

Abstract
Poly(ADP-ribose) polymerase-1 (PARP-1), has gained considerable attention as a target for therapeutic inhibitors in breast cancers. Previously we showed that PARP-1 localizes to active gene promoters to regulate histone methylation and RNA polymerase II activity (Pol II), altering the expression of various tumor-related genes. Here we report a role for PARP-1 in estrogen-dependent transcription in estrogen receptor alpha (ERα)-positive (ER+) breast cancers. Global nuclear run-on and sequencing (GRO-seq) analyses functionally linked PARP-1 to the direct control of estrogen-regulated gene expression in ER+ MCF-7 breast cancer cells by promoting transcriptional elongation by Pol II. Furthermore, ChIP-seq analyses revealed that PARP-1 regulates the estrogen-dependent binding of ERα and FoxA1 to a subset of genomic ERα binding sites, promoting active enhancer formation. Moreover, we found that the expression levels of the PARP-1- and estrogen-coregulated gene set are enriched in the luminal subtype of breast cancers, and high PARP-1 expression in ER+ cases correlates with poor survival. Finally, treatment with a PARP inhibitor or a transcriptional elongation inhibitor attenuated estrogen-dependent growth of multiple ER+ breast cancer cell lines. Taken together, our results show that PARP-1 regulates critical molecular pathways that control the estrogen-dependent gene expression program underlying the proliferation of ER+ breast cancer cells.
Keywords: Poly(ADP-ribose) polymerase-1 (PARP-1), estrogen signaling, transcription, estrogen receptor alpha (ERα), FoxA1, breast cancer
Introduction
Estrogen signaling regulates molecular events that have profound effects on the normal functioning of critical biological processes in normal and disease states. Upon binding to 17β-estradiol (E2), estrogen receptor alpha (ERα) localizes to regulatory regions across the genome to promote the formation of transcriptional enhancers that drive the E2-dependent expression of target genes (1,2). In many cases, the binding of pioneer factors, such as FoxA1, precedes E2-dependent binding of ERα, which is a critical step for E2-dependent transcription (3–5). Activation of ER-dependent transcription drives enhancer-promoter looping and transcription by RNA polymerase II (RNA Pol II) at target promoters (2,6–9).
Aberrant estrogen signaling plays a critical role in several pathophysiological conditions, including ERα-positive (ER+) breast cancers (10). Around 70% of breast cancers express ERα at the time of diagnosis and, therefore, respond well to antiestrogen therapies; however, the majority of them relapse and become refractory (10). Understanding how additional signaling pathways, regulatory cofactors, and regulatory post-translational modifications (PTMs) impact E2-signaling and ERα-dependent gene regulation may suggest new targets for treating ER+ breast cancers. Herein, we explore the role of one such cofactor, poly(ADP-ribose) polymerase-1 (PARP-1), and the PTM it mediates, ADP-ribosylation (ADPRylation), in ERα-dependent gene regulation in ER+ breast cancers.
PARP-1, the founding and most abundant member of the PARP family of enzymes, is an ADP-ribosyl transferase with many hundreds of known substrates (11,12). Yet, the precise nature of this modification, how it regulates its substrates’ functions, and its global effects on the genome and proteome are not well understood. PARP-1 is upregulated in a number of cancer cell lines, as well as in malignant tissues (13,14). In breast cancer cell lines, the basal activity of PARP-1 is highly variable and independent of DNA damage (15). In ovarian cancers, the levels and patterns of PARP-1-dependent ADPRylation have been shown to correlate well with clinical outcomes (16). Thus, accumulating evidence has brought PARP-1 to the forefront of clinical cancer research as an emerging therapeutic target in several cancers, including breast, ovarian, and prostate cancers (17).
In its historical role in DNA repair, inhibition of the catalytic activity of PARP-1 was shown to be efficient in treating BRCA1/2 mutant or homologous recombination (HR)-deficient cancers by inducing cell death through synthetic lethality (17). However, our recent findings indicate that the use of PARP inhibitors in the clinic could potentially be expanded to include a broader array of cancer types regardless of BRCA1/2 status, acting through alternate molecular pathways unrelated to DNA repair (16,18). For example, PARP-1 has been shown to regulate gene expression by modulating chromatin structure and acting as a transcriptional coregulator (19). Our previous studies have shown that PARP-1 localizes to the promoters of more than 90% of expressed genes in MCF-7 ER+ breast cancer cells (20) and regulates transcriptional elongation by RNA polymerase II (12). In this study, we interrogate the role of PARP-1 in E2-dependent transcription in ER+ breast cancer cells.
Materials and Methods
Additional details regarding the materials and methods are provided in the Supplementary Materials.
Antibodies
Details for the following antibodies used are provided in the Supplementary Materials: PARP-1, ERα, FoxA1, H3K27ac and β-actin.
Cell Culture and Treatments
MCF-7 cells were kindly provided by Benita S. Katzenellenbogen (University of Illinois, Urbana-Champaign) and T47D cells were obtained from the ATCC and used for the genomic and cell-based assays described herein. Prior to all experiments, the MCF-7 and T47D cells were grown for 3 days in phenol red-free MEM Eagle or RPMI medium supplemented with 5% charcoal-dextran-treated calf serum (CDCS) or 10% charcoal-dextran-treated FBS (CDFBS), respectively. For experiments, cells were treated with 100 nM 17β-estradiol (E2) or vehicle (ethanol) for 40 or 180 minutes. Fresh cell stocks were regularly replenished from the original stocks, verified for cell type identity using the GenePrint 24 system (Promega, B1870), and confirmed as mycoplasma-free every three months using a commercial testing kit.
Stable shRNA-Mediated Knockdown in MCF-7 Cells
Retroviruses were generated by transfection of pSUPER.retro vectors, each expressing a different shRNA sequence directed against the cognate target (Luciferase or PARP-1) (12). The resulting viruses were collected, filtered, and used to infect the parental MCF-7 cell line. Stably transduced cells were isolated under appropriate drug selection with 0.5 μg/mL puromycin or 800 μg/mL G418, expanded, and frozen in aliquots for future use.
Preparation of Cell Extracts and Western blotting
Preparation of whole cell lysates and determination of protein concentrations.
Cells were collected, washed with ice-cold PBS, resuspended in Whole Cell Lysis Buffer [50 mM Tris-HCl pH 7.5, 0.5 M NaCl, 1 mM EDTA, 1% NP-40, 10% glycerol, and 1x complete protease inhibitor cocktail (Roche, 11697498001)]. The cell extracts were collected, aliquoted, flash-frozen in liquid N2, and stored at −80 °C.
Western blotting.
Aliquots of the cell extracts were run on polyacrylamide-SDS gels, transferred to nitrocellulose membranes and blotted as described previously (5,7,8). The signals were detected using an ECL detection reagent (Thermo Fisher Scientific, 34077, 34095).
Cell Proliferation Assays
Cell proliferation was assessed using a crystal violet staining assay as described previously (21). MCF-7 or T47D cells were grown in CDCS or CDFBS medium, respectively, for 3 days and treated with vehicle or E2 (100 nM), with or without PARP-1 inhibitor (Niraparib, Olaparib, Talazoparib) and with or without Flavopiridol, as indicated. At selected time points, the cells were fixed with 10% formaldehyde and stained with 0.1% crystal violet, which was extracted using 10% glacial acetic acid read at 595 nm.
Preparation of Global Run-On (GRO)-Sequencing Libraries
Nuclei from MCF-7 cells with shRNA-mediated knockdown of luciferase (Luc; as a control) or PARP-1 were isolated and subjected to GRO-seq as described previously (12,22). Library quality was assessed using a 2200 TapeStation (Agilent Technologies).
Analysis of GRO-seq Data
The GRO-seq data were analyzed using software described previously (5,22) and the approaches described below. Software, scripts, and other information about the analyses can be obtained by contacting the corresponding author (W.L.K.).
Quality Control.
The GRO-seq data quality was assessed using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adapter contamination and polyA tails were removed from GRO-seq reads using the default parameters of Cutadapt (v1.9.1) software (23). Reads >32 bp long were retained for alignment.
Read Alignment and Gene Annotation.
Reads were aligned to the human reference genome (hg19), including autosomes, the X chromosome, one complete copy of an rDNA repeat (GenBank ID: U13369.1) using the BWA aligner (v 0.7.12) (24). Overlaps and redundancies were removed from the combined gene lists to eliminate the possibility of double counting.
Determining Gene Regulation and Generating Heatmaps.
