

Abstract
In type 1 diabetes (T1D) autoreactive CD8 T cells infiltrate pancreatic islets and destroy insulin-producing β cells. Progression to T1D onset is a chronic process, which suggests that the effector activity of β-cell autoreactive CD8 T cells needs to be maintained throughout the course of disease development. The mechanism that sustains diabetogenic CD8 T cell effectors during the course of T1D progression has not been completely defined. Here we used single-cell RNA sequencing to gain further insight into the phenotypic complexity of islet-infiltrating CD8 T cells in NOD mice. We identified two functionally distinct subsets of activated CD8 T cells, CD44highTCF1+CXCR6− and CD44highTCF1−CXCR6+, in islets of prediabetic NOD mice. Compared to CD44highTCF1+CXCR6− CD8 T cells, the CD44highTCF1−CXCR6+ subset expressed higher levels of inhibitory and cytotoxic molecules and was more prone to apoptosis. Adoptive cell transfer experiments revealed that CD44highTCF1+CXCR6− CD8 T cells, through continuous generation of the CD44highTCF1−CXCR6+ subset, were more capable than the latter population to promote insulitis and the development of T1D. We further showed that direct interleukin-27 (IL-27) signaling in CD8 T cells promoted the generation of terminal effectors from the CD44highTCF1+CXCR6− population. These results indicate that islet CD44highTCF1+CXCR6− CD8 T cells are a progenitor-like subset with self-renewing capacity and under an IL-27 controlled mechanism they differentiate into the CD44highTCF1−CXCR6+ terminal effector population. Our study provides new insight into the sustainability of the CD8 T cell response in the pathogenesis of T1D.
Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disease resulting in the destruction of insulin producing pancreatic β cells (1). Studies in both humans and the NOD mouse implicate CD8 T cells as the primary mediators of β-cell destruction in T1D. In human T1D patients, CD8 T cells are the dominant cell type within the islet infiltrate and in situ MHC tetramer staining has revealed their β-cell reactivity (2, 3). In NOD mice, CD8 T cells are required for islet inflammation and T1D development (4–6). Following initial activation in the pancreatic lymph nodes (PLN), autoreactive CD8 T cells migrate to the pancreas, recognize cognate antigen in association with MHC class I molecules expressed on the surface of β cells, and initiate cytotoxic programs (7–10). However, the detailed mechanisms that control autoreactive CD8 T cell differentiation and sustain their effector activity during the progression of T1D are not fully defined.
T1D is a polygenic disease in which genome wide association studies and meta-analyses have identified greater than 50 significantly linked loci in humans (11–17). Among the list of T1D candidate genes is IL27 (encoding the p28 subunit) (11, 18). Additionally, human genetic studies have suggested an inflammatory role for interleukin-27 (IL-27) in the pathogenesis of T1D (19, 20). IL-27, a heterodimeric cytokine composed of two noncovalently linked subunits Epstein-Barr virus-induced gene 3 (EBI3) and IL-27p28, has been shown to play a role in the differentiation of CD8 T cells (21–25). We previously demonstrated that IL-27 signaling was required for the development of T1D in the NOD mouse and that direct IL-27 signaling enhanced the diabetogenic activity of CD8 T cells (26). While direct IL-27 signaling did not have a significant effect on the phenotype of CD8 T cells in the spleen and PLN, it promoted their accumulation and expression of T-bet and IFNγ in the pancreatic islets (26). This is consistent with the previous observation that there is further differentiation of CD8 T cells within the local islet environment (9, 10). Recently, the presence of multiple CD8 T cell differentiation states have been shown to be critical in maintaining the anti-viral response during the setting of chronic viral infection (27–31). IL-27 signaling was shown to improve the anti-viral CD8 T cell response during chronic viral infection by enhancing the proliferation and maintenance of the self-renewing progenitor population (32). Collectively, these observations led us to hypothesize that multiple antigen-driven CD8 T cell differentiation states exist during the progression of T1D and that direct IL-27 signaling may regulate this differentiation process.
In this study, we characterized the heterogeneity of CD8 T cells in the pancreatic islets of prediabetic NOD mice. Our data reveal the presence of phenotypically and functionally distinct subsets of islet-infiltrating CD8 T cells and identify a renewable source of autoreactive CD8 T cells within islet infiltrate during the progression of T1D. Furthermore, we found that direct IL-27 signaling regulates the transition of islet-infiltrating CD8 T cells between functionally distinct subsets.
Materials and Methods
Mice
NOD/ShiLtJ (NOD), NOD.129S7(B6)-Rag1tm1Mom/J (NOD.Rag1−/−), and NOD.B6-Ptprcb/6908MrkTacJ (NOD.Cd45.2) mice were procured from The Jackson Laboratory and maintained at the Medical College of Wisconsin (MCW). NOD.Tnfrsf9−/− and NOD.Il27ra−/− mice have been previously described (26, 33).
Study approval
All mice were used in accordance with and approved by Institutional Animal Care and Use Committee guidelines at the MCW.
Generation of mixed bone marrow chimeras
Bone marrow (BM) was harvested from the tibias and femurs of 6–9-week-old NOD.Il27ra−/− and NOD.Cd45.2 females. T cell-depleted NOD.Il27ra−/− and NOD.Cd45.2 BM cells were mixed at a 1:1 ratio (2.5×106 cells each) and infused into lethally irradiated (1100 rads) 6–8-week-old (NOD × NOD.Cd45.2)F1 females.
10x Genomics library preparation and sequencing
Approximately 2,500 cells per sample were used to prepare individual single-cell RNA sequencing (scRNA-seq) libraries using the Chromium Single Cell 3’v2 Reagent Kit (10x Genomics). Libraries were quantified using the KAPA Library Quantification Kit (Roche) and then sequenced on an Illumina NextSeq using the NextSeq 500/550 High Output Kit v2 (150 cycles) (Illumina). Raw sequencing data was downloaded using the Python Run Downloader (Illumina). Reads were demultiplexed and converted to gene-barcode matrices with mm10 as the reference transcriptome using the Cell Ranger (version 1.3.1) mkfastq and count functions respectively. Each sample had a sufficient number of genes detected (>10,000), a high percentage of reads mapped to the genome (>80%), and >1000 cells detected.
Isolation of cells from the pancreatic islets
Pancreatic islets were isolated as previously described (34). In some experiments, islets were pooled from multiple mice during picking. Isolated islets were dispersed in non-enzymatic cell dissociation buffer (Gibco) to obtain a single cell suspension.
Cell sorting
For isolation of cells used in the scRNA-seq experiment, islet single-cell suspensions were washed once and incubated with 5μg/mL Fc block (anti-mouse CD16/CD32, clone 2.4G2, BioXCell). Cells were then stained with antibodies against CD45.1 (A20) for 30 minutes at 4°C, washed, and passed through a 30μm filter. Single CD45.1+ cells were sorted on a FACSAria III (BD Biosciences). For isolation of islet-infiltrating CD8 T cell subsets, islet single cell suspensions were washed with complete RPMI and incubated at 37°C for 1 hour, washed and incubated with 5μg/mL Fc block, and then stained for 30 minutes at 4°C with antibodies against CD45.1 (A20), CD8 (53–6.7), CD44 (IM7), SLAMF6 (330-AJ), CXCR6 (SA051D1) and lineage markers (Lin) B220 (RA3–6B2), CD11b (M1/70), and CD4 (RM4–5). Cells were washed and passed through a 30μm filter. Single CD45.1+Lin−CD8+CD44low, CD45.1+Lin−CD8+CD44highSLAMF6+CXCR6−, and CD45.1+Lin−CD8+CD44highSLAMF6−CXCR6+ cells were sorted on a FACSAria II (BD Biosciences).
Single-cell RNA sequencing dataset analyses
Data analysis was performed in R (version 3.6.2) using the package Seurat (version 3.1.5) (35). Cells with unique feature counts <100 or >5000 and percent of counts from mitochondrial genes >5% were removed. The count data was log-normalized and the 7- and 12-week samples were combined using the IntegrateData function. Principal component (PC) analysis was performed on the integrated dataset considering the top 2000 variably expressed genes. All cells were initially clustered with Shared Nearest Neighbor (SNN) clustering and visualized using t-distributed stochastic neighbor embedding (t-SNE). Canonical markers were used to classify cell types. T cell clusters were isolated and re-clustered using SNN clustering. CD8 T cell clusters were then further isolated and cells expressing Cd4 at a level > 0.25 were removed. (Figure S1). The remaining 632 CD8 T cells were re-clustered with SNN clustering (resolution of 0.3) and t-SNE visualization using the top 12 most statistically significant PCs. Heatmaps, violin plots, and t-SNE plots were generated using the package Seurat. Bar plots were generated using the R package ggplot2 (version 3.3.0). The volcano plot was generated using the R package EnhancedVolcano (version 1.4.0). Gene module scores were calculated using the AddModuleScore function in Seurat.
SCENIC analysis
Data analysis was performed in R using the package SCENIC (36). Log-normalized UMI counts were used as the input gene values. The algorithm identified 401 total regulons in our dataset. The regulons can be divided into two categories: main regulons are identified by using high confidence annotations only (inferred by orthology) and “extended” regulons are identified by including lower confidence annotations (inferred by motif similarly). For regulons identified having both a main and an extended version, we kept only the main version resulting in 256 unique regulons in our dataset. The activity of those 256 regulons were used to generate a t-SNE plot. Cells in the t-SNE were colored according to their Seurat cluster identities; however, these groupings were used as a visualization tool only and had no effect on the distribution of cells in the t-SNE. Differential expression analysis (two sample t-test) was performed to identify cluster-specific marker regulons (cluster identities from Seurat analysis). The results were visualized on a heatmap using the top 20 regulons with the highest average regulon activity and an FDR <0.05. Differential expression analysis (two sample t-test) was performed to compare regulon activity between cluster 2 and 3 (cluster identities from Seurat analysis). For this comparison we utilized both main and extended regulons to maximize the putative target genes. A volcano plot was generated using the R package EnhancedVolcano (version 1.9.13). The gene regulatory network models were generated using Cytoscape (version 3.8.2).
