

Abstract
Cytomegalovirus (CMV) infection is a significant complication after solid organ transplantation. We used single cell T cell receptor (TCR) αβ sequencing to determine how memory inflation impacts clonality and diversity of the CMV-responsive CD8 and CD4 T cell repertoire in the first year after transplantation in human subjects. We observed CD8 T cell inflation but no changes in clonal diversity, indicating homeostatic stability in clones. In contrast, the CD4 repertoire was diverse and stable over time, with no evidence of CMV-responsive CD4 T cell expansion. We identified shared CDR3 TCR motifs among patients, but no public CMV-specific TCRs. Temporal changes in clonality in response to transplantation and in the absence of detectable viral reactivation suggest changes in the repertoire immediately after transplantation followed by an expansion with stable clonal competition that may mediate protection.
Introduction
Cytomegalovirus (CMV) causes significant complications after solid organ transplantation (1–3). While an ongoing immune response controls CMV replication, the virus remains in a latent state and can periodically reactivate (4). Transplant immunosuppression impairs immune control of CMV (5).
In immunocompetent mice and humans, latent CMV infection contributes to a lifelong expansion of memory CD8 T cells. This expansion has been termed memory inflation, a process hypothesized to result from repeated exposure to CMV antigens, leading to the long-term expansion of CMV-reactive memory T cells (6, 7). In the elderly, CMV responsive T cells can comprise up to 40% of the entire CD8 T cell repertoire (8).
CMV dramatically shapes host T cell repertoire diversity. An important factor in how CMV shapes TCR diversity is clonal competition. Proliferating T cells are limited by the overall size of the T cell niche, and compete for survival (9, 10). In the context of chronic viral infection, successful competition of a clone requires continuous interaction with viral antigen (10). A subset of CMV gene products drive oligoclonal expansion of CMV-responsive CD8 T cells (11). These include immediate early 1 (IE-1), one of the first genes expressed during reactivation (12). Following hematopoietic stem cell transplantation, CMV reactivation has been associated with reduced diversity of the effector memory T cell repertoire (13). The majority of previous studies have relied on TCRβ chain sequencing (14, 15) which does not fully account for clonality of TCRαβ pairs.
Recent studies including our own have revealed expansion of CMV-responsive CD8 T cells in the first year after transplantation consistent with a more rapid form of memory inflation which we have termed “accelerated inflation” (16–18). These findings raise several questions. Do the expanding cells derive from pre-existing clones? Is pre-existing clonal competition maintained during the expansion? What further insight do we obtain from analysis of paired TCRαβ beyond that from TCRβ alone? To address these questions, we measured paired TCR αβ repertoire longitudinally from transplant recipients pre-transplant (baseline), and three and twelve months post-transplant. The analyses addressed homeostasis of clonal expansion and diversity of both CD4 and CD8 T cells responsive to CMV IE-1 epitopes.
Materials and Methods
Human Subjects
Six recipients of heart or kidney transplant were enrolled at the University of Pennsylvania pre-transplant as described (16). All subjects were CMV antibody seropositive (CMV+) at the time of transplant with standard of care immunosuppressive therapy controlled by treating physicians (Table SI). Blood samples analyzed were obtained pre-transplant baseline and approximately three and twelve months post-transplant. This study was approved under IRB protocol 817637 and all subjects gave informed consent. Identifiable information was blinded to those performing experiments. CMV viral load was monitored as described (16); no included subjects had evidence of CMV viremia during the study period. HLA typing of donors and recipients was completed pre-transplant.
Blood collection and processing
Blood was collected in vacutainer tubes with EDTA (Ethylenediaminetetraacetic acid) (19). Within 1–24 hours, peripheral blood mononuclear cells (PBMC) were isolated via Ficoll gradient and frozen as previously described (19).
Peptide libraries
The IE-1 peptide library (GenScript, Piscataway, NJ) consisted of 15 amino acid peptides with 11 amino acid overlap for the length of the immunodominant CMV polypeptide IE-1 (20).
Cell stimulation
Individual samples (1 subject, 1 time point) were thawed, rested, and stimulated as described (16, 21). Briefly, cells were rested overnight, then stimulated with the IE-1 library (0.8 μg/mL) at 37°C and 5% CO2 for five hours. Prior to rest and stimulation, cells were counted using a hemacytometer and trypan blue. Antibody to human CD107a (BioLegend, San Diego, CA) was added with the peptide library. Cells were stained using the interferon gamma (IFNγ) Secretion Assay (Miltenyi) following the manufacturer’s protocol.
Staining for sorting
Cells were surface stained as described (21) with antibodies to CD4 (RPA-T4), CD8 (SK1), CD3 (OKT3), CD14 (61D3), CD16 (3G8), CD19 (HIB19), CD57 (HNK1), CD45RA (HI100), and CD27 (O323) from BioLegend (San Diego, CA, USA) and Abcam (Cambridge, UK) and Zombie Aqua dye (BioLegend, San Diego, CA). For sorting, cells were resuspended in PBS with 0.5% bovine serum albumin and 2 mM EDTA, then filtered through 35 μm nylon mesh into 5 mL round bottom tubes (Corning, Corning, NY, USA).
Sorting
Samples were sorted on a BD FACSARIAIII (Franklin Lakes, NJ) in the VA Palo Alto Flow Cytometry Core. Sorter was calibrated and compensation completed as described (21). Cells were sorted into 96-well Hard-Shell plates (BioRad, Hercules, CA, USA) containing 12 μL of 1X One-Step RT-PCR buffer (Qiagen, Hilden, Germany) per well.
After sorting, plates were immediately covered in aluminum sealing foil (Corning) and centrifuged in an Allegra X-15R table top centrifuge (Beckman Coulter, Brea, CA, USA) at 300xg for 2 minutes and stored at −80° C immediately following centrifugation.
Nested PCR and sequencing
Reverse transcription, nested amplification, barcoding, and library preparation were completed as described (21, 22). Next generation sequencing (NGS) was completed using Illumina MiSeq (San Diego, CA, USA) in the Stanford Functional Genomics Facility. Data were processed as described (22).
Analysis of TCR sequencing data
The bottom 10% of total, TCRβ, and TCRα reads were removed from analysis. Index sort data were extracted using FlowJo 10 (Ashland, OR) and compiled into a spreadsheet with sequencing data. Clones were defined as follows using Python (v.3.7.4):
For all clones listed in the analysis, at least one cell with the clone detected had both CDR3β and CDR3α detected, and therefore both are listed for the clone. Furthermore,
If CDR3β is detected for a cell, then a clone will be defined as cells with identical CDR3β.
If CDR3α is detected and CDR3β is NOT detected for a cell, then a clone will be defined as cells with identical CDR3α (for either alpha chain detected, if there are two).
If neither CDR3α nor CDR3β are detected, the cell can be excluded from analysis.
TCR diversity metrics
Gini coefficient and Shannon entropy are two metrics of diversity. The Gini coefficient (23) measures inequality, with 0 representing a large number of non-expanded clones and 1 representing one large clone. Shannon entropy measures distribution, with a higher number representing greater diversity (24). Both were determined from the frequency represented by each clone using online calculators (http://shlegeris.com/gini, http://www.endmemo.com/bio/shannonentropy.php). For Shannon entropy, we normalized values to the total number of clones per sample. CDR3 length was defined as the number of amino acids in the CDR3β region beginning with a cysteine and ending with a phenylalanine. Moritisa-Horn Similarity Index was computed using the Divo package in R (25). Clustered heatmaps were prepared using the heatmap.2 function in R.
TCR motif sharing
Motif sharing analysis was conducted using the algorithm GLIPH2 (http://50.255.35.37:8080/) which analyzes Vβ motifs (26, 27). TCRs identified in this study were analyzed relative to all IE-1 specific and responsive TCRs in two databases as of 2/19/2020: VDJdb (https://vdjdb.cdr3.net/ and reference 28), and McPAS-TCR (http://friedmanlab.weizmann.ac.il/McPAS-TCR/ and reference 29). GLIPH2 was computed using two different HLA databases, recipient and donor HLA. GLIPH2 predicts HLA association based on the input files by statistical probability but does not define the association. Output was filtered as described in Figure S1. Motifs detected in multiple TCRs of the same CDR3 length with an HLA match between individuals were considered specific for the same HLA-peptide combination (27).
TCR V region sharing
The frequency of each Vαβ pair in each subject was determined using the count function in the R package tidyverse. Circos plots (30) were generated using the R package circlize (31) with values annotationTrack = “grid”, preAllocateTracks = 1, grid.col = grid.col, transparency = 0.5 values into chordDiagram function.
Data analysis and statistics
Sort was completed using FACSDiva software (BD, Franklin Lakes, NJ). Gating was completed using FlowJo 10. Graphs and statistics were completed in GraphPad Prism (San Diego, CA), with the exception of clustered heatmaps, which were completed in R (v. 3.6.3). Other analyses were completed in Microsoft Excel (Redmond, WA) unless otherwise indicated. Two-tailed testing and alpha of 0.05 were used unless otherwise noted. Pairwise comparisons were analyzed with the Wilcoxon matched-pairs signed rank test. Comparison of two unpaired samples was computed using Mann-Whitney. One-way comparison of three matched groups was computed using Kruskal-Wallis (values missing) with Dunn’s multiple comparisons test. Distributions were compared using Kolmgorov-Smirnov. Two-way comparison of three matched groups was computed using two-way analysis of variance (ANOVA) with Sidak correction.
