

Abstract
Elevated N-linked glycosylation of immunoglobulin G variable regions (IgG-VN-Glyc) is an emerging molecular phenotype associated with autoimmune disorders. To test the broader specificity of elevated IgG-VN-Glyc, we studied patients with distinct subtypes of myasthenia gravis (MG), a B cell-mediated autoimmune disease. Our experimental design focused on examining the B cell repertoire and total IgG. It specifically included adaptive immune receptor repertoire sequencing to quantify and characterize N-linked glycosylation sites in the circulating B cell receptor repertoire, proteomics to examine glycosylation patterns of the total circulating IgG, and an exploration of human-derived recombinant autoantibodies, which were studied with mass spectrometry and antigen binding assays to respectively confirm occupation of glycosylation sites and determine whether they alter binding. We found that the frequency of IgG-VN-Glyc motifs was increased in the total B cell receptor repertoire of MG patients when compared to healthy donors. The elevated frequency was attributed to both biased V gene segment usage and somatic hypermutation. IgG-VN-Glyc could be observed in the total circulating IgG in a subset of MG patients. Autoantigen binding, by four patient-derived MG autoantigen-specific monoclonal antibodies with experimentally confirmed presence of IgG-VN-Glyc, was not altered by the glycosylation. Our findings extend prior work on patterns of immunoglobulin variable region N-linked glycosylation in autoimmunity to MG subtypes.
Introduction
The vast diversity of immunoglobulin G variable regions (IgG-V) is critical for host immunity. This diversity arises through VDJ recombination and somatic hypermutation (SHM). Historically, IgG-V diversity has been represented by amino acid sequence alone with little focus on post-translational modifications. Recently, the presence of N-linked glycosylation in IgG-V (IgG-VN-Glyc) has been shown to contribute to diversity (1, 2). IgG-VN-Glyc is contingent upon the presence of the predictive amino acid motif N-X-S/T, where X can be any amino acid except for proline. This motif is most often introduced as a consequence of SHM (3). Less often it can be provided by the few germline gene segments (IGHV1-8, IGHV4-34, IGHV5-10-1, IGLV3-12, and IGLV5-37) in which it is encoded (4).
Higher frequencies of IgG-VN-Glyc than that which is found in healthy individuals have been observed in B cell malignancies (5–9) and in autoimmune diseases (10). Specifically, increased frequencies have been reported for ANCA-associated vasculitis (AAV) (11–13), rheumatoid arthritis (RA) (14–17), and primary Sjogren’s syndrome (pSS) (18, 19). The in vivo function of glycosylation in the IgG-V, a critical region of antigen contact, is not thoroughly understood. Follicular lymphomas may leverage N-linked glycosylation of their B cell receptor variable regions to activate antigen-independent signaling pathways that support survival (20). Antigen binding can also be influenced by IgG-VN-Glyc; this includes both increases and decreases in affinity and modulated functional activity. This is well highlighted by anti-citrullinated protein autoantibodies found in RA patients, where 80–100% of the autoantibodies include IgG-VN-Glyc, and antigen binding properties are consequently altered (15, 16, 21).
Myasthenia gravis (MG) is an autoimmune disorder affecting neuromuscular transmission. MG patients experience severe muscle weakness and increased fatigability (22, 23). The molecular immunopathology of MG is directly attributed to the presence of circulating IgG isotype autoantibodies specifically targeting extracellular domains of postsynaptic membrane proteins at the neuromuscular junction (NMJ) (23, 24). The most common subtype of autoantibody-mediated MG (approximately 85% of patients) is characterized by autoantibodies against the nicotinic acetylcholine receptor (AChR) (23). In many of the remaining patients, autoantibodies targeting the muscle-specific tyrosine kinase (MuSK) or lipoprotein receptor-related protein 4 (LRP4) are present (25–28). While MG autoantibodies cause disease, the underlying immune pathophysiology of the MG subtypes is distinct (29). AChR MG is governed primarily by IgG1 and IgG3 subclass autoantibodies (30, 31), which facilitate pathology through blocking acetylcholine, activating complement-mediated damage or initiating internalization of AChRs (32–35). LRP4 autoantibodies putatively disrupt Agrin-LRP4 signaling and primarily belong to theIgG1 and IgG2 subclass (27, 36). Conversely, the MuSK MG subtype is most often associated with IgG4 subclass autoantibodies, which are incapable of activating complement and other effector functions, but rather mediate pathology through blocking MuSK binding partners and its kinase activity (37–39).
Given that IgG isotype autoantibodies directly facilitate MG pathology and that their autoimmune mechanisms are divergent, we hypothesized that N-linked glycosylation might be differentially represented in the distinct AChR and MuSK MG subtypes. To that end we applied complementary sequencing and proteomic-based approaches to investigate variable region N-linked glycosylation patterns of the bulk B cell receptor repertoire and total circulating IgG in AChR and MuSK MG. Nucleotide-level sequencing was used to test for elevated IgG-VN-Glyc frequency in AChR and MuSK MG B cell receptor repertoires. Total IgG from sera was then evaluated with proteomic approaches to determine whether elevated IgG-VN-Glyc could be observed in the circulation. Finally, we explored whether N-linked glycans impact binding to pathogenic targets by using four patient-derived monoclonal autoantibodies with N-linked glycan occupancy validated by mass spectrometry. We show that IgG-VN-Glyc sequence motifs and total IgG-VN-Glyc are more frequent in both AChR and MuSK MG in comparison to healthy controls, and that the patterns differ between the two MG subtypes. However, the presence of IgG-VN-Glyc was not required for antigen binding by the four patient-derived pathogenic MG autoantibodies we tested.
Methods
Patient selection
This study was approved by Yale University’s Institutional Review Board (clinicaltrials.gov || NCT03792659). Informed written consent was received from all participating patients prior to inclusion in this study. Peripheral blood lymphocytes (PBMCs) and serum was collected from MG subjects at the Yale Myasthenia Gravis Clinic, New Haven, Connecticut, USA (40). BCR repertoire sequencing for the MuSK MG cohort (N=3) was derived from our previous study (41). AChR MG patients (N=11) and heathy control subjects (N=9) were selected for BCR-based adaptive immune receptor repertoire-sequencing (AIRR-Seq) using PBMC-derived RNA. Serum samples from all 3 MuSK MG subjects and 9 AChR MG subjects (8 overlapping with paired AIRR-Seq) were also investigated for the presence of VH gene glycosylation-specific signatures in the serum. For all patients with MuSK MG diagnoses and several patients with AChR MG diagnoses, longitudinal serum samples were collected. Detailed demographics and clinical data for the study cohort are presented in Supplemental Table 1.
A patient with rheumatoid arthritis (RA) (n = 1) was enrolled in an Institutional Review Board-approved research study at the University of California San Francisco (UCSF) for pathogen and autoantibody detection.
Protein electrophoresis and immunoblotting
For the initial screening processes patient sera samples were diluted 1:1 with 2X Storage Buffer (2X PBS, 20mM HEPES, 0.04% Sodium Azide, 20% Glycerol). The IgG from human sera or MuSK-specific mAb 4A3 and AChR-specific mAb 637 were captured with AG beads (Thermo Fisher) and then eluted by boiling at 95 degrees in 2X Laemmli buffer (with 10% beta-mercaptoethanol). For immunoblotting the gel was transferred to 0.45-micron nitrocellulose membrane and blotted with secondary anti-human IgG conjugated to IR800 dye (LICOR). Nitrocellulose blots were imaged with LICOR scanner and analyzed qualitatively by eye for presence of altered migration patterns in IgG. Potential IgG heavy chain migration phenotypes were qualitatively called by an experimenter blind to experimental conditions. For the three MuSK-specific (mAb MuSK1A, MuSK1B and MuSK3-28) and the AChR-specific mAb 637, Mini-PROTEAN® TGX Stain-Free™ Precast Gels (Biorad) and Laemmli Sample Buffer (Biorad) were used for SDS-page. Prior to electrophoresis the proteins were reduced with 0.1 M DTT (Thermo Fisher Scientific) and heat denatured at 95 °C for 5 min. After electrophoresis the gel was stained with Coomassie blue solution. Bands were visualized with the ChemiDoc™ Touch Imaging System (Biorad). Enzymatic assays for PNGase F and Endo S were performed according to manufacturer’s instructions (NEB). The effect of the enzymatic assays was either analyzed by Coomassie staining or immunoblotting.
BCR library preparation, pre-processing and analysis
First, RNA was isolated from frozen peripheral blood mononuclear cells using the RNAeasy Mini kit (Qiagen) per manufacturer’s instructions. Bulk libraries were prepared from RNA using reagents from New England Biolabs as part of the NEBNext® Immune Sequencing Kit as described previously (42, 43). Briefly, cDNA was reverse-transcribed by a template-switch reaction to add a 17-nucleotide unique molecular identifier (UMI) to the 5’ end with streptavidin magnetic bead purification. This was then followed by two rounds of PCR; the first round enriched for immunoglobulin sequences using IGHA, IGHD, IGHE, IGHG, and IGHM-specific 3’ primers and added a 5’ index primer. Libraries were purified with AMPure XP beads (Beckman) after which another round of PCR added Illumina P5 Adaptor sequences to each amplicon. The number of PCR cycles were selected based on real-time quantitative PCR to avoid the plateau phase. Libraries were then purified again with AMPure XP beads. Libraries were pooled in equimolar libraries and sequenced by 325 cycles for read 1 and 275 cycles for read 2 using paired-end sequencing with a 20% PhiX spike on the Illumina MiSeq platform according to manufacturer’s recommendations.
