

Abstract
Despite recent advancements in deep learning methods for protein structure prediction and representation, little focus has been directed at the simultaneous inclusion and prediction of protein backbone and sidechain structure information. We present SidechainNet, a new dataset that directly extends the ProteinNet dataset. SidechainNet includes angle and atomic coordinate information capable of describing all heavy atoms of each protein structure and can be extended by users to include new protein structures as they are released. In this paper, we provide background information on the availability of protein structure data and the significance of ProteinNet. Thereafter, we argue for the potentially beneficial inclusion of sidechain information through SidechainNet, describe the process by which we organize SidechainNet, and provide a software package (https://github.com/jonathanking/sidechainnet) for data manipulation and training with machine learning models.
Keywords: Deep learning, proteins, dataset, software, machine learning, protein structure
1 |. INTRODUCTION
The deep learning subfield of protein structure prediction and protein science has made considerable progress in the last several years. In addition to the success of several deep learning methods in the 2018 and 2020 Critical Assessment of protein Structure Prediction (CASP) competitions1;2;3, many novel deep learning-based methods have been developed for protein representation4;5, property prediction6;7, and structure prediction8;9;10;11. Such methods are demonstrably effective and extremely promising for future research. They have the potential to make complex inferences about proteins much faster than competing computational methods and at a cost several orders of magnitude lower than experimental methods.
1.1 |. Protein Structure Data Availability and Information Leakage
The availability of existing data is a notable challenge for applications of deep learning methods to protein science and, in particular, to protein structure prediction. This limitation is not equally present in other applications of deep learning. For instance, in the field of image recognition, the ImageNet12 dataset contains 14 million annotated and uniformly presented images. Similarly, in the field of natural language processing, linguistic databases and the web provide access to hundreds of millions of samples of annotated textual data and effectively limitless access to unannotated data. Though the Universal Protein Resource (UniProt) database now contains a whopping 156 million unique protein sequences13, there are currently only 174,014 protein structures in the Protein Data Bank14 (PDB), of which only 99,835 represent unique protein sequences. Furthermore, although protein structure data is accessible through the PDB in individual files, it is not otherwise preprocessed for machine learning tasks in a manner analogous to other machine learning domains.
In addition to data availability, biased or skewed data presents another major challenge for machine learning practitioners15. Something as simple as dividing the data into training and testing splits for model development and evaluation can introduce harmful biases. Failure to analyze the similarities or differences between training and evaluation splits can lead to “information leakage” in which trained models exhibit overly optimistic performance by learning non-generalizable features from the training set. One such example of this in structural biology research is the Database of Useful Decoys: Enhanced (DUD-E) dataset16. DUD-E remains a common benchmark for evaluating molecular docking programs and chemoinformatic-focused machine learning methods despite recent research17;18 uncovering dataset bias that explains misleading performance by many of the deep learning methods trained on it.
1.2 |. Treatment of Protein Backbone and Sidechain Information
A common way to describe the structure of a protein is to divide it into two separate components–the backbone and the sidechains that extend from it. The protein backbone is a linear chain of nitrogen, carbon, and oxygen atoms. The torsional angles (Φ, Ψ, and Ω) that connect these atoms form the overall shape of the protein. In contrast, protein sidechains are chemical groups of zero to ten heavy atoms connected to the central α-carbon of each amino acid residue. Each of the twenty distinct amino acids is defined by the unique structure and chemical composition of its sidechain component. Consequently, each protein is defined by the unique sequence of its constituent amino acids. The precise orientation of amino acid sidechains is critical to the biochemical function of proteins. Enzyme catalysis, drug binding, and protein-protein interactions all depend on a level of atomic precision that is not accounted for in backbone structure alone. Thus, the protein backbone and sidechain are both crucially important to protein structure and function.
Despite this, many predictive methods treat backbone structure prediction and complete sidechain structure prediction as distinct problems. For instance, one common approach is to predict the protein backbone alone and then add sidechains to the generated structure through a conformational or energy-minimizing search19. Another common method is to optimize the structure using backbone dependent rotamer libraries20. In both cases, sidechain placement is dependent on predicted backbone structure.