The effects of E2, PARP-1 knockdown, and co-treatment (E2 + PARP-1 knockdown) on the expression of coding and non-coding genes were analyzed using edgeR (25). A false discovery rate (FDR) cutoff of 1% was used to identify significantly regulated genes. Genes in the heatmaps were ordered using hierarchical clustering. We used custom R scripts to convert counts to transcripts per million (TPM) for each replicate, as well as each pool of replicates.
Metagene Analysis.
The average read densities of sense and anti-sense reads were computed on adjacent lines for an 8 kb window surrounding regulated gene TSS (±4 kb) using the metagene function in the groHMM package (26).
Analysis of Pausing Indices.
Pausing indices representing the base pair-normalized difference in read depth between the promoter proximal region (−100 – 300 bp) and the gene body (1–13 kb) were calculated using the pausing Index function in the groHMM package, as previously described (26,27).
Preparation of Chromatin Immunoprecipitation (ChIP)-Sequencing Libraries
ChIP was performed as previously described (5,8). Pre-cleared crosslinked chromatin-containing lysates were subjected to immunoprecipitation reactions with antibodies against ERα, FoxA1, or H3K27ac. Ten ng of ChIPed DNA for each condition was used to generate libraries for deep sequencing, as previously described (5,28). After quality control analyses, the libraries were sequenced using a HiSeq 2000 sequencer (Illumina; Single-end reads, 50 bp for all samples).
Analysis of ChIP-seq Data
Quality Control.
Quality control for the ChIP-seq data was performed using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
Read Alignment and Peak Calling.
The raw reads were aligned to the human reference genome (GRCh37/hg19) using default parameters in Bowtie (ver. 1.0.0) (29). The aligned reads were subsequently filtered for quality and uniquely mappable reads using Samtools (ver. 0.1.19) (30) and Picard (ver. 1.127; http://broadinstitute.github.io/picard/). Library complexity was measured using BEDTools (v2.17.0) (31) and met minimum ENCODE data quality standards (32). Relaxed peaks were called using MACS (v2.1.0) (33) with a p-value = 1 × 10−2 for each replicate, pooled replicates’ reads, and pseudoreplicates (Supplementary Table S1).
Effect of PARP-1 Knockdown on ERα and FoxA1 Binding.
To identify the ERα and FoxA1 binding sites affected by PARP-1 knockdown, we used BEDTools (v2.17.0) (31) to find the closest peaks from PARP-1 affected target genes upon E2 treatment.
ChIP-qPCR
ChIP-qPCR assays were performed as previously described (5,7,8). Pre-cleared crosslinked chromatin-containing lysates were subjected to immunoprecipitation reactions with antibodies against ERα and FoxA1. The ChIPed DNA was extracted and analyzed by quantitative PCR using the primers listed in the Supplementary Methods.
Preparation of polyA+ RNA-Sequencing Libraries
Total RNA isolation.
Estrogen-withdrawn MCF-7 cells were treated with ethanol or 100 nM E2 for 3 hours. Total RNA was isolated from MCF-7 cells using the RNeasy kit (Qiagen, 74136) according to the manufacturer’s instructions.
Library preparation and sequencing.
The total RNA samples were subjected to enrichment of polyA+ RNA and sequencing as described previously (34). After quality control analyses, the libraries were sequenced using a HiSeq 2000 sequencer (Illumina; Single-end reads, 50 bp for all samples). At least two biological replicates were sequenced for each condition for a minimum of roughly 20 million raw reads.
Analysis of RNA-seq Data
Quality control.
Quality control for the RNA-seq data was performed using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
Read alignment.
The reads were then mapped to the human reference genome (GRCh37/hg19) with previously described comprehensive gene annotation using the default parameters in Tophat (v2.0.12).
Differential gene expression.
Differences in gene expression between RNA-seq datasets were calculated using the cufflinks suite with a statistical threshold of FDR, 0.05 (35).
Pathway Analysis
Gene set enrichment analysis (GSEA) was performed by computing overlaps between pre-ranked genes with c2: curated gene sets (canonical pathways) obtained from the Broad Institute (http://software.broadinstitute.org/gsea/msigdb) (36).
Kaplan-Meier and Gene Expression Analyses in Breast Cancer Tumor Samples
Kaplan-Meier estimators (37,38) were generated using the Gene Expression-Based Outcome for Breast Cancer Online (GOBO) tool (http://co.bmc.lu.se/gobo/) (39). Gene expression levels in breast tumor samples were also obtained using the GOBO tool.
Genomic Data Set Availability
The new genomic data sets reported herein (GRO-seq, RNA-seq and ChIP-seq from MCF-cells) are available from the NCBI’s Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) using the following accession numbers: GSE74142, GSE173976, and GSE166168 (see Supplementary Materials).
Results
PARP-1 regulates estrogen-dependent transcription
To investigate the role of PARP-1 in estrogen-dependent gene regulation, we examined and quantified the direct transcriptional output in PARP-1-depleted ERα-positive MCF-7 cells treated with 17β-estradiol (E2) using global run-on coupled with deep sequencing (GRO-seq). MCF-7 cell lines were generated with stable shRNA-mediated knockdown (KD) of Luciferase (Luc) or PARP-1 (KD; LucKD control versus PARP-1KD), which did not alter ERα or FoxA1 expression (Fig. 1A). The cells were treated with vehicle or E2 for 40 minutes and collected for GRO-seq analysis (Fig. 1B).
Figure 1. PARP-1 modulates the estrogen-dependent transcriptional program in breast cancer cells.
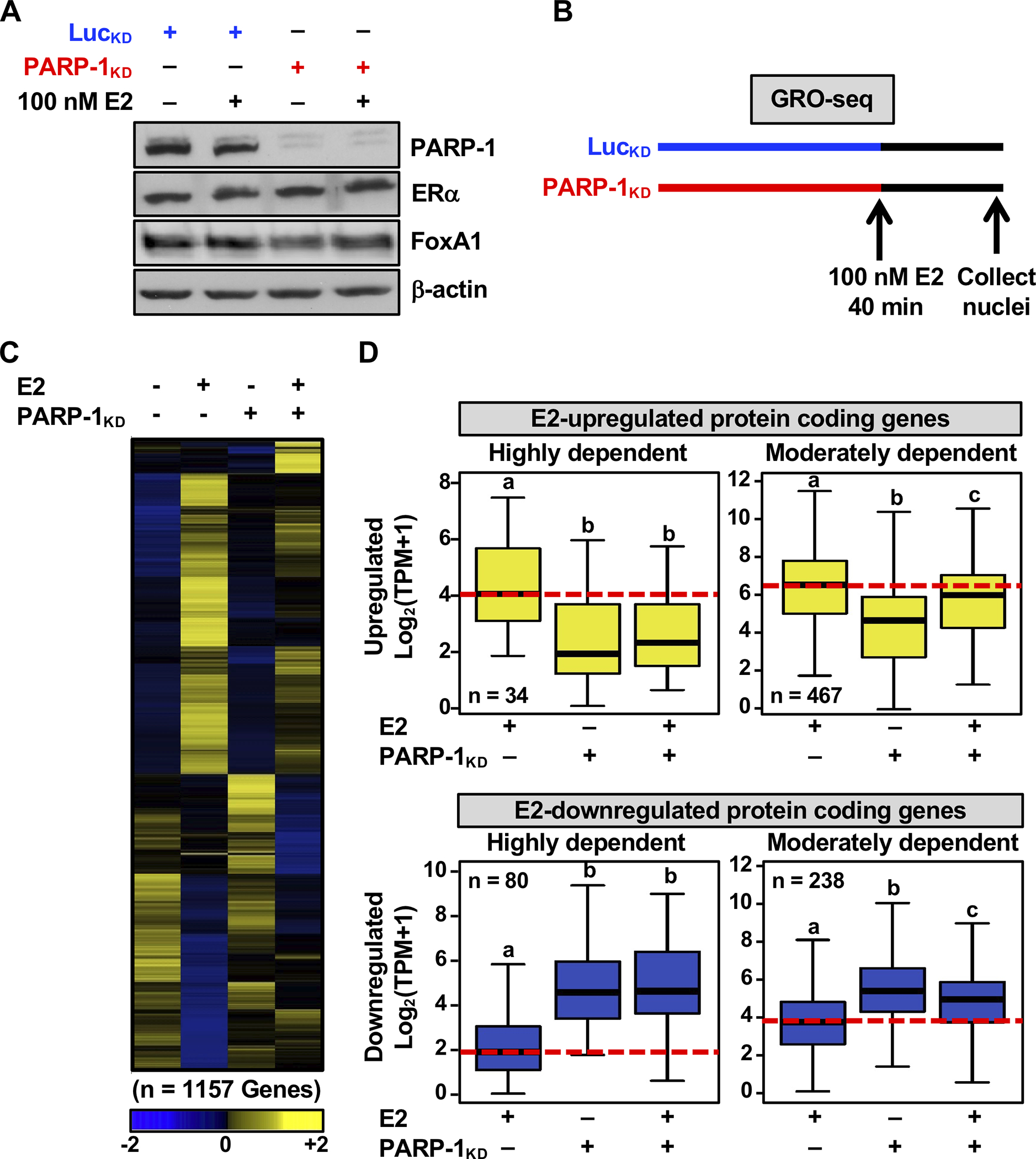
(A) Western blot showing the levels of PARP-1, ERα, and FoxA1 in MCF-7 whole cell lysates after shRNA-mediated knockdown (KD; LucKD control versus PARP-1KD).