Processing and analysis of bulk B cell receptor sequences was carried out using tools from the Immcantation framework as done previously (44). Preprocessing was performed using pRESTO. Briefly, sequences with a phred score below 20 were removed and only those that contained constant region and template switch sequences were preserved. UMI sequences were then grouped, and consensus sequences were constructed for each group and assembled into V(D)J sequences in a two-step process involving an analysis of overlapping sequences (<8 nucleotides) or alignment against the IMGT (the international ImMunoGeneTics information system®) IGHV reference (IMGT/GENE-DB v3.1.19; retrieved December 1, 2019) if no significant overlap was found. Isotypes were assigned by local alignment of the 3’ end of the V(D)J sequence to constant region sequences. Duplicate sequences were removed and only V(D)J sequences reconstructed from more than 1 amplicon were preserved. Primer sequences used for this analysis are available at: https://bitbucket.org/kleinstein/immcantation.
V(D)J germline genes were assigned to reconstructed V(D)J sequences using IgBLAST v.1.14.0 and also using the December 1, 2019 version of the IMGT gene database (45). V(D)J sequences with IGH associated V and J genes were then selected for further analysis and non-functional sequences were removed. Germline sequences were reconstructed for each V(D)J sequence with D segment and N/P regions masked (with Ns) using the CreateGermlines.py function within Change-O v1.0.0(46). VH gene nucleotides up to IMGT position 312 were translated from both the aligned sequence and germline reconstructed V(D)J sequence using BioPython v1.75. To quantify the frequency of N-X-S/T glycosylation motifs, matches to the regular expression pattern “N[^P][S,T]” were quantified for each translated sequence, including for translated CDR and FWR fragments of the VH gene sequence (defined by IMGT coordinates) (4). N-X-S/T glycosylation motifs in the CDR3 and FWR4 regions were similarly quantified separately and included for CDR and FWR distribution analyses.
To build the lineage tree in Supplemental Fig. 1A, B cells were first clustered into clones by partitioning based on common IGHV gene annotations, IGHJ gene annotations, and junction lengths. Within these groups, sequences differing from one another by a length normalized Hamming distance of 0.2 within the junction region were defined as clones by single-linkage clustering using Change-O v.1.0.1(46). The Hamming distance threshold was determined by manual inspection of the distance to the nearest sequence neighbor plot using SHazaM v1.0.2(47). Phylogenetic tree topology and branch lengths of an illustrative clonal lineage were estimated using the HLP19 model in IgPhyML v1.1.3 and visualized using ggtree v2.0.4 and custom R scripts (48, 49).
Data availability
Sequencing data were deposited in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA738368/).
Mass Spectrometry
High-resolution mass spectrometry (HRMS) was employed to confirm the change in glycosylation status between wild type and mutated variants. In order to reduce complexity at the intact mass level a “middle-down” approach was utilized (50, 51). Intact antibodies were incubated with IdeS protease, followed by reduction of disulfide bonds. This workflow is well known to break down antibodies into three ~25 kDa subunits – LC, Fc, and Fd – and thereby separate disease-associated glycosylation within the variable region from standard glycosylation in the constant region. The heavy chain variable region (VH) is located within the Fd subunit. Purified mAb (2 mg/mL in PBS) was treated with 1 unit of IdeS protease (Promega) per 1 μg of mAb, and the sample was incubated at 37°C in a shaking incubator for 1.5 hours. The digested sample was then diluted into 6 M guanidinium chloride to a final IgG concentration of 1 mg/mL, and Tris(2-carboxyethyl) phosphine hydrochloride (TCEP-HCl) was added for a final concentration of 30 mM. The sample was incubated at 37°C in a shaking incubator for 1.5 hours, then the reaction was quenched by the addition of trifluoroacetic acid (final TFA concentration of 0.1% v/v). The sample was desalted by buffer exchange into LC-MS buffer (5 rounds of buffer exchange with an Amicon Ultra-0.5 mL centrifugal filter unit, 10 kDa MWCO).
The digested and reduced antibody species were further desalted and separated on a monolithic C4 column (RP-5H, 100 mm, 0.5 mm i.d., Thermo Scientific) with an Ultimate 3000 RSLCnano system (Thermo Scientific) using a binary gradient. The gradient utilized solvent A: 95% water, 5% acetonitrile, and 0.2% formic acid, and solvent B: 5% water, 95% acetonitrile, and 0.2% formic acid.
Data were acquired on a Q Exactive HF instrument with an attached HESI source (sheath gas = 10, auxiliary gas = 2, spare gas = 2, spray voltage = 3500 V, S-lens RF level = 65). MS1 acquisition used a scan range window of 400 to 2,000 m/z with 1 microscan and an AGC target of 1e6, at a resolution of 15,000.
Site directed mutagenesis of glycosylation site
Glycosylation sites (N-X-S/T) present in the V regions of the monoclonal antibodies were removed by mutating the asparagine (N) either to a glutamine (Q) or a serine (S). This was performed with Q5® Site-Directed Mutagenesis Kit (NEB) according to manufacturer’s instructions. The primers were designed with NEBaseChanger. Sequences of all expression plasmids were verified by Sanger sequencing.
Recombinant expression of human monoclonal antibodies (mAbs)
The mAbs were produced as previously described (Takata et al., 2019). Briefly, HEK293A cells were transfected with equal amounts of the heavy and the corresponding light chain plasmid using linear PEI (Polysciences Cat# 23966). The media was changed after 24 h to basal media (50% DMEM 12430, 50% RPMI 1640, 1% antibiotic/antimycotic, 1% Na-pyruvate, 1% Nutridoma). After 6 days the supernatant was harvested and Protein G Sepharose® 4 Fast Flow beads (GE Healthcare) were used for antibody purification.
Live cell-based autoantibody assay
Cell-based assays for detection of AChR or MuSK antibody binding were performed as we have previously described (52). Briefly, the cDNA encoding human AChR α, β, δ, ε-subunits and rapsyn-GFP were each cloned into pcDNA3.1-hygro plasmid vectors (Invitrogen, CA) and cDNA encoding human full-length MuSK was cloned into pIRES2-EGFP plasmid vector (Clontech). AChR and MuSK vectors were kindly provided by Drs. D. Beeson and A. Vincent of the University of Oxford. HEK293T (ATCC® CRL3216™) cells were transfected with either MuSK-GFP, or the AChR domains together with rapsyn-GFP. On the day of the CBA, the mAbs were added to the transfected cells in a dilution series (10 – 0.02 μg/ml). The binding of each mAb was detected with Alexa Fluor®-conjugated AffiniPure Rabbit Anti-Human IgG, Fcγ (309-605-008, Jackson Immunoresearch) on a BD LSRFortessa® (BD Biosciences). FlowJo software (FlowJo, LLC) was used for analysis.
Statistics
R v4.0.3 was used for all statistical analysis. Data frame handling and plotting was performed using functions from the tidyverse v1.3.0 in R and pandas v0.24.2 in python v3.7.5. A significance threshold of p<0.05 was used and shown on plots with a single asterisk; double asterisks correspond to a p<0.01. Unpaired one-tailed Wilcoxon tests were used for comparisons with healthy controls in repertoire analysis; the alternative hypothesis was that the average count of glycosylation motifs for each V(D)J sequence in MG BCR repertoires would be higher.
Results
The frequency of IgG-VN-glyc is elevated in the bulk B cell repertoire of patients with MG.
N-linked glycosylation sites only occur at amino acid sequence positions with the motif (N-X-S/T, where X = not proline). Elevated IgG-VN-Glyc in MG could arise from the introduction of these sites by SHM or the use of germline sites found in a small subset of VH gene segments (IGHV1-8, IGHV4-34 and IGHV5-10-1). To quantify global differences in the glycosylation frequency of the B cell repertoire, we examined the encoded B cell receptor repertoire generated by adaptive immune receptor repertoire sequencing (AIRR-seq) from the mRNA of circulating PBMCs from healthy donors (HD) and MG patients. For the AIRR-seq the MuSK MG patient cohort (N=3) included 12 unique timepoint samples, the AChR (N=10) included 10 unique timepoint samples, and each HD (N=9) included a single time point (Supplemental Table 1). The AIRR-seq library included a total of 10,565,778 (heavy chain only) raw reads; after quality control and processing, a high-fidelity data set was generated that consisted of 764,644 unique error-corrected sequences, which was further filtered to include only IgG subclass sequences that consisted of 232,094 sequences.
We observed a statistically significant elevation in median IgG-VN-Glyc site frequency for AChR MG (13.0%; P=0.039, one-tailed Wilcoxon test) and MuSK MG (17.4%, P=0.018, one-tailed Wilcoxon test) in comparison to healthy controls (10.3%) (Fig 1A). To investigate if the increased frequency of N-linked glycosylation sites was generated through preferred use of the three VH-gene segments that encode an N-X-S/T motif or through SHM, we assessed the frequency of the motif in germline reversions of the VH-gene segments (Fig 1B). We observed no increases in the germline frequency of IgG-VN-Glyc sites when comparing healthy and AChR MG patients (P=0.55, one-tailed Wilcoxon test), while the MuSK MG cohort exhibited a significant elevation (P=0.05, one-tailed Wilcoxon test), thus reflecting increases in the usage of select V gene segments (IGHV4-34, IGHV1-8 and IGHV5-10-1, in descending order of usage frequency). An illustrative example of N-X-S/T motif acquisition and conservation through the SHM process is shown for a B cell clonal family present in a MuSK MG repertoire, which includes acquisition of two motifs (Supplemental Fig 1A).
Figure 1. Frequency of IgG isotype-specific VN-Glyc sites in the B cell receptor repertoire.