It is worth noting that some programs like Rosetta21 allow for the simultaneous optimization of protein backbone and sidechains. However, such methodology is not consistently adopted throughout the protein structure prediction literature. Relatedly, one notable result from CASP14, the most recent iteration of the CASP competition, was the success of AlphaFold 222, a deep learning protein structure prediction method. Though details of its inner workings have not yet been fully released, AlphaFold 2 does incorporate, to some extent, both protein sidechain and backbone information in its procedure.
Still, the methods frequently employed by the research community that asynchronously predict protein backbone and sidechain structures remain effective. A possible reason for separating backbone and sidechain information could be that the community believes adequate progress in backbone structure prediction has been made without accounting for sidechain conformations during training. Alternatively, it may be the case that such information is simply not readily available for training deep learning models. Nonetheless, the potentially positive effect of utilizing the complete backbone and sidechain structure at training time has not been adequately addressed.
2 |. METHODS
We propose SidechainNet, a protein sequence and structure dataset that addresses the concerns described in Sections 1.1 and 1.2. Namely, this work aims to account for data similarity across training splits, to improve upon existing datasets by adding features relevant to all-atom protein structure prediction, and, finally, to make such data available and easily accessible to researchers in both the machine learning and structural biology fields.
2.1 |. Addressing Protein Structure Data Availability and Information Leakage
SidechainNet is based on Mohammed AlQuraishi’s ProteinNet23. ProteinNet is a dataset designed to mimic the assessment methodology of CASP proceedings.
Under the CASP contest organization, participants develop predictive methods using any publicly available protein structures. Thereafter, predictive methods are assessed using a specific set of proteins selected by contest organizers and withheld from participants until assessment. Organizers select proteins that are challenging to predict and, in the case of the Free Modeling contest category, have minimal similarity to available protein structures. In effect, the CASP process results in a portion of data for method development and a separate portion for evaluation. AlQuraishi proposed that these subsets could be treated as training and testing sets and re-purposed for machine learning.
Since no validation sets are constructed by the CASP organizers, AlQuraishi used clustering methods to extract distinct protein sequences and structures from each training set to use for validation. For each CASP contest, AlQuraishi ultimately constructed seven different validation sets–each containing an increasing upper bound on the similarity between sequences in the given validation set and those in the training set. AlQuraishi’s construction allows users to evaluate a model’s performance on proteins that have high similarity to a given training set (akin to the Template-Based Modeling CASP category) or proteins that have low or zero similarity to a training set (akin to the Free Modeling CASP category).
As a result of the constraints imposed by the CASP contest structure and AlQuraishi’s validation set construction, ProteinNet accounts for protein sequence similarity across data splits. This prevents information leakage and minimizes misleading performance. For these reasons, we have replicated AlQuraishi’s training, testing, and validation set constructions in SidechainNet.
2.2 |. Unifying Protein Backbone and Sidechain Information
SidechainNet also extends ProteinNet by including all heavy atoms of each protein and all necessary torsional angles for atomic reconstruction. We are interested in determining whether the availability of complete sidechain conformation information can improve the performance of structure prediction methods. However, even if such models are not more accurate, they will be fundamentally more expressive as they will generate all-atom protein structures. These structures could be used without modification in downstream analyses for structure-based drug discovery and even molecular dynamics simulations.
2.3 |. SidechainNet Properties
To the maximum extent possible, SidechainNet replicates the features included in ProteinNet (Table 1). However, SidechainNet also adds support for several new features. More information about the new features is included below.
TABLE 1.
Differences between ProteinNet and SidechainNet.
| Entry | Dimensionality | ProteinNet | SidechainNet |
|---|---|---|---|
| Primary sequence | L × 1 | X | X |
| PSSM† + Information Content | L × 21 | X | X |
| Missing residue mask | L × l | X | X |
| Secondary structure labels | L × l | X | X |
| Backbone coordinates‡ | L × 4 × 3 | X | X |
| Backbone torsion angles | L × 3 | X | |
| Backbone bond angles | L × 3 | X | |
| Sidechain torsion angles | L × 6 | X | |
| Sidechain coordinates | L × 10 × 3 | X | |
| Crystallographic resolution | 1 | X | |
| Unmodified primary sequence§ | L × 1 | X |
L represents protein length.
