dataset of 588 structures was processed by removal of crystallographic copies and relaxation with constraint backbone atoms to be used as template database for all following benchmark studies. The template database can be found in Table S1 of the Supporting Material.
Template availability for antibody sequences varies for every loop region
In order to understand the current status of template availability for a random given human antibody sequence, we used antibody sequences from the Human Immunome Program (HIP) database, which contains multiple immune repertoires and represents therefore an ideal test case.6 A BLAST21 search was conducted against a randomized set of almost 5 million heavy chain sequences and almost 2 million light chain sequences from the HIP database and the highest similarity match with our structure database was recorded. To further understand which regions of the antibody have the highest template availability, this analysis was broken down in each respective CDR, determined by the Chothia numbering scheme9 (Figure 1). This analysis revealed that the expected coverage of a human HCDR3 by a template in our human antibody structure database is between 50 and 70 percent. This finding contrasts with the HCDR1, HCDR2, LCDR1, and LCDR2, which have expected coverages of 80 percent or higher, and the LCDR3 with an expected coverage of 70 percent or higher.
Figure 1:
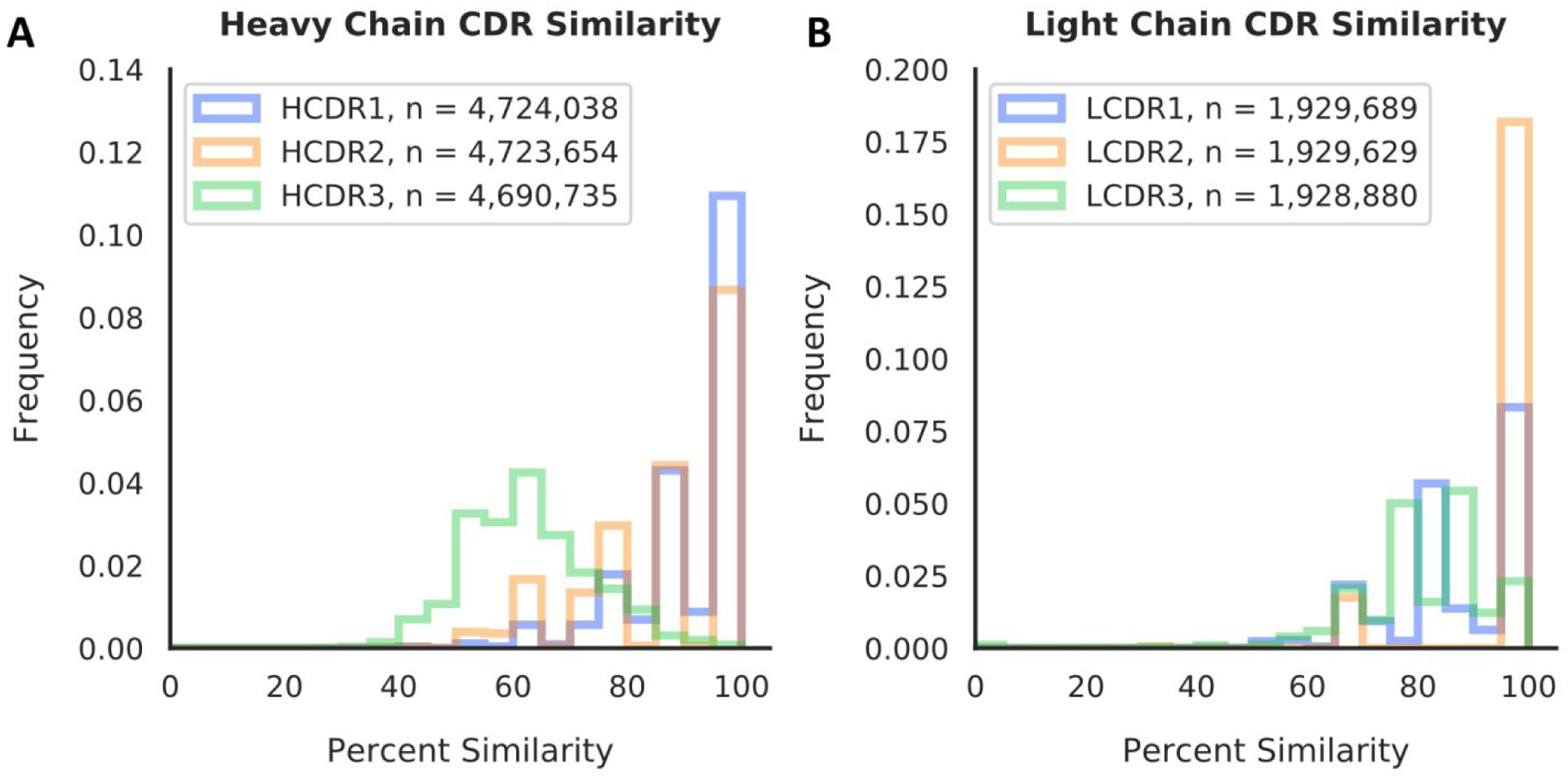
Sequence similarity between randomly chosen antibody sequences from the HIP database and our template database of human co-crystal structures. Over 4 million antibody heavy chains and almost 2 million antibody light chains were tested using a BLAST search and their highest percentage match was recorded for each CDR individually and binned in steps of 5 percent. A. distribution of template similarity for heavy chain CDRs. B. Distribution of template similarity for light chain CDRs.
The Rosetta software suite supports multiple protocols for homology modeling.
RosettaAntibody and AbPredict are two antibody-specific protocols. RosettaAntibody selects templates for HCDR1, HCDR2, LCDR1, LCDR2, and LCDR3 from a database of antibody structures and grafts these onto a template heavy and light chain framework based on their canonical loop conformations.10 It then models HCDR3 de novo and optimizes the heavy chain and light chain orientation.15,23 AbPredict, in contrast, selects templates for each of four regions: the heavy chain Fv (including HCDR1 and HCDR2), light chain Fv (including LCDR1 and LCDR2), the HCDR3, and the LCDR3 to mimic the method by which genomic recombination is used in the human body to produce a diversity of antibodies. Templates for the first four regions are grafted onto a reference structure, and a database is used to optimize the heavy chain-light chain orientation.24 Both RosettaAntibody and AbPredict allow selection of more than one template for each region, however they are recombined in a predetermined way. RosettaCM, the general framework for comparative modeling in Rosetta, assembles topologies from given sequence alignments through recombination of templated regions and the construction of untemplated regions de novo. A gap closure method is used to regularize junctions between segments. It can accept any number of input structures as templates and can be adapted for specific purposes like antibody modeling.19 In order to understand, which method allows for the best performance in the modeling of human antibodies in co-crystal structures and overcomes the lack of high sequence similarity templates for some regions of these antibodies, these methods need to be compared. A single template method in RosettaCM was chosen as a further control to estimate the gain of performance through availability of multiple templates. It used the structure with the overall highest sequence similarity as the only template. For RosettaCM a multi-template approach was chosen that collected five templates for each CDR and framework, however, the full antibody was used as template. The default template search was used for RosettaAntibody and AbPredict. As a control for the performance of all methods, the antibody modeled based on its own crystal structure using RosettaCM was used. The methods tested vary in how they select and use templates.
Antibody homology modeling methods were compared in a benchmark
As a benchmark set, 87 human antibodies from antibody-antigen co-crystal structures were collected to directly compare the quality of models produced by each antibody homology modeling method. As primary quality criterion, the backbone root mean square deviation (RMSD) was measured against the native antibody crystal structure. The full list of structures in the benchmark is provided in Table S2 in Supporting Materials. To simulate the modeling of human antibodies with unknown structures, we prevented each methodology from using the existing crystal structure as a template and chose the ten best models from each method using their Rosetta energy as selection criterion. The median RMSD of these ten models to the native antibody crystal structure was calculated and recorded over the entire antibody backbone as well as for each individual CDR (Figure 2 and Table 1).