Results
Clonal homeostasis of CMV-responsive T cell clones is maintained post-transplant
The three time points analyzed in this study span two distinct immunomodulatory stages. Between pre- and three months post-transplant (hereafter denoted as pre–3), the subject received an organ transplant, induction therapy, maintenance immunosuppression, and antiviral prophylaxis. These factors can each modulate T cell immunity to CMV (32–34). Induction in three of six subjects included T cell depletion with rabbit anti-thymocyte globulin (rATG), which leads to preferential reconstitution of highly differentiated CD8 T cells and skewing of the T cell repertoire (32). In contrast, three to 12 months post-transplant (3–12) represents post-transplant homeostasis in the context of maintenance immunosuppression without antiviral prophylaxis.
We focused on the IE-1 response due to its association with the early stages of CMV reactivation and with memory inflation (6, 35). In order to identify longitudinal changes in the TCR repertoire on a single cell level, we index sorted CMV IE-1-responsive T cells from PBMC of six CMV+ transplant recipients without evidence of CMV reactivation followed by nested PCR and sequencing of individual cells (Figure 1A and references 21, 22). To identify IE-1-responsive cells we stimulated PBMC with a peptide library consisting of overlapping 15mers spanning the entire IE-1 polypeptide (20). We then index sorted based on IFNγ production after five hours of stimulation (20, 21). All subsequent analyses are of T cells producing IFNγ in response to IE-1 stimulation.
Figure 1: Enrichment of differentiated phenotype in CMV-IE-1 responsive cells:
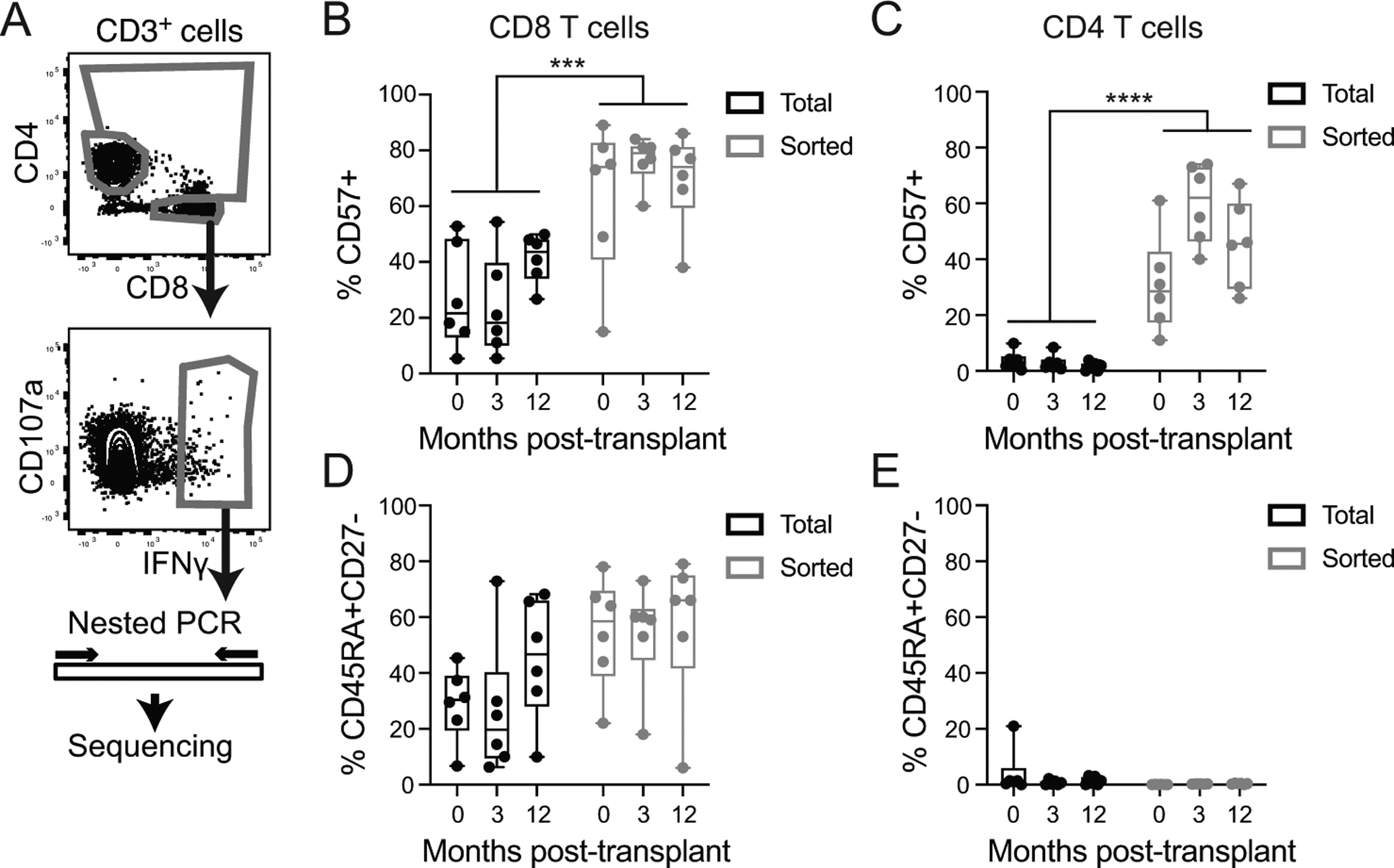
A) PBMC were stimulated with IE-1 peptide for 5 hours, stained for IFNγ capture and index sorted. Gates were on live singlet lymphocytes, CD3+ and CD14−CD16−CD19−, and as shown. TCR and other genes were amplified and sequenced. Protein expression data analyzed to identify the frequency of aged (CD57+, B and C) and TEMRA (CD45RA+CD27−, D and E) in total and sorted CD8 (B and D) and CD4 (C and E) T cells. Graphs include individual data points and box plot showing median and interquartile range. Statistics computed with two-way ANOVA with Sidak’s correction for multiple comparisons. *** represents p<0.001, **** represents p<0.0001. n=6 subjects.
In comparison with total T cells, the isolated IE-1 responsive population was highly enriched for cells expressing CD57, a marker associated with aging and terminal differentiation in both CD8 (Figure 1B, p=0.001) and CD4 (Figure 1C, p<0.0001) T cells. There was also a non-statistical trend towards enrichment of terminally differentiated effectors (CD45RA+CD27−) in sorted CD8 T cells (Figure 1D, p=0.08). CD4 T had a much smaller proportion of CD45RA+CD27− cells in both the total and sorted populations (Figure 1E), consistent with previous reports (36, 37). Thus, the sorted cells represent a highly differentiated population, with distinct phenotypes between CD8 and CD4 T cells.
We first compared the TCR repertoire of CMV-responsive CD8 (Figure 2A) and CD4 (Figure 2B) T cells from pre–3 and 3–12 months. Each subject had a pre-transplant CD8 T cell repertoire of IE-1 responsive clones that was distinct from other subjects. In each subject, the relative proportion of each clone evolved following transplantation (Figure 2A). The repertoire was highly oligoclonal at all three time points in five of six subjects. A single dominant clone comprising between 25% and 72% of the CD8 T cells was detected at twelve months post-transplant in those five subjects (Figure 2A). In four subjects, that clone was detectable at earlier time points. In two of the three subjects who did not receive lymphodepleting induction therapy (Table I), the dominant clone was already dominant pre-transplant (Figure 2A). In contrast, the clone dominating at month 12 was absent pre-transplant in two of the three subjects who received lymphodepleting induction, and less than 5% of CD8 T cells in the third. Clonal dynamics differed depending on the use of lymphodepletion at the time of transplant. Rare clones, defined as representing <2% of a subject’s CD8 T cells, comprised 3–79% of clones depending on the subject and time point (Figure 2A). In marked contrast to the oligoclonality which characterized the CD8 T cell repertoire, CD4 T cells were highly polyclonal at all time points. Only subject 4 had any CD4+ clones larger than 3% of all sorted CD4+ cells, and no subject had evidence of clonally expanded CD4 T cells comprising more than 5% at any time point (Figure 2B).