Position Specific Scoring Matrices (PSSMs) developed from multiple sequence alignments.
SidechainNet includes atomic coordinates for backbone oxygen atoms while ProteinNet does not.
During processing, a select number of non-standard residues are converted to their standard forms (e.g., selenomethionine is partially converted to methionine). The unmodified primary sequence entry retains the 3-letter amino acid codes for each of the original amino acids in the sequence, while the primary sequence data entry contains the standardized 1-letter amino acid codes.
2.3.1 |. Angle Information
One important component of protein structure data often utilized in structure prediction methods but absent from ProteinNet is the set of torsional angles that describe the orientation of the protein. The canonical backbone torsional angles (Φ, Ψ, and Ω) for each residue are provided in SidechainNet.
In the development of SidechainNet, we were surprised to observe that we incurred a noticeable amount of error when attempting to reconstruct a protein’s Cartesian coordinates given only its backbone torsional angles (Figure 1B). We determined that the error was due to our assumption that several important non-torsional angles in the protein backbone (C-N-CA, N-CA-C, and CA-C-N) could be fixed using reference values from AMBER force fields24 rather than being allowed to vary. Thus, even if a model perfectly predicted backbone torsional angles, it would still be subject to reconstruction error if such bond angles were held fixed. For that reason, we decided to include these angles in SidechainNet as well (Figures 1A and 1B).
FIGURE 1.
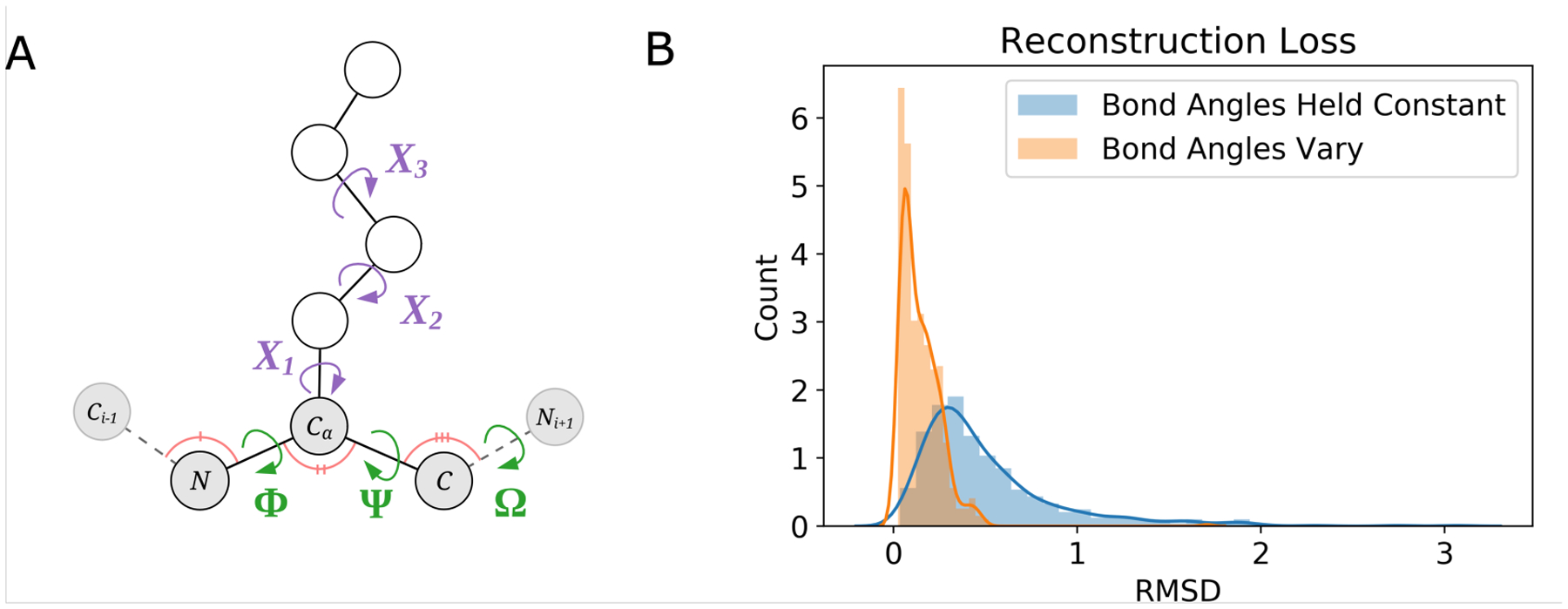
(A) A methionine residue annotated with the 3 categories of angles measured by SidechainNet: the canonical backbone torsional angles (Φ, Ψ, and Ω) in green, the rotatable sidechain-specific torsional angles (X1−3) in purple, and the 3-atom backbone bond angles in pink. Atoms considered part of the residue’s sidechain are colored white, while the backbone atoms are colored gray.