Figure 2:
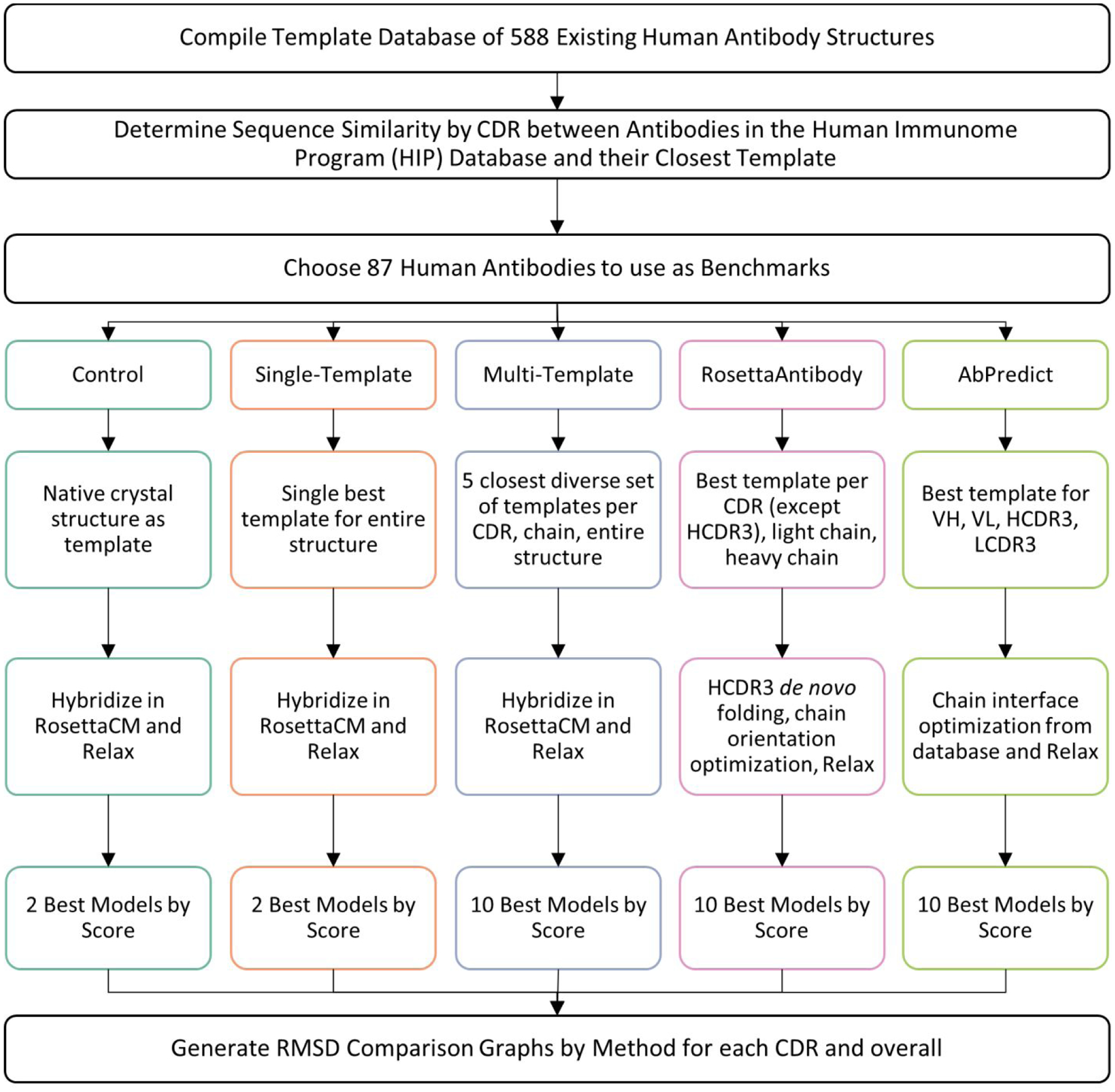
Workflow for benchmark set-up and protocol used to compare different antibody homology modeling methods in Rosetta.
Table 1:
Median backbone and HCDR3 RMSDs by method for each antibody tested from the benchmark set.
| PDB | Median Backbone RMSD to Native Crystal Structure of the Top 10 of Models by Energy Score | Median HCDR3 RMSD to Native Crystal Structure of the Top 10 of Models by Energy Score | HCDR3 Sequence Identity to the Closest HCDR3 Template | HCDR3 Length | ||||
|---|---|---|---|---|---|---|---|---|
| Multi-Template | Rosetta Antibody | AbPredict | Multi-Template | Rosetta Antibody | AbPredict | |||
| 1.25 | 2.41 | x† | 3.99 | x | x | 0.36 | ||
| 1.42 | 1.46 | 0.84 | 3.15 | 4.54 | 1.65 | 0.55 | ||
| 0.99 | 1.18 | 0.81 | 2.20 | 5.21 | 2.77 | 0.40 | ||
| 1.44 | 1.49 | 1.99 | 4.09 | 4.45 | 6.22 | 0.44 | ||
| 0.85 | 1.25 | 1.01 | 1.60 | 8.52 | 3.53 | 0.25 | ||
| 1.15 | 1.19 | 1.12 | 2.01 | 4.63 | 5.23 | 0.33 | ||
| 1.15 | 1.38 | 2.60 | 1.72 | 4.17 | 4.73 | 0.38 | ||
| 1.06 | x | 0.95 | 2.30 | x | 2.62 | 0.60 | ||
| 1.89 | 1.79 | 1.90 | 5.22 | 6.00 | 3.92 | 0.45 | ||
| 1.43 | 1.08 | x | 3.31 | 4.10 | x | 0.36 | ||
| 1.66 | 1.72 | 0.94 | 3.10 | 6.69 | 5.74 | 0.31 | ||
| 1.30 | 1.31 | x | 2.62 | 2.33 | x | 0.75 | ||
| 1.23 | 1.55 | 2.62 | 3.66 | 4.87 | 8.01 | 0.36 | ||
| 1.36 | 1.17 | 1.58 | 1.79 | 2.75 | 3.22 | 0.56 | ||
| 1.79 | 1.33 | 1.68 | 3.07 | 4.55 | 5.20 | 0.33 | ||
| 1.37 | 2.27 | 0.84 | 4.23 | 9.00 | 4.94 | 0.31 | ||
| 1.09 | 1.23 | 0.95 | 4.55 | 4.05 | 3.81 | 0.27 | ||
| 1.15 | 1.46 | 0.87 | 3.05 | 4.29 | 4.39 | 0.45 | ||
| 1.49 | 1.31 | x | 5.47 | 4.53 | x | 0.57 | ||
| 1.18 | 1.26 | 3.65 | 1.58 | 5.13 | 9.69 | 0.40 | ||
| 1.07 | 1.26 | 0.98 | 2.44 | 2.84 | 3.26 | 0.44 | ||
| 0.92 | 1.53 | x | 1.83 | 4.19 | x | 0.56 | ||
| 1.29 | 1.26 | 1.88 | 3.62 | 5.22 | 6.29 | 0.31 | ||
| 0.93 | 1.56 | 0.68 | 2.21 | 5.79 | 3.25 | 0.20 | ||
| 1.01 | 1.22 | 0.99 | 2.39 | 2.29 | 3.46 | 0.33 | ||
| 0.99 | 1.25 | 1.41 | 1.99 | 3.41 | 5.37 | 0.56 | ||
| 0.96 | 1.05 | 1.11 | 2.07 | 3.09 | 3.14 | 0.22 | ||
| 1.09 | x | x | 2.45 | x | x | 0.33 | ||
| 1.25 | 1.73 | 1.16 | 1.75 | 6.01 | 2.62 | 0.67 | ||
| 1.17 | 1.48 | x | 1.68 | 3.32 | x | 0.57 | ||
| 0.96 | 1.73 | 0.96 | 1.31 | 8.67 | 5.67 | 0.42 | ||
| 1.71 | 2.21 | 1.65 | 4.01 | 7.23 | 4.29 | 0.38 | ||
| 1.22 | 1.09 | 0.72 | 3.33 | 5.18 | 4.75 | 0.60 | ||
| 1.25 | 1.30 | x | 3.49 | 7.68 | x | 0.46 | ||
| 1.06 | 1.85 | 0.83 | 2.09 | 3.42 | 4.14 | 0.67 | ||
| 1.19 | 2.11 | 1.04 | 2.54 | 3.84 | 2.56 | 0.67 | ||
| 1.09 | 1.32 | 3.54 | 3.67 | 4.88 | 8.56 | 0.25 | ||
| 0.97 | 1.22 | 1.04 | 3.02 | 3.72 | 3.91 | 0.33 | ||
| 1.45 | 2.11 | 1.78 | 4.18 | 8.23 | 5.88 | 0.53 | ||
| 1.17 | 1.09 | 1.00 | 3.49 | 3.55 | 3.82 | 0.42 | ||
| 1.69 | 1.71 | x | 2.13 | 3.24 | x | 0.56 | ||
| 1.24 | 1.30 | 0.93 | 4.83 | 4.20 | 5.24 | 0.40 | ||
| 1.11 | 1.36 | 1.02 | 2.74 | 3.66 | 3.38 | 0.50 | ||
| 1.50 | 1.77 | 1.01 | 4.39 | 9.42 | 4.46 | 0.15 | ||
| 1.03 | 1.44 | 1.13 | 1.96 | 3.69 | 2.90 | 0.50 | ||
| 1.06 | x | 1.61 | 2.81 | x | 5.43 | 0.36 | ||
| 1.32 | 1.18 | 1.05 | 4.15 | 6.20 | 6.28 | 0.18 | ||
| 1.28 | 1.48 | 1.08 | 2.55 | 4.39 | 3.82 | 0.45 | ||
| 1.59 | 1.81 | x | 7.54 | 5.37 | x | 0.47 | ||
| 1.27 | 1.70 | 1.12 | 4.15 | 7.59 | 4.45 | 0.62 | ||
| 1.05 | x | 0.94 | 2.08 | x | 7.24 | 0.40 | ||
| 1.25 | 1.70 | 0.84 | 1.61 | 3.56 | 5.08 | 0.71 | ||
| 0.77 | 1.19 | 0.84 | 1.41 | 2.74 | 3.70 | 0.38 | ||
| 1.34 | 1.32 | 0.83 | 5.25 | 4.01 | 5.92 | 0.58 | ||
| 1.41 | x | 0.85 | 6.98 | x | 6.21 | 0.38 | ||
| 1.07 | 1.35 | 1.25 | 2.44 | 6.08 | 3.71 | 0.36 | ||
| 1.17 | 1.10 | 3.58 | 1.92 | 2.30 | 11.34 | 0.38 | ||
| 0.96 | 0.99 | 0.81 | 1.79 | 6.34 | 3.16 | 0.60 | ||
| 1.54 | 2.01 | 1.83 | 1.48 | 4.45 | 5.01 | 0.36 | ||
| 1.03 | 2.16 | 0.81 | 3.20 | 2.98 | 4.12 | 0.11 | ||
| 1.49 | 1.41 | 0.92 | 3.24 | 6.76 | 4.25 | 0.36 | ||
| 1.07 | 1.18 | 0.92 | 2.47 | 5.92 | 3.97 | 0.42 | ||
| 1.60 | 1.13 | 1.24 | 4.95 | 3.89 | 3.26 | 0.45 | ||
| 1.23 | 1.79 | 1.29 | 2.23 | 8.28 | 3.45 | 0.40 | ||
| 1.28 | x | 1.00 | 3.30 | x | 5.56 | 0.57 | ||
| 1.26 | 1.49 | x | 4.98 | 6.16 | x | 0.45 | ||
| 1.08 | 1.19 | x | 5.41 | 7.01 | x | 0.50 | ||
| 0.92 | x | 1.13 | 0.85 | x | 4.30 | 0.57 | ||
| 1.31 | 1.50 | 1.16 | 5.48 | 4.68 | 4.70 | 0.33 | ||