Flow cytometry
Islet single cell suspensions were resuspended in complete RPMI and incubated at 37°C for 1 hour. Cells were washed once with FACS buffer (PBS + sodium azide and 2% FBS) and stained for surface markers CD45.1 (A20), CD45.2 (104), CD3e (145–2C11), CD4 (RM4–5), CD8 (53–6.7), CD44 (IM7), SLAMF6 (Ly108) (330-AJ), CXCR6 (SA051D1), PD-1 (J43), LAG3 (eBIOC9B7W), and TIM3 (RMT3–23). All antibodies are from BD Biosciences, Biolegend, or eBioscience. Dead cells were discriminated using 7-aminoactinomycin (Sigma). For transcription factor analysis, islet single cell suspensions were labeled with BD Horizon fixable viability stain (BD Biosciences) and stained with cell surface markers. Cells were then fixed and permeabilized using the Foxp3/Transcription factor staining buffer set (eBioscience) and stained with the anti-TCF1 antibody (C63D9, Cell Signaling). For analysis of IGRP206–214-reactive CD8 T cells, cells were labeled with MHC class I (Kd) tetramers loaded with a mimotope peptide NRP-V7 (37) (obtained from the National Institutes of Health Tetramer Core Facility) for 15 minutes at room temperature prior to cell surface marker staining. For analysis of granzyme B expression, islet single cell suspensions were incubated at 37°C for 5 hours in the presence of 1X cell stimulation cocktail with protein transport inhibitor (eBioscience). Cells were labeled with fixable viability dye and stained with cell surface markers. Cells were then fixed and permeabilized using the Cytofix/Cytoperm fixation and permeabilization solution kit (BD Biosciences) and stained with anti-granzyme B (NGZB, eBioscience). For analysis of active caspase 3, islet single cell suspensions were resuspended in complete RPMI and incubated at 37°C for 5 hours. Cells were labeled with fixable viability dye and stained with cell surface markers. Cells were then fixed, permeabilized, and stained for active caspase 3 using the PE active caspase 3 apoptosis kit (BD Biosciences). Samples were run on the LSRII or LSRFortessa X20 cytometer (BD Biosciences). Data was analyzed with FlowJo software (Tree Star). Gating for active caspase 3 was based on isotype control antibody.
Adoptive cell transfer
Each CD8 T cell subset (1×104 cells) sorted from 12–16-week-old female donors was independently mixed with total splenocytes (2×106 cells) from 6-week-old NOD.Tnfrsf9−/− female mice and intravenously injected into NOD.Rag1−/− females.
Assessment of T1D and insulitis development
Mice were tested weekly for glycosuria (Bayer Diastix ®) and considered diabetic after two consecutive readings of >250 mg/dL. Pancreata from cell transfer recipients at time of diabetes onset or 16 weeks post-transfer were fixed in 10% neutral buffered formalin. Fixed pancreata were cut in 4μm sections with 60μm discarded between sections. The sections were stained with aldehyde fuchsin followed by hematoxylin and eosin (H&E) counterstain. At least 25 islets from 4 sections were scored per mouse. The insulitis score of each islet was determined as follows: 0-no infiltration, 1-leukocytes surrounding islet but no penetration, 2-estimated loss of up to 25% of the β cells, 3-estimated loss of up to 75% of the β cells, 4-end stage, less than 25% of the β cells remaining.
Cell culture
The sorted congenically labeled CD8 T cell subsets were mixed together with total splenocytes from NOD.Cd45.2 mice as depicted in Figure 5A. Each sample of pooled cells were then cultured for 3 days in complete RPMI alone (unstimulated) or in the presence of soluble anti-CD3 and anti-CD28 (1μg/mL each) with or without 10ng/mL recombinant mouse IL-27 (R&D Systems).
Figure 5. IL-27 signaling promotes the differentiation of islet-infiltrating CD8 T cells from the CD44highSLAMF6+CXCR6− progenitor-like subset to TCF1−CXCR6+ terminally differentiated effectors.

(A) Diagram depicting the experimental design. CD8 T cell subsets were sorted as in Figure 4A from the islets of 12–16-week-old non-diabetic females. (B) Representative flow cytometry plots depicting the phenotypes of sorted islet CD8 T cells after 3 days of culture. (C) CD8 T cell subset distributions summarized from 5 biological replicates in 3 independent experiments. Each symbol is an individual sample. The horizontal line depicts the mean. *p<0.05, **p<0.01 by Wilcoxon rank-sum test. (D) Cell expansion after 3 days of culture. Fold change in total cell number was calculated by dividing the total cell number on day 3 by the input cell number. The error bar represents the standard error of the mean. Data are pooled from 3 biological replicates in 2 independent experiments. *p<0.05, **p<0.01 by two-sample t test.
Statistics
Statistical tests were performed in R (version 3.6.2) for scRNA-seq analyses and in Graphpad Prism 8 (La Jolla, CA) for other comparisons. Adjusted p-values were calculated using Bonferroni correction for comparison of differentially expressed genes between cell clusters. Wilcoxon rank-sum test was used for comparison of gene module scores between cell clusters. Fisher’s exact test was used to compare the distribution of cells in different clusters between the two time points. Wilcoxon rank-sum test, Wilcoxon signed-rank test, Chi-squared test with Benjamini-Hochberg correction, or two-sample t test was used for comparison between two groups as indicated.
Results
Transcriptomic heterogeneity of islet-infiltrating CD8 T cells
CD8 T cells are important for T1D development but how they sustain their diabetogenic activity during disease progression has not been completely defined. To further address this question, we initially performed scRNA-seq to gain insight into the transcriptional heterogeneity of CD8 T cells in islets of NOD mice. T1D onset starts at 12–14 weeks of age and the incidence is higher than 80% by 30 weeks of age in females in our NOD colony. Thus, we sorted islet-infiltrating leukocytes (CD45+) pooled from five 7- or 12-week-old prediabetic NOD females (Figure S1A–B). Single cell cDNA libraries were generated for 7- and 12-week samples independently. After initial filtering, 1,087 cells from 7-week and 2,776 cells from 12-week samples were further characterized. Canonical correlation analysis was used to integrate the datasets (35). Subsequently, unsupervised graph-based clustering analysis identified 16 unique cell populations, including CD4 and CD8 T cells, cycling T cells, B cells, plasma cells, natural killer T cells, natural killer cells, conventional dendritic cells (DCs), plasmacytoid DCs, macrophages, and group 2 innate lymphoid cells (ILC2) with distinct marker gene expression (Figures S1A–C). We found all cell populations were present in 7- and 12-week samples with some proportional variations (Figure S1B). Hereafter, we focused on CD8 T cells to characterize their differentiation states at a higher resolution. A total of 632 CD8 T cells (177 from the 7-week and 455 from the 12-week samples) were identified (Figure S1D) and after re-clustering they formed four distinct clusters (Figure 1A). The distribution of cells among the four clusters was similar at both timepoints (Fisher’s exact test p = 0.8979) (Figure 1B). Cluster 1 displayed a gene expression pattern characteristic of naïve CD8 T cells, including increased expression of Sell, Lef1, Dapl1, Ccr7, and Il7r and minimal expression of Cd44 (Figures 1C–D and Table S1). Clusters 2, 3, and 4 expressed markers indicative of activated T cells, including Cd44 (Figures 1C–D and Table S1). Notably, clusters 2, 3, and 4 had increased expression of the transcription factor Tox (Figures 1C–D). Previous studies have shown that TOX is induced in CD8 T cells during chronic viral infection, and hence its expression in islet CD8 T cells is consistent with the presence of persistent β-cell autoantigen stimulation in NOD mice (38, 39). Uniquely, cluster 4 had high expression of cell proliferation-associated genes, including Cdk1, Birc5, Top2a, and Mki67, indicating that cells in this cluster represent actively cycling cells (40–43) (Figures 1C–D). To further assess the cell cycle status of cells in each cluster we used the CellCycleScoring feature in Seurat (44). Correspondingly, cells in clusters 1–3 were predominantly non-cycling cells and all cells in cluster 4 were actively cycling cells in the G2/M or S phase (Figure S1D). Subsequently, we focused on clusters 2 and 3 for additional analyses.
Figure 1. scRNA-seq reveals transcriptional heterogeneity of islet-infiltrating CD8 T cells.

(A) t-SNE plot of four distinct CD8 T cell clusters in pancreatic islets of NOD mice using cells from both 7- and 12-week-old animals. (B) Proportionality of islet-infiltrating CD8 T cell clusters. Tandem t-SNE plots depict four cell clusters present in the 7- or 12-week-old CD8 T cells (Left). The bar plot shows the frequency of cells within each cluster (Right). The number within each bar indicates the absolute cell number for the cluster. (C) The heatmap of the top 100 differentially expressed genes in each cell cluster. The color scale represents the z-score distribution. (D) Violin plots depicting the expression of select molecules. (E) Volcano plot of differentially expressed genes identified in cluster 2 compared to cluster 3. Red symbols represent genes of an average log fold-change >0.25 and an adjusted p-value <0.05. Labeled genes have an average log fold change ≥0.6 and adjusted p-value <0.01. (F) Violin plots illustrating enrichment (module scores) of virus-specific progenitor-like or terminally differentiated effector CD8 T cell gene signatures. Statistical significance was determined with Wilcoxon rank-sum test.
Interestingly, clusters 2 and 3 comprise transcriptionally distinct subsets of activated CD8 T cells with similarity to the antigen-experienced progenitor-like and terminally differentiated effectors found during chronic viral infection (27–29). Among the top genes significantly upregulated in cluster 2 compared to cluster 3 were Tcf7 (encoding TCF1), Slamf6, and Id3 (Figures 1D–E and Table S2). TCF1 expression is critical for the generation and persistence of the progenitor-like subset that sustains CD8 T cell immunity during chronic viral infection by giving rise to the terminally differentiated effectors (29). Slamf6 encodes a cell surface molecule that is also expressed by the TCF1+ progenitor-like CD8 T cells found during chronic viral infection (29, 45). Id3 encodes a transcriptional regulator that is expressed by long-lived memory CD8 T cells (46). Additionally, cluster 2 expressed Il7r and intermediate levels of inhibitory receptors Pdcd1 (encoding PD-1) and Lag3 (Figures 1D–E). However, consistent with the TCF1+ progenitor-like CD8 T cell phenotype in chronic viral infection, cluster 2 lacked expression of the inhibitory receptor Havcr2 (encoding TIM3) as well as cytotoxic molecules Gzmb and Fasl (29) (Figure 1D). Conversely, cluster 3 was distinguished by higher expression of the chemokine receptor Cxcr6. Additionally, cluster 3 shared multiple similarities with the terminally differentiated effectors found during chronic viral infection, including increased expression of transcription factors Eomes and Prdm1 (encoding BLIMP-1), the transcriptional regulator Id2, inhibitory receptors (Pdcd1, Lag3, and Havcr2), and effector and cytotoxic molecules (Ifng, Ccl3, Ccl4, Gzmb, and Fasl) (Figures 1D–E and Tables S1 and S2) (27, 47).