(B) Backbone bond angles (C-N-CA, N-CA-C, and CA-C-N; colored pink in 1A) are important for accurately reconstructing atomic coordinates from angles. When we assumed that these angles could be fixed during coordinate reconstruction, the result was greater reconstruction error (blue distribution). When we utilized the actual angle values during coordinate reconstruction, we achieved lower reconstruction error (orange distribution). Root-Mean-Square Deviation (RMSD) measures the difference between true atomic coordinates and coordinates reconstructed from recorded angles.
2.3.2 |. Sidechain Information
SidechainNet also includes sidechain-specific information that describes the orientation of each amino acid using both external (Cartesian) and internal (angular) coordinate systems. This information includes up to ten atomic coordinates and up to six torsional angles for the sidechain component of each residue.
The number of angles measured for an amino acid sidechain depends on the number of rotatable bonds within it. For residues with aromatic sidechains (e.g., tryptophan), the torsional angles describing the ring components of the residue are omitted since they can be inferred, but their complete atomic coordinates are still recorded.
3-atom bond angles (as opposed to 4-atom torsional angles) and bond lengths associated with sidechain structures are not recorded from structure files. Instead, we extracted reference values unique to each bond from the AMBER ff19SB force field24;25. When utilizing fixed angle values for sidechain 3-atom bond angles, the impact on reconstruction error is much smaller than the impact discussed in Figures 1 because the error does not accumulate between adjacent residues. Together with the recorded sidechain torsional angles, these angles and bond lengths can be used to generate all-atom sidechain structures.
2.4 |. Generation Procedure and Caveats
We constructed SidechainNet by, first, obtaining raw ProteinNet text records linked to in ProteinNet’s GitHub repository (https://github.com/aqlaboratory/proteinnet). Then, we parsed these records before saving them into Python dictionaries. Next, we re-downloaded all-atom protein structures (sequences, coordinates, and angles) for each protein described by ProteinNet from the PDB using the ProDy software package26. Finally, we combined the remaining data (PSSMs, Information Content, and secondary structure labels) into the final SidechainNet dataset by aligning the sequences observed during the re-downloading process with the sequences described by ProteinNet. We replaced the coordinate and missing residue information described in ProteinNet with the re-downloaded values to ensure consistency. Scripts and instructions for constructing SidechainNet data are available in our GitHub repository.
We did not include data for 468 (0.4%) of the 104,323 listed protein structures from ProteinNet’s CASP12 dataset. Some of these structures are of proteins that include D-amino acids. Such structures were purposely excluded despite being included in ProteinNet because they cannot be effectively modeled with L-amino acid sidechain structures. Other structures were excluded due to problematic file parsing, edge cases, and disagreement between the sequences we observed and those reported by ProteinNet that could not be resolved programmatically. Although we currently exclude these protein structures, we plan to resolve remaining issues in a subsequent release.
Furthermore, both ProteinNet and SidechainNet only consider single-molecule protein chains. Proteins from the PDB that contain multiple chains are divided into multiple independent entries, one for each chain. Any missing residue or atomic information is padded with vectors that contain sidechainnet.GLOBAL_PAD_CHAR and match the size of the corresponding data (e.g., 1×3 for each missing atom, 1×1 for each missing angle). In addition, detailed characteristics of structure data (e.g., b-factors, multiple or alternative sidechain locations) are ignored.
Lastly, structures are recorded directly from the PDB and have no guarantee to be energetically minimized. In the future, we plan to use AMBER to energetically minimize structures. This may make the data more consistent and amenable to training. It may also improve our accuracy when reconstructing protein atomic coordinates from angles.