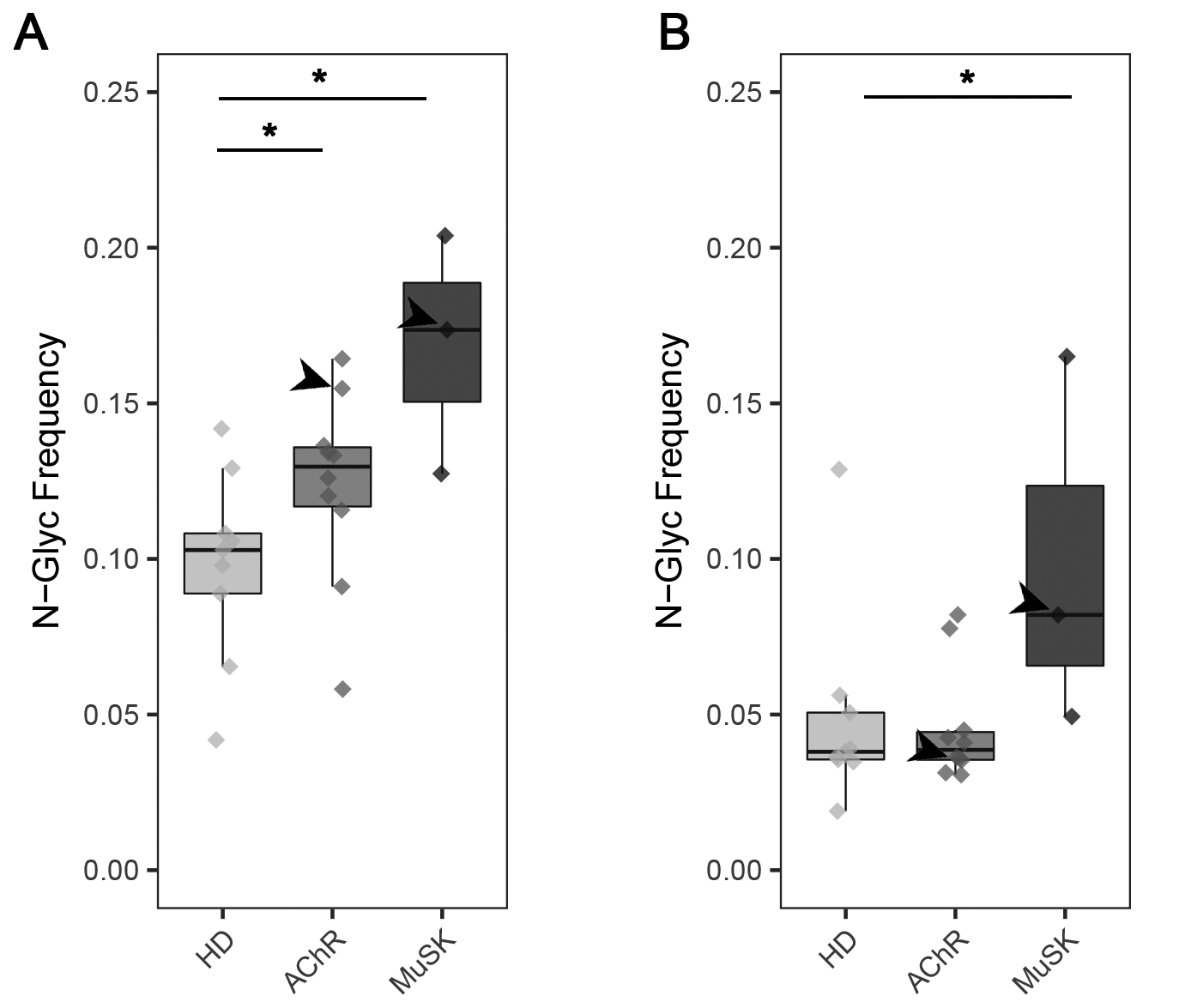
Analysis of adaptive immune receptor repertoire sequencing showing the number of N-linked glycosylation motifs in AChR and MuSK MG BCR repertoires relative to HDs. (A) The number of N-linked glycosylation motifs (N-X-S/T) per VH gene segment sequence in the repertoire derived from healthy controls, AChR and MuSK MG patients. (B) The number of N-linked glycosylation motifs (N-X-S/T) per germline reverted VH gene segment sequence in the BCR repertoire derived from healthy controls, AChR and MuSK MG patients. A significance threshold of p <0.05 was used and shown on plots with a single asterisk. Arrows point to AChR MG-1 or MuSK MG-1 depending on the boxplot in (A) and (B).
We then examined if the increases in glycosylation frequency were specific to complementarity-determining (CDR) regions, which are primarily responsible for antigen contact, or also included distribution in the framework regions (FWR), which maintain structural integrity of the variable domains. The motif could be found in all CDRs and FWRs in sequences from the HDs and MG patients with the exception of FWR2, in which the motif was absent in all sequences (Supplemental Fig 1B). The motif was most often observed in the FWR3 sequences from the HDs and MG patients. Sequences from the AChR MG patients revealed a significant increase in motif frequency only in the CDR2 region in comparison to HD (P=0.047, one-tailed Wilcoxon-test). Elevated frequencies were observed in comparisons of MuSK MG and HDs at all regions (Supplemental Fig 1B), and statistically significant increases were observed in the FWR1, CDR2, and FWR4 regions (P=0.009, P=0.0045, P=0.042, respectively - one-tailed Wilcoxon tests). Examining the location of the motifs within each region (Supplemental Fig 1C) showed that they were present throughout, but were not uniformly distributed, as some areas showed enriched accumulation. Those present in FWR1, although rare, were found close to the CDR1 (Supplemental Fig 1C).
In summary, the frequency of variable region N-linked glycosylation sites among IgG switched B cells differ when comparing healthy controls and AChR or MuSK MG patients. These differences result from SHM in AChR MG, while differences found in MuSK MG result from both SHM and elevated usage of V genes with germline encoded N-linked glycosylation sites.
Proteomic analysis demonstrates elevated IgG-VN-glyc in MG.
The heavy chains of total IgG isolated from serum of patients with autoimmune disease, such as RA and ANCA-associated vasculitis, migrate at a higher molecular weight (MW) than those of healthy controls due to the presence of a large fraction of IgG-VN-Glyc (13, 15). Having demonstrated that the B cell repertoire of both AChR and MuSK MG include elevated IgG-VN-Glyc site frequency, we next sought to investigate if total circulating IgG from patients with MG reflected this MW increase. To that end, we analyzed total IgG purified from serum samples from the MG cohort (MuSK MG, N=3; AChR MG, N=9) for the presence of IgG-VN-Glyc (Supplemental Table 1). Longitudinal samples were also included to evaluate the temporal stability of IgG-VN-Glyc patterns (Supplemental Table 1). The IgG-VN-Glyc presence was tested through the assessment of immunoglobulin heavy chain migration patterns by SDS-PAGE. Serum-derived IgG from a patient with RA was included as a positive control (Fig 2A). Total IgG migration patterns between healthy individuals and MG patients were compared (Fig 2B); differences were noted for one AChR patient (AChR MG-1) and one MuSK patient (MuSK MG-1). Longitudinal samples were assessed spanning a period of four years of clinical disease; the altered migration patterns remained consistent through all of the time points collected from these two subjects (Fig 2C). These two subjects also demonstrated an elevated frequency of IgG-VN-glyc in their B cell repertoire (Fig 1 arrows).
Figure 2. Proteomic analysis of total serum IgG glycosylation suggests elevated Fab N-linked glycosylation in MG.
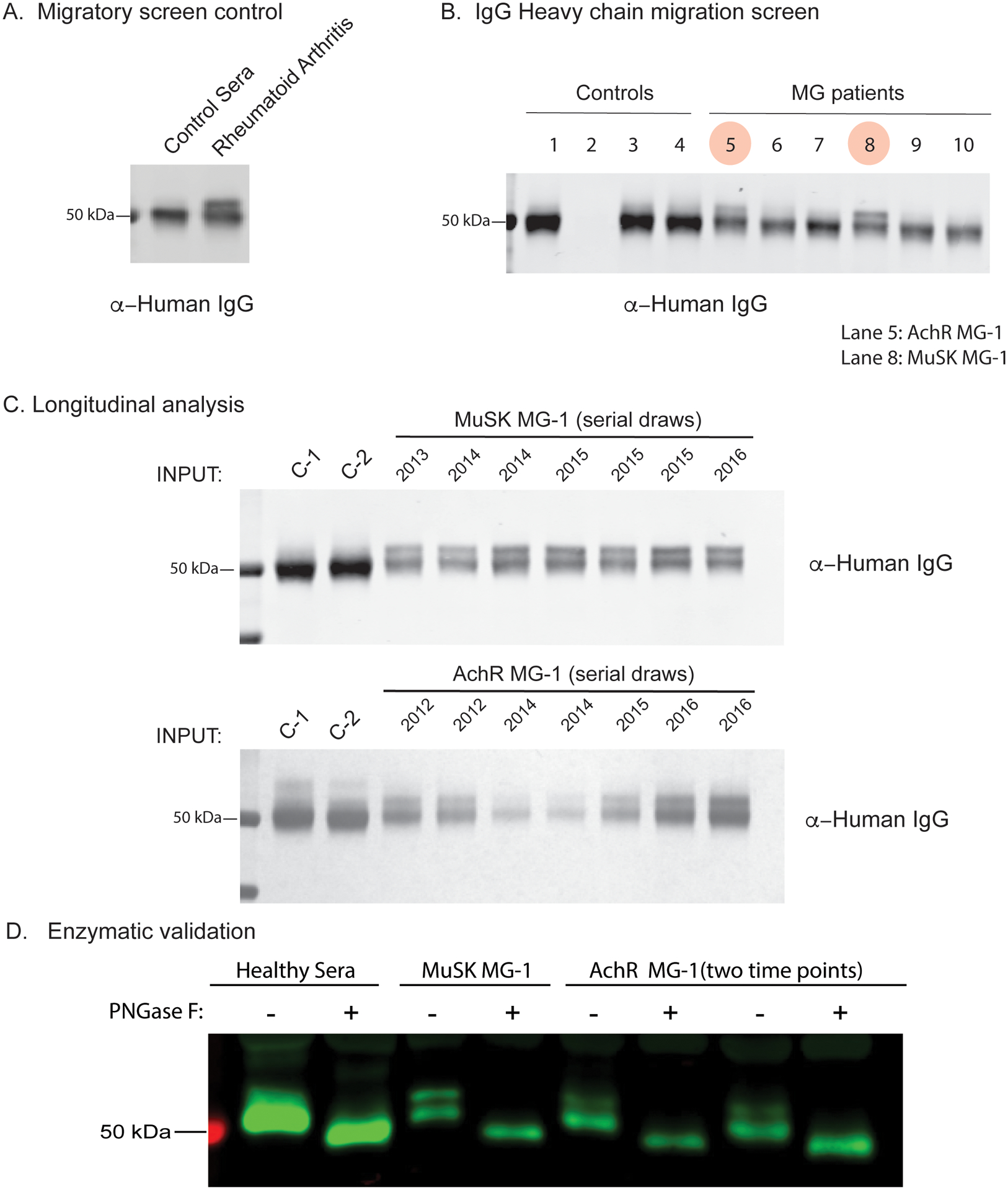
Total serum IgG heavy chain migratory patterns from SDS-PAGE and immunoblotting with anti-human secondary are shown for sub-panels. (A) Serum IgG heavy chain migration pattern from non-inflammatory control and a patient with RA shown by immunoblot. (B) Total serum IgG heavy chain migration patterns for non-inflammatory controls (Lane 1, 3, 4), A/G beads only (No antibody, Lane 2) and AChR MG (Lanes 5, 6, 7) or MuSK MG (Lanes 8, 9, 10) shown by immunoblot. Lane 5 corresponds to subject AChR MG-1 and lane 8 corresponds to subject MuSK MG-1. (C) Longitudinal serum IgG heavy chain patterns from subjects MuSK MG-1 and AChR MG-1 shown by immunoblotting with an anti-human IgG secondary antibody. (D) Enzymatic validation of N-linked glycosylation in total circulating IgG from a healthy individual, MuSK MG-1 and AChR MG-1 (two distinct serial draws), shown by immunoblot. Human IgG was incubated with or without PNGase F then subject to SDS-PAGE and immunoblotted with an anti-human IgG secondary antibody. Treatment with PNGase F results in loss of migration phenotype. RA = Rheumatoid Arthritis.