To further explore the parallelism between CD8 T cell differentiation states in T1D and chronic viral infection, we measured the enrichment of previously identified gene signatures in our scRNA-seq dataset (28). Expression of genes upregulated in progenitor-like CD8 T cells during chronic lymphocytic choriomeningitis virus (LCMV) infection were enriched in cluster 2; whereas those upregulated in terminally differentiated effector CD8 T cells were enriched in cluster 3 (Figure 1F). Overall, our scRNA-seq analysis identified transcriptionally distinct subsets of islet-infiltrating CD8 T cells and suggests that a progenitor-progeny relationship may exist between these subsets analogous to that seen in chronic LCMV infection.
Differential gene regulatory network activity in islet-infiltrating CD8 T cell subsets
To further understand the gene regulatory programs underlying the CD8 T cell differentiation states in T1D, we applied a computational method called single-cell regulatory inference and clustering (SCENIC) to our scRNA-seq dataset (36). The SCENIC analysis first identifies co-expression modules containing a transcription factor and possible gene targets within our scRNA-seq data, then potential direct targets of the transcription factor are identified using cis-regulatory motif analysis. The resulting transcription factor and putative targets defines a regulon. In this way, cells can be clustered based on overall similarities in cell-regulon activity, which considers not only transcription factor expression but also the expression of their target genes. Cell clustering based on the activity of the regulons detected in islet-infiltrating CD8 T cells closely corresponded to the four major subsets we identified by gene expression profiles (clusters 1–4 in Figure 1) (Figure 2A). This result indicates that distinct gene regulatory networks control the differentiation of islet-infiltrating CD8 T cells during the progression of T1D. Notably, cluster 1 cells displayed high activity of regulons consistent with their naive phenotype including Klf2, Lef1, Foxo1, and Foxp1 that are known to be important for thymic egress, naïve T cell homeostasis, and maintenance of the quiescent state (Figure 2B) (48–54). In contrast, cluster 4 cells had high activity of regulons associated with cell cycle progression including E2f1, E2f4, E2f7, E2f8, and Smc3, which is congruent with their actively cycling phenotype (Figure 2B) (55, 56). Interestingly, clusters 2 and 3 exhibited regulon activity with similarities and differences to the progenitor-like and terminally differentiated effector CD8 T cells found during chronic viral infection. Therefore, we focused subsequent analyses on these two subsets.
Figure 2. SCENIC analysis reveals differential gene regulatory network activity in islet-infiltrating CD8 T cell subsets.

scRNA-seq data from islet-infiltrating CD8 T cells was analyzed using SCENIC. (A) A t-SNE plot depicts the distribution of cells based on regulon activity. Cells are colored based on their membership in the 4 clusters defined in Figure 1 (Red = cluster 1, naive; Green = cluster 2, progenitor-like; Blue = cluster 3, terminally differentiated effector; Purple = cluster 4, cycling cells). (B) The heatmap of the top 20 differentially expressed regulons in each of the four cell clusters. The color scale represents the z-score distribution of regulon activity. The numbers in the parentheses represent the number of target genes in the regulon. The regulon names that include “_extended” denote transcription factors which have been linked to targets based on inclusion of lower confidence annotations inferred by motif similarity. (C) A volcano plot depicts differentially expressed regulons identified in cluster 2 compared to cluster 3. Blue symbols represent regulons upregulated in cluster 3 with an FDR <0.001. Green symbols represent regulons upregulated in cluster 2 with an FDR <0.001. The top 10 upregulated regulons in each cell cluster are labeled. For the purpose of determining the top 10 upregulated regulons in each cell cluster, the main and extended regulons of a particular transcription factor were considered as a single regulon. (D) Violin plots depict the regulon activity enrichment in each cell cluster for select regulons. (E-F) Gene regulatory network models depict the interaction between key transcription factors identified by the SCENIC analysis and their target genes also listed as cell subset signature genes in Table S1 (Up to 10 target genes per transcription factor are shown based on average log fold change in each cell subset). The key transcription factors for each CD8 T cell subset are highlighted in green (cluster 2, progenitor-like) and blue (cluster 3, terminally differentiated effector). The cell subset signature genes are highlighted in yellow. The transcription factor-target interactions are indicated by black arrows.
Of note, both clusters 2 and 3 had uniquely enriched activity of the Elf1 and Ikzf2 regulons (Figure 2B). Elf1 has been shown to regulate several T cell specific genes including promoting responsiveness to IL-2 via transcriptional control of Il2ra (57, 58). Ikzf2 (also known as Helios) has been shown to be expressed in a subset of terminal effector CD8 T cells that arise in patients with chronic untreated HIV-1 infection (59). Additionally, Ikzf2 has been associated with an exhausted CD8 T cell phenotype during chronic viral infection (60, 61). Therefore, Elf1 and Ikzf2 gene regulatory programs may play a role in the generation or maintenance of chronically activated CD8 T cells during the progression of T1D.
Reinforcing the conclusion that cluster 2 cells have a phenotype similar to the progenitor-like population, we observed that the Tcf7 regulon was among the top regulons with significantly increased activity in cluster 2 compared to cluster 3 (Figures 2C–D). Furthermore, TCF1 is predicted to directly promote the expression of cluster 2 signature genes Id3 and Traf1 (Figure 2E). Additionally, we found that the Tcf12, Elk4, Myc, Yy1, Bmyc, Ubtf, and Nrf1 regulons were also among the top regulons with increased activity in cluster 2 compared to cluster 3 suggesting that they may play a role in the generation or maintenance of the progenitor-like CD8 T cells in T1D (Figures 2C–D). Notably, TCF12 and ELK4 transcription factors were also predicted to directly promote the expression of Id3 (Figure 2E).
Supporting the conclusion that cluster 3 cells have a phenotype similar to the terminally differentiated effectors, we observed that the Runx2, Batf, Prdm1, Rora, Bhlhe40, and Eomes regulons were among the top regulons with significantly increased activity in cluster 3 compared to cluster 2 (Figures 2C–D). Additionally, we found the Trps1, E2f4, and Ets1 regulons were also among the top regulons with increased activity in cluster 3 compared to cluster 2 suggesting that they may play a role in the generation or maintenance of the effector CD8 T cells in the progression of T1D. Interestingly, we found that Batf, Bhlhe40, E2f4, Runx2, Ets1, and Rora are all predicted to reinforce the expression of the cluster signature marker Cxcr6 (Figure 2F). Furthermore, we found that the transcription factors representing the top regulons in cluster 3 compared to cluster 2 also promoted the expression of effector molecules including Ccl3, Ccl4, Ccl5, Ifng, Gzmb, inhibitory receptors including Pdcd1 and Lag3, and transcription factors with previously known roles in exhausted CD8 T cells including Tox and Nr4a2 (Figure 2F) (38, 39, 62). Collectively, these results indicate that the differentiation of islet-infiltrating CD8 T cells during the progression of T1D is controlled by distinct gene regulatory networks.
Differential expression of CD44, SLAMF6, TCF1, and CXCR6 distinguishes three subsets of islet-infiltrating CD8 T cells
We next asked whether surface markers identified by scRNA-seq could be used to detect CD8 T cell subsets via flow cytometry for the purpose of cell isolation. We analyzed islet-infiltrating CD8 T cells of 10 to 13-week-old non-diabetic NOD females for the expression of CD44 (a marker distinguishing activated cells from cluster 1), SLAMF6 (a cluster 2 marker), and CXCR6 (a cluster 3 marker). Analogous to the scRNA-seq data, flow cytometry revealed three major subsets of CD8 T cells including a CD44low population, a CD44highSLAMF6+CXCR6− population, and a CD44highSLAMF6−CXCR6+ population in the islet infiltrate (Figures 3A and S2A). To further characterize the phenotype of these CD8 T cell subsets, we analyzed the expression of transcription factor TCF1 (a cluster 2 marker that is highly expressed in memory cells (63, 64)) as well as inhibitory receptors PD-1, LAG3, and TIM3. Additionally, we analyzed granzyme B expression. Similar to the scRNA-seq result, the CD44low population expressed high levels of TCF1 but overall had low expression of PD-1, LAG3, TIM3, and granzyme B (Figures 3B–3F and S2B–S2F). The CD44highSLAMF6+CXCR6− population expressed high levels of TCF1 and intermediate levels of PD-1 and LAG3 but had limited expression of TIM3 and granzyme B (Figures 3B–3F and S2B–S2F). The CD44highSLAMF6−CXCR6+ population had minimal expression of TCF1 but expressed high levels of PD-1, LAG3, TIM3, and granzyme B (Figures 3B–3F and S2B–S2F). Thus, consistent with the scRNA-seq analysis, CD44, SLAMF6, and CXCR6 expression distinguishes three phenotypically distinct, and likely functionally different, subsets of islet-infiltrating CD8 T cells. Since high levels of both TCF1 and SLAMF6 were co-expressed in islet CD44highCXCR6− CD8 T cells, hereafter we use either CD44highTCF1+CXCR6− or CD44highSLAMF6+CXCR6− (i.e., SLAMF6 as a cell surface surrogate marker for TCF1) to define this subset.
Figure 3. Expression of CD44, SLAMF6, and CXCR6 defines three subsets of islet-infiltrating CD8 T cells.