| 1.26 | 1.67 | 0.81 | 4.45 | 8.45 | 5.67 | 0.42 | ||
| 2.09 | 1.77 | x | 4.19 | 5.33 | x | 0.62 | ||
| 1.16 | 1.13 | 1.09 | 5.01 | 8.18 | 4.28 | 0.33 | ||
| 1.06 | 0.94 | 1.20 | 1.80 | 2.48 | 4.78 | 0.57 | ||
| 1.65 | x | 2.07 | 6.42 | x | 7.83 | 0.38 | ||
| 1.27 | 1.77 | 0.91 | 2.82 | 3.36 | 3.44 | 0.50 | ||
| 0.93 | 1.62 | 1.89 | 2.72 | 5.22 | 4.05 | 0.33 | ||
| 1.61 | 3.41 | 1.49 | 2.91 | x | 5.78 | 0.25 | ||
| 1.10 | 1.42 | 0.88 | 7.60 | 5.12 | 6.00 | 0.20 | ||
| 0.98 | 1.14 | x | 3.00 | 4.08 | x | 0.33 | ||
| 1.81 | 1.84 | 1.32 | 2.78 | 3.69 | 4.86 | 0.50 | ||
| 1.40 | 1.97 | 1.18 | 4.23 | 6.63 | 4.92 | 0.38 | ||
| 1.21 | 1.87 | 2.17 | 2.28 | 10.10 | 6.48 | 0.31 | ||
| 1.84 | 2.02 | 3.94 | 7.28 | 7.59 | 10.92 | 0.27 | ||
| 0.94 | 1.41 | 0.98 | 1.64 | 5.21 | 3.78 | 0.44 | ||
| 1.33 | 1.44 | 1.57 | 3.02 | 4.41 | 3.75 | 0.45 | ||
| 1.31 | 1.52 | 1.08 | 3.30 | 7.86 | 4.27 | 0.29 | ||
X indicates a failure by the method represented to produce any models of sufficient quality
Multi-template modeling in RosettaCM matched the performance of RosettaAntibody and AbPredict for modeling the backbone
Backbone quality as defined by the backbone RMSD was measured to compare the overall antibody structure of the models generated by the methods tested to their respective crystal structure. The entire backbone of the ten best generated models by each method for each antibody in the benchmark set was analyzed. We found the control method to have a median RMSD of 0.56 Å, so an RMSD difference of 0.5 Å was chosen as the minimum difference to indicate a significant improvement between methods.
Multi-template modeling using RosettaCM was found to outperform RosettaAntibody for 15 of the 87 human antibodies tested. In addition, RosettaAntibody failed to model eight of the human antibodies in our benchmark set and never outperformed the multi-template method. Multi-template modeling outperformed AbPredict in modeling the backbone for eleven of the 87 human antibodies tested, with AbPredict failing to model an additional 14 human antibodies in our benchmark set. AbPredict outperformed multi-templating modeling for seven human antibodies in our benchmark set. RosettaAntibody outperformed AbPredict in modeling the entire backbone for six of the 87 human antibodies tested, while AbPredict outperformed RosettaAntibody for 19 of the human antibodies in our benchmark set (Figure 3). AbPredict modeled 29 antibodies with a backbone RMSD between 0.5 and 1 Å, an improvement over both the multi-template method (14 antibodies) and RosettaAntibody (two antibodies, compare SI Figure S6). However, the multi-template method modeled 60 antibodies with a backbone RMSD between 1.0 and 1.5 Å, besting both AbPredict (25 antibodies) and RosettaAntibody (45 antibodies). Therefore, the multi-template method modeled more antibodies at a backbone RMSD of less than 1.5 Å than AbPredict and RosettaAntibody. Conversely, the multi-template method only modeled one antibody at a backbone RMSD of greater than 2.0 Å, compared to nine antibodies for RosettaAntibody and eight antibodies for AbPredict (Figure 4). Therefore, the multi-template method more consistently produced models at a backbone RMSD of less than 2 Å to the native crystal structure when compared to both AbPredict and RosettaAntibody. For HCDR1 and HCDR2 as well as all three LCDRs, the multi-template method offered minor improvements in loop modeling performance compared to both AbPredict and RosettaAntibody, the results of which can be seen in Figures S1–S5 in the Supporting Material.
Figure 3:
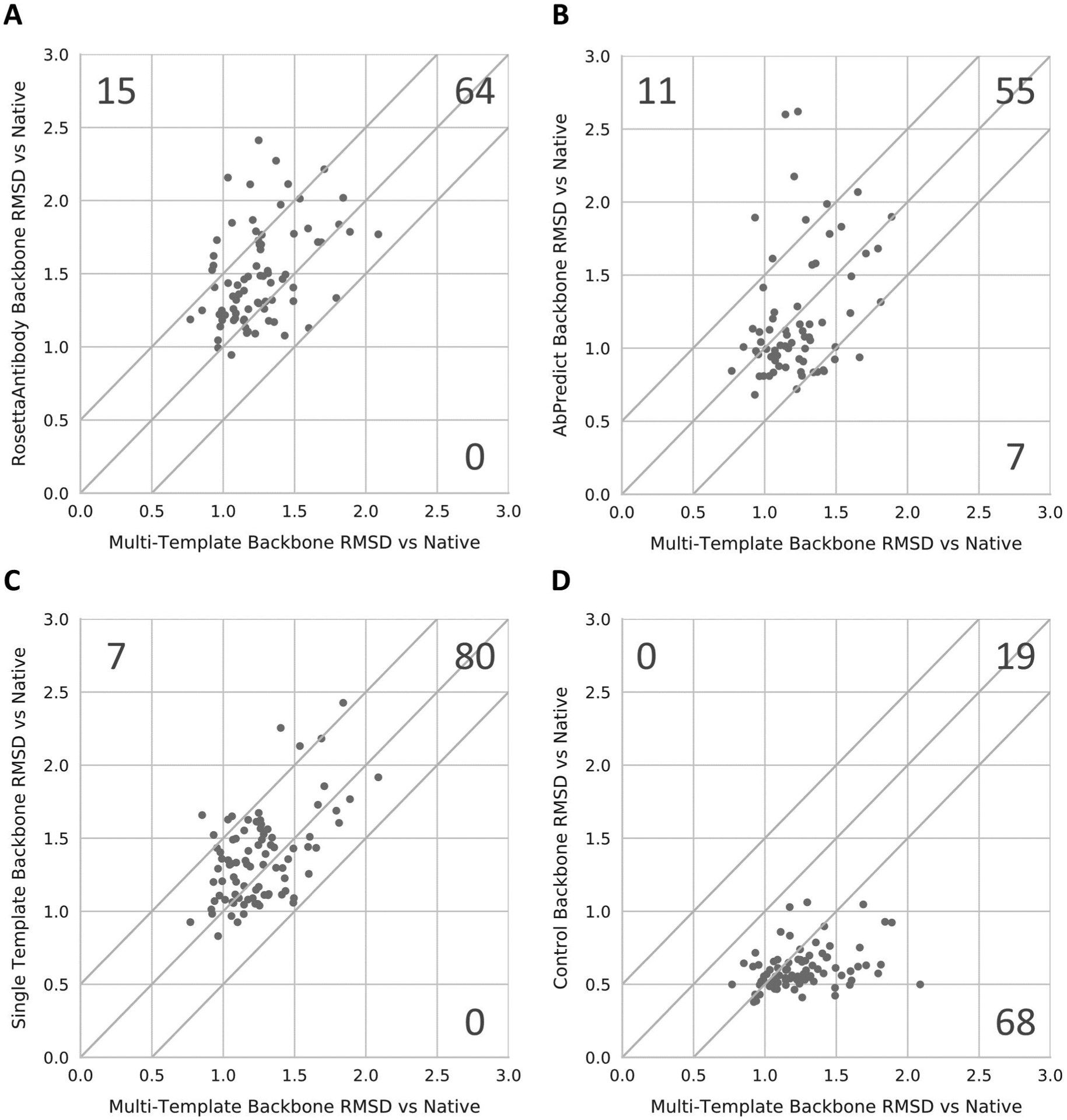
Comparison of antibody backbone modeling performance for the best models produced by each method to the best multi-template models. The performance of RosettaAntibody (A), AbPredict (B), Single Template (C), and our control method (D) were each individually compared to our multi-template method by graphing that method’s median RMSD against our multi-template method’s median RMSD for each antibody. Each chart has a line at y=x representing equal performance between methods. Points above the line represent cases where our multi-template method outperformed another method, whereas points below the line represent the opposite case.