To assess whether the altered migration patterns observed in MuSK MG-1 and AChR MG-1 reflect elevated IgG glycosylation as opposed to other possible modifications such as phosphorylation or ubiquitination, IgG was subjected to digestion with PNGase F or Endo S. Enzymatic digestion of MuSK MG-1 IgG with PNGase F, which non-specifically cleaves N-linked glycosylation units, resulted in a loss of the atypical IgG migration pattern (Fig 2D). By comparison, Endo S, which cleaves N-linked glycosylation at N297 of IgG constant region, caused a shift in gel mobility in both samples, but no change in the atypical pattern (Supplemental Fig 2). Removal of phosphates with CIP, a phosphatase enzyme, had no effect on migration (Supplemental Fig 2). These results suggest that a subset of patients with MG possess atypical IgG glycosylation specifically in the Fab region, likely due to IgG-VN-Glyc, which appears to be a stable feature over long periods of time (3–4 years).
MuSK and AChR human mAbs can contain occupied IgG-VN-Glyc sites
Having demonstrated that the bulk B cell receptor repertoire and total circulating IgG included elevated IgG-VN-Glyc frequencies, we next turned our attention to MG-specific autoantibodies. We had previously generated three human recombinant MuSK-specific mAbs that demonstrated in vitro pathogenic capacity (38, 53, 54). We found glycosylation motifs (N-X-S/T) in the variable region of all three MuSK mAbs, in either the heavy (MuSK1A and 3–28) or light chain (MuSK1B) (Fig 3A–C). Specifically, the motif was present in the heavy chain FWR3 of MuSK1A due to the use of IGHV1-8 where it is encoded in the germline. MuSK1B acquired the motif in the light chain (FWR1) through SHM, and the heavy chain lost the motif in the CDR2, which was present in the germline VH (IGHV4-34). MuSK3-28 acquired the motif in the heavy chain (CDR2) through SHM. We sought to test if these sites were occupied. Digestion with PNGase F reduced the MW of the heavy (MuSK1A and MuSK3-28) and light chain (MuSK2A) of the mAbs suggesting the presence of N-linked glycosylation on the antibodies (Supplemental Fig 3A–C). We then removed these putative glycosylation sites by mutagenesis and screened all constructs for variations in migratory pattern due to MW changes. Removal of glycosylation sites led to a change in gel mobility as expected in all three MuSK mAbs, which was also consistent with site-specific occupancy (Supplemental Fig 3D–F). Next, we performed intact mass spectrometry analysis to more precisely detect these glycosylation sites (Fig 3A–C). Differences in mass and mass spectra can be used to confirm the presence of IgG-VN-Glyc. All three MuSK autoantibodies were found to be glycosylated and the mutated variants were significantly less heterogeneous and lighter in mass by approximately 2 kDa (Fig 3A–C). Because N-glycans are extremely heterogenous molecular moieties, proteins containing IgG-VN-Glyc have elevated mass spectra heterogeneity; these findings confirm the presence of glycosylation, and that mutations were successful in disrupting the introduction of glycosylation in all three MuSK mAbs.
Figure 3. Mass spectrometry analysis of N-glycan occupancy in MuSK- and AChR-specific human monoclonal antibodies.
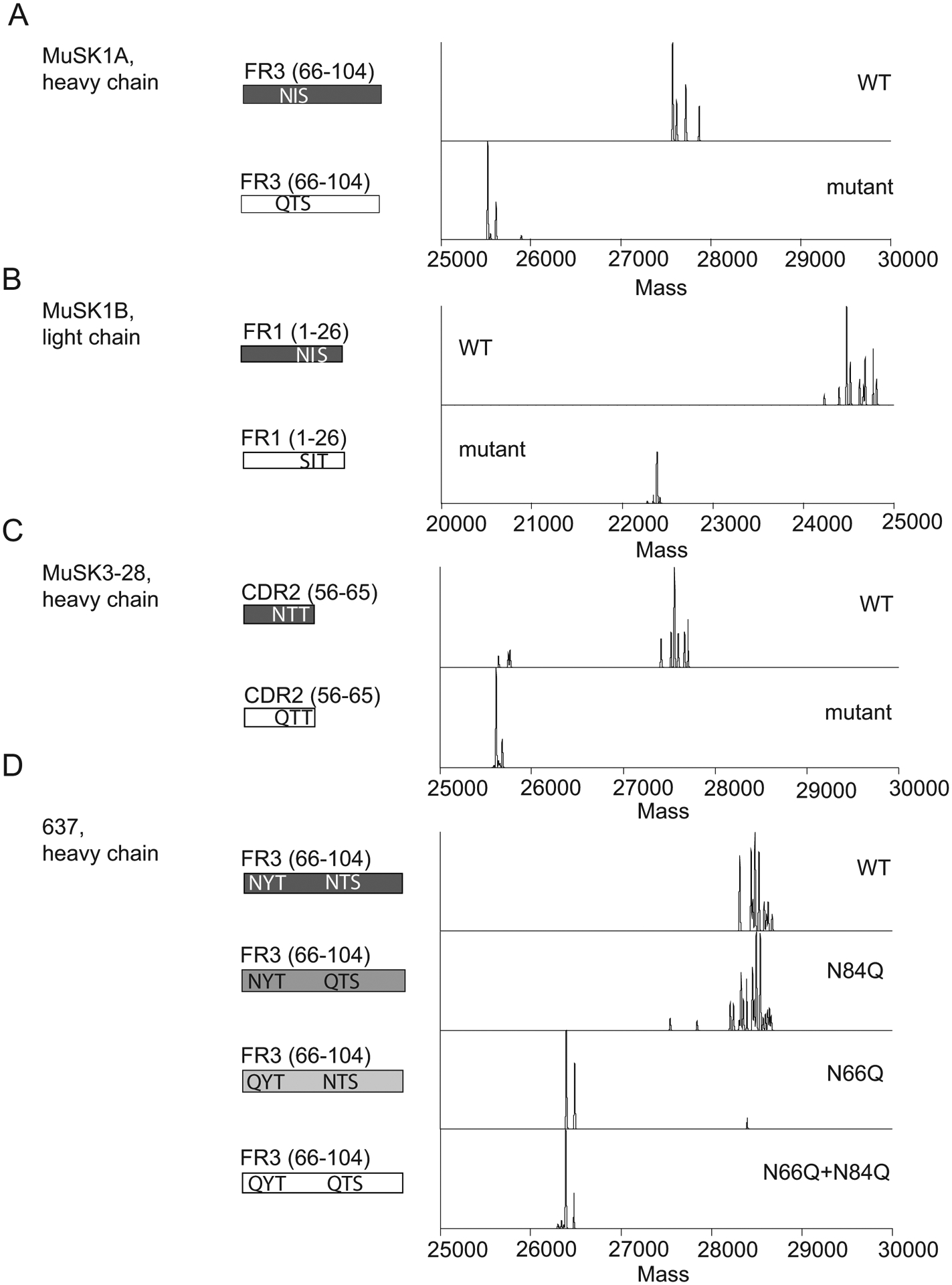
Validation of N-glycan variable region occupancy in three patient-derived monoclonal anti-MuSK antibodies (MUSK1A, MUSK1B, MUSK3-28) and one patient-derived monoclonal anti-AChR antibody (monoclonal antibody 637). Schematic of variable regions for anti-MuSK antibodies indicating regions (CDR or FWR) and localization of putative N-linked glycosylation amino acid motifs alongside deconvoluted mass spectra of the associated constructs (labels). This is shown for MUSK1A (A), MUSK1B (B), MUSK3-28 (C) and mAb 637 (D).