Figure 2: Clonal homeostasis of IE-1 responsive T cells post-transplant:
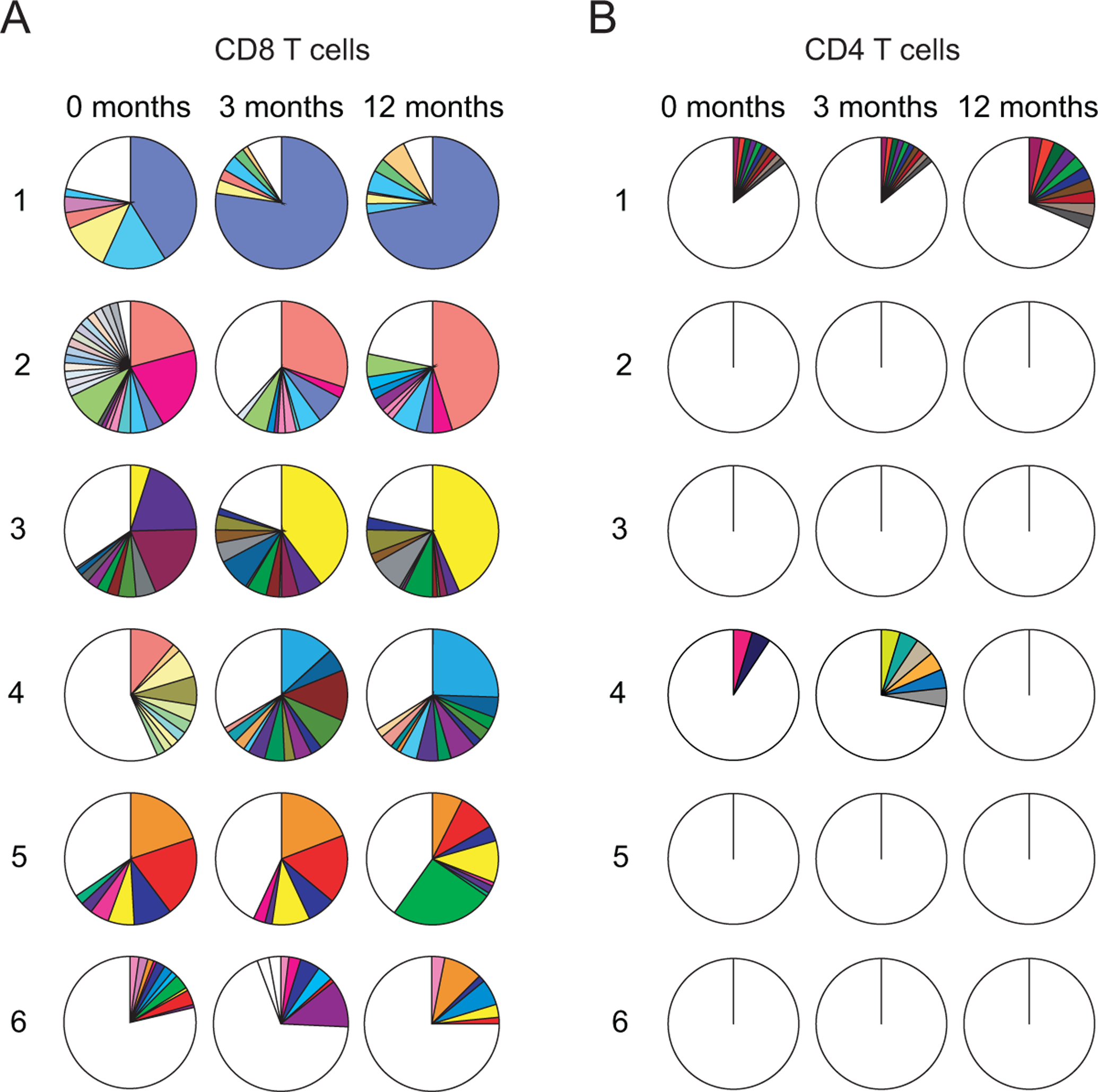
A) CD8 and B) CD4 T cell clonality measured pre- and three and twelve months post-transplant with each pie representing all sorted cells for an individual subject and time-point. Each color represents a single clone; white is a summation of all clones <2%. n=6 subjects.
Table I: Motifs detected by GLIPH associated with both donor and recipient HLA:
GLIPH2 computed on CDR3β from this sequencing data set, along with CDR3β from IE-1 associated TCRs from two publicly accessible databases. Hits filtered on final score (<10−8), whether there were shared HLAs detected across samples for the motif, and for same CDR3 length. In the column Sample, the number refers to the subject, and m_ refers to the month. Bold underline on amino acids indicates those amino acids are in the motif identified by GLIPH2.
| TcRß CDR3 | V | J | Sample | Frequency | Recipient HLA | Donor HLA |
|---|---|---|---|---|---|---|
| CASVPNYSNQPQHF | TRBV12-3 | TRBJ1-5 | 5 m12 | 1/143 | A*02:01, B*18:01, B*35:01, C*04 | A*24:02 |
| CASTPNYSNQPQHF | TRBV12-3 | TRBJ1-5 | 5 m12 | 27/143 | ||
| CASSPNYSNQPQHF | TRBV12-3 | TRBJ1-5 | 4 m3 | 1/98 | ||
| CASSPNYSNQPQHF | TRBV12-3 | TRBJ1-5 | 5 m12 | 1/143 | ||
| CASSPNFSNQPQHF | TRBV12-3 | TRBJ1-5 | 4 m3 | 3/98 | ||
| CASSPNFSNQPQHF | TRBV12-3 | TRBJ1-5 | 4 m12 | 8/104 | ||
| CASSPGGVTEAFF | TRBV11-2 | TRBJ1-1 | 4 m3 | 7/98 | A*02:01, B*18:01, B*35:01, C*04 | A*24:02 |
| CASSPGGVTEAFF | TRBV11-2 | TRBJ1-1 | 4 m12 | 6/104 | ||
| CASSPGGVAEAFF | TRBV11-2 | TRBJ1-1 | 5 m12 | 1/143 | ||
| CASSPGGVTEAFF | TRBV11-2 | TRBJ1-1 | 4 m3 | 7/98 | A*02:01, B*18:01, B*35:01, C*04 | A*24:02 |
| CASSPGGVTEAFF | TRBV11-2 | TRBJ1-1 | 4 m12 | 6/104 | ||
| CASSPGGYTEAFF | TRBV18 | TRBJ1-1 | 5 m0 | 1/171 | ||
| CSASSSDTQYF | TRBV20-1 | TRBJ2-3 | 5 m12 | 1/143 | A*02:01, B*18:01, B*35:01, C*04 | A*24:02 |
| CASSDSDTQYF | TRBV10-2 | TRBJ2-3 | 4 m12 | 1/104 | ||
| CSARDGTEDYGYTF | TRBV20-1 | TRBJ1-2 | 4 m0 | 1/151 | Not detected | A*24:02, B*44:01 |
| CSARDVTEDYGYTF | TRBV20-1 | TRBJ1-2 | 2 m3 | 1/154 | ||
| CASSDSDTQYF | TRBV10-2 | TRBJ2-3 | 4 m12 | 1/104 | A*02:01 | Not detected |
| CATSDGDTQYF | TRBV24-1 | TRBJ2-3 | 3 m0 | 1/235 |
Expansion of the CD8+ IE-1 responsive pool is driven by dominant clones
Analyses of clonal homeostasis addressed changes in the CMV-responsive population, but not the effect of those changes on the total T cell population. To evaluate this for CD8 T cells with evidence of accelerated inflation (16), we used IE-1 responsive cell frequencies determined by ICS of IFNγ on a separate aliquot of the same patient sample as published (16), as this approach is more sensitive than the IFNγ capture required for sorting unfixed cells. We determined the frequency of IE-1 responsive cells with each TCRαβ clone from sequence analysis of the sorted samples. We multiplied this number by the percentage IE-1 responsive of total CD8 T cells as determined by ICS. Expansion of the IE-1-responsive CD8 T cell population occurred from pre- to twelve months post-transplant in all subjects (Figure 3A).