Figure 4:
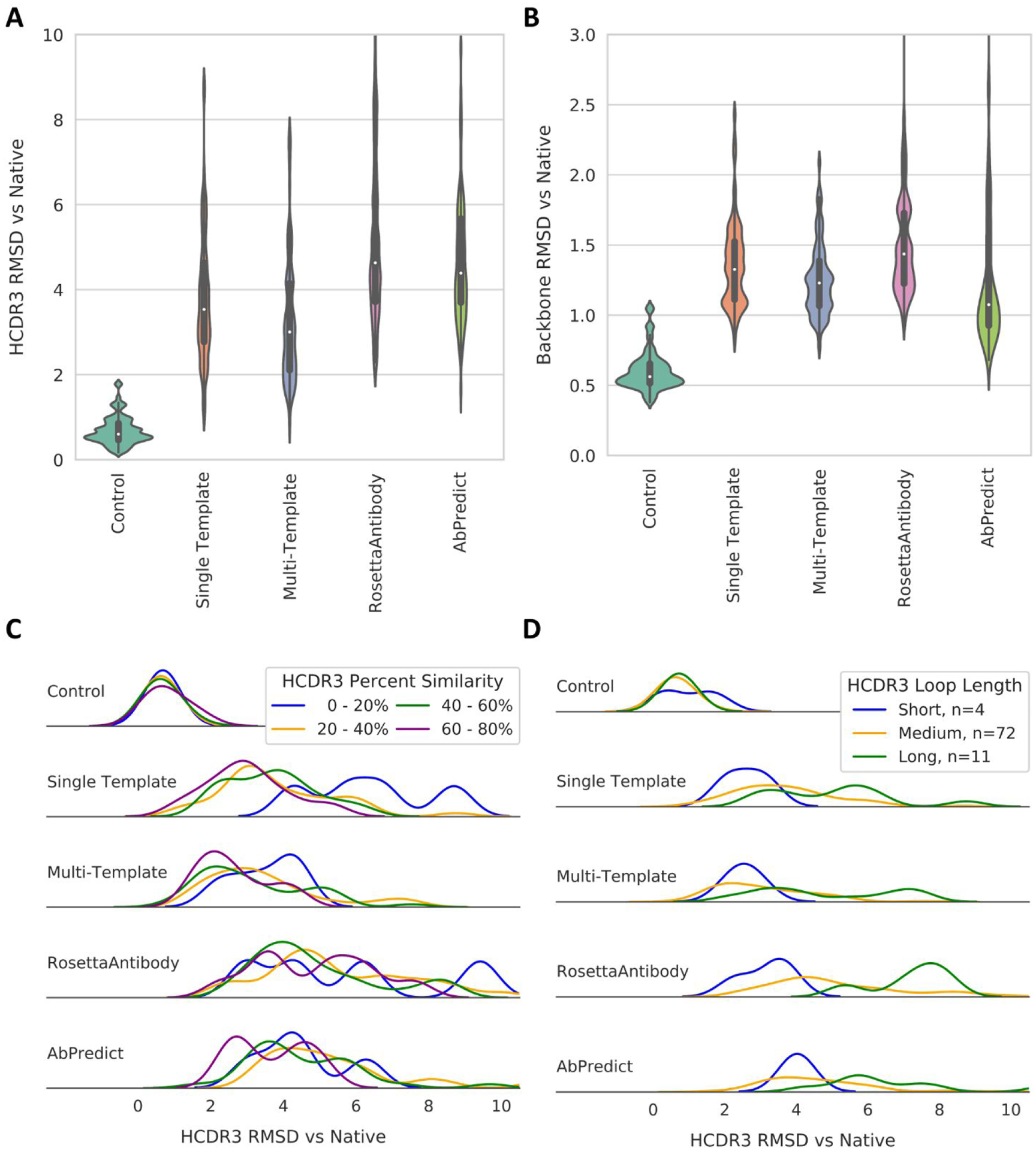
Comparison of antibody HCDR3 and backbone modeling performance for the best models produced by each method. 1000 models of each benchmark antibody were produced using the Multi-Template, Rosetta Antibody, and AbPredict protocols, and 200 models using the Single Template and Control protocols. All generated models were aligned to their respective native crystal structures and backbone RMSD was calculated between the HCDR3 loop of the native crystal structure and each corresponding model. The median RMSD for models of each benchmark antibody in the top 1% by score in Rosetta Energy Units for each method were recorded (A). The same technique was used to calculate RMSD of the backbone to the native structure (B). These models were also binned by HCDR3 sequence identity to the best template (C) and loop length (D). Short represents HCDR3 loop lengths of 1–6 amino acids (AA), medium of lengths 7 to 13 AA, and long of lengths 14 to 20 AA.
Multi-template modeling using RosettaCM modeled the HCDR3 loop more accurately than RosettaAntibody and AbPredict in most cases
HCDR3 as defined by the backbone RMSD was measured to compare the HCDR3 structure of the models generated by the methods tested to their respective native crystal structure. Analysis of the ten best models by total score for each method showed that multi-template modeling using RosettaCM produced models with a lower median HCDR3 RMSD to the native crystal structures than RosettaAntibody for 53 of the 87 human antibodies tested in our benchmark, defined as an RMSD improvement of at least 1 Å. In addition, RosettaAntibody failed to adequately model the HCDR3 loop, defined as an RMSD > 15 Å or an error thrown by the protocol that prevented any models from being generated, for ten of the human antibodies tested in our benchmark. Examples of errors thrown by RosettaAntibody included errors in grafting and incompatibility between loop flexibility and length. RosettaAntibody outperformed the multi-template method in modeling the HCDR3 loop for four of the human antibodies in our benchmark. Similarly, multi-template modeling outperformed AbPredict in modeling the HCDR3 loop for 44 of the human antibodies tested in our benchmark, with AbPredict failing to adequately model the HCDR3 loop for an additional 14 antibodies, mostly due to the lack of suitable templates with the same loop length. AbPredict outperformed the multi-template method in modeling the HCDR3 loop for four of the human antibodies in our benchmark (Figure 5). None of the RosettaCM-based methods, including multi-template modeling, ever failed to model the HCDR3 loop. The multi-template method modeled the HCDR3 loop at an RMSD of 0 to 2.5 Å for 34 antibodies in the benchmark set, a large improvement over both RosettaAntibody (four antibodies) and AbPredict (one antibody). In addition, the multi-template method modeled the HCDR3 loop of only eleven antibodies at an RMSD greater than 5 Å, fewer than both RosettaAntibody (35 antibodies) or AbPredict (27 antibodies).