(A) Representative flow cytometry plots depicting the gating strategy of three CD8 T cell subsets and the corresponding expression of TCF1 (B), LAG3 (C), PD-1(D), TIM3 (E), and granzyme B (F) in pancreatic islets of 10–13-week-old female NOD mice. The black line with grey shaded area shown in the histogram of panel (F) represents the isotype control staining of CD44high CD8 T cells. For granzyme B expression, the cells were stimulated ex vivo with PMA and ionomycin. Cells are gated on single viable CD45.1+CD3+CD4−CD8+ cells. Summarized geometric mean fluorescent intensity (MFI) results are from 2–3 independent experiments. Each symbol represents islet cells pooled from two mice (n=5 pools/group). The horizontal line indicates the mean. *p<0.05, **p<0.01 by Wilcoxon rank-sum test. (G) CD8 T cell subsets among NRP-V7 MHC-I tetramer positive and negative cells harvested from islets of 7–12-week-old female NOD mice. Cells are initially gated on single viable CD45.1+CD3+CD4−CD8+ cells. Representative flow cytometry plots depicting the CD8 T cell subset gating strategy are shown (Left). The relative proportion of TCF1+CXCR6− and TCF1−CXCR6+ CD8 T cells among CD44high MHC-I tetramer positive and negative cells is summarized (Right). Results are combined from 3 independent experiments. Each symbol represents a mouse, and the horizontal line is the mean. **p<0.01, ***p<0.001, ****p<0.0001 by Wilcoxon rank-sum test.
Next, we confirmed the existence of distinct subsets among β-cell specific CD8 T cells by analyzing IGRP206–214-reactive CD8 T cells in islet infiltrate. Cells were labeled with MHC class I (Kd) tetramers loaded with the mimotope peptide NRP-V7 (37). We found that IGRP206–214-reactive CD8 T cells in the pancreatic islets were exclusively CD44high and could be divided into TCF1+CXCR6− and TCF1−CXCR6+ subsets (Figure 3G). Interestingly, the relative proportion of TCF1−CXCR6+ cells was significantly higher among IGRP206–214-reactive than other CD44high CD8 T cells (Figure 3G). Collectively, these results provide further evidence to support the hypothesis that CD44highTCF1+CXCR6− and CD44highTCF1−CXCR6+ CD8 T cell subsets represent functionally distinct stages of β-cell-antigen-driven differentiation.
Islet CD44highTCF1+CXCR6− CD8 T cells give rise to the terminally differentiated TCF1−CXCR6+ effector subset to promote the development of insulitis and T1D
We next aimed to directly determine the lineage relationship between the 3 distinct subsets of islet-infiltrating CD8 T cells identified in Figure 3A by adoptive transfer experiments. We used SLAMF6 to sort islet CD8 T cell subsets and TCF1 to define them after adoptive transfer. The limited number of islet CD8 T cells that could be isolated from donor mice did not allow us to recover them for numerical and phenotypic analysis if transferred into lymphoreplete NOD mice. Thus, we isolated CD44low, CD44highSLAMF6+CXCR6−, and CD44highSLAMF6−CXCR6+ CD8 T cells from islets of 12–16-week-old non-diabetic (NOD × NOD.Cd45.2)F1 females and transferred them into sex-matched NOD.Rag1−/− mice (Figures 4A and S2G). To provide other lymphocyte populations, we co-transferred 1×104 sorted islet CD8 T cells with 2×106 total splenoctyes from 6-week-old NOD.Tnfrsf9−/− females (expressing only CD45.1). CD137 (encoded by Tnfrsf9) expression on β-cell autoreactive CD8 T cells is important for their accumulation and Tnfrsf9−/− CD8 T cells cannot induce diabetes in NOD.Rag1−/− recipients (65). Thus, NOD.Tnfrsf9−/− splenocytes can support, but not outcompete, the CD8 T cells isolated from (NOD × NOD.Cd45.2)F1 islets. At 7 weeks post-transfer, we analyzed the transferred (NOD × NOD.Cd45.2)F1 CD8 T cells in the spleens, PLN, and islets of the NOD.Rag1−/− recipients. The reconstitution levels of all three transferred CD8 T cell subsets were not statistically different in the spleen, albeit there was a tendency to be higher in the CD44highSLAMF6+CXCR6− recipients (Figure 4B). The absolute number of cells derived from transferred CD44highSLAMF6+CXCR6− CD8 T cells trended higher than the CD44low subset and was consistently higher than the CD44highSLAMF6−CXCR6+ subset in the PLN (Figure 4B). Strikingly, the transferred CD44highSLAMF6+CXCR6− CD8 T cells exhibited a robust capacity to accumulate in the islets compared to the CD44low and CD44highSLAMF6−CXCR6+ subsets (Figure 4B and S2H). The increased accumulation of the transferred CD44highSLAMF6+CXCR6− CD8 T cells in the PLN and islets suggests that these cells were undergoing antigen-driven expansion, although we cannot completely rule out the effect of homeostatic expansion that occurs in the lymphopenic NOD.Rag1−/− host. We further analyzed the phenotype of the transferred cells in the islets of the recipients and found that the transferred CD44highSLAMF6+CXCR6− CD8 T cells gave rise to both TCF1+ and TCF1− populations (Figure 4C). Whereas the transferred CD44low and CD44highSLAMF6−CXCR6+ CD8 T cells largely maintained their pre-transfer TCF1 expression pattern (TCF1+ and TCF1− respectively, Figure 4C).
Figure 4. Islet-infiltrating CD44highSLAMF6+CXCR6− progenitor-like CD8 T cells give rise to TCF1−CXCR6+ CD8 T cells to promote diabetes development.
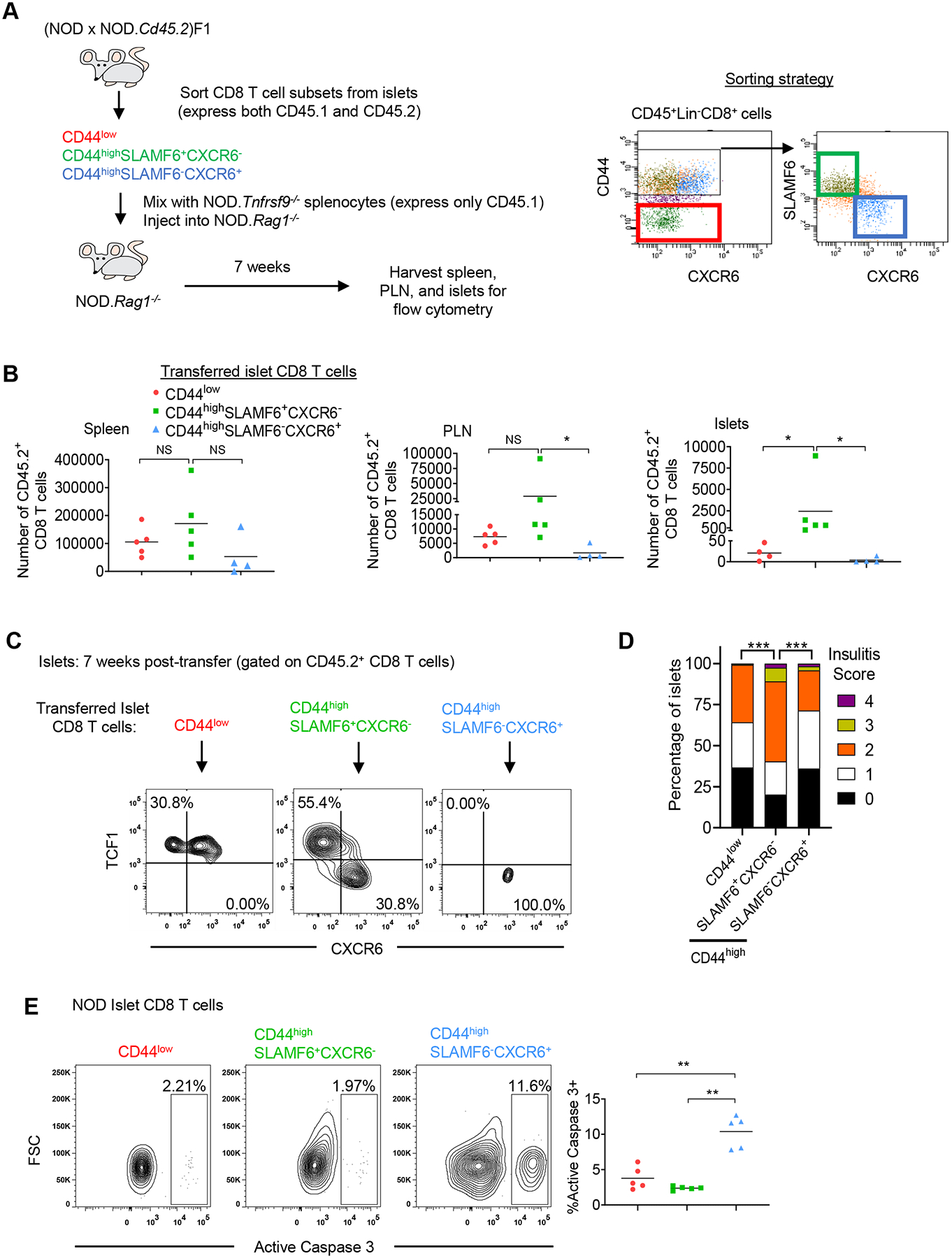
(A) Diagram depicting experimental design (Left) and the cell sorting strategy (Right). CD8 T cell subsets were sorted from 12–15-week-old non-diabetic (NOD × NOD.Cd45.2)F1 females, independently mixed at a 1:200 ratio with NOD.Tnfrsf9−/− splenocytes, and transferred into NOD.Rag1−/− recipients. (B) The absolute numbers of adoptively transferred CD45.2+ CD8 T cells in the spleen, PLN, and islets of recipient mice at 7 weeks post-transfer. Results are summarized from 3 independent experiments. Each symbol represents an individual recipient mouse, and the horizontal line depicts the mean (n=4–5/group). *p<0.05, **p<0.01, NS: not significant by Wilcoxon rank-sum test. (C) The phenotype of the adoptively transferred CD45.2+ CD8 T cells in the islets of recipient mice at 7 weeks post-transfer. Representative flow cytometry plots of 3 independent experiments are shown (n=4–5/group). (D) Histological analysis of pancreata in the non-diabetic recipient mice at 16 weeks post-transfer. Pancreatic islets were scored for insulitis: 0: no infiltration, 1: peri-insulitis, 2: ≤25% β cell loss, 3: between 25–75% β cell loss, and 4: >75% β cell loss. Summarized results depict the proportion of islets of each insulitis score in recipient mice. Results are from 3 independent transfer experiments (n=4 mice per group). The total number of islets analyzed in CD44low, CD44highSLAMF6+CXCR6−, and CD44highSLAMF6−CXCR6+ islet CD8 T cell recipients are 120, 119, and 119 respectively. ***p<0.0005 by Chi-squared test with Benjamini-Hochberg correction. (E) Representative flow cytometry plots depicting active caspase 3 expression in the CD8 T cell subsets in pancreatic islets of 10–12-week-old female NOD mice. Summarized frequency results are from 2 independent experiments. Each symbol represents islet cells pooled from two mice (n=5 pools/group). The horizontal line indicates the mean. *p<0.05, **p<0.01 by Wilcoxon rank-sum test.