Figure 3: Expansion of IE-1 responsive CD8 T cells dominated by expanded clones:
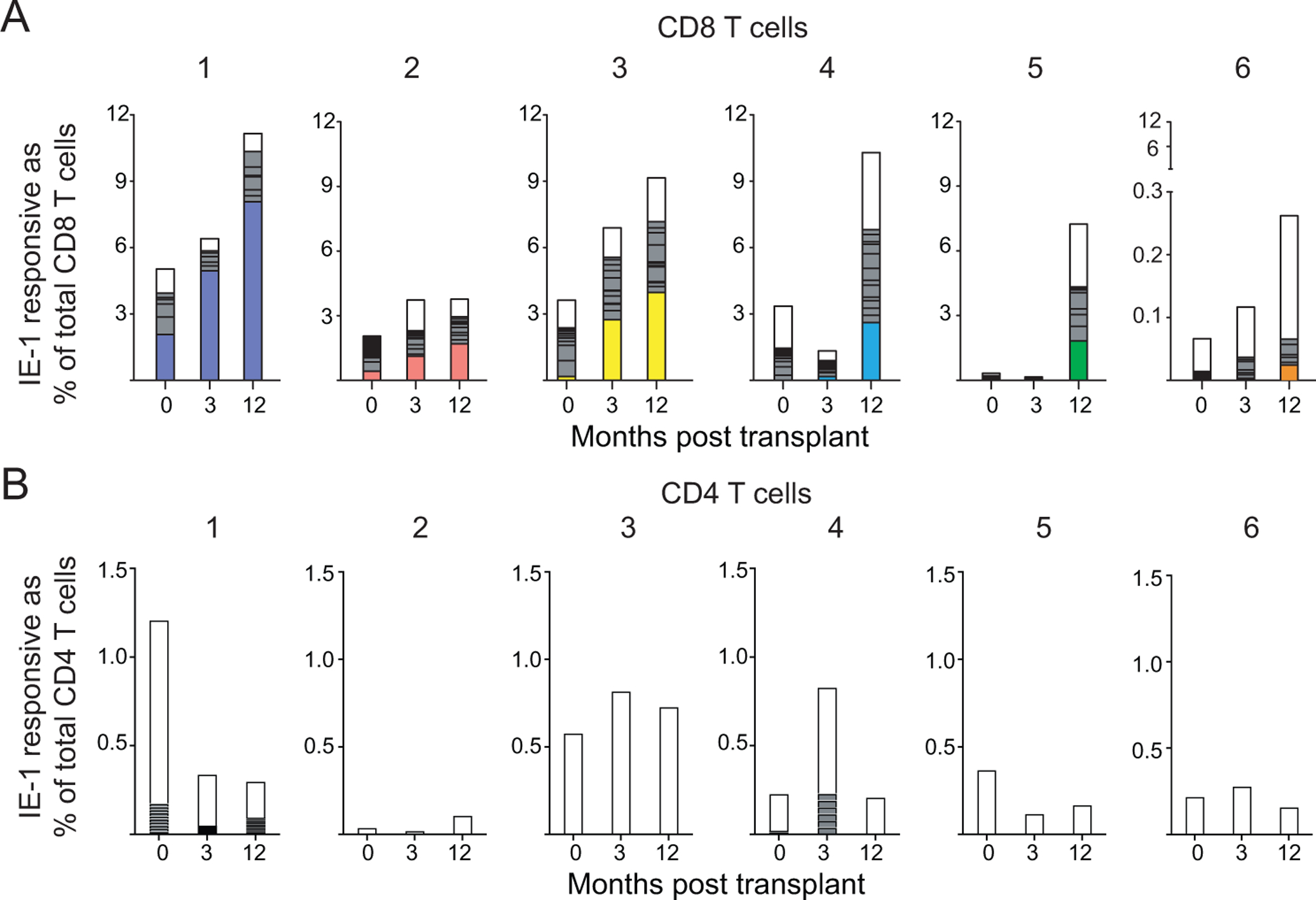
Clonality of A) CD8 or B) CD4 T cells measured pre- and three and twelve months post-transplant back-calculated as frequency of total A) CD8 or B) CD4 T cells based on % IFNγ+ in ICS data previously described (16). Each column is subdivided by the proportion of individual clones. The largest CD8 T cell clone at 12 months is colored and shown in that color for each time point for that subject. White = clones <2% of sample, gray = clones ≥2% of sample. n=6 subjects.
We next analyzed the changes in clonal dominance specifically in CD8 T cells. As noted above, five of six subjects had a single dominant CD8 T cell clone (Figure 3A). For subjects 1, 2, and 3, the dominant clone was present pre-transplant and clonally expanded post-transplant (Figure 3A, left). For subject 4, the dominant clone was first detected at three months post-transplant, and it expanded ten-fold by twelve months (Figure 3A, middle right). For subjects 5 and 6, the dominant clone was not detected prior to twelve months post-transplant (Figure 3A, right). Thus, these few clones drove the expansion observed at 12 months, either through expansion of previously detectable clones (subjects 1–3) or through new post-transplant clonal expansions (subjects 4–6). Subjects 5 and 6 received CMV+ organs, which suggests that the dominant clones appearing only at month 12 may have expanded in response to exposure to virus of donor origin. Neither lymphodepletion nor donor CMV serostatus accounted for differences in clonal expansion in this cohort. In contrast with CD8 T cell expansion and consistent with the overall clonality data for CD4 T cells, we detected no oligoclonality or clonal expansion (Figure 3B). Thus, the development of clonal dominance was CD8 T cell-specific.
Sharing of Vαβ pairs is higher in CD8 than CD4 T cells and is not affected by shared HLA
To determine if there were public TCRs, we next analyzed Vα and Vβ region usage. To visualize the relationship between Vα and Vβ pairing across subjects we utilized Circos plots (30). For both CD8 and CD4 T cells, the frequencies of each individual Vα, Vβ, and Vαβ pair varied across subjects. Of the 41 Vα and 42 Vβ detected in CD8 T cells in the sequencing data, we detected over 80% of each in every subject, indicating broad use of individual TCR V regions in CMV IE-1 responsive CD8 T cells (Figure 4A). The largest clones in each sample were represented by distinct Vαβ pairs. Regarding CD4 T cells, of the 40 Vα and 41 Vβ we detected over 75% or 65% respectively in every subject (Figure 4B). Sharing of Vαβ pairs was analyzed using the Morisita-Horn Similarity Index (25). Very few CD8 Vαβ pairs were shared between subjects (Figure 4C). Sharing was detected between subjects 1 and 3 (Morisita-Horn Index 0.17), 3 and 6 (0.16), and 4 and 5 (0.21). In contrast, very few CD4 Vαβ pairs were shared at all, even between different time points within subject (Figure 4D), consistent with a lack of oligoclonal expansion. We then sought to determine whether recipients with shared HLA class I were more likely to share CD8 Vαβ pairs using the Morisita-Horn Similarity Index to quantify overlap of Vαβ pairs. There was no statistical difference in sharing between pairs of subjects with at least one HLA match versus those with no match (Figure 4E, p>0.99). The overall sharing of Vαβ pairs was much lower in CD4 T cells, though the minimal sharing within subjects was statistically greater than the sharing between subjects (Figure 4F). Thus, in this cohort, no public motifs were identified on the basis of Vαβ sharing.
Figure 4: CD8 T cell Vαβ pairs are shared within but not between subjects:
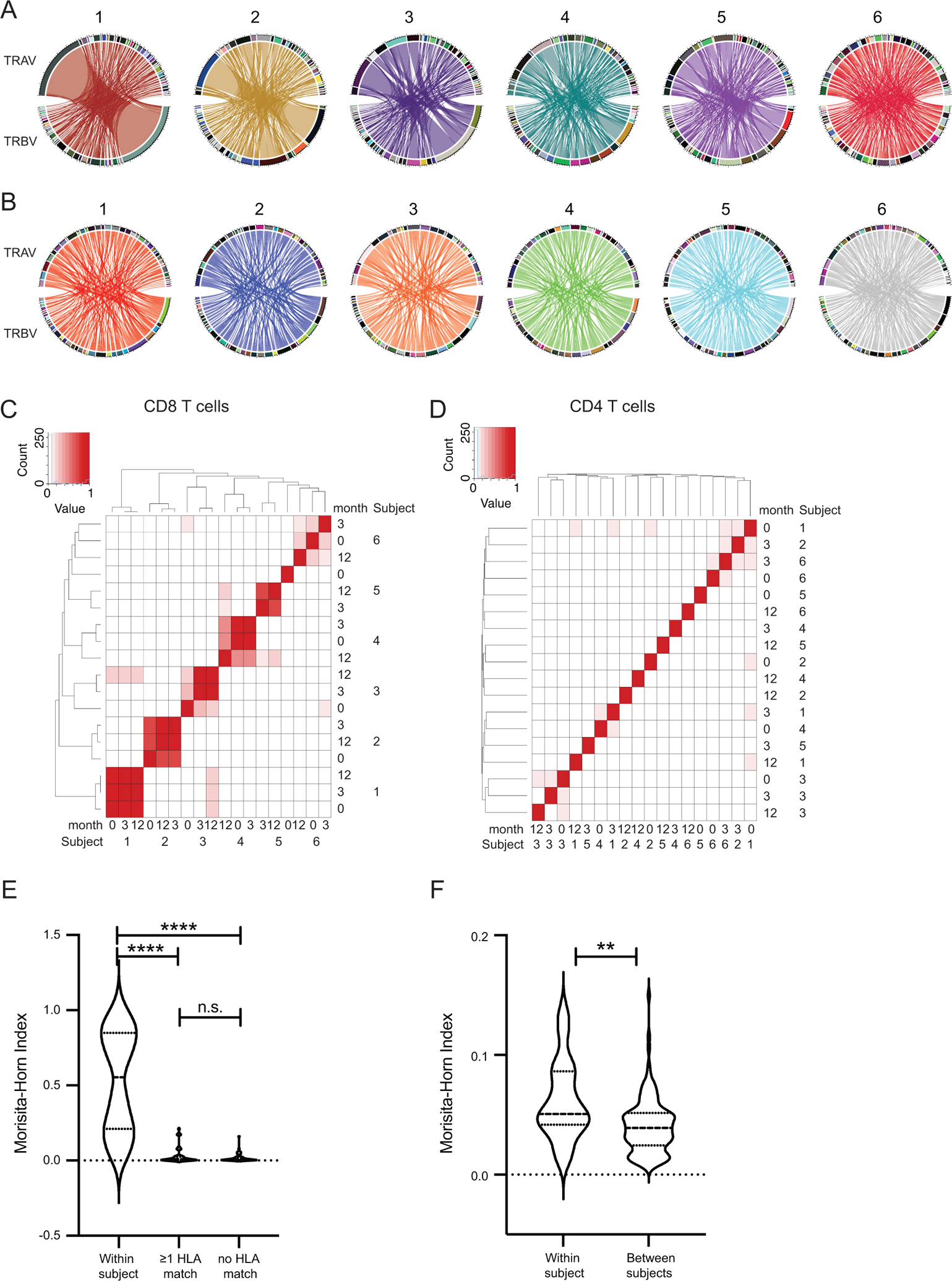
Circos plots of Vαβ pairing of A) CD8 T cells and B) CD4 T cells in all 6 subjects averaged across time points. Heatmap of Morisita-Horn Index of Similarity calculated for sharing of Vαβ pairs across samples for C) CD8 and D) CD4 T cells. Quantification of Morisita-Horn Index within and between subjects for E) CD8 and F) CD4 T cells. Statistics computed using (C-E) Kruskal-Wallis test with Dunn’s multiple comparisons test or F) Mann-Whitney test. ** = p<0.01, **** = p<0.0001. n=6 subjects.