Next, to extend these findings to AChR MG, we evaluated the patient-derived AChR-specific mAb 637 (55) as its sequence shows two predicted N-linked glycosylation sites (N66 and N84) within the variable region of the FR3 of heavy chain, which were acquired through SHM (Fig 3D). The heavy chain of mAb 637 migrated at a lower MW when treated with PNGase F in comparison to untreated mAb (Supplemental Fig 3G). We subsequently performed mutagenesis and produced several different constructs disrupting the two predicted glycosylation sites—at N66 and N84. Mutation of N66 alone or N66 and N84 together resulted in a construct that migrated at lower MW, while mutation of only the N84 did not affect migration (Supplemental Fig 3H, I). We further explored this result using intact mass spectrometry analysis of the Fd from the WT and three mutants (Fig 3D). The WT construct and the construct containing a mutation at position N84 had a complex mixture of proteoforms clustered between 28.3 – 28.8 kDa, whereas constructs containing mutations at N66 (including a mutant at both N66 and N84) were less heterogeneous, with proteoforms clustering closer to 26.4 kDa (Fig 3D). The lighter mass and simplified proteoform signature of variants containing a mutation at N66 suggests that N66 is the main site of glycosylation in mAb 637 and not N84. Additionally, removal of the glycosylation site N66 did not shift glycosylation to the second predicted site at N84. In summary, autoantigen-specific mAbs in MG can contain occupied N-glycosylation of their IgG variable regions.
The effect of glycosylation on MG-derived monoclonal antibody binding.
We then sought to test the contribution of IgG-VN-Glyc to MG mAb binding. Given that these mAbs were previously validated for their capacity to bind AChR or MuSK in live cell-based assays (38, 53, 54), we tested the contribution of IgG-VN-Glyc sites to binding using the same approach (Fig 4A–D). When tested over a wide range of concentrations (10 – 0.02 μg/mL), we found that loss of IgG-VN-Glyc did not affect binding of the three anti-MuSK mAbs or the anti-AChR mAb to their respective targets.
Figure 4. The effect of glycosylation on MuSK and AChR-specific human monoclonal antibody binding properties.
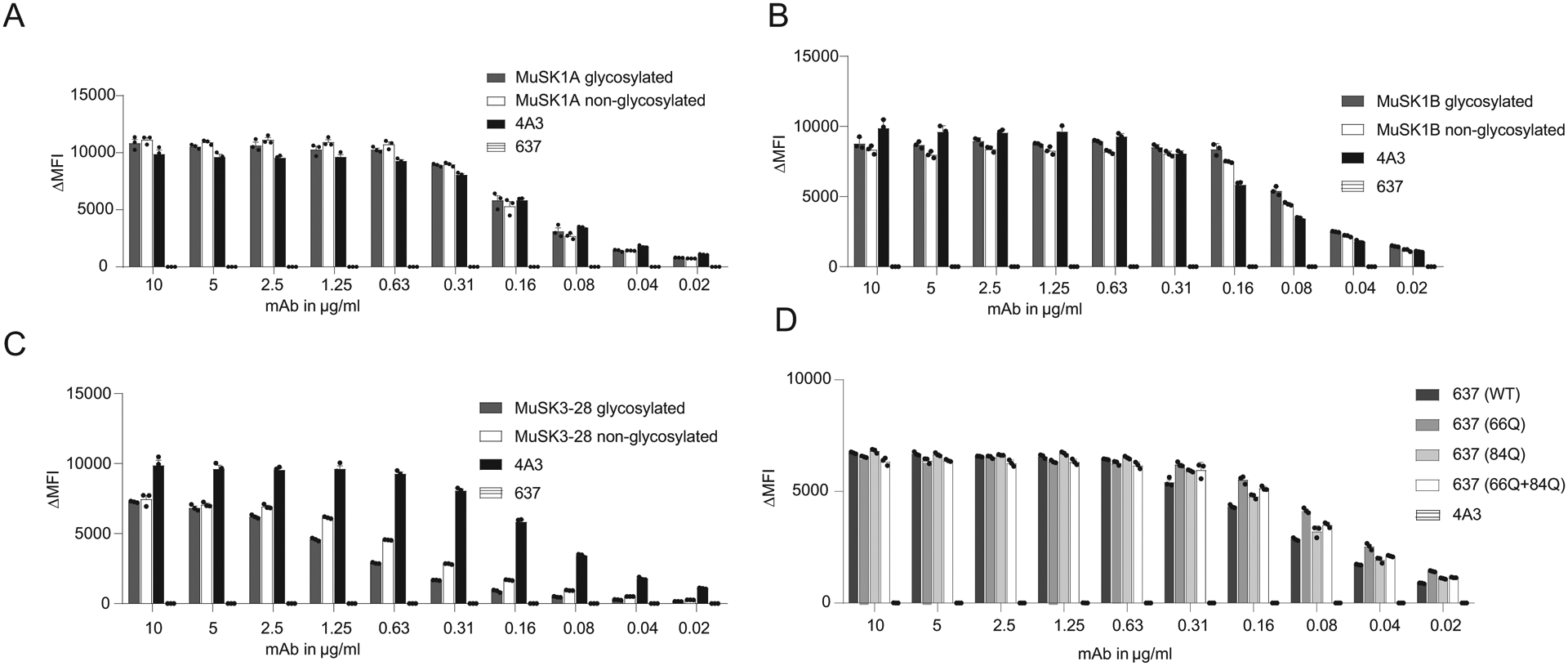
Antigen binding of three MuSK monoclonal antibodies (mAb) and one AChR-specific human mAb is not affected by the presence of variable region N-linked glycans. Wildtype MuSK and AChR mAbs and their glycovariants were tested for surface binding to MuSK or AChR with live cell-based assays (CBA) using MuSK-GFP-transfected (A-C) or AChR and rapsyn-GFP-transfected (D) HEK293T cells. All wildtype mAbs and their variants were analyzed in a two-fold dilution series. In the MuSK CBA (A-C), humanized MuSK-specific mAb 4A3 was used as the positive control and human AChR-specific mAb 637 as the negative control. In the AChR CBA (D), the MuSK-specific mAb 4A3 was used as the negative control. Each data point represents the mean value from three independent experiments, and error bars represent SDs. The ΔMFI was calculated by subtracting the signal from non-transfected cells from that of the transfected cells.
Discussion
The percentage of IgG in healthy individuals that includes V-region glycosylation is estimated to be between 10–25% (1). The wide range reflects different approaches of measurement, particularly whether the measurement was derived from genomic sequencing or analysis of immunoglobulins (1). Nucleotide level sequencing demonstrates that the frequency of V-region glycosylation motifs in healthy individuals is 9–12% (8, 19, 21, 56). Our BCR sequencing data of the IgG heavy chain VH gene segment from healthy controls identified approximately 0.1 glycosylation motifs per sequence. As the motif is rarely present more than once in each sequence, this indicates that approximately 10% of VH gene segments carry a glycosylation motif, which is consistent with these previous studies. We used this baseline value to establish that elevated numbers of the IgG-VN-Glyc motif are found in the IgG VH gene segment sequences derived from MG disease subtypes. Two mechanisms are thought to contribute to increased IgG-VN-Glyc motif frequency. The first is enriched usage, at the naïve B cell stage, of the five germline VH or VL gene segments that contain N-linked glycosylation motifs (IGHV1-8, IGHV4-34, IGHV5-10-1, IGVL3-12 and IGVL5-37). The second mechanism is selection, during affinity maturation, of B cell clones that acquire N-linked glycosylation motifs through the SHM process. The germline encoded motifs in the three heavy chains are found in CDR2 of IGHV4-34 and in the FWR3 of IGHV1-8 and IGHV5-10-1. Our sequence analysis showed that these motifs, when they are acquired through SHM, are distributed throughout the variable region with the exception of FWR2. The distribution mirrors SHM patterns in that mutations accumulate preferentially in CDRs and FWR3. While replacement mutations can be observed in FWR2, our data (and that of others (21)) suggest that a glycosylation motif is not tolerated in this region, suggesting that such alterations are constrained by the role of the FWRs in conserving the overall structure of the antibody. Similarly, motifs found in FWR1 were restricted to regions near the flexible CDR loops that it flanks. These collective findings indicate that the acquisition of the motif may be driven by positive selection. It is also possible that the motifs could be selectively neutral but arise as a consequence of SHM. If so, MG repertoires could have more motifs than the HD repertoires simply by having more SHM, and the motifs could be concentrated around the CDRs due to the presence of known hotspot motifs in those regions.
SHM appears to be a major contributor to the increased frequency of the IgG-VN-Glyc sites in the AChR MG patients we studied. Positive selection leading to enriched N-linked glycosylation DNA motifs has been observed in the parotid gland of patients with pSS, a structure known to contain ectopic lymphoid follicles in these patients (19). Similarly, the thymus in a subset of MG patients includes germinal centers, which are thought to contribute to the generation and maturation of AChR autoantibody-producing B cells (57–59). Thus, positive selection of N-linked glycosylation motifs may occur in this compartment, and support for this possibility is provided by our previous study where we found that the IgG-switched BCR sequences in MG thymus were enriched in N-linked glycosylation motifs (43). The current study was not designed to investigate whether thymic lymphofollicular hyperplasia or the surgical removal of the thymus is associated with the frequency of IgG-VN-Glyc frequency in the periphery. However, it is reasonable to speculate that the heterogeneity of IgG-VN-Glyc frequency found in the AChR MG patients studied here may relate to MG disease subtypes, some of which are defined by thymus-related pathology.
Both V gene usage and the SHM process contributed to the elevated frequencies we observed in the MuSK MG patients. Defects in B cell tolerance checkpoints can skew the developing repertoire (42). Such defects are known to exist in both AChR and MuSK MG (60), and thus may contribute to enrichment of the V genes containing N-linked glycosylation motifs, which we observed in some patients with MuSK MG. However, the accumulation of additional motifs through SHM suggests that, in the MuSK disease subtype, antigen-driven positive selection also plays a role in the conspicuously elevated IgG-VN-Glyc frequency. It remains possible that this selection is an antigen-independent process. Examples of this mechanism include interactions between glycosylated B cell receptors and lectins (20), which are thought to drive proliferation in B cell malignancies and some autoimmune diseases (19).