3 |. RESULTS
SidechainNet was originally developed for Python and the PyTorch machine learning framework due to their ease of use and popularity within the research community. Here we discuss how researchers may use our Python package to load SidechainNet and efficiently train models with it.
3.1 |. SidechainNet as a Python dictionary
In its simplest form, SidechainNet is stored as a Python dictionary organized by the same training, validation, and testing splits described in ProteinNet. There are multiple validation sets included therein.
Within each of SidechainNet’s training, validation, and testing splits is another dictionary mapping data types (seq, ang, etc.) to a list containing this data for everyprotein. Shown below, seq{i}, ang{i}, etc. refer to the ith protein in the dataset.

By default, the load function downloads the data from the web into the current directory and loads it as a Python dictionary. If the data already exists locally, it reads it from disk. Other than the requirement that the data must be loaded using Python, this method of data loading is agnostic to any downstream analyses.

3.2 |. SidechainNet as PyTorch DataLoaders
The load function can also be used to load SidechainNet data as a dictionary of torch.utils.data.DataLoader objects. PyTorch DataLoaders make it simple to iterate over dataset items for training machine learning models. This method is recommended for using SidechainNet data with PyTorch. For a complete description of the data available to the user when using SidechainNet’s DataLoaders, see Table 2.
TABLE 2.
Data attributes available to the user when utilizing SidechainNet’s custom DataLoaders.
| Batch Attribute | Description |
|---|---|
| pids | Tuple of ProteinNet/SidechainNet IDs |
| seqs | Tensor of sequences† |
| int_seqs | Tensor of sequences represented as integers |
| str_seqs | Tuple of sequences represented as strings |
| msks | Tensor of missing residue masks (redundant with padding in data) |
| evos | Tensor of Position Specific Scoring Matrix + Information Content |
| secs | Tensor of secondary structure labels† |
| angs | Tensor of angles |
| crds | Tensor of coordinates |
| seq_evo_sec | Tensor with concatenated values of seqs, evos, and secs |
| resolutions | Tuple of crystallographic resolutions (Å), when available |
| is_modified | Tensor of modified residue bit-vectors. Each entry is a bit-vector where a 1 signifies that the residue at that position has been modified to match a standard residue supported by SidechainNet (e.g., selenomethionine to methionine) |
May be formatted as integer or one-hot sequences depending on value of scn.load(…seq_as_onehot=…).
By default, the provided DataLoaders use a custom batching method that randomly generates batches of proteins of similar length. For efficient GPU usage, it generates larger batches when the average length of proteins in the batch is small and smaller batches when the proteins are large. The probability of selecting small-length batches is decreased so that each protein in SidechainNet is included in a batch with equal probability. See dynamic_batch_size and collate_fn arguments for more information on modifying this behavior.

3.3 |. Converting Angle Representations into All-Atom Structures
An important component of this work is the inclusion of both angular and 3D coordinate representations of each protein. Researchers developing methods that rely on angular representations may be interested in converting this information into Cartesian coordinates. For this reason, SidechainNet provides tools to convert angles into coordinates.
In the below example, angles is a NumPy matrix or Torch Tensor following the same organization as the NumPy angle matrices provided in SidechainNet. sequence is a string representing the protein’s amino acid sequence. Both of these are obtainable from the SidechainNet data structures described in Sections 3.1 and 3.2.

3.4 |. Visualizing All-Atom Structures with PDB, py3Dmol, and gLTF Formats
SidechainNet makes it easy to visualize both existing and predicted all-atom protein structures, as well as structures represented as either angles or coordinates. These visualizations are available as PDB files, py3Dmol.view objects, and Graphics Library Transmission Format (gLTF) files. Examples of each are included below.
The PDB format is a typical format for representing protein structures and can be opened in software tools like PyMOL27. py3Dmol (built on 3Dmol.js28) enables users to visualize and interact with protein structures on the web and in Jupyter Notebooks via an open-source, object-oriented, and hardware-accelerated Javascript library (Figure 2). Finally, gLTF files, despite their larger size, can be convenient for visualizing proteins on the web or in contexts where other protein visualization tools are not supported.
FIGURE 2.