(B) Schematic representation showing a timeline of Luc or PARP-1 knockdown (LucKD control, PARP-1KD), E2 treatment, and cell collection for the GRO-seq experiment.
(C) Heatmap showing the transcription regulation of all protein-coding genes under each treatment condition, as revealed by GRO-seq. The gene expression values (TPM) shown in the heatmap were z-score normalized for each gene.
(D) Box plots showing changes in GRO-seq signals for E2-regulated protein-coding (E2-upregulated, top; E2-downregulated, bottom) genes that are highly (left) or moderately (right) dependent on PARP-1. Bars marked with different letters are significantly different from each other (Wilcoxon rank sum test, p < 2.5 × 10−7).
Sequencing of newly synthesized transcripts identified a subset of the E2-regulated protein-coding genes that are dependent on PARP-1 for efficient expression (Fig. 1C and Supplementary Fig. S1A). Within this subset, E2-upregulated protein-coding genes that were highly or moderately dependent on PARP-1 were suppressed upon PARP-1 KD (Fig. 1D, upper panels, and Supplementary Fig. S1B, upper panels). In contrast, E2-downregulated protein-coding genes dependent on PARP-1 were de-repressed upon PARP-1 KD (Fig. 1D, lower panels, and Supplementary Fig. S1B, lower panels). Importantly, PARP-1 KD had no significant effect on all the total set of expressed genes in MCF-7 cells, indicating that PARP-1 selectively regulates E2- regulated genes (Supplementary Fig. S1C). A large fraction of the E2-regulated genes controlled by PARP-1 at the transcriptional level (as assessed by GRO-seq at 40 min. of E2 treatment) were similarly affected at the steady-state RNA level (as assessed by RNA-seq at 3 hours of E2) (Supplementary Fig. S1D). We also identified a third category comprising a small number of synergistically up- or down-regulated genes in response to E2 and PARP-1 KD (Supplementary Fig. S2, A and B). Similar regulation patterns were also observed for E2-regulated lncRNA genes (Supplementary Fig. S2, C and D).
PARP-1 regulates estrogen-dependent transcription by promoting release of Pol II into active elongation
Our previous studies have shown that the estrogen-dependent transcriptional response in breast cancer cells is rapid and dynamic, and involves a regulatory step the releases promoter-proximal paused Pol II into active elongation (22,27). Furthermore, our studies have shown that PARP-1 regulates transcriptional elongation by ADPRylating the RNA polymerase II (Pol II)-associated complex negative elongation factor (NELF) (12), which is also a target of inhibition by the P-TEFb kinase complex (40). Therefore, we explored the potential role of PARP-1 in regulating estrogen-dependent Pol II activity, including loading, pausing, and elongation genome-wide. For this, we analyzed GRO-seq data across the PARP-1- and E2-coregulated gene sets (Fig. 2). The GRO-seq read density at transcription start sites (TSSs) or in gene bodies is an indicator of Pol II activity (40). The change in read density indicates the effect of PARP-1 depletion on E2-dependent transcription.
Figure 2. PARP-1 positively regulates estrogen-dependent Pol II loading and elongation at E2-upregulated genes.
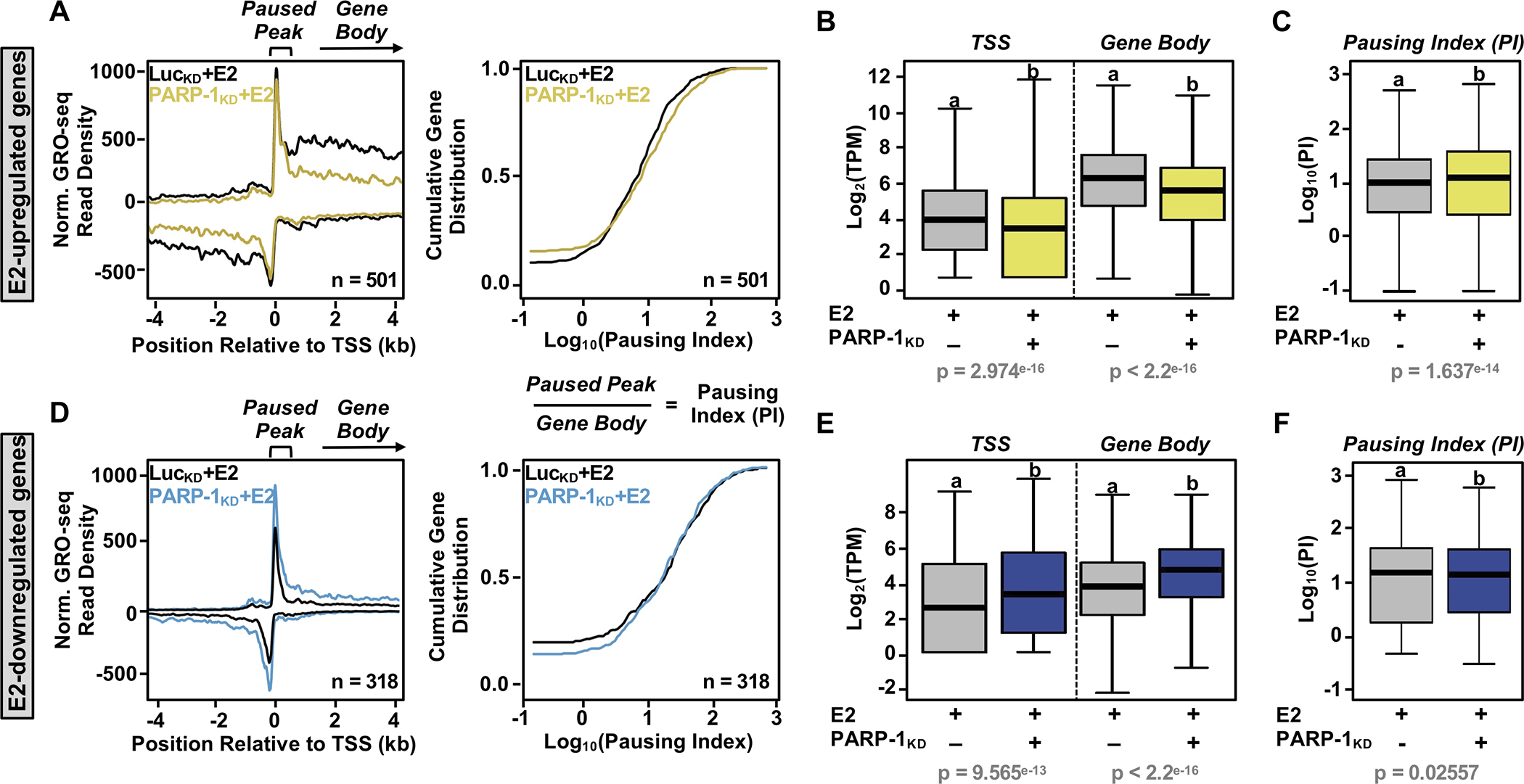
(A and D) Metagenes of GRO-seq read density at gene promoter regions within −4 to +4 kb of transcription start site (TSS) (left) and cumulative gene distribution plots of the pausing indices (right) of E2-upregulated (A) and E2-downregulated (D) protein-coding genes that are highly or moderately dependent on PARP-1. The data are from E2-treated MCF-7 cells subjected to shRNA-mediated knockdown (KD; LucKD control versus PARP-1KD).
(B and E) Box plots showing changes in GRO-seq signal (TPM) for PARP-1-dependent E2-upregulated (B) and E2-downregulated (E) genes around the promoter (100 bp to + 300 bp relative to the TSS) (left) and gene body (+300 to 13 kb relative to the TTS) (right).
(C and F) Box plots showing changes in the pausing index (PI) for PARP-1-dependent E2-upregulated (C) and E2-downregulated (F) genes.
In panels B, C, E, and F, bars marked with different letters are significantly different from each other (Wilcoxon rank sum test, p < 2.5 × 10−7 or 0.02557).