The antigen binding of the four human mAbs that we studied was not disturbed by N-linked glycosylation. Although a limited number of mAbs were available for study, these results align well with data derived from other human MuSK-binding mAbs; some of which include glycosylation motifs and others that do not, indicating that the glycan is not essential for binding (39). These collective results indicate that selection of the IgG-VN-Glyc in human MuSK autoantibodies may not have been driven by MG specific self-antigen positive selection. This is somewhat unexpected given that the IgG-VN-Glyc sites could be found in regions responsible for antigen contact (CDRs). However, it remains possible that the positions of the IgG-VN-Glyc in the human MuSK-binding mAbs we studied were not essential for antigen-autoantibody contact. Structural studies of MuSK-mAb complexes would be required to test this possibility. Similarly, the variable regions of anti-citrullinated protein autoantibodies (ACPA) from patients with RA are consistently glycosylated, but their binding is not influenced by the modification (17). However, other investigations suggest that binding can be modulated as a consequence of their presence (15, 16, 21). These findings suggest that an autoantigen-independent selection mechanism may influence the IgG-VN-Glyc motif frequency in the autoimmune repertoire in some, but not all, autoimmune diseases.
Our proteomic analysis of the serum-derived total IgG from two of the study subjects (one from each of the AChR and MuSK MG cohorts) showed a higher molecular weight band in the electrophoresis studies. These results indicate that a large fraction of circulating IgG included variable region N-linked glycosylation. The higher molecular weight bands are not attributable to only the AChR- or MuSK-specific IgG, given that these autoantibodies are present at very low concentrations in the circulation. While a number of the MG patients were observed to have elevated IgG-VN-Glyc motif frequency in the B cell repertoire sequencing data, the serum-derived IgG from some of the same patients did not include a higher molecular weight band in the proteomic analysis. Moreover, the intensity of the higher molecular weight bands in some samples suggests that the fraction of IgG-VN-Glyc is higher than the B cell sequencing data indicate. These results align well with empirically-derived models that describe a lack of clonal overlap between the serum IgG and circulating B cell repertoire highlighting the discordance between serum antibody repertoire (humoral immunity) and the VH gene repertoire (61–63). Rather, these findings may reflect that much of the circulating IgG is derived from long-lived plasma cells residing in the bone marrow.
One of two possible glycosylation sites in an autoantibody known to be specific for AChR, mAb 637, was shown to be unoccupied. We speculate that this is indicative of context specific N-glycosylation (local amino acid sequence containing the motif or cellular environment) or inherent selectivity for one site over the other possibly due to conformation or solvent accessibility. Nevertheless, this site did not appear to contribute to binding activity, similar to our observations obtained by testing the MuSK mAbs. We recognize, as a study limitation, that the in vitro expression of these mAbs may not emulate the glycosylation occupancy in vivo. We did use a mammalian expression system (human embryonic kidney cells), to achieve the best approximation of the in vivo status, and we experimentally confirmed occupancy for the antigen binding studies. It remains to be investigated whether variables such as the stage of B cell activation or tissue residence could alter the occupancy.
Finally, a consensus on the function of IgG-VN-Glyc in health or disease is unclear. Several possibilities have been described, including perturbation of antibody-antigen interactions (binding affinity, specificity), altered metabolism of B cells or IgG in vivo (half-life, clearance), mis-localization of IgG to host tissue, redemption of autoreactive B cells, and inappropriate selection/expansion of autoreactive B cells in germinal centers (1, 2, 64). Elimination of these motifs or removal of the glycosyl moiety itself have been observed to impair antigen binding—such as in anti-adalimumab/infliximab antibodies derived from patients treated for RA (21). However other studies have suggested a more nuanced picture; a study of anti-CCP (cyclic citrullinated protein) autoantibodies showed no contribution of N-linked glycans to binding (17). Here we show unequivocal evidence that the presence of N-linked glycans is not required for binding in the case of four MG autoantibodies. This finding is in agreement with a number of studies focusing on human autoantibodies, which demonstrated that IgG-VN-Glyc can alter binding or activity but is not required for these properties (15, 21, 65).
Our study is not without limitations. MG is a heterogeneous disease; a number of AChR MG subtypes are well documented. Accordingly, we recognize the limited number of patients included in this investigation and thus encourage caution in generalizing our findings. Furthermore, we acknowledge that the investigation of several human mAbs is unlikely to precisely represent the circulating MG autoantibody repertoire. Thus, we cannot conclude that AChR or MuSK autoantibodies are universally glycosylated. Other investigations (13, 15) of human autoimmune disease, which showed the conspicuous presence of IgG-VN-Glyc by electrophoresis, specifically focused on antigen-enriched autoantibodies rather than the B cell receptor repertoire and total circulating IgG, which was the focus of our study. While we studied a small set of specific autoantibodies in the form of human-derived mAbs, we recognize that the mAbs, serve only to represent a fraction of the total circulating autoantibody repertoire. While challenging, it will be important to isolate serum-derived MuSK and AChR-specific autoantibodies in order to investigate both the frequency and properties of the variable region glycans in this broad population of circulating MG autoantibodies.
In summary, IgG-VN-Glyc of the total circulating IgG and bulk B cell receptor repertoire is elevated in a subset of AChR and MuSK MG patients. These findings are consistent with those of a previous study (56) that showed elevated (albeit not statistically significant) V region N-glycosylation sites in the B cell receptor repertoire of MG patients compared to healthy controls. Our findings extend this molecular phenotype beyond RA, pSS, SLE, and AAV. IgG-VN-Glyc did not affect AChR or MuSK autoantibody binding in the limited number of human mAbs we tested. We speculate an elevation in N-linked glycosylation motifs containing V gene sequences may be driven by the presence of dysregulated germinal centers that contribute to B cell selection defects observed in the disease. Our findings contribute to efforts to understand the basic biology of IgG-VN-Glyc and its association with disease.
Supplementary Material
Key Points.
IgG-VN-Glyc sequence motif frequency is elevated in the MG B cell receptor repertoire
The fraction of total IgG-VN-Glyc in MG serum is elevated
The presence of IgG-VN-Glyc did not alter binding of several MG patient-derived mAbs
Acknowledgments
The authors thank Karen Boss for expert copy editing and proofreading, Dr. Bailey Munro-Sheldon for verifying autoantibody titers, and Charlotte A. Gurley for assisting with manuscript and reference formatting.
Disclosures
KCO has received research support from Ra Pharma and is a consultant and equity shareholder of Cabaletta Bio. KCO is the recipient of a sponsored research subaward from the University of Pennsylvania, the primary financial sponsor of which is Cabaletta Bio. KCO has received speaking and advising fees from Alexion and Roche. MLF has received research support from Grifols. RJN has received research support from Genentech, Alexion Pharmaceuticals, argenx, Annexon Biosciences, Ra Pharmaceuticals, Momenta, Immunovant, and Grifols. RJN has served as consultant/advisor for Alexion Pharmaceuticals, argenx, CSL Behring, Grifols, Ra Pharmaceuticals, Immunovant, Momenta and Viela Bio. SHK receives consulting fees from Northrop Grumman. MRW has received research support from Roche/Genentech. KBH receives consulting fees from Prellis Biologics.
Funding support
KCO is supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH through awards R01-AI114780 and R21-AI142198, (NIH) through the Rare Diseases Clinical Research Consortia of the NIH (award number U54-NS115054) and by a Neuromuscular Disease Research program award from the Muscular Dystrophy Association (MDA) under award number MDA575198. RJ is supported by the NIAID award number F31-AI154799. SHK is supported by the NIAID under award number R01-AI104739. MLF is a recipient of the James Hudson Brown — Alexander Brown Coxe Postdoctoral Fellowship in the Medical Sciences and the research of MLF has further been supported through a DFG Research fellowship (FI 2471/1-1). NLK acknowledges support from the National Institute of General Medical Sciences under award number P41 GM108569 for the National Resource for Translational and Developmental Proteomics. CMB is funded by The Emiko Terasaki Foundation (Project 7027742 / Fund B73335) and by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health (award 1K99NS117800-01). SEV is funded by the National Institute of Diabetes and Digestive and Kidney Diseases of the NIH (award 1F30DK123915-01). JDL is funded by a grant from Chan Zuckerberg Biohub. JDL, MRW and CMB are funded by the National Institute of Mental Health (NIMH) of the NIH (award 1R01MH122471-01).