Figure 5:
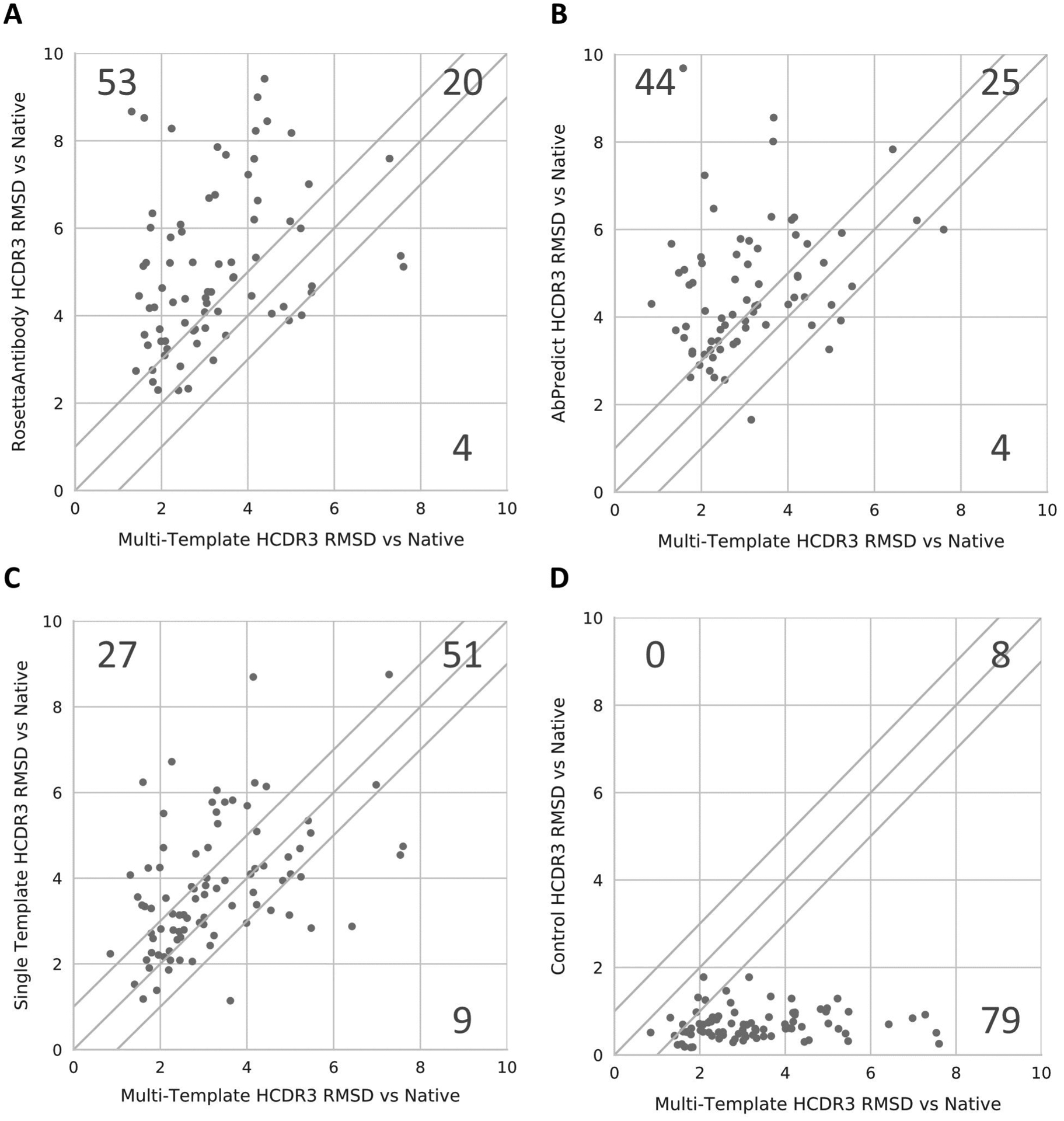
Comparison of antibody HCDR3 modeling performance for the best models produced by each method to the best multi-template models. The performance of RosettaAntibody (A), AbPredict (B), Single Template (C), and our control method (D) were each individually compared to our multi-template method by graphing that method’s median RMSD against our multi-template method’s median RMSD for each antibody. Each chart has a line at y=x representing equal performance between methods. Points above the line represent cases where our multi-template method outperformed another method, whereas points below the line represent the opposite case.
Multi-template modeling using RosettaCM showed the greatest improvement in HCDR3 loop modeling for longer loops and loops with lower template sequence identity
The performance of the different modeling methods also was analyzed based on HCDR3 loop length, categorized as short (1 to 6 amino acids in length), medium (7 to 13 amino acids in length), and long (14 to 20 amino acids in length). Of the 87 antibodies in the benchmark set, four were categorized as having short HCDR3 loops, 72 as having medium-length loops, and eleven as having long HCDR3 loops. For every method excluding the control, modeling performance decreased as loop length increased. For the multi-template method, median RMSD to the native structure for short HCDR3 loops was 2.5 Å, 2.8 Å for medium-length loops, and increased to 4.2 Å for long loops. AbPredict experienced a similar degradation in HCDR3 modeling performance with increasing loop length with a median RMSD to the native structure of 4.0 Å for short HCDR3 loops, 4.3 Å for medium-length loops, and 6.0 Å for long HCDR3 loops. The performance of RosettaAntibody saw an even greater amount of performance degradation for modeling the HCDR3 loop with increasing loop length. Median RMSD to the native structure was 3.4 Å for short HCDR3 loops, increasing to 4.5 Å for medium-length loops and 7.6 Å for long HCDR3 loops. Meanwhile, HCDR3 modeling performance for the control method saw little variation at different loop lengths with a median HCDR3 RMSD to the native structure of 0.9 Å for short loops, 0.6 Å for medium-length loops, and 0.7 Å for long loops.
HCDR3 modeling performance of the different methods was also assessed at different levels of template similarity, defined as the highest HCDR3 template sequency identity of any template used by the multi-template method to guide antibody structure prediction. The modeling performance of the multi-template method improved with increasing HCDR3 template sequence identity. Between 0 and 20 percent template sequence identity, median HCDR3 RMSD to the native structure was 3.7 Å, decreasing to 3.1 Å between 20 and 40 percent, 2.8 Å between 40 and 60 percent, and 2.4 Å between 60 and 80 percent. AbPredict also experienced a similar trend, with a median HCDR3 RMSD of 4.3 Å between 0 and 20 percent template sequence identity, 4.9 Å between 20 and 40 percent, 4.0 Å between 40 and 60 percent, and 3.7 Å between 60 and 80 percent. However, RosettaAntibody exhibited little variation in performance with differing HCDR3 template sequence identity, with the best performance at 40 to 60 percent template sequence identity (4.4 Å median HCDR3 RMSD) and the worst at 0 to 20 percent template sequence identity (5.3 Å median HCDR3 RMSD), as seen in Figure 4.
Specific examples from benchmarking revealed method-dependent advantages and disadvantages in HCDR3 modeling
Examples were investigated for cases where the multi-template approach significantly outperformed both RosettaAntibody and AbPredict in modeling the HCDR3 loop and for cases where either RosettaAntibody and AbPredict significantly outperformed the multi-template approach in modeling the HCDR3 loop (Figure 6). For each example, the ten best models by score for the methods concerned were aligned onto the native crystal structure of the antibody to help visually determine the modes of success and failure of the different methods in modeling the HCDR3 loop. In the case of the Peptostreptococcus magnus protein L antibody (PDB: 1HEZ)25, each of the top ten models produced by the multi-template method closely matched the HCDR3 loop of the native crystal structure. Half of the top ten models produced by AbPredict approximated the general shape of the HCDR3 loop, while the other half deviated more significantly from the native crystal structure, suggesting a lesser degree of consistency among models produced by AbPredict when compared to the multi-template method. None of the top ten models produced by Rosetta Antibody approximated the general shape of the HCDR3 loop and displayed a greater degree of variability amongst themselves when compared to both the multi-template method and AbPredict. These observations remained when investigating examples in which either AbPredict or RosettaAntibody significantly outperformed the multi-template method. AbPredict significantly outperformed the multi-template method in modeling an anti-HIV-1 capsid protein antibody (PDB: 1AFV)26, but the multi-template method showed a greater structural consistency among the top ten models by score whereas some of the top AbPredict models had additional kinks not present in the other models. The even greater degree of variability present among RosettaAntibody models continued in the modeling of an Zika virus envelope protein-specific antibody (PDB:5KVF)27, where although the top ten models by score had a lower RMSD to the native crystal structure than the multi-template models, none of the models by either method closely approximated the shape of the native HCDR3 loop. The multi-template models once again showed a great degree of structural consistency while none of the RosettaAntibody models shared a similar loop shape. The greater degree of structural inconsistency amongst the top models of RosettaAntibody when compared to AbPredict was also shown in the modeling of an human vascular endothelial growth factor-binding antibody (PDB: 2QR028), where AbPredict significantly outperformed RosettaAntibody in modeling the HCDR3 loops. The top ten models produced by AbPredict each approximated the shape of the native HCDR3 loop, while none of the top models produced by RosettaAntibody approximated the shape of the native HCDR3 loop and exhibited a great degree of structural inconsistency amongst themselves.