py3Dmol enables the user to interactively inspect all-atom protein structures from SidechainNet within Jupyter/iPython Notebooks.

3.5 |. Extending SidechainNet with User-Specified Data
Finally, we have considered the fact that users may be interested in utilizing SidechainNet’s functionality without strictly conforming to SidechainNet’s original organization and composition. To aid researchers with potentially different goals than the authors, we allow users to (1) specify which proteins to include in SidechainNet, and/or (2) customize their data split assignment. This functionality, found in sidechainnet. create_custom, enables users to work around the predetermined list of proteins and dataset splits inherited from ProteinNet. Importantly, this also allows SidechainNet to expand to include new protein structures as they are released. That said, information directly acquired from ProteinNet (e.g., Position Specific Scoring Matrices and secondary structure information) may be omitted if users’ specified proteins were not previously included in ProteinNet.
4 |. DISCUSSION
SidechainNet builds upon the strong foundation of ProteinNet to provide a protein structure dataset with minimal bias and maximum information content. By including many of the angles necessary to accurately reconstruct atomic Cartesian coordinates from an angle-only protein representation, we also enable future users to develop methods that utilize either or both internal and external coordinate systems with a previously inaccessible level of atomic detail and model complexity. In addition, the tools developed for SidechainNet’s programmatic construction may also be adapted for the generation of similar all-atom datasets with alternative methods of data clustering (e.g., datasets based on CATH29).
Methods developed to explicitly include the orientation and atomic coordinates of protein sidechains may have the upper hand in tasks such as structure-based drug discovery or in the analysis of specific enzyme activities, where such information is necessary. Although the impact of including sidechain information on existing predictive methods has yet to be studied, we hope SidechainNet makes such research more accessible.
Additionally, SidechainNet may find use in applications beyond protein structure prediction by making highly organized and uniformly preprocessed protein structure and sequence data easily accessible via Python. Protein science is currently experiencing a critical moment with respect to the development of new machine learning methods. The data that SidechainNet provides may prove to be a useful tool for developing methods to study protein representation, predict protein structure-property relationships, or to predict protein-protein and protein-ligand interactions.
Source code and example machine learning workflows using SidechainNet can be found at https://github.com/jonathanking/sidechainnet. To install, use the pip command-line program (pip install sidechainnet).
ACKNOWLEDGMENTS
Many thanks to Mohammed AlQuraishi for the inspiring work on protein structure prediction and for developing ProteinNet, without which the development of SidechainNet would not have been possible. Thanks, also, to Jeppe Hallgren for the development of a ProteinNet text record parser, which we have used in part here.
Funding information
University of Pittsburgh Center for Research Computing; National Institute of General Medical Sciences, Grant: R01GM108340; National Institute of Health, HHMI-NIBIB Interfaces Initiative, T32 Training Grant: EB009403
Footnotes
CONFLICT OF INTEREST
The authors have declared no conflict of interest.
REFERENCES
- [1].Hutson Matthew. AI protein-folding algorithms solve structures faster than ever. Nature, July2019. doi: 10.1038/d41586-019-01357-6. URL 10.1038/d41586-019-01357-6. [DOI] [PubMed] [Google Scholar]