We observed that PARP-1 depletion reduced the density of elongating Pol II in the gene bodies of E2-upregulated genes, but only modestly affected the density of Pol II in the proximal promoter region (Fig. 2A, left panel). As a consequence, there was a modest increase in the pausing indices (i.e., GRO-seq reads in the paused peak/reads in the gene body) upon PARP-1 knockdown (Fig. 2A, right panel; rightward shift of the curve). In contrast, PARP-1 depletion increased the density of both gene body and promoter-proximal Pol II for the E2-downregulated genes (Fig. 2D, left panel), resulting in no observable change in the pausing indices (Fig. 2D, right panel). A more quantitative assessment of these analyses are shown in the boxplots in Fig. 2, B, C, E, and F, which highlight these observations. The effects of PARP-1 depletion on the pausing index was much greater for a selected set of E2-regulated genes that are highly dependent on PARP-1 (Supplementary Fig. S3A), as well as a selected set of E2-upregulated genes in various PARP-1-regulated pathways (Supplementary Fig. S3, B and C). Together, these results demonstrate that PARP-1 regulates the estrogen-dependent gene expression program at the transcriptional level, in part by acting to promote release of paused Pol II into productive elongation in response to estrogen signaling.
PARP-1 regulates estrogen-dependent binding of ERα and FoxA1 to their target sites
Distally located ERα-bound regions in the genome can function as enhancers to control target gene expression by establishing contact with neighboring promoters through chromatin looping (Fig. 3A). E2-dependent ERα binding in breast cancer cells is directly regulated by the ‘pioneer’ transcription factor FoxA1 (3,4), and ERα binding sites are marked by acetylated histone H3 lysine 27 (H3K27ac, an indicator of active enhancers) (8). To assess whether PARP-1 regulates E2-dependent binding of ERα or FoxA1 to cognate sites located near estrogen target genes, we performed ChIP-seq analyses for ERα or FoxA1 upon PARP-1 knockdown with or without E2 treatment (Fig. 3A). We observed a global decrease in ERα and FoxA1 binding with an increase in the distance between E2-upregulated target gene promoters and the nearest significant ERα or FoxA1 peak (Fig. 3B, rightward shift of the curve), and a significant increase in the distance to the closest ERα or FoxA1 peaks from the promoters of E2-upregulated protein-coding genes that are highly or moderately dependent on PARP-1 (Fig. 3C).
Figure 3. Role of PARP-1 in estrogen-dependent binding of ERα and FoxA1 near estrogen upregulated genes.
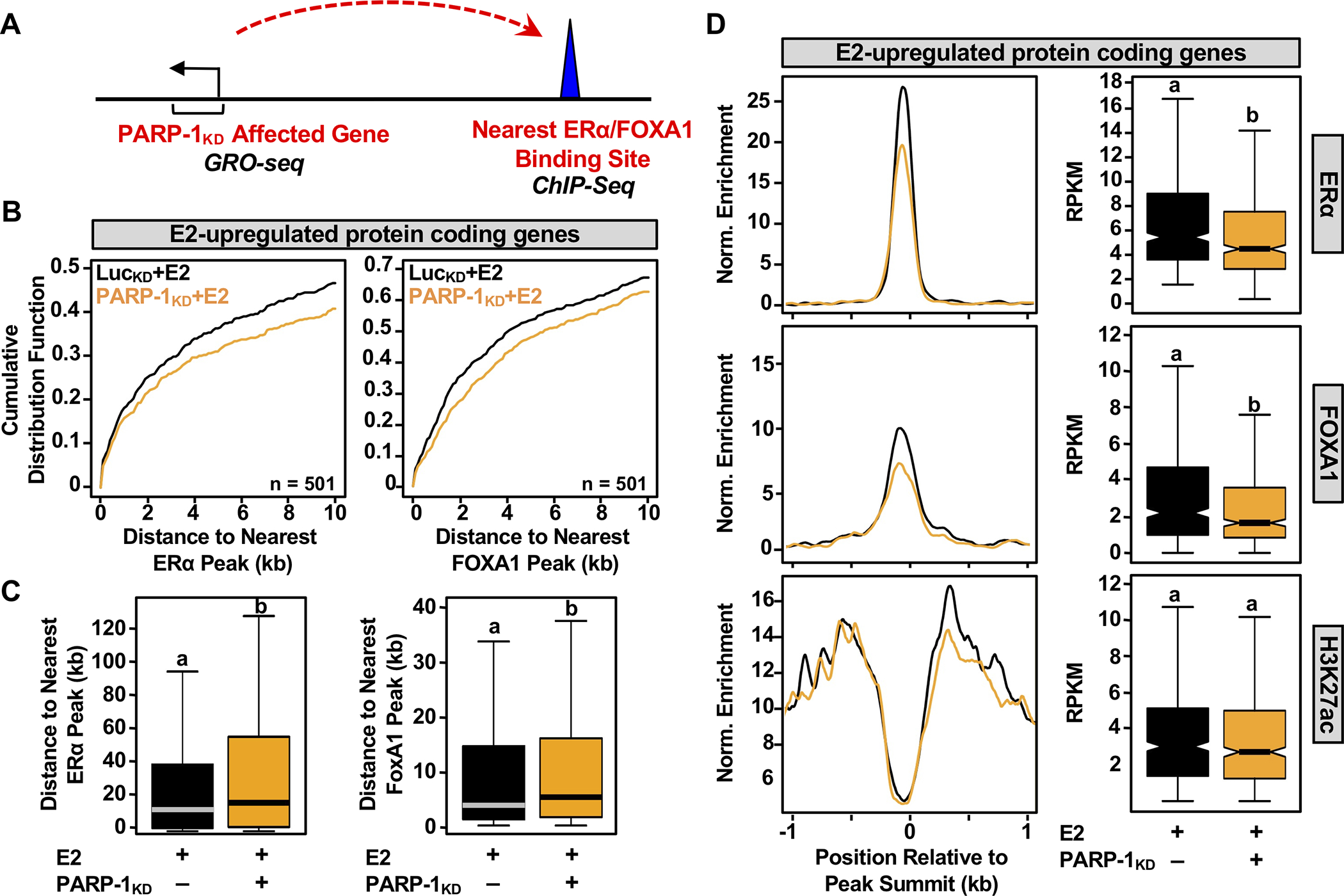
(A) Schematic overview of the pipeline for integrating ChIP-seq and GRO-seq data to link ERα and FoxA1 binding to PARP-1 affected target genes in MCF-7 cells upon E2 treatment.
(B) Cumulative distribution plots of distance to closest ERα or FoxA1 peaks from E2-upregulated protein-coding genes that are highly or moderately dependent on PARP-1.
(C) Box plots showing the distance to the closest ERα or FoxA1 peaks from the promoters of E2-upregulated protein-coding genes that are highly or moderately dependent on PARP-1. Bars marked with different letters are significantly different from each other (Wilcoxon rank sum test, p < 2.5 × 10−7).
(D) Metagene plots and boxplots showing a positive correlation between PARP-1 knockdown-mediated suppression at ERα binding sites within −1 and +1 kb with known co-binding markers. (Left) Metagene plots of ChIP-seq read counts in E2-treated MCF-7 cells subjected to shRNA-mediated knockdown for luciferase (black) or PARP-1 (yellow). (Right) Box plot representations of the corresponding ChIP-seq data at ERα binding sites. Bars marked with different letters are significantly different from each other (Wilcoxon rank sum test, p < 2.92 × 10−8).
From related metagene analyses and box plot quantifications, we observed that PARP-l knockdown significantly reduced E2-dependent ERα and FoxA1 binding to target sites neighboring E2-upregulated genes (Fig. 3D). We did not, however, observe a significant effect of PARP-1 depletion on H3K27ac enrichment (Fig. 3D, bottom panels), likely because the sites retained significant, albeit reduced, ERα and FoxA1 binding. The effects of PARP-1 depletion on ERα binding occurred regardless of whether there was co-binding with FoxA1 at the enhancer (Supplementary Fig. S4), suggesting that PARP-1 may act through multiple mechanisms, one targeting FoxA1 (which would also affect ERα) and one targeting ERα directly. The key features of these observations were evident in browser tracks of genomic data covering key E2 target genes (e.g., GREB1 and NRIP1; Fig. 4A and Supplementary Fig. 5, respectively). The effects of PARP-1 knockdown on ERα and FoxA1 binding were recapitulated using the PARP-1 inhibitor Niraparib, pointing to a role for PARP-1 catalytic activity (Fig. 4, B and C),.
Figure 4. PARP-1 catalytic activity supports estrogen-dependent binding of ERα and FoxA1 to enhancers.