We next asked whether there was a difference in the ability of purified islet CD44low, CD44highSLAMF6+CXCR6−, and CD44highSLAMF6−CXCR6+ CD8 T cells to induce insulitis and diabetes. We performed a series of five independent transfer experiments similar to the transfer strategy depicted in Figure 4A. Each transfer experiment included 1–2 NOD.Rag1−/− recipients of each CD8 T cell subset. The recipients were followed weekly for diabetes development. In two out of five transfer experiments the recipients of CD44highSLAMF6+CXCR6− CD8 T cells became diabetic at 10- and 14-weeks post-transfer respectively. In contrast, none of the CD44low or CD44highSLAMF6−CXCR6+ CD8 T cell recipients developed diabetes. At 16 weeks post-transfer, the recipient pancreata from transfer experiments where none of the mice became diabetic were histologically analyzed for insulitis. Histological analysis revealed that CD44highSLAMF6+CXCR6− CD8 T cell recipients had the highest level of insulitis compared to CD44highSLAMF6−CXCR6+ and CD44low CD8 T cell recipients (Figure 4D). The inability of CD44highSLAMF6−CXCR6+ CD8 T cells to exhibit long-term accumulation in the PLN and islets of NOD.Rag1−/− recipients suggests that these cells are terminally differentiated and prone to apoptosis, which likely precludes them from inducing T1D despite having an effector phenotype. Consistent with this interpretation, a significantly higher proportion of CD44highSLAMF6−CXCR6+ CD8 T cells from the islets of NOD mice expressed active caspase 3 than the other subsets (Figure 4E). Together these experiments indicate that β-cell-antigen-driven differentiation of CD8 T cells occurs in a stepwise process, whereby the CD44highTCF1+CXCR6− progenitor-like subset is capable of self-renewal and can give rise to the CXCR6+ effector population. These results also indicate that the CD44highTCF1+CXCR6− progenitor-like subset is required to continuously replenish the effector cell pool and maintain the autoreactive CD8 T cell response over time. In addition, the results provide evidence to support the idea that the majority of CD44low naïve-like CD8 T cells found in the pancreatic islets at later stages of disease progression are not β-cell antigen specific.
IL-27 signaling promotes the generation of islet CD44highTCF1−CXCR6+ CD8 T cells from the CD44highTCF1+CXCR6− progenitor-like population
We previously demonstrated that IL-27 signaling in CD8 T cells promotes T1D progression in NOD mice (26). Direct IL-27 signaling enhanced the accumulation of islet-infiltrating CD8 T cells and promoted their expression of T-bet and IFNγ (26). Thus, we hypothesized that IL-27 signaling promotes the generation of CD44highTCF1−CXCR6+ terminally differentiated CD8 T effectors in the pancreatic islets of NOD mice. To initially test this idea, we used an in vitro culture system to analyze the effect of IL-27 on the lineage relationship between the CD8 T cell subsets isolated from pancreatic islets. We used SLAMF6 to sort islet CD8 T cell subsets and TCF1 to define them after in vitro differentiation. We independently sorted the CD8 T cell subsets (2×104 each) from the islets of NOD (expressing CD45.1) and (NOD × NOD.Cd45.2)F1 mice, and then mixed them with NOD.Cd45.2 splenocytes (feeder cells) as depicted in Figure 5A. Mixed cells were stimulated for three days with anti-CD3 and anti-CD28 in the presence or absence of recombinant murine IL-27. Congenically marked islet CD44highSLAMF6+CXCR6− and CD44highSLAMF6−CXCR6+ CD8 T cells were cultured together to directly compare their differentiation potential.
In the absence of IL-27, CD44highSLAMF6+CXCR6− cells gave rise to both TCF1+CXCR6− and TCF1−CXCR6+ cells, whereas the CD44highSLAMF6−CXCR6+ and CD44low subsets primarily maintained their TCF1−CXCR6+ and TCF1+CXCR6− expression patterns respectively (Figures 5B–C). Interestingly, addition of exogenous IL-27 to the culture resulted in a significantly increased proportion of CD44highSLAMF6+CXCR6− cells acquiring the TCF1−CXCR6+ phenotype (Figures 5B–C). The presence of IL-27 also promoted CD44low cells to downregulate TCF1 and upregulate CXCR6, while the CD44highSLAMF6−CXCR6+ subset maintained the TCF1−CXCR6+ phenotype (Figures 5B–C). These results indicate that IL-27 signaling promotes downregulation of TCF1 and upregulation of CXCR6 in non-terminally differentiated CD8 T cells and suggest a role for IL-27 signaling in promoting the generation of CD44highTCF1−CXCR6+ CD8 T cells from the CD44highTCF1+CXCR6− population. IL-27 signaling may promote the generation of both progenitor-like and effector CD8 T cells from the CD44low population; however, we cannot rule out the possibility that naïve CD8 T cells can directly become effectors and bypass the progenitor-like state in the presence of IL-27 signaling. Additionally, these results further support the conclusion that islet-infiltrating CD44highTCF1+CXCR6− progenitor-like CD8 T cells give rise to the CD44highTCF1−CXCR6+ terminally differentiated effector population.
Recently, it has been reported that IL-27 signaling promotes the proliferation and maintenance of the antigen-experienced TCF1+ progenitor CD8 T cell population during chronic viral infection (32). Therefore, we asked if IL-27 signaling differentially affects the expansion of cultured CD44highSLAMF6+CXCR6− and CD44highSLAMF6−CXCR6+ islet CD8 T cell subsets. We analyzed the change in total cell numbers after three days in culture with or without IL-27. Consistent with the idea that the CD44highSLAMF6+CXCR6− CD8 T cell subset isolated from islets resemble the self-renewing progenitor-like cells, they underwent significantly higher expansion compared to CD44highSLAMF6−CXCR6+ CD8 T cells in the absence of IL-27 (Figure 5D). The limited expansion of the CD44highSLAMF6−CXCR6+ CD8 T cells is consistent with our observation that these cells are prone to apoptosis (Figure 4E). Addition of IL-27 dramatically enhanced the expansion of sorted CD44highSLAMF6+CXCR6− CD8 T cells, but the effect was modest for the CD44highSLAMF6−CXCR6+ subset (Figure 5D). Together, these results suggest that IL-27 signaling promotes the expansion of the islet-infiltrating CD44highTCF1+CXCR6− CD8 T cells and simultaneously drives the differentiation of terminal effectors.
CD8 T cell-intrinsic IL-27 signaling promotes the generation of the CD44highTCF1−CXCR6+ CD8 T cell subset within pancreatic islets
To further confirm the effect of IL-27 on the differentiation of CD44highTCF1−CXCR6+ CD8 T cells in vivo, we generated mixed bone marrow (BM) chimeric mice to directly compare wildtype and Il27ra−/− CD8 T cell subsets. We reconstituted lethally irradiated (NOD × NOD.Cd45.2)F1 recipients with an equal number of wildtype NOD.Cd45.2 and NOD.Il27ra−/− (expressing only CD45.1) BM cells and analyzed islet infiltrate in prediabetic chimeras at 10–12 weeks post-reconstitution (Figure 6A). Consistent with our previous result, the frequency of total Il27ra−/− CD8 T cells was significantly reduced in the pancreatic islets compared to the wildtype counterpart, but no difference was seen in the spleen (26). Interestingly, we found that IL-27Rα deficiency significantly reduced the frequency of CD44high CD8 T cells in the pancreatic islets of the mixed BM chimeras (Figures 6B–C). In this experiment, SLAMF6 was used as a surrogate marker for TCF1 to further define the two CD44high subsets. The proportion of SLAMF6−CXCR6+ cells among CD44high CD8 T cells was found significantly reduced in Il27ra−/− compared to wildtype CD8 T cells in the islets (Figures 6B and 6D). This result indicates that CD8 T cell-intrinsic IL-27 signaling promotes the generation of terminally differentiated CD44highSLAMF6−TCF1−CXCR6+ CD8 T effectors within the pancreatic islets.
Figure 6. CD8 T cell-intrinsic IL-27 signaling promotes the generation of the terminally differentiated CD44highSLAMF6−CXCR6+ subset in pancreatic islets.