IE-1 responsive T cells contain motifs that recognize antigen in the context of donor or recipient MHC
Vα and Vβ regions contain conserved CDR1 and CDR2 regions that contribute to HLA specificity, but the CDR3 region is the part that directly binds peptide-MHC (38, 39). To more specifically detect motifs within CDR3 regions, we analyzed antigen specificity of expanded clones using the grouping of lymphocyte interactions by paratope hotspots 2 (GLIPH2) computational approach as described in Figure S1. GLIPH2 can predict whether clonally expanded CMV-responsive T cells bind to donor or recipient HLA, and therefore whether they can be predicted to respond to pre-existing antigen in the recipient or new antigen exposure derived from the donor. We focused specifically on CD8 T cells due to the dominance of CD8 T cells in response to IE-1 antigen (20). We identified four TCRs that could be associated with either donor or recipient HLA, one TCR associated only with donor HLA, and one associated only with recipient HLA (Table I). The TCRs with shared V and J regions and CDR3 length are indicative of highly conserved sequence that may represent public motifs. TCRs associated with donor HLA were found in recipients of both CMV+ and CMV− organs, and therefore cannot be fully explained by presentation of donor virus. These analyses demonstrate that patients 4 and 5 share at least two motifs for common TCR binding to CMV antigen.
To further understand motif sharing, we also analyzed those motifs detected by GLIPH2 as shared between at least one subject in our study and one or both of the two databases analyzed. This analysis identified 8 shared motifs that were not detected in the original GLIPH2 analysis (Table SII).
Clonal diversity of T cells was maintained from pre to 12 months post-transplant
In order to determine how the clonality data correlate to diversity, we assessed clonal diversity and homeostasis. Gini coefficient (23) and Shannon entropy (24) are measures of diversity based on frequency represented by each clone. CDR3 length distribution is a measure of changes in the identity of clones present (40). The Morisita-Horn Similarity Index quantifies the overlap of clones in two populations (25).
First we calculated diversity of CD8 T cell clones. There was a non-statistical trend toward decreasing diversity from pre-3 (Wilcoxon matched-pairs signed rank test, p=0.094) based on Gini coefficient only. Shannon entropy from pre-3 showed no statistical change (p=0.16). There were no statistical differences in either Gini coefficient or Shannon entropy from 3–12 (Figure 5A, B, p>0.68), indicating stability in clonal diversity. We also assessed homeostasis of individual clones through CDR3 length, which did not change statistically between time points (Figure 5C). Morisita-Horn Similarity was 45% for subject 5, 62% for subject 4, and >85% for subjects 1, 2, and 3. Subject 6 had similarity of 5%. Thus, despite allogeneic transplantation, immunosuppression, and significant expansion of the IE-1 responsive CD8 population, each of these four measures demonstrated stability of clonal competition.
Figure 5: Clonal diversity of IE-1 responsive T cells maintained post-transplant:
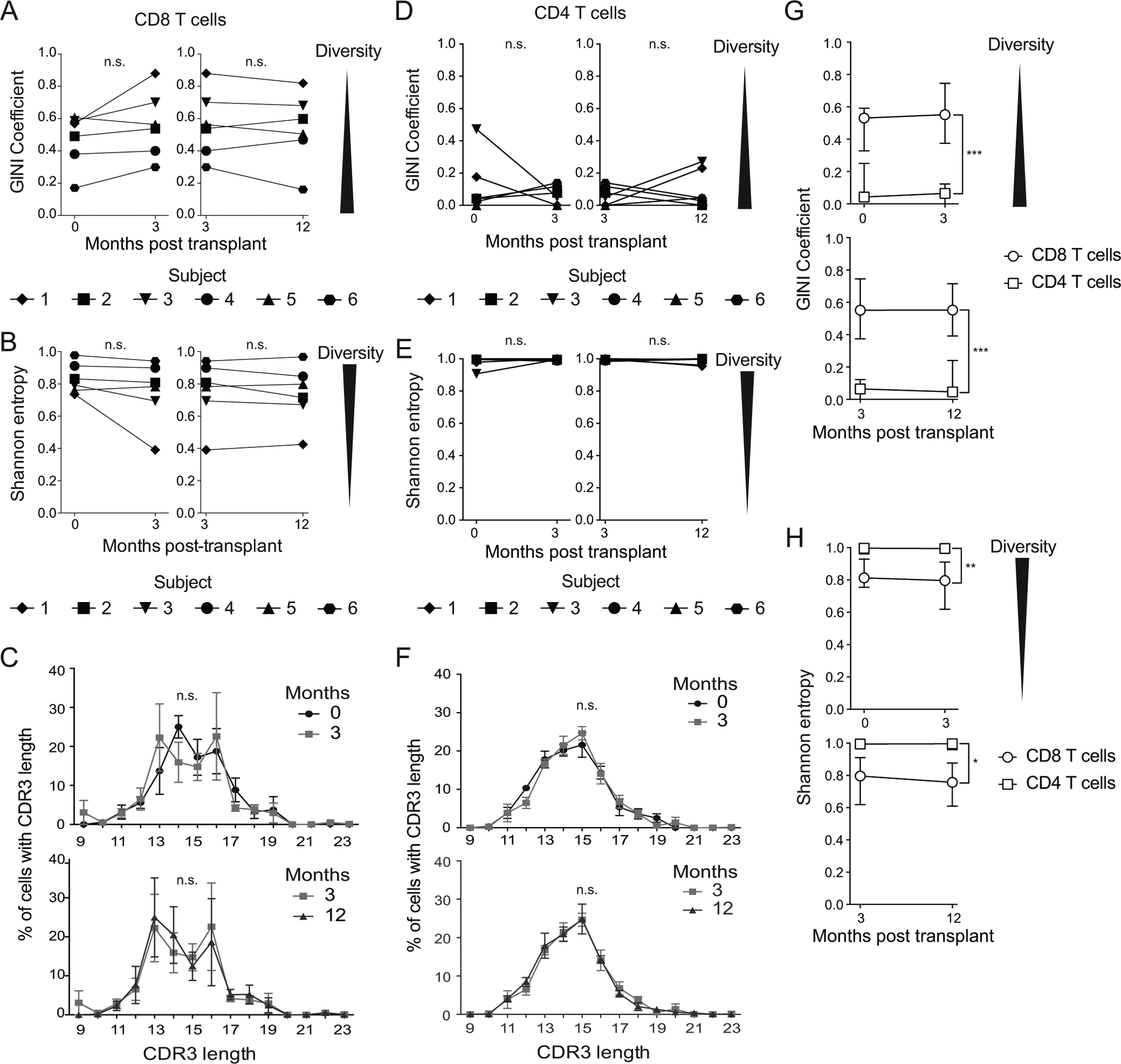
Gini coefficient (A and D) and Shannon entropy (B and E) calculated for the diversity of each CD8 (A and B) and CD4 (D and E) T cell sample. Shannon entropy value normalized to the size of the sample. CDR3β length at each time point measured and averaged across patients for C) CD8 and F) CD4 T cells. Data shown in C and F are mean and standard error of the mean. Median G) Gini and H) Shannon values for CD8 and CD4 T cells at time points indicated. Error bars represent interquartile range. n=6 subjects. Wilcoxon matched-pairs signed rank test (A, B, D, and E), Kolmogorov-Smirnov test (C and F), or (G and H) two-way ANOVA used to compute statistics. * = p<0.05, ** = p<0.01, *** = p<0.001. n=6 subjects.
We also analyzed diversity of CD4 T cell clones using identical statistical approaches. When compared with CD8 T cells, clonal diversity was higher pre-transplant by both Gini coefficient and Shannon entropy. Diversity remained high post-transplant among CD4 T cells with no statistical change (Figure 5D, E, p>0.81). In addition, CD4 T cells did not show any significant changes in CDR3 length post-transplant (Figure 5F, p>0.99). Morisita-Horn Similarity was <5% for all subjects for CD4 T cells. Overall, CD4 T cells had a high degree of clonal diversity at all time points assessed.
These analyses demonstrate distinct patterns of TCR diversity in CD8 and CD4 T cells. We compared the Gini coefficient (Figure 5G) and Shannon entropy (Figure 5H) across patients within time point. Consistent with the difference in clonality (Figure 2), diversity was consistently higher in CD4s across time points (Figure 5G, H, p<0.05). There was no statistical change in diversity over time (p>0.26). Further evidence of higher diversity in CD4 T cells come from comparing CDR3 length, which is more skewed to shorter lengths in CD8 than CD4 T cells at all time points (Figure 5C, F). Thus, the metrics overall demonstrate much greater diversity of CD4 clones.