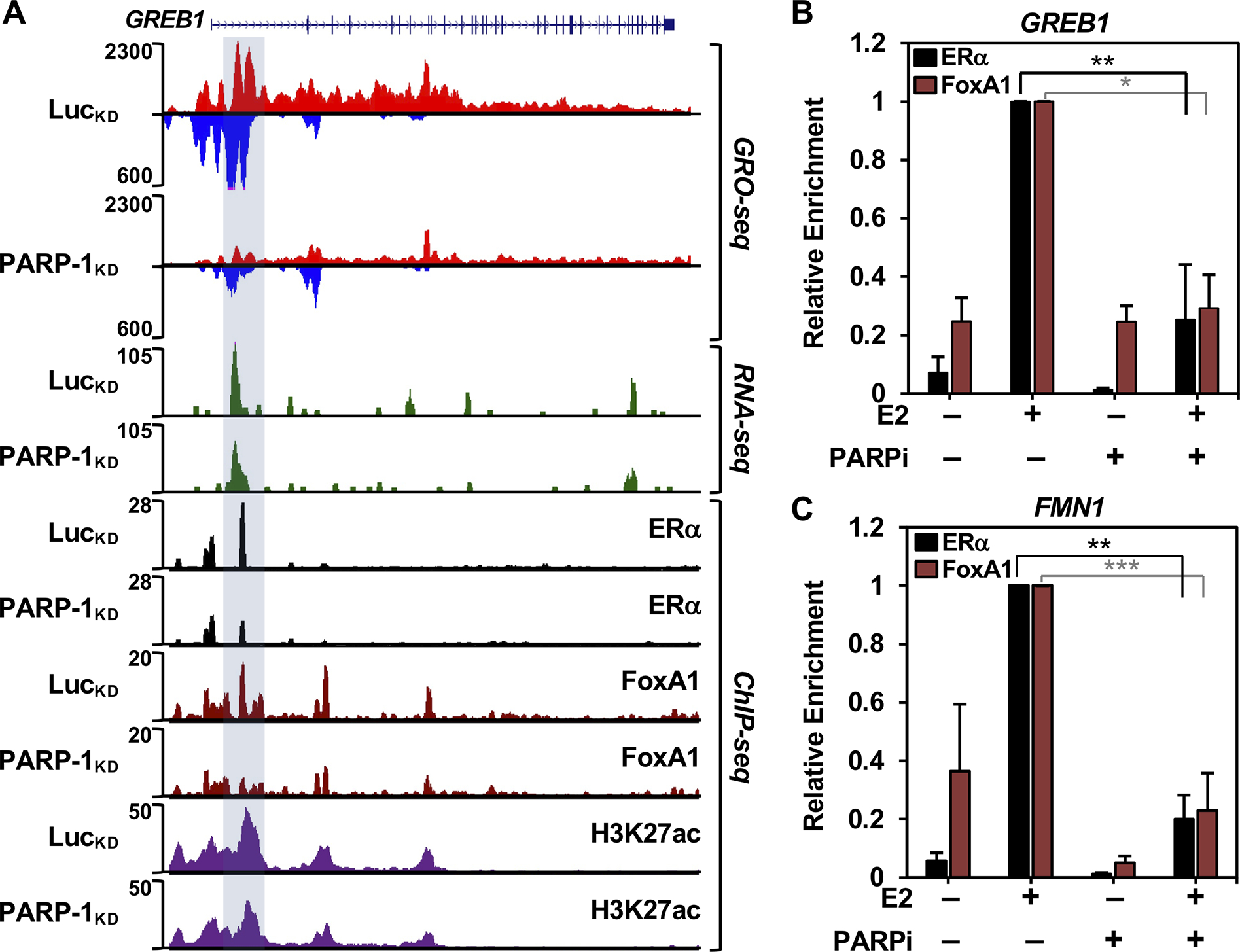
(A) Genome browser tracks of GRO-seq, RNA-seq, and ChIP-seq data at the PARP-1-dependent E2-upregulated GREB1 gene in LucKD vs. PARP-1KD MCF-7 cells upon E2 treatment.
(B and C) ChIP-qPCR analysis showing ERα and FoxA1 binding in the presence of E2 and/or Niraparib (PARP inhibitor; PARPi) to the distal ERα enhancer of the GREB1 gene (region highlighted in panel A) (B) and the FMN1 gene (C). Asterisks indicate significant differences assessed by unpaired t-tests (p-values: *<0.05, **<0.005, ***<0.0005).
Similar results were observed for FoxA1 with E2-downregulated genes (Supplementary Fig. S6A; ERα binding is negligible for these genes, so this assessment cannot be made). PARP-l knockdown also significantly reduced E2-dependent FoxA1 binding to target sites neighboring E2-downregulated genes (Supplementary Fig. S6B; again, ERα binding is negligible for these genes, so this assessment cannot be made). Together, these results indicate that PARP-1 regulates E2-dependent transcription, in part, by controlling the binding of ERα and FoxA1 to regulatory sites in the genome.
PARP-1 expression predicts clinical outcomes in ER+ breast cancers
To further examine the biological significance of PARP-1-mediated regulation of E2-dependent gene expression, we performed gene set enrichment analysis (GSEA) to identify enriched functional pathways. GSEA identified relevant terms associated with breast biology and estrogen-dependent gene regulation, including ‘nuclear estrogen receptor alpha’ and ‘FoxA1 transcription factor’ networks (Fig. 5A), as well as the term ‘pathways in cancer’ (Fig. 5A), suggesting that these genes are essential players in the development of breast cancer. We defined a “PARP-1 Signature Gene Set” (PSGS), which contains the PARP-1-dependent E2-upregulated protein coding genes from Fig. 1. As expected, expression of PARP1 mRNA is directly correlated with expression of the PSGS across ER+ breast cancers (Supplementary Fig. S7). We examined the expression of the PSGS in patient breast tumor samples stratified by molecular subtype (PAM50) (Fig. 5B, left panel). Expression of the genes in this set was significantly elevated in the luminal subtype of breast cancer, which is characterized by ER⍺ expression. Indeed, the stratification of patient samples by ER⍺ status confirms the differential expression of these genes in ER– versus ER+ breast cancers (Fig. 5B, right panel). A similar analysis of the PARP-1-independent E2-upregulated protein coding genes revealed that although the magnitude of the expression is comparable to PARP-1-dependent E2-upregulated genes, their expression does not exhibit the same stratification across breast cancer types (Fig. 5B versus Supplementary Fig. S8A).
Figure 5. PARP-1 regulates estrogen signaling, and predicts ER-positive luminal subtypes and poorer overall survival of ER-positive breast cancer patients.