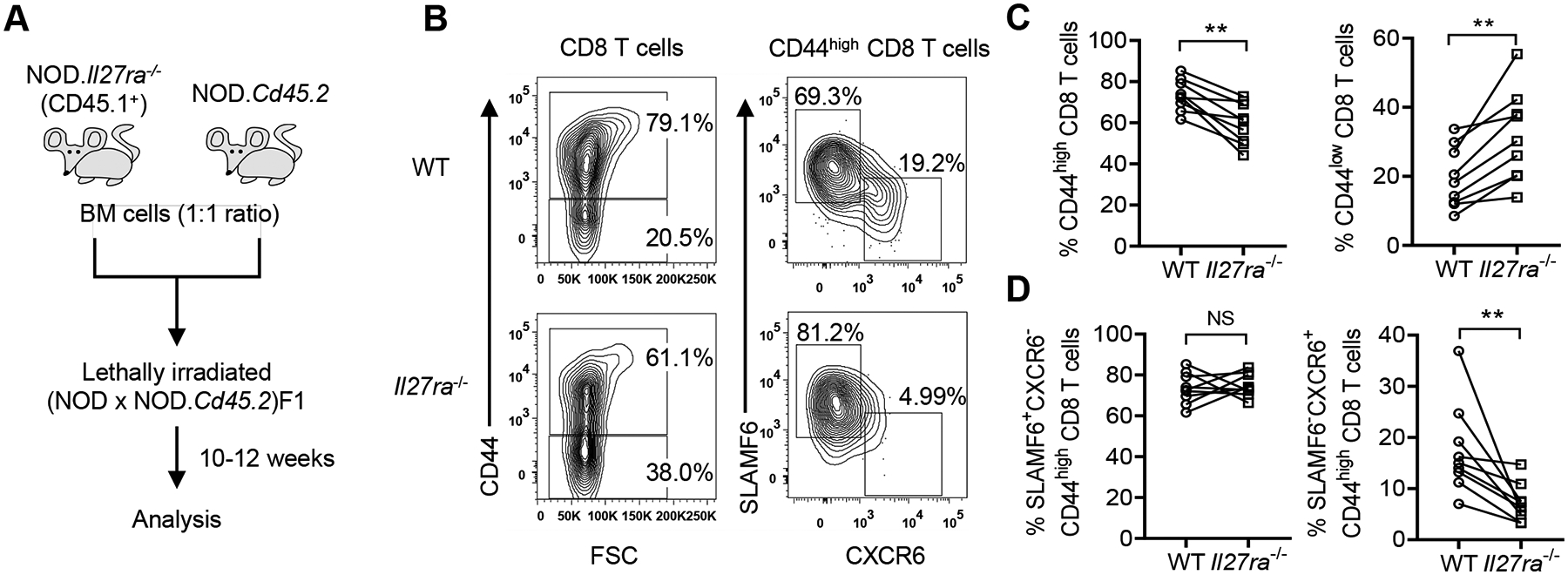
(A) Diagram depicting the experimental design. (B) Representative flow cytometry profiles of wildtype (WT) and Il27ra−/− CD8 T cells. Cells are initially gated on single, viable, CD45.2+ (WT) or CD45.1+ (Il27ra−/−) CD3+CD8+ cells. (C) The proportions of CD44high (Left) and CD44low (Right) among WT and Il27ra−/− CD8 T cells. (D) The frequencies of SLAMF6+CXCR6− (Left) and SLAMF6−CXCR6+ (Right) subsets among WT and Il27ra−/− CD44high CD8 T cells. Summarized data are from 2 independent experiments (n=9/group). **p<0.01, NS: not significant by Wilcoxon signed-rank test.
Discussion
Our results have revealed transcriptional, phenotypic, and functional heterogeneity within islet-infiltrating CD8 T cells. We identified three major subsets of CD8 T cells infiltrating the pancreatic islets, including a CD44low population, a CD44highTCF1+CXCR6− population, and a CD44highTCF1−CXCR6+ population, in NOD mice with established insulitis. The CD44low population had a transcriptional profile similar to naïve cells and was unable to accumulate to a significant level in the islets and cause diabetes upon transfer into NOD.Rag1−/− hosts. Therefore, the CD44low population isolated from the islets of NOD mice at later stages of T1D progression is likely primarily composed of β-cell antigen non-specific bystander cells that are recruited to the islets due to ongoing inflammation. Recruitment of bystander T cells to the islets during the progression of T1D has been previously reported (66, 67), although their role in T1D pathogenesis is not clear. The CD44highTCF1+CXCR6− and CD44highTCF1−CXCR6+ populations had transcriptional profiles and gene regulatory networks respectively similar to the progenitor-like and terminally differentiated effectors that are found during chronic viral infection. The CD44highTCF1+CXCR6− population had a robust ability to accumulate in the islets upon transfer into the NOD.Rag1−/− host, which led to significant insulitis and the development of T1D. Furthermore, the CD44highTCF1+CXCR6− population gave rise to the CD44highTCF1−CXCR6+ effector population in the NOD.Rag1−/− hosts, whereas the CD44highTCF1−CXCR6+ population maintained their TCF1− pre-transfer phenotype. Thus, our study demonstrates that the differentiation of islet-infiltrating CD8 T cells occurs in a stepwise manner.
The identification of the CD44highTCF1+CXCR6− progenitor population within islet-infiltrating CD8 T cells has important implications for β-cell autoimmunity. Studies in NOD mice have revealed that thymic development of β-cell autoreactive T cells is limited to a narrow postnatal period (68) and that priming in PLN is required at 3 weeks but dispensable for T1D development after 10 weeks of age (7). Therefore, some autoreactive CD8 T cells retain their proliferative potential over time to sustain the autoimmune response. We demonstrated here, utilizing both in vitro and in vivo experiments, that the islet-infiltrating CD44highTCF1+CXCR6− CD8 T cells efficiently expand and generate terminally differentiated effectors. These results indicate that the CD44highTCF1+CXCR6− population is a source of self-renewing antigen-experienced β-cell specific CD8 T cells critical for maintaining the autoreactive response at the site of autoimmune attack. Future studies will be important to determine the factors that regulate the generation and persistence of these cells.
Our gene regulatory network analysis revealed multiple transcription factors potentially controlling the differentiation of islet-infiltrating CD8 T cells during the progression of T1D. The CD44highTCF1+CXCR6− progenitors had high activity of Tcf12. TCF12 (also known as HEB) is a transcriptional regulator that binds to DNA at specific E box sites and has been shown to play a role in the development of memory-precursor CD8 T cells during viral infection. Specifically, a deficiency of both TCF12 and TCF3 (also known as E2A) led to an increase in the generation of short-lived effector CD8 T cells but a decrease in the formation of the memory-precursor CD8 T cells during viral infection (69). Furthermore, we have recently shown that TCF3 is an important epigenetic regulator in the formation of memory CD8 T cells during viral infection (70). Therefore, TCF12 may play a role, alone or in conjunction with TCF3, in the generation of islet-reactive progenitor-like CD8 T cells during T1D development. Our SCENIC analysis also identified that the islet-infiltrating CD44highTCF1−CXCR6+ effector CD8 T cells had high activity of Batf. BATF expression has been shown to promote the CD8 T cell exhaustion program during chronic viral infection and is required for maintenance of the effector CD8 T cell response (71–73). The role of BATF in the pathogenesis of T1D is unknown, however it is possible that BATF is important for the maintenance of effector CD8 T cells during the long-term exposure to β cell antigens in the progression of T1D. Future studies are warranted to investigate the roles of TCF12, BATF, and potentially other transcription factors identified in our SCENIC analysis in the development of the autoreactive CD8 T cell response in T1D.
Our analysis of islet-infiltrating CD8 T cells revealed that both the CD44highTCF1+CXCR6− progenitors and CD44highTCF1−CXCR6+ effectors expressed inhibitory receptors. Sustained expression of high levels of inhibitory receptors is associated with an altered T cell differentiation state called exhaustion (74). Nevertheless, we demonstrate that islet CD44highTCF1+CXCR6− CD8 T cells remain functional as they efficiently expand and give rise to the CD44highTCF1−CXCR6+ effector population to promote T1D development. In addition, IL-27Ra-deficient CD8 T cells have decreased capacity to differentiate into CD44highTCF1−CXCR6+ effecter cells (Figure 6) and exhibit reduced diabetogenic activity (26). Thus, both CD44highTCF1+CXCR6− and CD44highTCF1−CXCR6+ are important for T1D development. Interestingly, we observed that some CD44highTCF1−CXCR6+ CD8 T cells expressed very high levels of inhibitory receptors PD-1, LAG3, and TIM3. Furthermore, at the transcript level, Havcr2 (encoding TIM3) expressing cells were only found in the islets of 12- but not 7-week-old mice (data not shown). These observations raise the possibility that exhaustion of β-cell autoreactive CD8 T cells may represent a counteracting mechanism to restrain their effector function during T1D progression. Nevertheless, this mechanism eventually fails in NOD mice progressing to diabetes onset. This possibility is further supported by the rapid T1D onset caused by PD-1/PD-L1 or TIM3 blockade in NOD mice (75, 76). A recent study analyzing β cell-specific CD8 T cells in peripheral blood of patients with T1D reported a correlation between the frequency of an exhausted population and the rate of disease progression (77), demonstrating that exhaustion may also be a counteracting mechanism present in human T1D. However, it remains to be determined whether this type of self-regulation is intrinsic to CD8 T cells or is imposed by other extrinsic regulatory mechanisms.
We have previously shown that IL-27 signaling is required for T1D development in NOD mice and promoted the accumulation and effector phenotype of CD8 T cells within the pancreatic islets (26). In our current study, we expanded this research and discovered that IL-27 signaling promoted the expansion of the islet-infiltrating CD44highTCF1+CXCR6− CD8 T cells and simultaneously drove their differentiation into the CD44highTCF1−CXCR6+ terminal effector population. This result combined with our previous report showing that IL-27Rα-deficient CD8 T cells have a reduced ability to transfer T1D (26) further indicates that the CD44highTCF1−CXCR6+ effector population is important for the development of T1D. Previous literature studying the differentiation of effector CD8 T cells in the context of acute LCMV infection have shown that effector cell differentiation and the self-renewal of progenitors are coupled to cell proliferation (78). Therefore, IL-27 signaling may promote the proliferation of the islet-infiltrating CD44highTCF1+CXCR6− CD8 T cells thereby hastening their arrival at the terminally differentiated state. Both IL-27 and IL-6 utilize the gp130 receptor (24). IL-6 has been shown to stimulate CD8 T cell proliferation through a STAT3 mediated pathway (79, 80). Similarly, IL-27 also induces CD4 T cell, and likely CD8 T cell, proliferation through a STAT3 dependent mechanism (22, 81, 82). However, we have previously shown that IL-6-deficient NOD mice develop T1D at a rate identical to wildtype NOD mice (26). Therefore, additional mechanisms unique to IL-27 signaling are likely responsible for the effects of IL-27 on islet-infiltrating CD8 T cells.