Discussion
Using in-depth analysis of the TCRαβ repertoire of CMV-responsive T cells in the first year post-transplant, we demonstrated that clonal homeostasis and diversity of these populations are stably maintained. CD8 T cells clonally expanded during the first three months and maintained clonality from 3–12 months post-transplant, while CD4 T cells were stably polyclonal throughout the time course. We identified TCR motifs indicative of shared specificity, but no public TCRs. Our most striking finding was that in both CD8 and CD4 T cell populations, regardless of degree of oligoclonality, clonal diversity was stably maintained throughout the time course.
The distinct patterns of clonality from pre-3 and 3–12 months post-transplant represent different immunological pressures. The period from pre-3, representing transplantation and associated changes, is characterized by increased clonality and a trend towards decreased diversity. These changes are consistent with the new antigen exposure and therapies defining this period. In contrast, during the immunosuppressive maintenance period from 3–12 months post-transplant, clonality and diversity are likewise maintained. This finding suggests that once homeostasis of the TCR repertoire is re-established post-transplant, it remains consistent, at least within the first year after transplantation and initiation of immunosuppression. The contrast between these periods illustrates the importance of understanding the full immunological context in interpreting TCR repertoire.
Maintenance of TCR homeostasis suggests that, at least during the first year after transplantation, memory inflation of CMV-responsive T cells does not impact the specificity of the response. However, while none of these subjects had viremia detected during the study period, the clinical tests did not rule out viremia below the threshold of detection, or viral replication localized to sites other than the blood. Thus, it is unknown whether this pattern is independent of or associated with specific patterns of subclinical viremia. Indeed, a study of CMV viral load and CMV-specific T cells in tissues from organ donors demonstrated virus persistence in blood as well as varying distributions of CMV-specific T cells across tissues (41). As our results identify clonal homeostasis in the blood compartment, comparison of clonality between blood and tissue sites will be important to determine if these findings apply to tissue resident T cells as well.
Our finding of stable TCR repertoire maintenance confirms and extends previous work in this area. CMV-responsive CD4 T cells were analyzed in rhesus macaques in a study that found that the clonality of the repertoire was stable over time (45). In addition, several studies of memory inflation in mouse CMV-ovalbumin models demonstrated striking stability of CD8 TCR clones over time during the inflationary period (42–44). These studies suggest that clonal dominance is determined by early differentiation after infection (42) and that clonal stability is maintained throughout expansion in response to stochastic exposure to CMV antigen (44). This finding on stochasticity may explain why clonality changed from pre- to post-transplant and was stable thereafter: transplantation and immunosuppression are both sources of stochastic changes that could alter the repertoire, but once the patient stabilizes post-transplant, the clonality becomes less variable.
Studies in humans provide further context for our results. In CMV seropositive healthy adults, dominant CD8 TCRβ clones persisted over the course of four years (46). A study of CMV-specific T cells in HIV-infected children and adults found that dominant clones were stably maintained over 10 or more years (47). These studies demonstrate the stability of CMV-specific T cells in untransplanted individuals. In the context of transplantation, one study of primary CMV responses after transplantation with CMV seropositive kidney grafts found that the CMV-specific CD8 T cell TCRβ repertoire was stably maintained for 5 years after the primary response (48). Another study found that dominant CMV-specific CD8 TCRβ clones present pre-transplant in CMV seropositive kidney recipients were largely maintained a year post-transplant (49). Our work confirms these findings, and further indicates modulation of the repertoire in the early post-transplant period, prior to the extended period of maintenance. This modulation was greater for subjects who received lymphodepleting induction therapy than for subjects who did not. This is not surprising given that lymphodepleting induction therapy significantly decreases the T cell population within the first month post-transplant (50), but suggests that CMV-responsive TCR repertoire stability is impacted by this therapy. The number of subjects in the study was too limited for a statistical comparison between induction therapies, so this will be an important area for future study. Our study also builds upon known clonal stability in transplant and non-transplant populations by including three time points spanning pre-transplant to a year after, and by incorporating analysis of both CD4 and CD8 T cells. Further, ours is the first longitudinal study in transplant recipients of paired TCRαβ, which more definitively defines clones than the TCRβ focused studies described above.
Another commonly observed feature of CMV-responsive T cells is public TCRs. A study of public repertoires of MCMV-specific T cells demonstrated that dominant H2kb-restricted clones were largely public, but that the proportion public decreased late post-infection (43). Studies of human CMV-specific TCR repertoires have also identified public TCRs at varying frequencies and affinities (51, 52). The relative lack of public TCRs in our study may be expected from the HLA disparate nature of the cohort. Specifically, our study found no public CDR3 sequences, but use of GLIPH2 did identify eight public TCRβ motifs shared with CMV-specific TCRs in databases, as well as six shared between subjects in our study. Further, four of the latter six motifs detected were shared between subjects 4 and 5, who were matched for 4 of 6 HLA-A, B, and C alleles within recipient. One GLIPH2 motif was shared between subjects 3 and 4, who were matched for HLA-A*02. The final motif was shared between subjects 2 and 4, who had no matches within recipient, but two matches between one recipient and the other donor: HLA-A2*02 shared between 4 and the donor for 2, and HLA-A2*24 shared between 2 and the donor for 4. Intriguingly, the motif shared between subjects 2 and 4 was detected at month 0 in subject 4, and month 3 in subject 2. If this motif can be shown to be HLA-A*02 restricted, that would be evidence of a TCR that appeared in subject 2 post-transplant due to exposure to antigen restricted to donor HLA-A*02. Our analysis of Vαβ sharing between subjects also identified sharing between subjects 4 and 5, as well as between subjects 1 and 3, who were matched for HLA-B*07. These findings are consistent with the fact that studies of public TCRs typically identify TCRs binding a specific HLA type (51–53). Thus, our study extends these findings by demonstrating that public TCR sharing is largely dependent on analysis within MHC restriction. Further the decrease in proportion public late after MCMV infection (44) suggests the hypothesis that stochastic changes in exposure to CMV antigen can lead to expansion of private TCRs over time. Particularly given the small number of patients, this could explain the limited number of public TCR specificities.
The accelerated expansion of CMV-responsive T cells post-transplant provides a unique opportunity for longitudinal study of the T cell repertoire of memory inflating T cells in humans. Due to the long-term nature of memory inflation, it is most commonly studied in the murine CMV model (12) and across age groups of different individuals (8). Matched samples from individual patients allowed temporal comparison of clonality and diversity. Accelerated inflation in this patient cohort enabled analysis of TCR in a population known to be inflating on a time scale feasible for study. Furthermore, inclusion of a pre-transplant time point allowed us to establish a baseline missing from studies that focus only on post-transplant events.
Our usage of a computational approach to analyze IE-1-responsive TCR motifs is also novel. Previous analyses of CMV-responsive T cell repertoire have focused on T cells that respond to specific peptides or bind to MHC-tetramer. GLIPH2 has been validated as a computational approach to identify TCR motifs (26, 27). This study has a unique approach with use of all possible epitopes for the IE-1 gene, and identification of associated motifs. In particular, this approach is one of the few that incorporates analysis of a wide variety of HLA types, instead of the few most commonly studied ones.
An important consideration for understanding the findings of this study is that the use of IFNγ to isolate CMV-responsive T cells excluded the minority of cells responding to CMV by expressing different cytokines and IE-1 specific cells that do not mount any response due to anergy or exhaustion (54, 55). Thus, these repertoire analyses do not fully recapitulate the CMV-specific T cell repertoire. However, because this approach excludes those CMV-specific T cells that no longer produce cytokine due to exhaustion or anergy, our analyses focused on those CMV-responsive T cells most likely to contribute to post-transplant changes in immunity.
Overall, this study demonstrates stable maintenance of clonality and TCR diversity from 3–12 months post-transplant. This stability is striking given the changes in clonality from pre-transplant to three months and the changes in population size from 3–12 months. These findings suggest that the repertoire maintains the same capacity to recognize CMV throughout the 3–12 months post-transplant period, even in the context of significant population expansion. In addition, repertoire analysis of CMV-responsive T cells at three months can most likely be used to predict the repertoire at 12 months, which may be important to use of T cell immunity in diagnostics for personalized therapies.
Supplementary Material
Key Points:
CMV-responsive CD4 and CD8 T cell clonality is stable up to a year post-transplant.
The CD8 repertoire becomes more clonal in the first three months after transplant.
Common CDR3 TCRβ motifs were detected among subjects with matched HLA type.
Acknowledgments:
We thank Drs Sheri Krams and Holden Maecker for advice on experimental design, Dr. Xuhuai Ji for assistance with sequencing library preparation, Dr. Weiqi Wang for assistance running the data processing pipeline, Dr. Jennifer Trofe-Clark for provision of clinical data, Dr. Jane Buckner for helpful discussions, Dawn Bruffert for advice on analysis in Excel, the Palo Alto Veteran’s Affair (VA) Flow Cytometry Core for FACSAria III usage, and the Stanford Functional Genomics Facility for bioanalyzer and MiSeq usage.