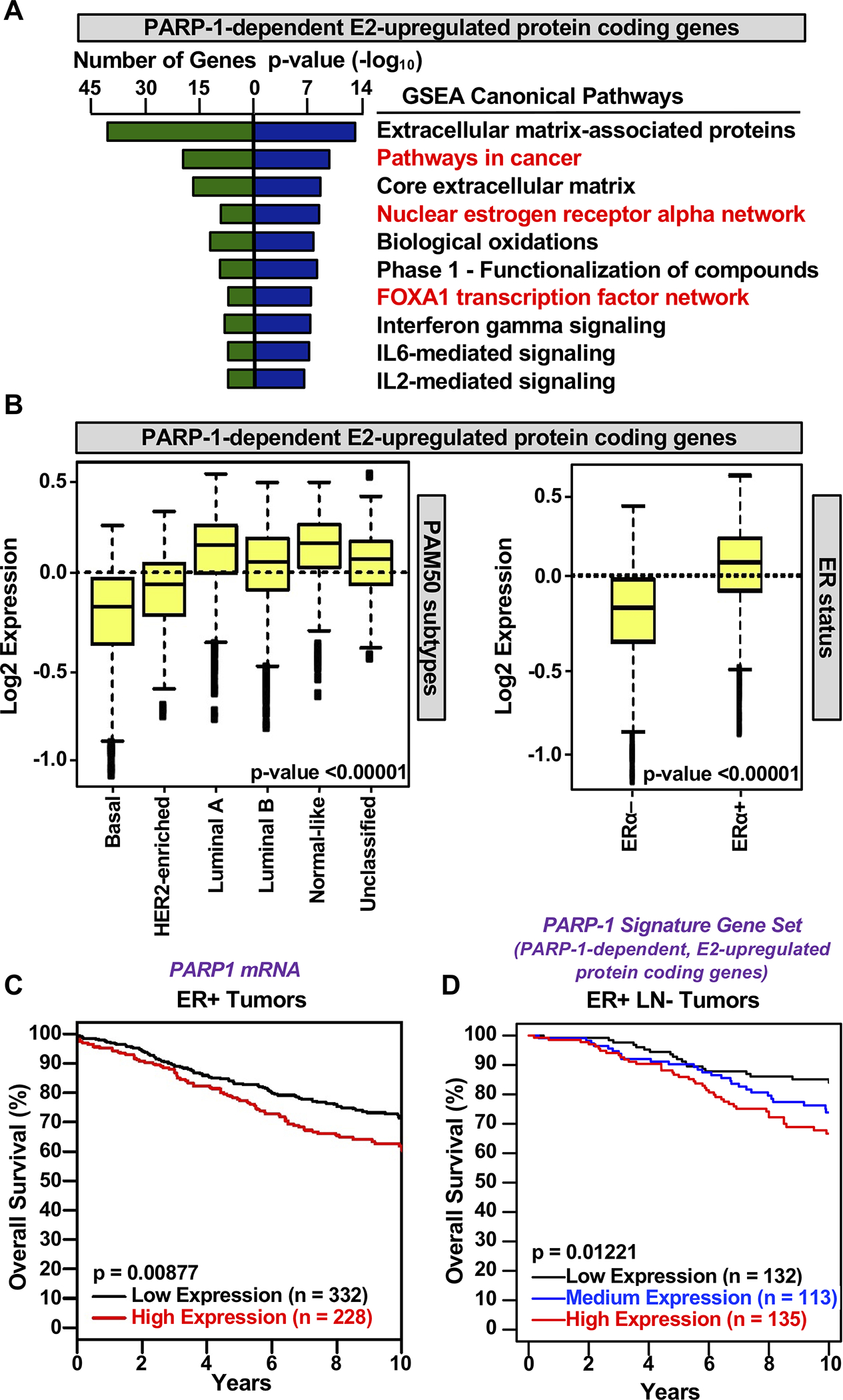
(A) GSEA enriched pathways for PARP-1 dependent E2-regulated protein coding genes. The number of genes represented in each term and the p-value (-log10) are shown.
(B) Box plots of expression values for PARP-1-dependent E2-upregulated protein coding genes suppressed by PARP-1 knockdown in patient breast tumor samples stratified by subtype (PAM50; left), confirming the differential expression of these genes in ER-positive breast cancer patients (ER status; right). Observed differences are significant as determined by an ANOVA comparison of the means (p-value < 0.00001).
(C) Kaplan-Meier survival analyses for breast cancer based on PARP-1 mRNA expression. High expression of PARP-1 mRNA is predictive of poor outcome.
(D) Kaplan-Meier survival analyses for breast cancer based on expression of the PARP-1 signature gene set (PARP-1-dependent, E2-upregulated protein coding genes).
To determine the potential clinical utility of PARP-1 as a target in ER+ breast cancers, we stratified patient samples based on expression of PARP1 mRNA or expression of the PSGS. Kaplan-Meier survival analysis indicated that high expression of PARP1 mRNA (Fig. 5C) or high expression of the PSGS (Fig. 5D) is predictive of poor outcomes. Importantly, a similar correlation was not observed with a PARP-1-independent E2-upregulated gene set (Supplementary Fig. S8B), confirming the specific connection to PARP-1. Poor outcomes related to high expression of the PSGS was also observed in data from The Cancer genome Atlas (TGCA) (Supplementary Fig. S8C). Similar analyses of the PARP-1-dependent E2-downregulated gene set identified cancer-relevant terms (by GSEA; Supplementary Fig. S9A) and stratification of their expression across breast cancer types (Supplementary Fig. S9B). Together, these results indicate that PARP-1-dependent E2-regulated genes are associated with luminal breast cancer and that expression of PARP1 mRNA or the PSGS predicts clinical outcomes in ER+ cancer patients.
PARP-1 regulates estrogen-dependent growth in ER+ breast cancer cells
Having defined some mechanisms for PARP-1-mediated regulation of E2-dependent transcription, as well as the potential of PARP-1-regulated genes as predictive markers in ER+ breast cancers, we sought to connect the mechanisms to the biology. Our previous results identified a role for PARP-1 in regulating transcriptional elongation by Pol II through the direct ADP-ribosylation and inhibition of NELF (12) (Fig. 6A). NELF is also targeted for inhibition by the P-TEFb kinase complex (40) (Fig. 6A). Interestingly, Flavopiridol, a chemical inhibitor of P-TEFb, also inhibits PARP-1-mediatd ADP-ribosylation of NELF (12). Thus, both PARP-1 and P-TEFb inhibitors work to enhance NELF activity and reduce transcriptional elongation, similar to the effects of PARP-1 depletion shown in Fig. 2, A and C. Thus, we surmised that Flavopiridol would show similar growth inhibitory effects as PARP inhibitor. To test this, we performed cell proliferation assays in the presence of vehicle or E2 treatment, with or without FDA-approved PARP inhibitors (Niraparib, Olaparib, or Talazoparib) with or without Flavopiridol in MCF-7 and T47D ER+ breast cancer cells (Fig. 6B). We observed that both Flavopiridol and the PARP inhibitors inhibited E2-mediated cell proliferation. These results suggest that transcriptional regulatory mechanisms involving the regulation of transcriptional elongation by Pol II play important roles in E2-mediated cell proliferation.
Figure 6. PARP-1 and P-TEFb inhibition attenuate estrogen-stimulated cell growth.
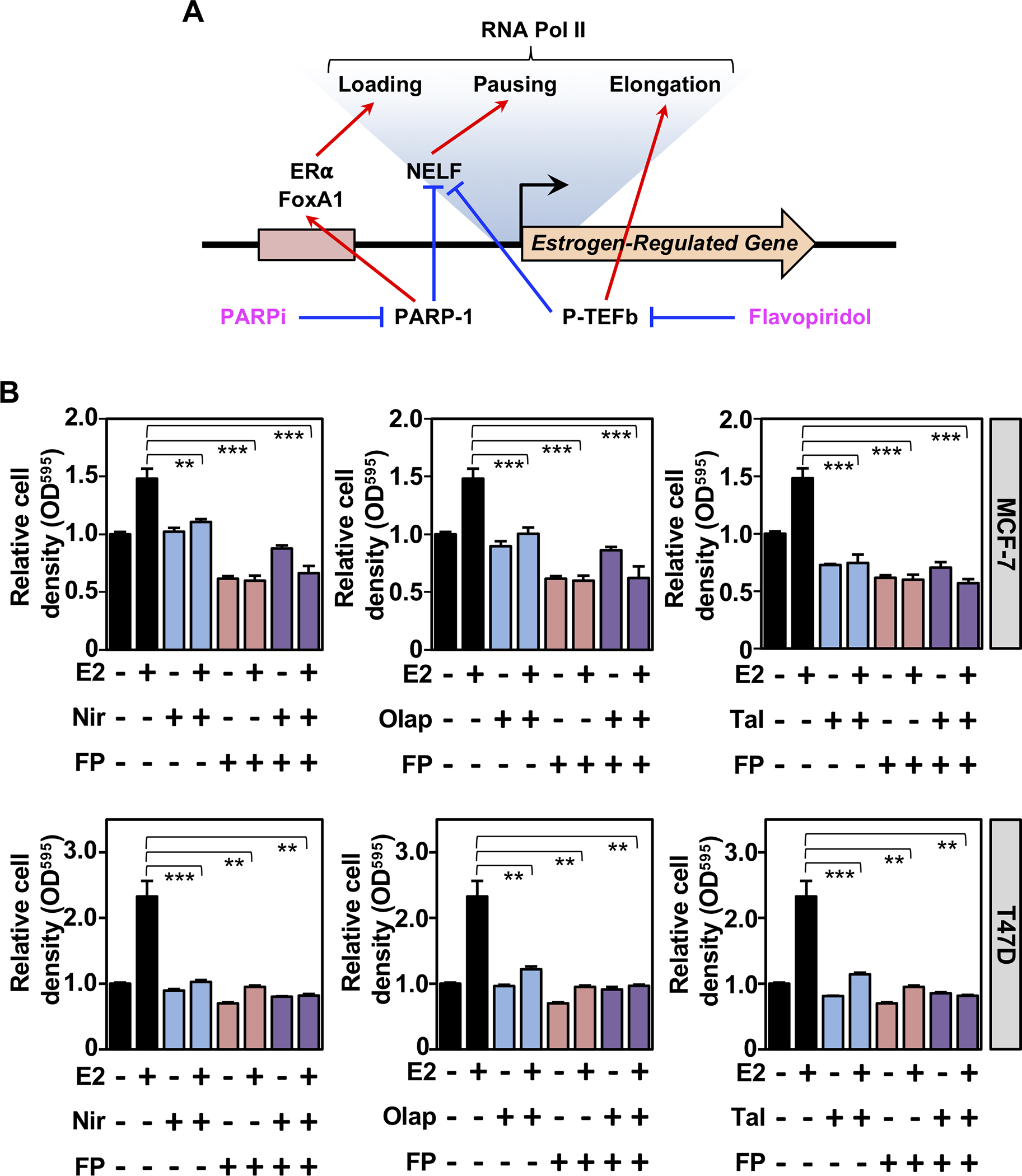
(A) Model depicting the roles of PARP-1 and P-TEFb in estrogen-dependent transcription and effects of their cognate inhibitors.