Previous in vitro studies have shown that IL-27 signaling through STAT1 in CD8 T cells promoted synergistic upregulation of IFNγ production with IL-12 via upregulation of IL-12Rβ2 expression (23). Interestingly, in the context of systemic inflammation, it has been shown that downregulation of TCF1 during CD8 T cell cycling facilitates effector differentiation and is mediated via IL-12Rβ2/STAT4 signaling (83). Therefore, it is plausible that direct IL-27 signaling via STAT1 in islet-infiltrating CD8 T cells leads to downregulation of TCF1 expression via enhanced IL-12Rβ2 signaling. Although STAT1-deficient NOD mice are completely protected from T1D, NOD mice globally deficient in IL-12 still develop T1D (84, 85). Therefore, a novel mechanism likely underlies the effect of direct IL-27 signaling on the phenotype and function of islet-infiltrating CD8 T cells. Interestingly, a recent study analyzing the gene expression profiles of CD8 T cells stimulated in the presence or absence of IL-27 reported that Batf and Prdm1 were among the transcriptions factors significantly upregulated in response to IL-27 stimulation, whereas Tcf7 and Elk4 were among the transcription factors that were significantly downregulated in response to IL-27 stimulation (86). Given that we identified Batf and Prdm1 as signature regulons underlying the effector CD8 T cell state and Tcf7 and Elk4 as signature regulons underlying the progenitor-like CD8 T cell state in the pancreatic islets, IL-27 signaling may promote the differentiation of CD8 T cells during the progression of T1D by directly targeting key transcription factors. Future studies aimed at elucidating the specific signaling pathways downstream of IL-27 in autoreactive CD8 T cells are warranted.
Recently, two other studies utilizing scRNA-seq to analyze islet-infiltrating immune cells in NOD mice also described transcriptional heterogeneity among CD8 T cells (87, 88). Similar to our finding, the authors identified naïve, cycling, effector, and memory CD8 T cells as well as “exhausted” TCF1+ and TCF1− populations expressing inhibitory receptors. Our in vitro and in vivo studies provide additional information by showing the lineage relationship of functionally distinct CD44highTCF1+CXCR6− progenitors and CD44highTCF1−CXCR6+ terminally differentiated effectors, and that the presence of both populations is important for T1D progression. As CD8 T cell differentiation is a continuous process, it is most likely that cells within our CD44highTCF1+CXCR6− and CD44highTCF1−CXCR6+ populations can be further phenotypically and functionally refined. A limitation in these scRNA-seq studies is the lack of information regarding T cell specificity. Our flow cytometry data confirmed the existence of the CD44highTCF1+CXCR6− progenitors and CD44highTCF1−CXCR6+ terminally differentiated effectors among IGRP206–214-reactive CD8 T cells in the pancreatic islets. Additionally, we found that the relative proportion of CD44highTCF1−CXCR6+ effector cells was significantly higher among the IGRP206–214-reactive compared to other CD44high CD8 T cells. This result supports the likelihood that another layer of control of differentiation states within the islet-infiltrating CD8 T cells is conferred by antigen specificity.
In conclusion, we identified functional heterogeneity among islet-infiltrating CD8 T cells. Furthermore, we revealed a hierarchical structure of islet-infiltrating CD8 T cell differentiation and uncovered an important role for IL-27 signaling in this process. Completely defining the mechanisms underlying islet autoreactive CD8 T cell differentiation will have important implications for the development of targeted immunotherapies for T1D.
Supplementary Material
Key Points.
Functionally distinct subsets of CD8 T cells infiltrate islets in type 1 diabetes.
Self-renewing islet CD44highTCF1+ CD8 T cells maintain the autoreactive response.
IL-27 modulates the differentiation of islet-infiltrating CD8 T cell subsets.
Acknowledgements:
We are grateful to Caren Crumer for excellent mouse colony management. We thank the molecular biology core lab at the Blood Research Institute for scRNA-seq technical assistance. We thank the Children’s Research Institute flow cytometry and histology cores for technical assistance. We thank the National Institutes of Health Tetramer Core Facility for providing MHC class I tetramers.
This work is supported by NIH grants DK107541, AI144360, and DK121747 (to Y.-G.C), AI125741 and AI148403 (to W.C.), DK118786 (to A.E.C.), DK108557 (to D.M.S), DK127526 (to M.Y.K), and HL126166 and HL139008 (to W.R.D.). D.M.S. and M.Y.K are members of the Medical Scientist Training Program at the Medical College of Wisconsin, which is partially supported by a training grant from NIGMS (T32-GM080202).
Footnotes
Competing interests: The authors have no financial conflicts of interest.
Data and code availability:
The scRNA-seq data generated in this paper is available in the GEO database under accession number GSE155595. URL: https://urldefense.com/v3/__https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155595__;!!H8mHWRdzp34!qrtUxGRWeXWbeLMYOt3UoFJSSo_Iwhnt04fkUtBdKYv1DqKTLcOZy3SQdAU6poA$
References:
- 1.Serreze DV, and Leiter EH. Genes and cellular requirements for autoimmune diabetes susceptibility in nonobese diabetic mice. Curr Dir Autoimmun. 2001;4:31–67. [DOI] [PubMed] [Google Scholar]
- 2.Codina-Busqueta E, Scholz E, Munoz-Torres PM, Roura-Mir C, Costa M, Xufre C, et al. TCR bias of in vivo expanded T cells in pancreatic islets and spleen at the onset in human type 1 diabetes. J Immunol. 2011;186(6):3787–97. [DOI] [PubMed] [Google Scholar]
- 3.Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. 2012;209(1):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christianson SW, Shultz LD, and Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42(1):44–55. [DOI] [PubMed] [Google Scholar]
- 5.Serreze DV, Leiter EH, Christianson GJ, Greiner D, and Roopenian DC. Major histocompatibility complex class I-deficient NOD-B2mnull mice are diabetes and insulitis resistant. Diabetes. 1994;43(3):505–9. [DOI] [PubMed] [Google Scholar]
- 6.Wicker LS, Leiter EH, Todd JA, Renjilian RJ, Peterson E, Fischer PA, et al. Beta 2-microglobulin-deficient NOD mice do not develop insulitis or diabetes. Diabetes. 1994;43(3):500–4. [DOI] [PubMed] [Google Scholar]
- 7.Gagnerault MC, Luan JJ, Lotton C, and Lepault F. Pancreatic lymph nodes are required for priming of beta cell reactive T cells in NOD mice. J Exp Med. 2002;196(3):369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamilton-Williams EE, Palmer SE, Charlton B, and Slattery RM. Beta cell MHC class I is a late requirement for diabetes. Proc Natl Acad Sci U S A. 2003;100(11):6688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham KL, Krishnamurthy B, Fynch S, Mollah ZU, Slattery R, Santamaria P, et al. Autoreactive cytotoxic T lymphocytes acquire higher expression of cytotoxic effector markers in the islets of NOD mice after priming in pancreatic lymph nodes. Am J Pathol. 2011;178(6):2716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graham KL, Krishnamurthy B, Fynch S, Ayala-Perez R, Slattery RM, Santamaria P, et al. Intra-islet proliferation of cytotoxic T lymphocytes contributes to insulitis progression. Eur J Immunol. 2012;42(7):1717–22. [DOI] [PubMed] [Google Scholar]
- 11.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradfield JP, Qu HQ, Wang K, Zhang H, Sleiman PM, Kim CE, et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011;7(9):e1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evangelou M, Smyth DJ, Fortune MD, Burren OS, Walker NM, Guo H, et al. A method for gene-based pathway analysis using genomewide association study summary statistics reveals nine new type 1 diabetes associations. Genet Epidemiol. 2014;38(8):661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fortune MD, Guo H, Burren O, Schofield E, Walker NM, Ban M, et al. Statistical colocalization of genetic risk variants for related autoimmune diseases in the context of common controls. Nat Genet. 2015;47(7):839–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39(7):857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet. 2015;47(4):381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper JD, Smyth DJ, Smiles AM, Plagnol V, Walker NM, Allen JE, et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet. 2008;40(12):1399–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergholdt R, Brorsson C, Palleja A, Berchtold LA, Floyel T, Bang-Berthelsen CH, et al. Identification of novel type 1 diabetes candidate genes by integrating genome-wide association data, protein-protein interactions, and human pancreatic islet gene expression. Diabetes. 2012;61(4):954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45(10):1238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasela S, Kisand K, Tserel L, Kaleviste E, Remm A, Fischer K, et al. Pathogenic implications for autoimmune mechanisms derived by comparative eQTL analysis of CD4+ versus CD8+ T cells. PLoS Genet. 2017;13(3):e1006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity. 2002;16(6):779–90. [DOI] [PubMed] [Google Scholar]
- 22.Schneider R, Yaneva T, Beauseigle D, El-Khoury L, and Arbour N. IL-27 increases the proliferation and effector functions of human naive CD8+ T lymphocytes and promotes their development into Tc1 cells. Eur J Immunol. 2011;41(1):47–59. [DOI] [PubMed] [Google Scholar]
- 23.Morishima N, Owaki T, Asakawa M, Kamiya S, Mizuguchi J, and Yoshimoto T. Augmentation of effector CD8+ T cell generation with enhanced granzyme B expression by IL-27. J Immunol. 2005;175(3):1686–93. [DOI] [PubMed] [Google Scholar]
- 24.Morishima N, Mizoguchi I, Okumura M, Chiba Y, Xu M, Shimizu M, et al. A pivotal role for interleukin-27 in CD8+ T cell functions and generation of cytotoxic T lymphocytes. J Biomed Biotechnol. 2010;2010:605483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pennock ND, Gapin L, and Kedl RM. IL-27 is required for shaping the magnitude, affinity distribution, and memory of T cells responding to subunit immunization. Proc Natl Acad Sci U S A. 2014;111(46):16472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ciecko AE, Foda B, Barr JY, Ramanathan S, Atkinson MA, Serreze DV, et al. Interleukin-27 Is Essential for Type 1 Diabetes Development and Sjogren Syndrome-like Inflammation. Cell Rep. 2019;29(10):3073–86 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537(7620):417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol. 2016;1(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity. 2016;45(2):415–27. [DOI] [PubMed] [Google Scholar]