Funding:
This work was supported by awards to J.S.M. from the American Heart Association (13IRG13640042) and the Veterans Administration (1I01CX001971). L.E.H. was supported by grants from Enduring Hearts and the American Heart Association (17POST33660597), the National Institutes of Health [K01 1K01DK123196], the Stanford Translational Research and Applied Medicine Program and support from National Institutes of Health T32 AI07290 and DK007357-31. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. PK is funded by the Bill and Melinda Gates Foundation (OPP1113682), the National Institute of Allergy and Infectious Diseases (NIAID) grants 1U19AI109662, U19AI057229, and 5R01AI125197, Department of Defense contracts W81XWH-18-1-0253 and W81XWH1910235, and the Ralph & Marian Falk Medical Research Trust outside of the work presented here.
Abbreviations:
- ANOVA
Analysis of variance
- CDR3
Complementarity Determining Region 3
- CMV
Cytomegalovirus
- CMV+
Cytomegalovirus seropositive
- DBSCAN
Density-based spatial clustering of applications with noise
- DMSO
dimethyl sulfoxide
- EDTA
Ethylenediaminetetraacetic acid
- FBS
fetal bovine serum
- GLIPH
grouping of lymphocyte interactions by paratope hotspots
- HLA
Human Leukocyte Antigen
- IE-1
immediate early 1
- IFNγ
interferon gamma
- ICS
intracellular cytokine staining
- NGS
Next generation sequencing
- PBMC
peripheral blood mononuclear cells
- TCR
T cell receptor
- TEMRA
Terminally differentiated effector re-expressing CD45RA
- TNF
Tumor necrosis factor alpha
Footnotes
Competing interests: JSM has a family member who is employed by and has an equity interest in Genentech/Roche. SS is currently employed by Notable Labs. ML is currently employed by Quell Therapeutics. No patents have been filed pertaining to the results presented in this paper.
Data and materials availability:
Sequencing data were deposited to the Sequence Read Archive (BioProject accession number PRJNA752378, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA752378).
References:
- 1.Kotton CN 2013. CMV: prevention, diagnosis, and therapy. Am. J. Transplant 13: 24–40. [DOI] [PubMed] [Google Scholar]
- 2.Razonable RR, Humar A, and ASTInfectiousDiseasesCommunityofPractice. 2013. Cytomegalovirus in solid organ transplantation. Am. J. Transplant 13: 93–106. [DOI] [PubMed] [Google Scholar]
- 3.Leeaphorn N, Garg N, Thamcharoen N, Khankin EV, Cardarelli F, and Pavlakis M. 2018. Cytomegalovirus mismatch still negatively affects patient and graft survival in the era of routine prophylactic and preemptive therapy: A paired kidney analysis. Am. J. Transplant 19: 573–584. [DOI] [PubMed] [Google Scholar]
- 4.Jarvis MA, and Nelson JA. 2002. Mechanisms of human cytomegalovirus persistence and latency. Front. Biosci 7: d1575–1582. [DOI] [PubMed] [Google Scholar]
- 5.Eid AJ, and Razonable RR. 2010. New developments in the management of cytomegalovirus infection after solid organ transplantation. Drugs 70: 965–981. [DOI] [PubMed] [Google Scholar]
- 6.Klenerman P 2018. The (gradual) rise of memory inflation. Immunol. Rev 283: 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Hara GA, Welten SPM, Klenerman P, and Arens R. 2012. Memory T cell inflation: understanding cause and effect. Trends in Immunology 33: 84–90. [DOI] [PubMed] [Google Scholar]
- 8.Crough T, and Khanna R. 2009. Immunobiology of human cytomegalovirus: from bench to bedside. Clin. Microbiol. Rev 22: 76–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leitão C, Freitas AA, and Garcia S. 2009. The role of TCR specificity and clonal competition during reconstruction of the peripheral T cell pool. J Immunology 182: 5232–5239. [DOI] [PubMed] [Google Scholar]
- 10.Shin H, Blackburn SD, Blattman JN, and Wherry EJ. 2007. Viral antigen and extensive division maintain virus-specific CD8 T celsl during chronic infection. J. Exp. Med 204: 941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schober K, Buchholz VR, and Busch DH. 2018. TCR repertoire evolution during maintenance of CMV-specific T-cell populations. Immunol. Rev 283: 113–128. [DOI] [PubMed] [Google Scholar]
- 12.Karrer U, Sierro S, Wagner M, Oxenius A, Hengel H, Koszinowski UH, Phillips RE, and Klenerman P. 2003. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J. Immunol 170: 2022–2029. [DOI] [PubMed] [Google Scholar]
- 13.Suessmuth Y, Mukherjee R, Watkins B, Koura DT, Finstermeier K, Desmarais C, Stempora L, Horan JT, Langston A, Qayed M, Khoury HJ, Grizzle A, Cheeseman JA, Conger JA, Robertson J, Garrett A, Kirk AD, Waller EK, Blazar BR, Mehta AK, Robins HS, and Kean LS. 2015. CMV reactivation drives posttransplant T-cell reconstitution and results in defects in the underlying TCRb repertoire. Blood 125: 3835–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindau P, Mukherjee R, Gutschow MV, Vignali M, Warren EH, Riddell SR, Makar KW, Turtle CJ, and Robins HS. 2019. Cytomegalovirus exposure in the elderly does not reduce CD8 T cell repertoire diversity. J Immunology 202: 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schober K, Voit F, Grassmann S, Müller TR, Eggert J, Jarosch S, Weißbrich B, Hoffman P, Borkner L, Nio E, Fanchi L, Clouser CR, Radhakrishnan A, Mihatsch L, Lückemeier L, Leube J, Dössinger G, Klein L, Neuenhahn M, Oduro JD, Cicin-Sain L, Buchholz VR, and Busch DH. 2020. Reverse TCR repertoire evolution toward dominant low-affinity clones during chronic CMV infection. Nat. Immunol 21: 434–441. [DOI] [PubMed] [Google Scholar]
- 16.Higdon LE, Trofe-Clark J, Liu S, Margulies KB, Sahoo MK, Blumberg E, Pinsky BA, and Maltzman JS. 2017. Cytomegalovirus responsive CD8+ T cells expand after solid organ transplantation in the absence of CMV disease. Am. J. Transplant 17: 2045–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar D, Chin-Hong P, Kayler L, Wojciechowski D, Limaye AP, Gaber AO, Ball S, Mehta AK, Cooper M, Blanchard T, MacDougall J, and Kotton CN. 2019. A prospective multicenter observational study of cell-mediated immunity as a predictor for cytomegalovirus infection in kidney transplant recipients. Am. J. Transplant 19: 2505–2516. [DOI] [PubMed] [Google Scholar]
- 18.Martín-Gandul C, Pérez-Romero P, Mena-Romo D, Molina-Ortega A, González-Roncero FM, Suñer M, Bernal G, Cordero E, and t. S. N. f. R. i. I. D. (REIPI). 2018. Kinetic of the CMV-specific T-cell immune response and CMV infection in CMV-seropositive kidney transplant recipients receiving rabbit anti-thymocyte globulin induction therapy: a pilot study. Transpl. Infect. Dis 20: e12883. [DOI] [PubMed] [Google Scholar]
- 19.Higdon LE, Lee K, Tang Q, and Maltzman JS. 2016. Virtual global transplant laboratory standard operating procedures for blood collection, PBMC isolation, and storage. Transplantation Direct 2: e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bunde T, Kirchner A, Hoffmeister B, Habedank D, Hetzer R, Cherepnev G, Proesch S, Reinke P, Volk H-D, Lehmkuhl H, and Kern F. 2005. Protection from cytomegalovirus after transplantation is correlated with immediate early 1-specific CD8 T cells. J. Exp. Med 201: 1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higdon LE, Cain CJ, Colden MA, and Maltzman JS. 2019. Optimization of single-cell plate sorting for high throughput sequencing applications. J. Immunol. Methods 466: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han A, Glanville J, Hansmann L, and Davis MM. 2014. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat. Biotechnol 32: 684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ceriani L, and Verme P. 2012. The origins of the Gini index: extracts from Variabilità e Mutabilità (1912) by Corrado Gini. The Journal of Economic Inequality 10: 421–443. [Google Scholar]
- 24.Shannon CE 1948. A mathematical theory of communication. The Bell System Technical Journal XXVII: 379–423. [Google Scholar]
- 25.Rempala G, and Seweryn M. 2013. Methods for diversity and overlap analysis in T-cell receptor populations. J. Math. Biol 67: 1339–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, Ji X, Han A, Krams SM, Pettus C, Haas N, Lindestam Arlehamm CS, Sette A, Boyd SD, Scriba TJ, Martinez OM, and Davis MM. 2017. Identifying specificity groups in the T cell receptor repertoire. Nature 547: 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang H, Wang C, Rubelt F, Scriba TJ, and Davis MM. 2020. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat. Biotechnol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shugay M, Bagaev D, Zvyagin I, Vroomans R, Crawford J, Dolton G, Komech E, Sycheva A, Koneva A, Egorov E, Eliseev A, Van Dyk E, Dash P, Attaf M, Rius C, Ladell K, McLaren J, Matthews K, Clemens E, Douek D, Luciani F, van Baarle D, Kedzierska K, Kesmir C, Thomas P, Price D, Sewell A, and Chudakov D. 2018. VDJdb: a curated database of T-cell receptor sequences with known antigen specficity. Nucleic Acids Res 46: D419–D427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tickotsky N, Sagiv T, Prilusky J, Shifrut E, and Friedman N. 2017. McPAS-TCR: A manually-curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics: 2924–2929. [DOI] [PubMed] [Google Scholar]