- [2].Senior Andrew W., Evans Richard, Jumper John, Kirkpatrick James, Sifre Laurent, Green Tim, Qin Chongli, Žídek Augustin, Nelson Alexander W. R., Bridgland Alex, Penedones Hugo, Petersen Stig, Simonyan Karen, Crossan Steve, Kohli Pushmeet, Jones David T., Silver David, Kavukcuoglu Koray, and Hassabis Demis. Improved protein structure prediction using potentials from deep learning. Nature, 577(7792):706–710, January2020. doi: 10.1038/s41586-019-1923-7. URL 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- [3].Kryshtafovych Andriy, Schwede Torsten, Topf Maya, Fidelis Krzysztof, and Moult John. Critical assessment of methods of protein structure prediction (CASP)—round XIII. Proteins: Structure, Function, and Bioinformatics, 87(12):1011–1020, October2019. doi: 10.1002/prot.25823. URL 10.1002/prot.25823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Alley Ethan C., Khimulya Grigory, Biswas Surojit, Mohammed AlQuraishi, and George M. Church. Unified rational protein engineering with sequence-based deep representation learning. Nature Methods, 16(12):1315–1322, October2019. doi: 10.1038/s41592-019-0598-1. URL 10.1038/s41592-019-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ingraham John, Garg Vikas K, Barzilay Regina, and Jaakkola Tommi. Generative models for graph-based protein design. In Advances in Neural Information Processing Systems, 2019. [Google Scholar]
- [6].Rifaioglu Ahmet Sureyya, Doğan Tunca, Martin Maria Jesus, Cetin-Atalay Rengul, and Atalay Volkan. DEEPred: Automated protein function prediction with multi-task feed-forward deep neural networks. Scientific Reports, 9(1), May2019. doi: 10.1038/s41598-019-43708-3. URL 10.1038/s41598-019-43708-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Seo Seokjun, Oh Minsik, Park Youngjune, and Kim Sun. DeepFam: deep learning based alignment-free method for protein family modeling and prediction. Bioinformatics, 34(13):i254–i262, June2018. doi: 10.1093/bioinformatics/bty275. URL 10.1093/bioinformatics/bty275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xu Jinbo. Distance-based protein folding powered by deep learning. Proceedings of the National Academy of Sciences, 116(34):16856–16865, August2019. doi: 10.1073/pnas.1821309116. URL 10.1073/pnas.1821309116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yang Jianyi, Anishchenko Ivan, Park Hahnbeom, Peng Zhenling, Ovchinnikov Sergey, and Baker David. Improved protein structure prediction using predicted interresidue orientations. Proceedings of the National Academy of Sciences, 117(3):1496–1503, January2020. doi: 10.1073/pnas.1914677117. URL 10.1073/pnas.1914677117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].AlQuraishi Mohammed. End-to-end differentiable learning of protein structure. Cell Systems, 8(4):292–301.e3, April2019. doi: 10.1016/j.cels.2019.03.006. URL 10.1016/j.cels.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ingraham John, Riesselman Adam, Sander Chris, and Marks Debora. Learning protein structure with a differentiable simulator. In International Conference on Learning Representations, 2019. URL https://openreview.net/forum?id=Byg3y3C9Km. [Google Scholar]
- [12].Russakovsky Olga, Deng Jia, Su Hao, Krause Jonathan, Satheesh Sanjeev, Ma Sean, Huang Zhiheng, Karpathy Andrej, Khosla Aditya, Bernstein Michael, Berg Alexander C., and Fei-Fei Li. ImageNet Large Scale Visual Recognition Challenge. International Journal of Computer Vision (IJCV), 115(3):211–252, 2015. doi: 10.1007/s11263-015-0816-y. [DOI] [Google Scholar]
- [13].The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Research, 47(D1):D506–D515, November2018. doi: 10.1093/nar/gky1049. URL 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Berman HM. The protein data bank. Nucleic Acids Research, 28(1):235–242, January2000. doi: 10.1093/nar/28.1.235. URL 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jones David T.. Setting the standards for machine learning in biology. Nature Reviews Molecular Cell Biology, 20(11):659–660, September2019. doi: 10.1038/s41580-019-0176-5. URL 10.1038/s41580-019-0176-5. [DOI] [PubMed] [Google Scholar]