References
- 1.van de Bovenkamp FS, Hafkenscheid L, Rispens T, and Rombouts Y. 2016. The Emerging Importance of IgG Fab Glycosylation in Immunity. J Immunol 196: 1435–1441. [DOI] [PubMed] [Google Scholar]
- 2.Kanyavuz A, Marey-Jarossay A, Lacroix-Desmazes S, and Dimitrov JD. 2019. Breaking the law: unconventional strategies for antibody diversification. Nat Rev Immunol 19: 355–368. [DOI] [PubMed] [Google Scholar]
- 3.Dunn-Walters D, Boursier L, and Spencer J. 2000. Effect of somatic hypermutation on potential N-glycosylation sites in human immunoglobulin heavy chain variable regions. Mol Immunol 37: 107–113. [DOI] [PubMed] [Google Scholar]
- 4.Lefranc MP 2001. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res 29: 207–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, and Stevenson FK. 2002. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood 99: 2562–2568. [DOI] [PubMed] [Google Scholar]
- 6.Radcliffe CM, Arnold JN, Suter DM, Wormald MR, Harvey DJ, Royle L, Mimura Y, Kimura Y, Sim RB, Inoges S, Rodriguez-Calvillo M, Zabalegui N, de Cerio AL, Potter KN, Mockridge CI, Dwek RA, Bendandi M, Rudd PM, and Stevenson FK. 2007. Human follicular lymphoma cells contain oligomannose glycans in the antigen-binding site of the B-cell receptor. J Biol Chem 282: 7405–7415. [DOI] [PubMed] [Google Scholar]
- 7.Odabashian M, Carlotti E, Araf S, Okosun J, Spada F, Gribben JG, Forconi F, Stevenson FK, Calaminici M, and Krysov S. 2020. IGHV sequencing reveals acquired N-glycosylation sites as a clonal and stable event during follicular lymphoma evolution. Blood 135: 834–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koning MT, Quinten E, Zoutman WH, Kielbasa SM, Mei H, van Bergen CAM, Jansen P, Vergroesen RD, Willemze R, Vermeer MH, Tensen CP, and Veelken H. 2019. Acquired N-Linked Glycosylation Motifs in B-Cell Receptors of Primary Cutaneous B-Cell Lymphoma and the Normal B-Cell Repertoire. J Invest Dermatol 139: 2195–2203. [DOI] [PubMed] [Google Scholar]
- 9.Zabalegui N, de Cerio AL, Inoges S, Rodriguez-Calvillo M, Perez-Calvo J, Hernandez M, Garcia-Foncillas J, Martin-Algarra S, Rocha E, and Bendandi M. 2004. Acquired potential N-glycosylation sites within the tumor-specific immunoglobulin heavy chains of B-cell malignancies. Haematologica 89: 541–546. [PubMed] [Google Scholar]
- 10.Visser A, Hamza N, Kroese FGM, and Bos NA. 2018. Acquiring new N-glycosylation sites in variable regions of immunoglobulin genes by somatic hypermutation is a common feature of autoimmune diseases. Ann Rheum Dis 77: e69. [DOI] [PubMed] [Google Scholar]
- 11.Holland M, Yagi H, Takahashi N, Kato K, Savage CO, Goodall DM, and Jefferis R. 2006. Differential glycosylation of polyclonal IgG, IgG-Fc and IgG-Fab isolated from the sera of patients with ANCA-associated systemic vasculitis. Biochim Biophys Acta 1760: 669–677. [DOI] [PubMed] [Google Scholar]
- 12.Xu PC, Gou SJ, Yang XW, Cui Z, Jia XY, Chen M, and Zhao MH. 2012. Influence of variable domain glycosylation on anti-neutrophil cytoplasmic autoantibodies and anti-glomerular basement membrane autoantibodies. BMC Immunol 13: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lardinois OM, Deterding LJ, Hess JJ, Poulton CJ, Henderson CD, Jennette JC, Nachman PH, and Falk RJ. 2019. Immunoglobulins G from patients with ANCA-associated vasculitis are atypically glycosylated in both the Fc and Fab regions and the relation to disease activity. PLoS One 14: e0213215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youings A, Chang SC, Dwek RA, and Scragg IG. 1996. Site-specific glycosylation of human immunoglobulin G is altered in four rheumatoid arthritis patients. Biochem J 314 (Pt 2): 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rombouts Y, Willemze A, van Beers JJ, Shi J, Kerkman PF, van Toorn L, Janssen GM, Zaldumbide A, Hoeben RC, Pruijn GJ, Deelder AM, Wolbink G, Rispens T, van Veelen PA, Huizinga TW, Wuhrer M, Trouw LA, Scherer HU, and Toes RE. 2016. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann Rheum Dis 75: 578–585. [DOI] [PubMed] [Google Scholar]
- 16.Hafkenscheid L, Bondt A, Scherer HU, Huizinga TW, Wuhrer M, Toes RE, and Rombouts Y. 2017. Structural Analysis of Variable Domain Glycosylation of Anti-Citrullinated Protein Antibodies in Rheumatoid Arthritis Reveals the Presence of Highly Sialylated Glycans. Mol Cell Proteomics 16: 278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lloyd KA, Steen J, Amara K, Titcombe PJ, Israelsson L, Lundstrom SL, Zhou D, Zubarev RA, Reed E, Piccoli L, Gabay C, Lanzavecchia A, Baeten D, Lundberg K, Mueller DL, Klareskog L, Malmstrom V, and Gronwall C. 2018. Variable domain N-linked glycosylation and negative surface charge are key features of monoclonal ACPA: Implications for B-cell selection. Eur J Immunol 48: 1030–1045. [DOI] [PubMed] [Google Scholar]
- 18.Visser A, Doorenspleet ME, de Vries N, Spijkervet FKL, Vissink A, Bende RJ, Bootsma H, Kroese FGM, and Bos NA. 2018. Acquisition of N-Glycosylation Sites in Immunoglobulin Heavy Chain Genes During Local Expansion in Parotid Salivary Glands of Primary Sjogren Patients. Front Immunol 9: 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamza N, Hershberg U, Kallenberg CG, Vissink A, Spijkervet FK, Bootsma H, Kroese FG, and Bos NA. 2015. Ig gene analysis reveals altered selective pressures on Ig-producing cells in parotid glands of primary Sjogren’s syndrome patients. J Immunol 194: 514–521. [DOI] [PubMed] [Google Scholar]
- 20.Coelho V, Krysov S, Ghaemmaghami AM, Emara M, Potter KN, Johnson P, Packham G, Martinez-Pomares L, and Stevenson FK. 2010. Glycosylation of surface Ig creates a functional bridge between human follicular lymphoma and microenvironmental lectins. Proc Natl Acad Sci U S A 107: 18587–18592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van de Bovenkamp FS, Derksen NIL, Ooijevaar-de Heer P, van Schie KA, Kruithof S, Berkowska MA, van der Schoot CE, H IJ, van der Burg M, Gils A, Hafkenscheid L, Toes REM, Rombouts Y, Plomp R, Wuhrer M, van Ham SM, Vidarsson G, and Rispens T. 2018. Adaptive antibody diversification through N-linked glycosylation of the immunoglobulin variable region. Proc Natl Acad Sci U S A 115: 1901–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, and Tzartos S. 2016. Myasthenia gravis - autoantibody characteristics and their implications for therapy. Nat Rev Neurol 12: 259–268. [DOI] [PubMed] [Google Scholar]
- 23.Vincent A 2002. Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol 2: 797–804. [DOI] [PubMed] [Google Scholar]
- 24.Yi JS, Guptill JT, Stathopoulos P, Nowak RJ, and O’Connor KC. 2018. B cells in the pathophysiology of myasthenia gravis. Muscle & nerve 57: 172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, and Vincent A. 2001. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 7: 365–368. [DOI] [PubMed] [Google Scholar]
- 26.McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, Newsom-Davis J, and Vincent A. 2004. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol 55: 580–584. [DOI] [PubMed] [Google Scholar]
- 27.Higuchi O, Hamuro J, Motomura M, and Yamanashi Y. 2011. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 69: 418–422. [DOI] [PubMed] [Google Scholar]
- 28.Zhang B, Tzartos JS, Belimezi M, Ragheb S, Bealmear B, Lewis RA, Xiong WC, Lisak RP, Tzartos SJ, and Mei L. 2012. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 69: 445–451. [DOI] [PubMed] [Google Scholar]
- 29.Fichtner ML, Jiang R, Bourke A, Nowak RJ, and O’Connor KC. 2020. Autoimmune Pathology in Myasthenia Gravis Disease Subtypes Is Governed by Divergent Mechanisms of Immunopathology. Front Immunol 11: 776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodgaard A, Nielsen FC, Djurup R, Somnier F, and Gammeltoft S. 1987. Acetylcholine receptor antibody in myasthenia gravis: predominance of IgG subclasses 1 and 3. Clin Exp Immunol 67: 82–88. [PMC free article] [PubMed] [Google Scholar]
- 31.Lefvert AK, Cuenoud S, and Fulpius BW. 1981. Binding properties and subclass distribution of anti-acetylcholine receptor antibodies in myasthenia gravis. J Neuroimmunol 1: 125–135. [DOI] [PubMed] [Google Scholar]
- 32.Drachman DB, Adams RN, Josifek LF, and Self SG. 1982. Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med 307: 769–775. [DOI] [PubMed] [Google Scholar]
- 33.Drachman DB, Angus CW, Adams RN, Michelson JD, and Hoffman GJ. 1978. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med 298: 1116–1122. [DOI] [PubMed] [Google Scholar]
- 34.Sterz R, Hohlfeld R, Rajki K, Kaul M, Heininger K, Peper K, and Toyka KV. 1986. Effector mechanisms in myasthenia gravis: end-plate function after passive transfer of IgG, Fab, and F(ab’)2 hybrid molecules. Muscle Nerve 9: 306–312. [DOI] [PubMed] [Google Scholar]
- 35.Howard FM Jr., Lennon VA, Finley J, Matsumoto J, and Elveback LR. 1987. Clinical correlations of antibodies that bind, block, or modulate human acetylcholine receptors in myasthenia gravis. Ann N Y Acad Sci 505: 526–538. [DOI] [PubMed] [Google Scholar]
- 36.Zisimopoulou P, Evangelakou P, Tzartos J, Lazaridis K, Zouvelou V, Mantegazza R, Antozzi C, Andreetta F, Evoli A, Deymeer F, Saruhan-Direskeneli G, Durmus H, Brenner T, Vaknin A, Berrih-Aknin S, Frenkian Cuvelier M, Stojkovic T, DeBaets M, Losen M, Martinez-Martinez P, Kleopa KA, Zamba-Papanicolaou E, Kyriakides T, Kostera-Pruszczyk A, Szczudlik P, Szyluk B, Lavrnic D, Basta I, Peric S, Tallaksen C, Maniaol A, and Tzartos SJ. 2014. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J Autoimmun 52: 139–145. [DOI] [PubMed] [Google Scholar]