- 30.Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, et al. CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity. 2019;51(6):1028–42 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity. 2020;52(5):825–41 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Z, Zak J, Pratumchai I, Shaabani N, Vartabedian VF, Nguyen N, et al. IL-27 promotes the expansion of self-renewing CD8(+) T cells in persistent viral infection. J Exp Med. 2019;216(8):1791–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen YG, Forsberg MH, Khaja S, Ciecko AE, Hessner MJ, and Geurts AM. Gene targeting in NOD mouse embryos using zinc-finger nucleases. Diabetes. 2014;63(1):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foda BM, Ciecko AE, Serreze DV, Ridgway WM, Geurts AM, and Chen YG. The CD137 Ligand Is Important for Type 1 Diabetes Development but Dispensable for the Homeostasis of Disease-Suppressive CD137(+) FOXP3(+) Regulatory CD4 T Cells. J Immunol. 2020;204(11):2887–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–902 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trudeau JD, Kelly-Smith C, Verchere CB, Elliott JF, Dutz JP, Finegood DT, et al. Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J Clin Invest. 2003;111(2):217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao C, Sun HW, Lacey NE, Ji Y, Moseman EA, Shih HY, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol. 2019;20(7):890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019;571(7764):265–9. [DOI] [PubMed] [Google Scholar]
- 40.Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448(7155):811–5. [DOI] [PubMed] [Google Scholar]
- 41.Xing Z, Conway EM, Kang C, and Winoto A. Essential role of survivin, an inhibitor of apoptosis protein, in T cell development, maturation, and homeostasis. J Exp Med. 2004;199(1):69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nielsen CF, Zhang T, Barisic M, Kalitsis P, and Hudson DF. Topoisomerase IIalpha is essential for maintenance of mitotic chromosome structure. Proc Natl Acad Sci U S A. 2020;117(22):12131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, and Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133(4):1710–5. [PubMed] [Google Scholar]
- 44.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dragovich MA, Adam K, Strazza M, Tocheva AS, Peled M, and Mor A. SLAMF6 clustering is required to augment T cell activation. PLoS One. 2019;14(6):e0218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. 2011;12(12):1221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity. 2019;51(6):1043–58 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Surh CD, and Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29(6):848–62. [DOI] [PubMed] [Google Scholar]
- 49.Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature. 2006;442(7100):299–302. [DOI] [PubMed] [Google Scholar]
- 50.Hamilton SE, and Jameson SC. CD8 T cell quiescence revisited. Trends Immunol. 2012;33(5):224–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, et al. Tcf1 and Lef1 transcription factors establish CD8(+) T cell identity through intrinsic HDAC activity. Nat Immunol. 2016;17(6):695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feng X, Wang H, Takata H, Day TJ, Willen J, and Hu H. Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat Immunol. 2011;12(6):544–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao X, Shan Q, and Xue HH. TCF1 in T cell immunity: a broadened frontier. Nat Rev Immunol. 2021. [DOI] [PubMed] [Google Scholar]
- 54.Ouyang W, Beckett O, Flavell RA, and Li MO. An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity. 2009;30(3):358–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sumara I, Vorlaufer E, Gieffers C, Peters BH, and Peters JM. Characterization of vertebrate cohesin complexes and their regulation in prophase. J Cell Biol. 2000;151(4):749–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bancos S, Cao Q, Bowers WJ, and Crispe IN. Dysfunctional memory CD8+ T cells after priming in the absence of the cell cycle regulator E2F4. Cellular immunology. 2009;257(1–2):44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serdobova I, Pla M, Reichenbach P, Sperisen P, Ghysdael J, Wilson A, et al. Elf-1 contributes to the function of the complex interleukin (IL)-2-responsive enhancer in the mouse IL-2 receptor alpha gene. J Exp Med. 1997;185(7):1211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi HJ, Geng Y, Cho H, Li S, Giri PK, Felio K, et al. Differential requirements for the Ets transcription factor Elf-1 in the development of NKT cells and NK cells. Blood. 2011;117(6):1880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Naluyima P, Lal KG, Costanzo MC, Kijak GH, Gonzalez VD, Blom K, et al. Terminal Effector CD8 T Cells Defined by an IKZF2(+)IL-7R(−) Transcriptional Signature Express FcgammaRIIIA, Expand in HIV Infection, and Mediate Potent HIV-Specific Antibody-Dependent Cellular Cytotoxicity. J Immunol. 2019;203(8):2210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heim K, Binder B, Sagar, Wieland D, Hensel N, Llewellyn-Lacey S, et al. TOX defines the degree of CD8+ T cell dysfunction in distinct phases of chronic HBV infection. Gut. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, and Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37(6):1130–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A. 2019;116(25):12410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, and Held W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci U S A. 2010;107(21):9777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, and Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33(2):229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Forsberg MH, Ciecko AE, Bednar KJ, Itoh A, Kachapati K, Ridgway WM, et al. CD137 Plays Both Pathogenic and Protective Roles in Type 1 Diabetes Development in NOD Mice. J Immunol. 2017;198(10):3857–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Calderon B, Carrero JA, Miller MJ, and Unanue ER. Entry of diabetogenic T cells into islets induces changes that lead to amplification of the cellular response. Proc Natl Acad Sci U S A. 2011;108(4):1567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sandor AM, Lindsay RS, Dyjack N, Whitesell JC, Rios C, Bradley BJ, et al. CD11c(+) Cells Are Gatekeepers for Lymphocyte Trafficking to Infiltrated Islets During Type 1 Diabetes. Front Immunol. 2019;10:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He Q, Morillon YM 2nd, Spidale NA, Kroger CJ, Liu B, Sartor RB, et al. Thymic development of autoreactive T cells in NOD mice is regulated in an age-dependent manner. Journal of immunology. 2013;191(12):5858–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.D’Cruz LM, Lind KC, Wu BB, Fujimoto JK, and Goldrath AW. Loss of E protein transcription factors E2A and HEB delays memory-precursor formation during the CD8+ T-cell immune response. Eur J Immunol. 2012;42(8):2031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schauder DM, Shen J, Chen Y, Kasmani MY, Kudek MR, Burns R, et al. E2A-regulated epigenetic landscape promotes memory CD8 T cell differentiation. Proc Natl Acad Sci U S A. 2021;118(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xin G, Schauder DM, Lainez B, Weinstein JS, Dai Z, Chen Y, et al. A Critical Role of IL-21-Induced BATF in Sustaining CD8-T-Cell-Mediated Chronic Viral Control. Cell Rep. 2015;13(6):1118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grusdat M, McIlwain DR, Xu HC, Pozdeev VI, Knievel J, Crome SQ, et al. IRF4 and BATF are critical for CD8(+) T-cell function following infection with LCMV. Cell Death Differ. 2014;21(7):1050–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Y, Zander RA, Wu X, Schauder DM, Kasmani MY, Shen J, et al. BATF regulates progenitor to cytolytic effector CD8(+) T cell transition during chronic viral infection. Nat Immunol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wherry EJ, and Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198(1):63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4(11):1093–101. [DOI] [PubMed] [Google Scholar]
- 77.Wiedeman AE, Muir VS, Rosasco MG, DeBerg HA, Presnell S, Haas B, et al. Autoreactive CD8+ T cell exhaustion distinguishes subjects with slow type 1 diabetes progression. J Clin Invest. 2020;130(1):480–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin WW, Nish SA, Yen B, Chen YH, Adams WC, Kratchmarov R, et al. CD8(+) T Lymphocyte Self-Renewal during Effector Cell Determination. Cell Rep. 2016;17(7):1773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gagnon J, Ramanathan S, Leblanc C, Cloutier A, McDonald PP, and Ilangumaran S. IL-6, in synergy with IL-7 or IL-15, stimulates TCR-independent proliferation and functional differentiation of CD8+ T lymphocytes. J Immunol. 2008;180(12):7958–68. [DOI] [PubMed] [Google Scholar]
- 80.Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, and Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161(9):4652–60. [PubMed] [Google Scholar]
- 81.Owaki T, Asakawa M, Morishima N, Mizoguchi I, Fukai F, Takeda K, et al. STAT3 is indispensable to IL-27-mediated cell proliferation but not to IL-27-induced Th1 differentiation and suppression of proinflammatory cytokine production. J Immunol. 2008;180(5):2903–11. [DOI] [PubMed] [Google Scholar]
- 82.Brender C, Tannahill GM, Jenkins BJ, Fletcher J, Columbus R, Saris CJ, et al. Suppressor of cytokine signaling 3 regulates CD8 T-cell proliferation by inhibition of interleukins 6 and 27. Blood. 2007;110(7):2528–36. [DOI] [PubMed] [Google Scholar]
- 83.Danilo M, Chennupati V, Silva JG, Siegert S, and Held W. Suppression of Tcf1 by Inflammatory Cytokines Facilitates Effector CD8 T Cell Differentiation. Cell Rep. 2018;22(8):2107–17. [DOI] [PubMed] [Google Scholar]
- 84.Kim S, Kim HS, Chung KW, Oh SH, Yun JW, Im SH, et al. Essential role for signal transducer and activator of transcription-1 in pancreatic beta-cell death and autoimmune type 1 diabetes of nonobese diabetic mice. Diabetes. 2007;56(10):2561–8. [DOI] [PubMed] [Google Scholar]
- 85.Trembleau S, Penna G, Gregori S, Chapman HD, Serreze DV, Magram J, et al. Pancreas-infiltrating Th1 cells and diabetes develop in IL-12-deficient nonobese diabetic mice. J Immunol. 1999;163(5):2960–8. [PubMed] [Google Scholar]
- 86.Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature. 2018;558(7710):454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zakharov PN, Hu H, Wan X, and Unanue ER. Single-cell RNA sequencing of murine islets shows high cellular complexity at all stages of autoimmune diabetes. J Exp Med. 2020;217(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hu H, Zakharov PN, Peterson OJ, and Unanue ER. Cytocidal macrophages in symbiosis with CD4 and CD8 T cells cause acute diabetes following checkpoint blockade of PD-1 in NOD mice. Proc Natl Acad Sci U S A. 2020;117(49):31319–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNA-seq data generated in this paper is available in the GEO database under accession number GSE155595. URL: https://urldefense.com/v3/__https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155595__;!!H8mHWRdzp34!qrtUxGRWeXWbeLMYOt3UoFJSSo_Iwhnt04fkUtBdKYv1DqKTLcOZy3SQdAU6poA$