- [16].Mysinger Michael M., Carchia Michael, Irwin John. J., and Shoichet Brian K.. Directory of useful decoys, enhanced (DUD-e): Better ligands and decoys for better benchmarking. Journal of Medicinal Chemistry, 55(14):6582–6594, July2012. doi: 10.1021/jm300687e. URL 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen Lieyang, Cruz Anthony, Ramsey Steven, Dickson Callum J., Duca Jose S., Hornak Viktor, Koes David R., and Kurtzman Tom. Hidden bias in the DUD-e dataset leads to misleading performance of deep learning in structure-based virtual screening. PLOS ONE, 14(8):e0220113, August2019. doi: 10.1371/journal.pone.0220113. URL 10.1371/journal.pone.0220113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sieg Jochen, Flachsenberg Florian, and Rarey Matthias. In need of bias control: evaluating chemical data for machine learning in structure-based virtual screening. Journal of chemical information and modeling, 59(3):947–961, 2019. [DOI] [PubMed] [Google Scholar]
- [19].Bhuyan Md Shariful Islam and Gao Xin. A protein-dependent side-chain rotamer library. BMC Bioinformatics, 12(S14), December 2011. doi: 10.1186/1471-2105-12-s14-s10. URL 10.1186/1471-2105-12-s14-s10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shapovalov Maxim V. and Dunbrack Roland L.. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure, 19(6): 844–858, June 2011. doi: 10.1016/j.str.2011.03.019. URL 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Raman Srivatsan, Vernon Robert, Thompson James, Tyka Michael, Sadreyev Ruslan, Pei Jimin, Kim David, Kellogg Elizabeth, DiMaio Frank, Lange Oliver, Kinch Lisa, Sheffler Will, Kim Bong-Hyun, Das Rhiju, Grishin Nick V., and Baker David. Structure prediction for CASP8 with all-atom refinement using rosetta. Proteins: Structure, Function, and Bioinformatics, 77(S9): 89–99, 2009. doi: 10.1002/prot.22540. URL 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jumper John, Evans Richard, Pritzel Alexander, Green Tim, Figurnov Michael, Tunyasuvunakool Kathryn, Ronneberger Olaf, Bates Russ, Žídek Augustin, Bridgland Alex, Meyer Clemens, Kohl Simon A A; Potapenko Anna, Andrew J Ballard Andrew Cowie, Bernardino Romera-Paredes Stanislav Nikolov, Jain Rishub, Adler Jonas, Back Trevor, Petersen Stig, Reiman David, Steinegger Martin, Pacholska Michalina, Silver David, Vinyals Oriol, Senior Andrew W, Kavukcuoglu Koray, Kohli Pushmeet, and Hassabis Demis. High accuracy protein structure prediction using deep learning, December2020. URL https://predictioncenter.org/casp14/doc/CASP14_Abstracts.pdf.
- [23].AlQuraishi Mohammed. ProteinNet: a standardized data set for machine learning of protein structure. BMC Bioinformatics, 20(1), June2019. doi: 10.1186/s12859-019-2932-0. URL 10.1186/s12859-019-2932-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Case David A., Cheatham Thomas E., Darden Tom, Gohlke Holger, Luo Ray, Merz Kenneth M., Onufriev Alexey, Simmerling Carlos, Wang Bing, and Woods Robert J.. The amber biomolecular simulation programs. Journal of Computational Chemistry, 26(16):1668–1688, 2005. doi: 10.1002/jcc.20290. URL 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tian Chuan, Kasavajhala Koushik, Belfon Kellon A. A., Raguette Lauren, Huang He, Migues Angela N., Bickel John, Wang Yuzhang, Pincay Jorge, Wu Qin, and Simmerling Carlos. ff19sb: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. Journal of Chemical Theory and Computation, 16(1):528–552, November2019. doi: 10.1021/acs.jctc.9b00591. URL 10.1021/acs.jctc.9b00591. [DOI] [PubMed] [Google Scholar]
- [26].Bakan A, Meireles LM, and Bahar I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics, 27(11):1575–1577, April2011. doi: 10.1093/bioinformatics/btr168. URL 10.1093/bioinformatics/btr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Schrödinger, LLC. The PyMOL molecular graphics system, version 1.8 November2015.
- [28].Rego N and Koes D. 3dmol.js: molecular visualization with WebGL. Bioinformatics, 31(8): 1322–1324, December 2014. doi: 10.1093/bioinformatics/btu829. URL 10.1093/bioinformatics/btu829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sillitoe Ian, Dawson Natalie, Lewis Tony E, Das Sayoni, Lees Jonathan G, Ashford Paul, Tolulope Adeyelu, Scholes Harry M, Senatorov Ilya, Bujan Andra, Ceballos Rodriguez-Conde Fatima, Dowling Benjamin, Thornton Janet, and Orengo Christine A. CATH: expanding the horizons of structure-based functional annotations for genome sequences. Nucleic Acids Research, 47(D1): D280–D284, November2018. doi: 10.1093/nar/gky1097. URL 10.1093/nar/gky1097. [DOI] [PMC free article] [PubMed] [Google Scholar]