- 37.Koneczny I, Cossins J, Waters P, Beeson D, and Vincent A. 2013. MuSK myasthenia gravis IgG4 disrupts the interaction of LRP4 with MuSK but both IgG4 and IgG1-3 can disperse preformed agrin-independent AChR clusters. PLoS One 8: e80695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takata K, Stathopoulos P, Cao M, Mane-Damas M, Fichtner ML, Benotti ES, Jacobson L, Waters P, Irani SR, Martinez-Martinez P, Beeson D, Losen M, Vincent A, Nowak RJ, and O’Connor KC. 2019. Characterization of pathogenic monoclonal autoantibodies derived from muscle-specific kinase myasthenia gravis patients. JCI Insight 4: e127167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huijbers MG, Vergoossen DL, Fillie-Grijpma YE, van Es IE, Koning MT, Slot LM, Veelken H, Plomp JJ, van der Maarel SM, and Verschuuren JJ. 2019. MuSK myasthenia gravis monoclonal antibodies: Valency dictates pathogenicity. Neurol Neuroimmunol Neuroinflamm 6: e547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anil R, Kumar A, Alaparthi S, Sharma A, Nye JL, Roy B, O’Connor KC, and Nowak RJ. 2020. Exploring outcomes and characteristics of myasthenia gravis: Rationale, aims and design of registry - The EXPLORE-MG registry. J Neurol Sci 414: 116830. [DOI] [PubMed] [Google Scholar]
- 41.Jiang R, Fichtner ML, Hoehn KB, Pham MC, Stathopoulos P, Nowak RJ, Kleinstein SH, and O’Connor KC. 2020. Single-cell repertoire tracing identifies rituximab-resistant B cells during myasthenia gravis relapses. JCI Insight 5: e136471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vander Heiden JA, Stathopoulos P, Zhou JQ, Chen L, Gilbert TJ, Bolen CR, Barohn RJ, Dimachkie MM, Ciafaloni E, Broering TJ, Vigneault F, Nowak RJ, Kleinstein SH, and O’Connor KC. 2017. Dysregulation of B Cell Repertoire Formation in Myasthenia Gravis Patients Revealed through Deep Sequencing. J Immunol 198: 1460–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang R, Hoehn KB, Lee CS, Pham MC, Homer RJ, Detterbeck FC, Aban I, Jacobson L, Vincent A, Nowak RJ, Kaminski HJ, Kleinstein SH, and O’Connor KC. 2020. Thymus-derived B cell clones persist in the circulation after thymectomy in myasthenia gravis. Proc Natl Acad Sci U S A 117: 30649–30660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupta NT, Adams KD, Briggs AW, Timberlake SC, Vigneault F, and Kleinstein SH. 2017. Hierarchical Clustering Can Identify B Cell Clones with High Confidence in Ig Repertoire Sequencing Data. J Immunol 198: 2489–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ye J, Ma N, Madden TL, and Ostell JM. 2013. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 41: W34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, and Kleinstein SH. 2015. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31: 3356–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yaari G, Vander Heiden JA, Uduman M, Gadala-Maria D, Gupta N, Stern JN, O’Connor KC, Hafler DA, Laserson U, Vigneault F, and Kleinstein SH. 2013. Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front Immunol 4: 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoehn KB, Vander Heiden JA, Zhou JQ, Lunter G, Pybus OG, and Kleinstein SH. 2019. Repertoire-wide phylogenetic models of B cell molecular evolution reveal evolutionary signatures of aging and vaccination. Proc Natl Acad Sci U S A 116: 22664–22672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu G, Lam TT, Zhu H, and Guan Y. 2018. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol Biol Evol 35: 3041–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Srzentic K, Fornelli L, Tsybin YO, Loo JA, Seckler H, Agar JN, Anderson LC, Bai DL, Beck A, Brodbelt JS, van der Burgt YEM, Chamot-Rooke J, Chatterjee S, Chen Y, Clarke DJ, Danis PO, Diedrich JK, D’Ippolito RA, Dupre M, Gasilova N, Ge Y, Goo YA, Goodlett DR, Greer S, Haselmann KF, He L, Hendrickson CL, Hinkle JD, Holt MV, Hughes S, Hunt DF, Kelleher NL, Kozhinov AN, Lin Z, Malosse C, Marshall AG, Menin L, Millikin RJ, Nagornov KO, Nicolardi S, Pasa-Tolic L, Pengelley S, Quebbemann NR, Resemann A, Sandoval W, Sarin R, Schmitt ND, Shabanowitz J, Shaw JB, Shortreed MR, Smith LM, Sobott F, Suckau D, Toby T, Weisbrod CR, Wildburger NC, Yates JR 3rd, Yoon SH, Young NL, and Zhou M. 2020. Interlaboratory Study for Characterizing Monoclonal Antibodies by Top-Down and Middle-Down Mass Spectrometry. J Am Soc Mass Spectrom 31: 1783–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fornelli L, Srzentic K, Huguet R, Mullen C, Sharma S, Zabrouskov V, Fellers RT, Durbin KR, Compton PD, and Kelleher NL. 2018. Accurate Sequence Analysis of a Monoclonal Antibody by Top-Down and Middle-Down Orbitrap Mass Spectrometry Applying Multiple Ion Activation Techniques. Anal Chem 90: 8421–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stathopoulos P, Chastre A, Waters P, Irani S, Fichtner ML, Benotti ES, Guthridge JM, Seifert J, Nowak RJ, Buckner JH, Holers VM, James JA, Hafler DA, and O’Connor KC. 2019. Autoantibodies against Neurologic Antigens in Nonneurologic Autoimmunity. J Immunol 202: 2210–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stathopoulos P, Kumar A, Nowak RJ, and O’Connor KC. 2017. Autoantibody-producing plasmablasts after B cell depletion identified in muscle-specific kinase myasthenia gravis. JCI Insight 2: e94263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fichtner ML, Vieni C, Redler RL, Kolich L, Jiang R, Takata K, Stathopoulos P, Suarez PA, Nowak RJ, Burden SJ, Ekiert DC, and O’Connor KC. 2020. Affinity maturation is required for pathogenic monovalent IgG4 autoantibody development in myasthenia gravis. J Exp Med 217: e20200513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Graus YF, de Baets MH, Parren PW, Berrih-Aknin S, Wokke J, van Breda Vriesman PJ, and Burton DR. 1997. Human anti-nicotinic acetylcholine receptor recombinant Fab fragments isolated from thymus-derived phage display libraries from myasthenia gravis patients reflect predominant specificities in serum and block the action of pathogenic serum antibodies. J Immunol 158: 1919–1929. [PubMed] [Google Scholar]
- 56.Koers J, Derksen NIL, Ooijevaar-de Heer P, Nota B, van de Bovenkamp FS, Vidarsson G, and Rispens T. 2019. Biased N-Glycosylation Site Distribution and Acquisition across the Antibody V Region during B Cell Maturation. J Immunol 202: 2220–2228. [DOI] [PubMed] [Google Scholar]
- 57.Cron MA, Maillard S, Villegas J, Truffault F, Sudres M, Dragin N, Berrih-Aknin S, and Le Panse R. 2018. Thymus involvement in early-onset myasthenia gravis. Ann N Y Acad Sci 1412: 137–145. [DOI] [PubMed] [Google Scholar]
- 58.Weis CA, Schalke B, Strobel P, and Marx A. 2018. Challenging the current model of early-onset myasthenia gravis pathogenesis in the light of the MGTX trial and histological heterogeneity of thymectomy specimens. Ann N Y Acad Sci 1413: 82–91. [DOI] [PubMed] [Google Scholar]
- 59.Marx A, Pfister F, Schalke B, Saruhan-Direskeneli G, Melms A, and Strobel P. 2013. The different roles of the thymus in the pathogenesis of the various myasthenia gravis subtypes. Autoimmun Rev 12: 875–884. [DOI] [PubMed] [Google Scholar]
- 60.Lee JY, Stathopoulos P, Gupta S, Bannock JM, Barohn RJ, Cotzomi E, Dimachkie MM, Jacobson L, Lee CS, Morbach H, Querol L, Shan JL, Vander Heiden JA, Waters P, Vincent A, Nowak RJ, and O’Connor KC. 2016. Compromised fidelity of B-cell tolerance checkpoints in AChR and MuSK myasthenia gravis. Ann Clin Transl Neurol 3: 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen J, Zheng Q, Hammers CM, Ellebrecht CT, Mukherjee EM, Tang HY, Lin C, Yuan H, Pan M, Langenhan J, Komorowski L, Siegel DL, Payne AS, and Stanley JR. 2017. Proteomic Analysis of Pemphigus Autoantibodies Indicates a Larger, More Diverse, and More Dynamic Repertoire than Determined by B Cell Genetics. Cell Rep 18: 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lavinder JJ, Horton AP, Georgiou G, and Ippolito GC. 2015. Next-generation sequencing and protein mass spectrometry for the comprehensive analysis of human cellular and serum antibody repertoires. Curr Opin Chem Biol 24: 112–120. [DOI] [PubMed] [Google Scholar]
- 63.Lavinder JJ, Wine Y, Giesecke C, Ippolito GC, Horton AP, Lungu OI, Hoi KH, DeKosky BJ, Murrin EM, Wirth MM, Ellington AD, Dorner T, Marcotte EM, Boutz DR, and Georgiou G. 2014. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc Natl Acad Sci U S A 111: 2259–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sabouri Z, Schofield P, Horikawa K, Spierings E, Kipling D, Randall KL, Langley D, Roome B, Vazquez-Lombardi R, Rouet R, Hermes J, Chan TD, Brink R, Dunn-Walters DK, Christ D, and Goodnow CC. 2014. Redemption of autoantibodies on anergic B cells by variable-region glycosylation and mutation away from self-reactivity. Proc Natl Acad Sci U S A 111: E2567–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jacquemin M, Radcliffe CM, Lavend’homme R, Wormald MR, Vanderelst L, Wallays G, Dewaele J, Collen D, Vermylen J, Dwek RA, Saint-Remy JM, Rudd PM, and Dewerchin M. 2006. Variable region heavy chain glycosylation determines the anticoagulant activity of a factor VIII antibody. J Thromb Haemost 4: 1047–1055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data were deposited in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA738368/).