Figure 6:
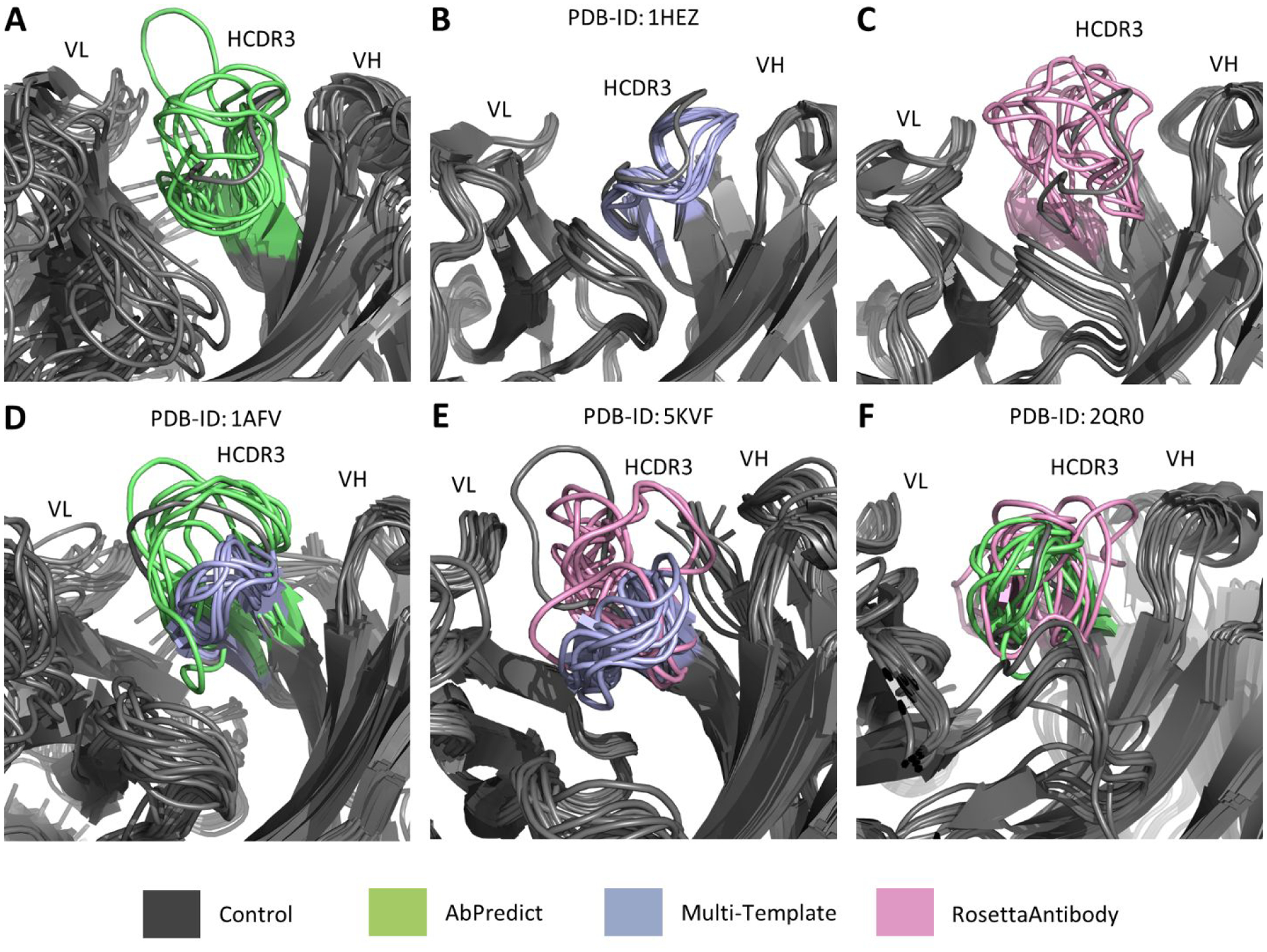
Examples of significant differences in HCDR3 modeling performance between multi-template modeling and AbPredict/RosettaAntibody. The top 10 models by score in Rosetta Energy Units for the two methods being compared in each example were aligned onto the corresponding antibody’s relaxed native crystal structure (grey). Our multi-template modeling method, displayed in purple (B), significantly outperformed AbPredict, displayed in green (A) and RosettaAntibody, displayed in pink (C), in modeling the HCDR3 loop of a Peptostreptococcus magnus protein L-binding human antibody (PDB ID 1HEZ)25. AbPredict only significantly outperformed our multi-template method in modeling the HCDR3 loop (D) when modeling an HIV-1 capsid protein-binding human antibody (PDB ID 1AFV)26. RosettaAntibody significantly outperformed our multi-template method in modeling the HCDR3 loop (E) when modeling the Zika virus envelope protein-binding human antibody (PDB ID 5KVF)27. AbPredict significantly outperformed RosettaAntibody in modeling the HCDR3 loop (F) when modeling a human vascular endothelial growth factor-binding antibody (PDB ID 2QR0)28.
Discussion
In summary, we constructed a database of human antibody structures to use as templates for antibody modeling and we compared the coverage of these templates against a large database of human antibody sequences. We modeled 87 human antibodies with known structures using the Rosetta template-based antibody modeling techniques RosettaAntibody and AbPredict as well as three methods of our own design based on RosettaCM (multi-template modeling, single template modeling, and a control method). We selected the best models produced by each method for each test antibody using their Rosetta energy score and we compared these models to their corresponding native crystal structures by calculating the backbone RMSD over the entire variable region as well as each CDR to their native crystal structure. We found that our multi-template method had similar performance to AbPredict and our single-template method when modeling the entire variable region backbone and offered minor improvements in backbone quality over RosettaAntibody. In addition, both our single template and multi-template methods never failed to model the entire antibody backbone as opposed to AbPredict and RosettaAntibody, both of which had errors in CDR identification and template selection. Our multi-template method offered significant improvements over AbPredict and RosettaAntibody in modeling HCDR3 in model quality, reliability, and robustness.
Many assessments of antibody modeling in the past, have mostly been evaluating apo-structures of antibodies. We asked if these methods are capable of sampling the bound conformation of an antibody. Therefore, we decided to only use antibodies as templates and in our benchmark set that were derived from co-crystal structures of antibodies with their respective antigens. One rationale behind that is that apo-crystal structures sometimes contain artificial contacts or conformations for the loops, dependent whether the loop form crystal contacts in the interfaces.29
One important question we considered was whether or not we were warranted in using multiple homology templates for each CDR. We compared sequences in our template database to sequences in the HIP database to test this hypothesis and found that HCDR3 likely had the most to gain from the use of multiple templates, as indicated by the expected template coverage of 50 to 70%. Although other CDRs had higher expected template coverage, a distribution in sequence coverage was observed for all CDRs, suggesting that multiple templates for each region could help for more unique CDR sequences. This observation prompted the design of our multi-template method using multiple templates for each CDR region.
The performance of AbPredict in modeling the entire variable region backbone was comparable to our multi-template method, an interesting result given the large difference between the two protocols in modeling each of the CDRs. The modeling approach used by AbPredict helps explain this observation, as a single template is used for the entire heavy chain excluding HCDR3 and a second template is used for the entire light chain excluding LCDR3. The grafted intermediate is optimized using a Monte-Carlo search over all conformational degrees of freedom for minimization of strain over the side chain and backbone,18,30 representing a similar technique as that used by RosettaCM on which the single template and multi-template methods are based. This finding matches the observations outlined by the developers of AbPredict describing a deviation of less than 1.2 Å for the top ranked models over backbone-carbonyl atoms.18 RosettaCM’s selection of the backbone was only guided by the weight of the template in the protocol based on sequence similarity, but it cannot be ruled out that similar template stretches were selected during the modeling process.
Meanwhile, the larger difference in the modeling performance by RosettaAntibody compared to the multi-template method over the entire variable region backbone can likewise be explained by RosettaAntibody’s modeling approach. Each CDR (expect for HCDR3) is grafted onto a single heavy chain and a single light chain template. Following this step, the only optimizations over the entire backbone are removal of grafting anomalies, optimization of the heavy chain/light chain orientation, and the final all-atom refinement step with coordinate constraints to backbone atoms in RosettaRelax.16 Since it lacks the Monte-Carlo-based all-atom search for minimization shared by AbPredict and the RosettaCM-based methods, RosettaAntibody has a greater dependence on backbone template similarity over the entire heavy chain and light chain. Given the diversity of human antibodies tested in our benchmark, it is unlikely that every antibody had a close template match for the entire heavy chain and light chain regions. This approach helps demonstrate the greater versatility of the other methods in adopting a Monte-Carlo-based all-atom minimization search.
The multi-template method saw significant improvement in HCDR3 modeling performance over AbPredict. This enhanced performance likely can be attributed to both the template selection and subsequent modeling approach used by AbPredict when compared to the multi-template method. While the multi-template selects five templates for the HCDR3 loop, optimizing for sequence similarity and diversity, AbPredict randomly selects all available HCDR3 fragments with similar loop length for use as templates. Owing to the underlying RosettaCM framework, the multi-template method is able to choose structural information from all five of its HCDR3 templates simultaneously, as well as any other templates selected for other use in other regions, to guide modeling, as well as incorporating information from templates used elsewhere in the antibody and fragment libraries, for all 1,000 models produced. In contrast, AbPredict proceeds by random recombination of each HCDR3 template with three templates used for the other regions of the antibody variable domain. Given the same 1,000 models, AbPredict must pursue fewer modeling trajectories for each random recombinational template pairing.18 Consequently, the best models produced by AbPredict show a greater variation amongst themselves when compared to the multi-template method and these models have fewer opportunities to approach the global energy minima. However, this can be overcome by producing higher number of models.