- 30.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, SJ J, and Marra M. 2009. Circos: an information aesthetic for comparative genomics. Genome Research 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu Z, Gu L, Eils R, Schlesner M, and Brors B. 2014. circlize implements and enhances circular visualization in R. Bioinformatics 30: 2811–2812. [DOI] [PubMed] [Google Scholar]
- 32.Havenith SHC, Remmerswaal EBM, Bemelman FJ, Yong SL, von KAMI Donselaar-van der Pant, van Lier RAW, and ten Berge IJM. 2012. Rapid T cell repopulation after rabbit anti-thymocyte globulin (rATG)treatment is driven mainly by cytomegalovirus. Clin. Exp. Immunol 169: 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welzl K, Weinberger B, Kronbichler A, Sturm G, Kern G, Mayer G, Grubeck-Loebenstein B, and Koppelstaetter C. 2014. How immunosuppressive therapy affects T cells from kidney transplanted patients of different age: the role of latent cytomegalovirus infection. Clin. Exp. Immunol 176: 112–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fehr T, Cippa PE, and Mueller NJ. 2015. Cytomegalovirus post kidney transplantation: prophylaxis versus pre-emptive therapy? Transpl. Int 28: 1351–1356. [DOI] [PubMed] [Google Scholar]
- 35.Mason R, Groves IJ, Wills MR, Sinclair JH, and Reeves MB. 2020. Human cytomegalovirus major immediate early transcripts arise predominantly from the canonical major immediate early promoter in reactivating progenitor-derived dendritic cells J. Gen. Virol 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian Y, Babor M, Lane J, Schulten V, Patil VS, Seumois G, Rosales SL, Fu Z, Picarda G, Burel J, Zapardiel-Gonzalo J, Tennekoon RN, De Silva AD, Premawansa S, Premawansa G, Wijewickrama A, Greenbaum JA, Vijayanand P, Weiskopf D, Sette A, and Peters B. 2017. Unique phenotypes and clonal expansions of human CD4 effector memory T cells re-expressing CD45RA. Nature Communications 8: 1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Mitri D, Azevedo RI, Henson SM, Libri V, Riddell NE, Macaulay R, Kipling D, Soares MVD, Battistini L, and Akbar AN. 2011. Reversible Senescence in Human CD4+CD45RA+CD27− Memory T Cells. J Immunology 187: 2093–2100. [DOI] [PubMed] [Google Scholar]
- 38.Marrack P, Scott-Brown J, Dai S, Gapin L, and Kappler J. 2008. Evolutionarily conserved amino acids that control TCR-MHC interaction. Annu. Rev. Immunol 26: 171–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia K, Adams J, Feng D, and Ely LK. 2009. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat. Immunol 10: 143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miqueu P, Guillet M, Degauque N, Doré J-C, Soulillou J, and Brouard S. 2007. Statistical analysis of CDR3 length distributions for the assessment of T and B cell repertoire biases. Mol. Immunol 44: 1057–1064. [DOI] [PubMed] [Google Scholar]
- 41.Gordon CL, Miron M, Thome JJC, Matsuoka N, Weiner J, Rak MA, Igarashi S, Granot T, Lerner H, Goodrum F, and Farber DL. 2017. Tissue reservoirs of antiviral T cell immunity in persistent human CMV infection. J. Exp. Med 214: 651–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grassmann S, Mihatsch L, Mir J, Kazeroonian A, Rahimi R, Flommersfeld S, Schober K, Hensel I, Leube J, Pachmayr LO, Kretschmer L, Zhang Q, Jolly A, Chaudhry MZ, Schiemann M, Cicin-Sain L, Höfer T, Busch DH, Flossdorf M, and Buchholz VR. 2020. Early emergence of T central memory precursors programs clonal dominance during chronic viral infection. Nat. Immunol 21: 1563–1573. [DOI] [PubMed] [Google Scholar]
- 43.Schober K, Fuchs P, Mir J, Hammel M, Fanchi L, Flossdorf M, and Busch DH. 2020. The CMV-Specific CD8+ T Cell Response Is Dominated by Supra-Public Clonotypes with High Generation Probabilities. Pathogens 9: 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith CJ, Venturi V, Quigley MF, Turula H, Gostick E, Ladell K, Hill BJ, Himelfarb D, Quinn KM, Greenaway HY, Dang THY, Seder RA, Douek DC, Hill AB, Davenport MP, Price DA, and Snyder CM. 2020. Stochastic Expansions Maintain the Clonal Stability of CD8+T Cell Populations Undergoing Memory Inflation Driven by Murine Cytomegalovirus. The Journal of Immunology 204: 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price DA, Bitmansour AD, Edgar JB, Walker JM, Axthelm MK, Douek DC, and Picker LJ. 2008. Induction and Evolution of Cytomegalovirus-Specific CD4+T Cell Clonotypes in Rhesus Macaques. The Journal of Immunology 180: 269–280. [DOI] [PubMed] [Google Scholar]
- 46.Iancu EM, Corthesy P, Baumgaertner P, Devevre E, Voelter V, Romero P, Speiser DE, and Rufer N. 2009. Clonotype Selection and Composition of Human CD8 T Cells Specific for Persistent Herpes Viruses Varies with Differentiation but Is Stable Over Time. The Journal of Immunology 183: 319–331. [DOI] [PubMed] [Google Scholar]
- 47.Attaf M, Roider J, Malik A, Rius Rafael C, Dolton G, Prendergast AJ, Leslie A, Ndung’u T, Kløverpris HN, Sewell AK, and Goulder PJ. 2020. Cytomegalovirus-Mediated T Cell Receptor Repertoire Perturbation Is Present in Early Life. Front. Immunol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klarenbeek PL, Remmerswaal EBM, Ten Berge IJM, Doorenspleet ME, Van Schaik BDC, Esveldt REE, Koch SD, Ten Brinke A, Van Kampen AHC, Bemelman FJ, Tak PP, Baas F, De Vries N, and Van Lier RAW. 2012. Deep Sequencing of Antiviral T-Cell Responses to HCMV and EBV in Humans Reveals a Stable Repertoire That Is Maintained for Many Years. PLoS Pathog 8: e1002889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Remmerswaal EBM, Klarenbeek PL, Alves NL, Doorenspleet ME, Schaik B. D. C. v., Esveldt REE, Idu MM, Leeuwen E. M. M. v., Bom-Baylon N. v. d., Kampen A. H. C. v., Koch SD, Pircher H, Bemelman FJ, Brinke A. t., Baas F, Berge I. J. M. t., Lier R. A. W. v., Vries N. d., and Frueh K. 2015. Clonal Evolution of CD8+ T Cell Responses against Latent Viruses: Relationship among Phenotype, Localization, and Function. J. Virol 89: 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gurkan S, Luan Y, Dhillon N, Allam SR, Montague T, Bromberg JS, Ames S, Lerner S, Ebcioglu Z, Nair V, Dinavahi R, Sehgal V, Heeger P, Schroppel B, and Murphy B. 2010. Immune Reconstitution Following Rabbit Antithymocyte Globulin. Am. J. Transplant 10: 2132–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huth A, Liang X, Krebs S, Blum H, and Moosmann A. 2019. Antigen-Specific TCR Signatures of Cytomegalovirus Infection. The Journal of Immunology 202: 979–990. [DOI] [PubMed] [Google Scholar]
- 52.Trautmann L, Rimbert M, Echasserieau K, Saulquin X, Neveu B, Dechanet J, Cerundolo V, and Bonneville M. 2005. Selection of T Cell Clones Expressing High-Affinity Public TCRs within Human Cytomegalovirus-Specific CD8 T Cell Responses. The Journal of Immunology 175: 6123–6132. [DOI] [PubMed] [Google Scholar]
- 53.Toya T, Taguchi A, Kitaura K, Misumi F, Nakajima Y, Otsuka Y, Konuma R, Adachi H, Wada A, Kishida Y, Konishi T, Nagata A, Yamada Y, Marumo A, Noguchi Y, Yoshifuji K, Mukae J, Inamoto K, Igarashi A, Najima Y, Kobayashi T, Kakihana K, Ohashi K, Suzuki R, Nagamatsu T, and Doki N. 2020. T-cell receptor repertoire of cytomegalovirus-specific cytotoxic T-cells after allogeneic stem cell transplantation. Sci. Rep 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maecker HT, Ghanekar SA, Suni MA, He X-S, Picker LJ, and Maino VC. 2001. Factors affecting the efficiency of CD8+ T cell cross-priming with exogenous antigens. J Immunology 166: 7268–7275. [DOI] [PubMed] [Google Scholar]
- 55.Sester U, Presser D, Dirks J, Gärtner B, Köhler H, and Sester M. 2008. PD-1 expresson and IL-2 loss of cytomegalovirus-specific T cells correlates with viremia and reversible functional anergy. Am. J. Transplant 8: 1486–1497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data were deposited to the Sequence Read Archive (BioProject accession number PRJNA752378, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA752378).