(B) Crystal violet proliferation assays after 6 days of growth in the presence of vehicle, E2, PARP inhibitor (Niraparib, Nir; Olaparib, Olap; or Talazoparib, Tal), and/or Flavopiridol (FP) as indicated in MCF-7 (top) and T47D (bottom) cells. Relative cell density based on OD595 was normalized to cells growing without E2, PARP inhibitor, or FP at day 6. Asterisks indicate observed differences are significant as assessed by unpaired t-tests (p-values **<0.05, ***<0.005).
Discussion
In this study, we explored a role for PARP-1 in regulating estrogen-dependent transcription, as well as downstream biological processes that impact cell growth and potentially clinical outcomes in breast cancer patients. Our study reveals PARP-1’s role in hormone-dependent gene expression and expands previous findings showing that PARP-1 is required for chromatin remodeling, transcription factor binding, and transcriptional regulation in breast cancer cells (12,20,41,42).
PARP-1 plays a critical role in regulating estrogen-dependent gene expression through ERα in breast cancer cells
In this study, we sought to understand the role of PARP-1 in estrogen-dependent transcription. GRO-seq in PARP-1-depeleted cells demonstrated a key role for PARP-1 in regulating E2-dependent gene expression (Fig. 1 and Supplementary Figs. S1 and S2), including effects on productive elongation by Pol II (Fig. 2 and Supplementary Fig. S3) and by modulating the binding of ERα and FoxA1 (Fig. 3 and Supplementary Fig. S4). However, H3K27ac enrichment was unaffected by PARP-1 depletion despite the decreases in ERα or FoxA1 recruitment (Fig. 3D) likely because the ERα binding sites retain significant, albeit reduced, ERα and FoxA1 binding. Furthermore, we have shown previously that the levels of H3K27ac are only modestly dependent on the presence of liganded ERα bound at an enhancer (8). Moreover, other chromatin modulators, such as RING1B, alter ERα recruitment without effecting H3K27ac enrichment at E2-regulated de novo super enhancers (43). Thus, we would not necessarily expect a change in H3K27ac enrichment with reduced ERα or FoxA1.
Our observations are consistent with a previous study examining the role of PARP-1 in androgen-dependent gene regulation through androgen receptor (AR), which showed a role for PARP-1 in AR binding to chromatin, AR-dependent transcription, and androgen-dependent proliferation in prostate cancer cells (44). However, another recent study showed that PARP-2, but not PARP-1, interacts with FoxA1 to facilitate AR recruitment to enhancer regions in prostate cancer cells (45). The reasons for the difference with our results on PARP-1 and FoxA1 are unclear, but could be due to intrinsic differences in the nuclear receptors, the cell types, or the genomic localization or activities of the PARP proteins.
Regarding the mechanism of PARP-1 effects on ERα and FoxA1 binding, we and others have previously shown that PARP-1 can affect some, but not all, transcription factors through (1) chromatin structural effects independent of its catalytic activity (46) and (2) direct ADPRylation of the transcription factors (47). Regarding the latter, previous studies have shown that both ERα (48) and FoxA1 (49) are ADPRylated proteins. The extent to which ADPRylation affects the activity of ERα and FoxA1 in breast cancer has yet to be determined, but it represents a potential regulatory mechanism. PARP-1 activity also supports transcriptional elongation by Pol II on estrogen-regulated genes. In this regard, we found that both PARP-1 and P-TEFb inhibitors work to enhance NELF activity and reduce transcriptional elongation (Fig. 6), similar to the effects of PARP-1 depletion. Collectively, these studies are a step forward toward establishing PARP-1 as a key player in signal-dependent transcription in hormone-dependent cancers.
Clinical utility of PARP-1 in ERα-positive breast cancers
PARP-1 has been shown to be an effective target for therapeutic intervention in BRCA1/2- or HR-defective cancers, including breast, ovarian, and prostate (17). Blocking DNA damage repair by PARP inhibitors renders these cancers sensitive to the endogenous BRCA1/2- or HR-defects, resulting in cell death via synthetic lethality. FDA-approved PARP-1 inhibitors, such as Olaparib, Rucaparib, Niraparib, Talazoparib, and Veliparib are currently being used clinically to treat BRCA1/2 mutant cancers (17). Although ~70% of breast cancers are ER+ at the time of diagnosis (10), analysis of TCGA (The Cancer Genome Atlas) suggests that only a small fraction of these breast cancers are BRCA1/2-mutant (~5%). The evidence presented here, along with previous studies from breast and ovarian cancers (18,50–53), supports the rationale for expanding the use of PARP inhibitors to ER+ breast cancers with wild-type BRCA1/2.
Our results demonstrate that PARP-1 controls cancer-related pathways, including those relevant to ER+ breast cancers (Fig. 5A), by regulating the gene expression program in breast cancer cells. Furthermore, PARP1 mRNA expression directly correlates with clinical outcomes in (i.e., overall survival) of ER+ breast cancer patients (Fig. 5, C and D). Interestingly, AR inhibitors can promote “BRCAness” (i.e., reduced expression of HR genes, including BRCA1, RAD54L, and RMI2) in castration-resistant prostate cancers, which is synthetically lethal with PARP inhibitors (54). Whether such a mechanism might also be functional in hormone-resistant breast cancer is unknown. Taken together, our results suggest a potential use of PARP inhibitors in the treatment of luminal breast cancers irrespective of BRCA1/2 status. Perhaps the most significant clinical utility of PARP-1 inhibitors in the treatment of ER+ breast cancers would be if they exhibit efficacy in Tamoxifen-resistant cancers. Interestingly, many therapy-induced Tamoxifen-resistant breast cancers arise as a result of activating mutations in ERα that turn the receptor into a constitutive activator even in the presence of Tamoxifen (55). We posit that PARP inhibitors could be effective in these cases, perhaps by mechanisms similar to those that are used to attenuate the activity of E2-activated ERα: effects on ERα binding, effects on FoxA1 binding, and effects on transcriptional elongation of ERα-regulated genes.
Supplementary Material
Implications.
PARP-1 regulates the estrogen-dependent genomic binding of ERα and FoxA1 to regulate critical gene expression programs by RNA polymerase II that underlie the proliferation of ERα-positive breast cancers, providing a potential therapeutic opportunity for PARP inhibitors in estrogen-responsive breast cancers.
Acknowledgments
The authors would like to thank members of the Kraus lab for their careful review and helpful suggestions on this work; the UT Southwestern Next Generation Sequencing Core, under the direction of Ralf Kittler; and Rosemary Plagens and Kristine Hussey for assistance with the genomic assays. S.S.G. is a CPRIT scholar in cancer research and is supported by a First-time Faculty Recruitment Award from the Cancer Prevention and Research Institute of Texas (CPRIT; RR170020). This work was supported by grants from the Cancer Prevention and Research Institute of Texas (RP190236) and the NIH/NIDDK (R01 DK069710) to W.L.K., funds from the Cecil H. and Ida Green Center for Reproductive Biology Sciences Endowment to W.L.K., and a postdoctoral fellowship from the Susan G. Komen Foundation (PDF12230441) to S.S.G.
Financial support:
This work was supported by grants from the Cancer Prevention and Research Institute of Texas (RP190236) and the NIH/NIDDK (R01 DK069710) to W.L.K., funds from the Cecil H. and Ida Green Center for Reproductive Biology Sciences Endowment to W.L.K., and a postdoctoral fellowship from the Susan G. Komen Foundation (PDF12230441) to S.S.G.
Footnotes
Disclosure: W.L.K. is a founder, consultant, and member of the Scientific Advisory Board for Ribon Therapeutics, Inc. and ARase Therapeutics, Inc. He is also a co-holder of U.S. Patent 9,599,606, covering a set of ADP-ribose detection reagents, which have been licensed to and are sold by EMD Millipore.
Disclaimer
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the article; or in the decision to publish the results.