RosettaAntibody adopts an almost exact opposite approach to modeling the HCDR3 loop as AbPredict, yet this also contributes to its inferior HCDR3 modeling performance when compared to the multi-template method for the majority of antibodies in our benchmark set. RosettaAntibody exclusively uses de novo modeling for the HCDR3 loop. The HCDR3 loop is modeled with simultaneous optimization of the heavy chain/light chain orientation.16 The advantages and disadvantages of this approach can be seen in the results, as RosettaAntibody approaches the HCDR3 modeling performance of the multi-template method for short loops. However, as loop length increases, RosettaAntibody exhibited the greatest performance degradation of any modeling method tested. This loss of performance can be expected since the number of possible conformations increases with loop length in accordance with Levinthal’s paradox,31 but the number of explored conformations remains constant to produce 1,000 models, so the correct conformation has a lower probability of being sampled. Although the multi-template method also experienced performance degradation with increasing loop length, the template-based approach helps reduce the conformational search space, affording the multi-template method a higher probability of sampling the correct conformation when compared to RosettaAntibody. RosettaAntibody did model the HCDR3 loop of 4 antibodies at a lower median RMSD than our multi-template method. In each case, the median RMSD for RosettaAntibody was greater than 3.5 Å and showed much greater variation amongst themselves compared to the multi-template method, suggesting that the multi-template method was biased towards a set of conformations that did not allow it to sample near the correct conformation.
Despite the template-dependent nature of the multi-template method, although increasing template sequence identity improved HCDR3 model quality, the multi-template method offered significant improvements over both AbPredict and RosettaAntibody at even the lowest band of sequence identity (0% to 20%). However, the approach adopted by the multi-template method prevents it from sampling as large of a conformational space as the other two methods. Therefore, if a template of high sequence identity is present but adopts a different conformation than the antibody being modeled, the multi-template method can be biased to produce inaccurate models.
Conclusions
Antibody modeling is still challenging despite the availability of many methods even in the same software framework, such as Rosetta. For the modeling of human antibodies from co-crystal structures we constructed a benchmark to compare available methods in Rosetta. A multi-template method based on RosettaCM adds to the already available toolset for antibody structure prediction, RosettaAntibody and AbPredict, especially when approaching the structurally diverse and difficult to model HCDR3 loop. This advantage is especially present for longer HCDR3 loops and structures with little to no template availability, where the recombination of sparsely available templates together with fragment insertion offer an improvement in contrast to the choice of a single template or pure de novo modeling. For antibodies with shorter HCDR3 loops and when high-similarity templates are available, all methods tested performed comparably well. Noteworthy, is the performance of AbPredict for the modeling of the overall backbone. The choice of method for antibody modeling tasks heavily depends on the available template and input structures. RosettaAntibody has been well-tested in the past, contains in-depth documentation and performs well if the antibody to be modeled has a good template match in the canonical structural clusters as defined by North et al.23 In cases, where there are less templates available a combinatory method such as RosettaCM offers advantages in the overall modeling performance. However, when the chosen templates contain no useful template information, the method will not overcome the lack of a template through conformational sampling. RosettaCM has been well-documented in the past and tutorials are available.19 AbPredict, despite performing well in the modeling of the backbone, heavily depends on the HCDR3 template, which when sampling the correct conformation performs well, otherwise the deviation is high. AbPredict was less robust in our hands. Overall, the choice of method for the modeling of a human antibody, as observed in antibody-antigen co-crystal structures, depends on the template availability. In some instances, obtaining models from multiple protocols as a consensus approach might be in order.
Methods
Construction of a template database of human antibody structures from the PDB
Antibodies with complete heavy chain and light chain sequences without chain breaks in the variable the domains were chosen from the PDB.32 If multiple structures were found in the PDB with identical heavy chain and light chain sequences, the structure with the lowest resolution was chosen for the database. Structures were renumbered and trimmed to the variable heavy chain/light chain pair. The trimmed structure underwent all-atom refinement with coordinate constraints on the backbone atoms using RosettaRelax33. A table of all antibody template structures can be found in Supporting Table S1 and further material is available for download under: https://github.com/pranavkodali/RosettaCMAntibodyBenchmark/tree/main.
Construction of a benchmark set of human antibodies from the PDB
The structural antibody database (SAbDab)34 was used to provide information on all human antibodies in the PDB. A list of every human antibody in complex with a protein antigen was compiled. The list was refined to only include non-engineered structures obtained by X-ray diffraction with a resolution of 3.5 Å or better. The 87 antibodies in the PDB that satisfied these requirements were selected as the test set. A table of all antibody-antigen structures can be found in Supporting Table S2.
Comparison of an immune repertoire to available structural templates
An antibody heavy chain sequence was randomly selected from the Human Immunome Program (HIP) Database35. The three heavy chain CDRs were identified from sequence. Blast36 identified human antibodies in the PDB with the highest sequence identity to each CDR and results were filtered to only include structures in our database of human antibody-antigen structures. The highest sequence identity was recorded, and the process was repeated more than four million heavy chain sequences. The process was also repeated almost two million times for antibody light chain sequences in the HIP database but modified to identify and run Blast against the three light chain CDRs for each sequence. The heavy chain and light chain data were graphed separately as histograms in Seaborn 0.9.1 with sequence identity binned at five percent intervals.
Antibody structure prediction using RosettaCM
The sequence of the target antibody was downloaded from the PDB.32 A script derived from RosettaAntibody was used to identify the three heavy chain CDRs and the three light chain CDRs from the sequence23. Blast36 identified human antibodies in the PDB with the highest sequence identity to each CDR, heavy chain, light chain, and entire antibody, and results were filtered to only include structures in our template database of human antibody structures with the structure corresponding to the input sequence excluded from the database. The fifteen best matches for each region were narrowed down to the five templates that maximized coverage of each region using ClustalW37 and repeated templates between regions were for a total of 28 templates. Each template file was aligned individually with target sequence in ClustalO38. Each template structure underwent all-atom refinement using RosettaRelax with coordinate constraints for the backbone atoms33. The sequence of the target antibody was threaded onto each relaxed template structure in RosettaCM19. Each template was assigned a weight in the hybridize mover corresponding to sequence identity with the target identity to amplify the influence of more similar templates. 1000 models were generated using RosettaCM19 and underwent all-atom refinement with coordinate constraints to backbone coordinates in RosettaRelax33. A final ensemble of 10 models were selected based on Rosetta all-atom score. This process was repeated for every antibody in the benchmark set.
Antibody structure prediction using RosettaAntibody
The sequence of the target antibody was downloaded from the PDB.32 The sequence was passed to RosettaAntibody23, and the protocol was executed in accordance with the published RosettaAntibody Protocol with several notable changes23: RosettaAntibody was recompiled with an additional flag to allow for the exclusion of the structure corresponding to the input sequence from template selection. The first grafted model was used as a starting point for 1,000 refined models as described in the original protocol, but additional grafted models were not used to match the number of models generated by the RosettaCM protocol. An additional all-atom refinement step with coordinate constraints to backbone atoms in RosettaRelax33 was added at the end of the RosettaAntibody Protocol for the 1,000 generated refined models. A final ensemble of 10 models were selected based on Rosetta all-atom score. This process was repeated for every antibody in the benchmark set.
Antibody structure prediction with AbPredict
The sequence of the target antibody was downloaded from the PDB.32 The sequence was formatted in accordance with the published AbPredict protocol18,39. VL, VH, LCDR3, and HCDR3 lengths were determined in accordance with the published AbPredict protocol and used to select fragments of the appropriate length from a database containing templates for each VL, VH, LCDR3 and HCDR3 length.39 A anti-meningococcal subtype P1.4 PorA antibody (PDB: 2BRR)40 is used by the protocol as backbone of the model assembly, followed by a random combination of the VL, VH, LCDR3 and HCDR3 backbone fragments. The protocol threads the target sequence onto this arbitrary backbone and executes a simulated-annealing Monte Carlo search. The process was done 40 times for each of 25 fragment combinations. The obtained models underwent all-atom refinement with coordinate constraints to backbone atoms in RosettaRelax33. A final ensemble of 10 models were selected based on Rosetta all-atom score. This process was repeated for every antibody in the benchmark set.
Antibody structure prediction using a single template
The RosettaCM protocol was followed with the following changes. Blast identified the human antibody in the PDB with the highest sequence identity to the entire antibody to be used as the template, and results were filtered as described for RosettaCM. 200 models were generated using RosettaCM19 and underwent all-atom refinement with coordinate constraints to backbone coordinates in RosettaRelax33. A final ensemble of two models were selected based on Rosetta all-atom score. This process was repeated for every antibody in the benchmark set.
Modeling the antibody structure based on its own crystal structure serves as positive control
The single template protocol was executed with the following change: The structure of the target antibody was downloaded from the PDB and used as the template and the sequence derived from the structure was aligned with the sequence of the target sequence. This process was repeated for every antibody in the benchmark set.
Data analysis
The native structure of the target antibody was downloaded from the PDB32 and trimmed to the to the variable heavy chain/light chain pair. The trimmed structure underwent all-atom refinement with coordinate constraints to backbone atoms in RosettaRelax33 The three heavy chain CDRs and the three light chain CDRs were identified from sequence with an adapted script from the RosettaAntibody framework23. The relaxed native structure and each relaxed generated model were aligned and the RMSD between the two overall structures was calculated in PyMol 2.3.4. The findseq PyMOL script was used to identify each CDR and calculate the RMSD of the CDR between the relaxed native structure and each relaxed generated model. Score versus RMSD plots were generated in Seaborn 0.9.1 for each antibody in the test set and plots representing results from the entire test set.
Example scripts and commands for all executed protocols can be found in the Supporting Material.
Supplementary Material
Acknowledgement
We thank Dr. Nina G. Bozhanova and Dr. Amandeep K. Sangha for help with the construction of the antibody template database.
Funding Sources
This work was supported by a grant from the National Institute of Health (NIH) (R01 GM073151) and the RosettaCommons. Work in the Meiler laboratory is supported by the NIH (U01 AI150739 and U19 AI117905).
Conflict of Interest
J.E.C. has served as a consultant for Takeda Vaccines, Sanofi-Aventis U.S., Pfizer, Novavax, Lilly and Luna Biologics, is a member of the Scientific Advisory Boards of CompuVax and Meissa Vaccines and is Founder of IDBiologics. The Crowe laboratory at Vanderbilt University Medical Center has received sponsored research agreements from IDBiologics. All other authors declare no conflict of interest.
References
- 1.Nybakken GE, Oliphant T, Johnson S, Burke S, Diamond MS, Fremont DH. Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature. 2005;437(7059):764–769. doi: 10.1038/nature03956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hashiguchi T, Fusco ML, Bornholdt ZA, et al. Structural Basis for Marburg Virus Neutralization by a Cross-Reactive Human Antibody. Cell. 2015;160(5):904–912. doi: 10.1016/J.CELL.2015.01.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hong M, Lee PS, Hoffman RMB, et al. Antibody recognition of the pandemic H1N1 Influenza virus hemagglutinin receptor binding site. J Virol. 2013;87(22):12471–12480. doi: 10.1128/JVI.01388-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014;32(2):158–168. doi: 10.1038/nbt.2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glanville J, Zhai W, Berka J, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci U S A. 2009;106(48):20216–20221. doi: 10.1073/pnas.0909775106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soto C, Bombardi RG, Branchizio A, et al. High frequency of shared clonotypes in human B cell receptor repertoires. Nature. 2019;566(7744):398–402. doi: 10.1038/s41586-019-0934-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kabat EA, Wu TT. Construction of a three-dimensional model of the polypeptide backbone of the variable region of kappa immunoglobulin light chains. Proc Natl Acad Sci U S A. 1972;69(4):960–964. doi: 10.1073/PNAS.69.4.960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chi X, Li Y, Qiu X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunology. 2020;160(3):233–247. doi: 10.1111/imm.13176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chothia C, Lesk AM, Tramontano A, et al. Conformations of immunoglobulin hypervariable regions. Nature. 1989;342(6252):877–883. doi: 10.1038/342877a0 [DOI] [PubMed] [Google Scholar]
- 10.North B, Lehmann A, Dunbrack RL. A New Clustering of Antibody CDR Loop Conformations. J Mol Biol. 2011;406(2):228–256. doi: 10.1016/J.JMB.2010.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foote J, Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol. 1992;224(2):487–499. doi: 10.1016/0022-2836(92)91010-M [DOI] [PubMed] [Google Scholar]
- 12.Koehler Leman J, Weitzner BD, Renfrew PD, et al. Better together: Elements of successful scientific software development in a distributed collaborative community. Schneidman-Duhovny D, ed. PLOS Comput Biol. 2020;16(5):e1007507. doi: 10.1371/journal.pcbi.1007507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leman JK, Weitzner BD, Lewis SM, et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat Methods. 2020;17(7):665–680. doi: 10.1038/s41592-020-0848-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adolf-Bryfogle J, Xu Q, North B, Lehmann A, Dunbrack RL Jr. PyIgClassify: a database of antibody CDR structural classifications. Nucleic Acids Res. 2015;43(Database issue):D432–8. doi: 10.1093/nar/gku1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sivasubramanian A, Sircar A, Chaudhury S, Gray JJ. Toward high-resolution homology modeling of antibody F v regions and application to antibody-antigen docking. doi: 10.1002/prot.22309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weitzner BD, Kuroda D, Marze N, Xu J, Gray JJ. Blind prediction performance of RosettaAntibody 3.0: grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins. 2014;82(8):1611–1623. doi: 10.1002/prot.24534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunbar J, Fuchs A, Shi J, Deane CM. ABangle: Characterising the VH-VL orientation in antibodies. Protein Eng Des Sel. 2013;26(10):611–620. doi: 10.1093/protein/gzt020 [DOI] [PubMed] [Google Scholar]
- 18.Lapidoth G, Parker J, Prilusky J, Fleishman SJ. AbPredict 2: a server for accurate and unstrained structure prediction of antibody variable domains. Bioinformatics. 2018;35(9):1591–1593. doi: 10.1093/bioinformatics/bty822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song Y, DiMaio F, Wang RY-R, et al. High-Resolution Comparative Modeling with RosettaCM. Structure. 2013;21(10):1735–1742. doi: 10.1016/J.STR.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bender BJ, Cisneros A, Duran AM, et al. Protocols for Molecular Modeling with Rosetta3 and RosettaScripts. Biochemistry. 2016;55:4763. doi: 10.1021/acs.biochem.6b00444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 22.Almagro JC, Teplyakov A, Luo J, et al. Second Antibody Modeling Assessment (AMA-II). Proteins Struct Funct Bioinforma. 2014;82(8):1553–1562. doi: 10.1002/prot.24567 [DOI] [PubMed] [Google Scholar]
- 23.Weitzner BD, Jeliazkov JR, Lyskov S, et al. Modeling and docking of antibody structures with Rosetta. Nat Protoc. 2017;12(2):401–416. doi: 10.1038/nprot.2016.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Norn CH, Lapidoth G, Fleishman SJ. High-accuracy modeling of antibody structures by a search for minimum-energy recombination of backbone fragments. Proteins Struct Funct Bioinforma. 2017;85(1):30–38. doi: 10.1002/prot.25185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graille M, Stura EA, Housden NG, et al. Complex between Peptostreptococcus magnus protein L and a human antibody reveals structural convergence in the interaction modes of Fab binding proteins. Structure. 2001;9(8):679–687. doi: 10.1016/S0969-2126(01)00630-X [DOI] [PubMed] [Google Scholar]
- 26.Momany C, Kovari LC, Prongay AJ, et al. Crystal structure of dimeric HIV-1 capsid protein. Nat Struct Biol. 1996;3(9):763–770. doi: 10.1038/nsb0996-763 [DOI] [PubMed] [Google Scholar]
- 27.Zhao H, Fernandez E, Dowd KA, et al. Structural Basis of Zika Virus-Specific Antibody Protection. Cell. 2016;166(4):1016–1027. doi: 10.1016/j.cell.2016.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fellouse FA, Esaki K, Birtalan S, et al. High-throughput Generation of Synthetic Antibodies from Highly Functional Minimalist Phage-displayed Libraries. J Mol Biol. 2007;373(4):924–940. doi: 10.1016/j.jmb.2007.08.005 [DOI] [PubMed] [Google Scholar]
- 29.Sung KH, Josewski J, Dübel S, Blankenfeldt W, Rau U. Structural insights into antigen recognition of an anti-β-(1,6)-β-(1,3)-D-glucan antibody. Sci Rep. 2018;8(1):1–10. doi: 10.1038/s41598-018-31961-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lapidoth GD, Baran D, Pszolla GM, et al. AbDesign: An algorithm for combinatorial backbone design guided by natural conformations and sequences. Proteins Struct Funct Bioinforma. 2015;83(8):1385–1406. doi: 10.1002/prot.24779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levinthal C ARE THERE PATHWAYS FOR PROTEIN FOLDING ? Vol 65.; 1968. [Google Scholar]
- 32.Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nivón LG, Moretti R, Baker D. A Pareto-Optimal Refinement Method for Protein Design Scaffolds. PLoS One. 2013;8(4). doi: 10.1371/journal.pone.0059004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunbar J, Krawczyk K, Leem J, et al. SAbDab: the structural antibody database. doi: 10.1093/nar/gkt1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crowe JE, Koff WC. Deciphering the human immunome. Expert Rev Vaccines. 2015;14(11):1421–1425. doi: 10.1586/14760584.2015.1082427 [DOI] [PubMed] [Google Scholar]
- 36.Madden T The BLAST Sequence Analysis Tool. In: The NCBI Handbook [Internet]. National Center for Biotechnology Information (US); 2003. [Google Scholar]
- 37.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lapidoth G, Fleishman S. AbPredict. RosettaCommons http://new.rosettacommons.org/docs/latest/application_documentation/structure_prediction/AbPredict2.PublishedOctober26, 2019. AccessedApril 8, 2021.
- 40.Oomen CJ, Hoogerhout P, Kuipers B, Vidarsson G, Van Alphen L, Gros P. Crystal structure of an anti-meningococcal subtype P1.4 PorA antibody provides basis for peptide-vaccine design. J Mol Biol. 2005;351(5):1070–1080. doi: 10.1016/j.jmb.2005.06.061 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.