References
- 1.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of estrogen action. Physiol Rev 2001;81(4):1535–65 doi 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 2.Vasquez YM, Kraus WL. The estrogen-regulated transcriptome: rapid, robust, extensive, and transient. In: Z X, editor. Estrogen Receptor and Breast Cancer Cancer Drug Discovery and Development: Humana Press; 2019. p 95–127. [Google Scholar]
- 3.Jozwik KM, Carroll JS. Pioneer factors in hormone-dependent cancers. Nat Rev Cancer 2012;12(6):381–5 doi 10.1038/nrc3263. [DOI] [PubMed] [Google Scholar]
- 4.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet 2011;43(1):27–33 doi 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franco HL, Nagari A, Kraus WL. TNFα signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol Cell 2015;58(1):21–34 doi 10.1016/j.molcel.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foulds CE, Feng Q, Ding C, Bailey S, Hunsaker TL, Malovannaya A, et al. Proteomic analysis of coregulators bound to ERalpha on DNA and nucleosomes reveals coregulator dynamics. Mol Cell 2013;51(2):185–99 doi 10.1016/j.molcel.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murakami S, Nagari A, Kraus WL. Dynamic assembly and activation of estrogen receptor alpha enhancers through coregulator switching. Genes Dev 2017;31(15):1535–48 doi 10.1101/gad.302182.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 2013;23(8):1210–23 doi 10.1101/gr.152306.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009;462(7269):58–64 doi 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lumachi F, Brunello A, Maruzzo M, Basso U, Basso SM. Treatment of estrogen receptor-positive breast cancer. Curr Med Chem 2013;20(5):596–604 doi 10.2174/092986713804999303. [DOI] [PubMed] [Google Scholar]
- 11.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 2012;13(7):411–24 doi 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 12.Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, et al. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 2016;353(6294):45–50 doi 10.1126/science.aaf7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. Upregulation of poly (ADP-Ribose) polymerase-1 (PARP1) in triple-negative breast cancer and other primary human tumor types. Genes Cancer 2010;1(8):812–21 doi 10.1177/1947601910383418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaremba T, Ketzer P, Cole M, Coulthard S, Plummer ER, Curtin NJ. Poly(ADP-ribose) polymerase-1 polymorphisms, expression and activity in selected human tumour cell lines. Br J Cancer 2009;101(2):256–62 doi 10.1038/sj.bjc.6605166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krukenberg KA, Jiang R, Steen JA, Mitchison TJ. Basal activity of a PARP1-NuA4 complex varies dramatically across cancer cell lines. Cell Rep 2014;8(6):1808–18 doi 10.1016/j.celrep.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conrad LB, Lin KY, Nandu T, Gibson BA, Lea JS, Kraus WL. ADP-ribosylation levels and patterns correlate with gene expression and clinical outcomes in ovarian cancers. Mol Cancer Ther 2020;19(1):282–91 doi 10.1158/1535-7163.MCT-19-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sachdev E, Tabatabai R, Roy V, Rimel BJ, Mita MM. PARP inhibition in cancer: an update on clinical development. Target Oncol 2019;14(6):657–79 doi 10.1007/s11523-019-00680-2. [DOI] [PubMed] [Google Scholar]
- 18.Kim DS, Camacho CV, Nagari A, Malladi VS, Challa S, Kraus WL. Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol Cell 2019;75(6):1270–85 e14 doi 10.1016/j.molcel.2019.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 2017;31(2):101–26 doi 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 2008;319(5864):819–21 doi 10.1126/science.1149250. [DOI] [PubMed] [Google Scholar]
- 21.Murakami S, Li R, Nagari A, Chae M, Camacho CV, Kraus WL. Distinct roles for BET family members in estrogen receptor alpha enhancer function and gene regulation in breast cancer cells. Mol Cancer Res 2019;17(12):2356–68 doi 10.1158/1541-7786.MCR-19-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 2011;145(4):622–34 doi 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011;17 doi 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 24.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1):139–40 doi 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chae M, Danko CG, Kraus WL. groHMM: a computational tool for identifying unannotated and cell type-specific transcription units from global run-on sequencing data. BMC Bioinformatics 2015;16:222 doi 10.1186/s12859-015-0656-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, et al. Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Mol Cell 2013;50(2):212–22 doi 10.1016/j.molcel.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, et al. A large genome center’s improvements to the Illumina sequencing system. Nat Methods 2008;5(12):1005–10 doi 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009;10(3):R25 doi 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25(16):2078–9 doi 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26(6):841–2 doi 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 2012;22(9):1813–31 doi 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc 2012;7(9):1728–40 doi 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong S, Joung JG, Zheng Y, Chen YR, Liu B, Shao Y, et al. High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb Protoc 2011;2011(8):940–9 doi 10.1101/pdb.prot5652. [DOI] [PubMed] [Google Scholar]
- 35.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28(5):511–5 doi 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dinse GE, Lagakos SW. Nonparametric estimation of lifetime and disease onset distributions from incomplete observations. Biometrics 1982;38(4):921–32. [PubMed] [Google Scholar]
- 38.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Amer Stat Assoc 1958;53(282):457–81. [Google Scholar]
- 39.Ringner M, Fredlund E, Hakkinen J, Borg A, Staaf J. GOBO: gene expression-based outcome for breast cancer online. PLoS One 2011;6(3):e17911 doi 10.1371/journal.pone.0017911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Core LJ, Lis JT. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science 2008;319(5871):1791–2 doi 10.1126/science.1150843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frizzell KM, Gamble MJ, Berrocal JG, Zhang T, Krishnakumar R, Cen Y, et al. Global analysis of transcriptional regulation by poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in MCF-7 human breast cancer cells. J Biol Chem 2009;284(49):33926–38 doi 10.1074/jbc.M109.023879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krishnakumar R, Kraus WL. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell 2010;39(5):736–49 doi 10.1016/j.molcel.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Chan HL, Garcia-Martinez L, Karl DL, Weich N, Slingerland JM, et al. Estrogen induces dynamic ERalpha and RING1B recruitment to control gene and enhancer activities in luminal breast cancer. Sci Adv 2020;6(23):eaaz7249 doi 10.1126/sciadv.aaz7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL, et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov 2012;2(12):1134–49 doi 10.1158/2159-8290.CD-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gui B, Gui F, Takai T, Feng C, Bai X, Fazli L, et al. Selective targeting of PARP-2 inhibits androgen receptor signaling and prostate cancer growth through disruption of FOXA1 function. Proc Natl Acad Sci U S A 2019;116(29):14573–82 doi 10.1073/pnas.1908547116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Z, Kraus WL. Catalytic-independent functions of PARP-1 determine Sox2 pioneer activity at intractable genomic loci. Mol Cell 2017;65(4):589–603 e9 doi 10.1016/j.molcel.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo X, Ryu KW, Kim DS, Nandu T, Medina CJ, Gupte R, et al. PARP-1 controls the adipogenic transcriptional program by PARylating C/EBPbeta and modulating its transcriptional activity. Mol Cell 2017;65(2):260–71 doi 10.1016/j.molcel.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang F, Wang Y, Wang L, Luo X, Huang K, Wang C, et al. Poly(ADP-ribose) polymerase 1 is a key regulator of estrogen receptor alpha-dependent gene transcription. J Biol Chem 2013;288(16):11348–57 doi 10.1074/jbc.M112.429134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhen Y, Zhang Y, Yu Y. A cell-line-specific atlas of PARP-mediated protein Asp/Glu-ADP-ribosylation in breast cancer. Cell Rep 2017;21(8):2326–37 doi 10.1016/j.celrep.2017.10.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390(10106):1949–61 doi 10.1016/S0140-6736(17)32440-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med 2016;375(22):2154–64 doi 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- 52.Wu XH, Zhu JQ, Yin RT, Yang JX, Liu JH, Wang J, et al. Niraparib maintenance therapy in patients with platinum-sensitive recurrent ovarian cancer using an individualized starting dose (NORA): a randomized, double-blind, placebo-controlled phase 3 trial. Ann Oncol 2021. doi 10.1016/j.annonc.2020.12.018. [DOI] [PubMed] [Google Scholar]
- 53.Keung MY, Wu Y, Badar F, Vadgama JV. Response of breast cancer cells to PARP inhibitors is independent of BRCA status. J Clin Med 2020;9(4) doi 10.3390/jcm9040940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li L, Karanika S, Yang G, Wang J, Park S, Broom BM, et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal 2017;10(480) doi 10.1126/scisignal.aam7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lei JT, Gou X, Seker S, Ellis MJ. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J Cancer Metastasis Treat 2019;5 doi 10.20517/2394-4722.2019.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.