

Abstract
Arsenic exposure in contaminated drinking water is a global health issue, as more than 200 million people are affected globally. Arsenic has been known to cause skin, liver, lung, bladder and prostate cancers. Accordingly, it has been categorized as a group I human carcinogen by the International Agency for Research on Cancer (IARC). Various natural and anthropogenic activities lead to the release of arsenic in the environment, contaminating air, water and food sources. Traditionally, genetic mutations have been the center of cancer research. However, emerging studies have now focused on the importance of epigenetics, metabolism and endoplasmic reticulum (ER) stress in cancer. Arsenic is highly capable of inducing stress in the cells via the generation of free radicals causing oxidative stress, epigenetic and genetic alterations, mitochondrial dysfunction, activation of intracellular signaling pathways, and impairment of autophagy and DNA repair systems. The cancer cells are able to utilize the unfolded protein response (UPR) to overcome these internal stresses in various stages of arsenic-induced carcinogenesis, from cancer growth to immune responses. The UPR is an evolutionarily conserved stress response that has both survival and apoptotic outcomes. PERK, IRE1α and ATF6α are the three ER stress sensors that are activated to maintain cellular proteostasis, which can also promote apoptosis on prolonged ER stress. The dual nature of UPR in different cancer types and stages is a challenge for researchers. We must investigate the role and the connections among ER stress-associated UPR, mitochondrial dysfunction and autophagy in arsenic malignancies to identify key targets for cancer prevention and therapeutics.
1. Introduction
Arsenic, believed to be originated from the Greek word “arsenikos,” meaning potent, is one of the most common elements of the Earth’s crust. Arsenic is naturally found in the air, soil, rocks, water bodies, and living beings, as arsine gas, organic or inorganic compounds [1, 2]. Organic compounds are a combination of arsenic with carbon and hydrogen, such as monomethyarsonic acid, dimethylarsinic acid, arsenobetaine, and arsenosugars. Inorganic arsenic is found as a combination of arsenic with elements like chlorine, sulfur, sodium, oxygen forming arsenic trichloride, orpiment, sodium arsenite, arsenic trioxide, and other compounds, respectively. Generally, the inorganic form of arsenic is more toxic compared to the organic forms [2].
Long-term exposure of arsenic has been found to cause cancers of the lung, skin and bladder, developmental defects, diabetes, pulmonary and cardiovascular diseases [3]. The International Agency for Research on Cancer (IARC) has classified arsenic as a group I human carcinogen. The United States Environmental Protection Agency (US EPA) has established the permissible limit of 10ppb (10ug/L) of arsenic in drinking water. However, higher concentrations of arsenic are found in various regions of the world. It is recently estimated about 94 million to 220 million people are potentially exposed to high arsenic concentrations in groundwater, including USA, China, India, Bangladesh, Mexico, Mongolia, Chile, Argentina, etc. [4, 5]. Drinking water contamination is a significant source of arsenic exposure and toxicity in the world. According to a study by the United States Geographical Survey, 2.1 million people in the USA are exposed to high levels of arsenic by drinking water from contaminated wells [6]. Developing countries primarily depend on groundwater as an essential source of drinking water, which contains arsenic concentration higher than 10ppb. Elution of arsenic from minerals surfaces, reduction of iron/arsenic oxides, oxidation of arsenic minerals caused by excessive lowering of the water table elevates groundwater arsenic levels. Consumption of foods containing arsenic, such as rice, seafood, milk, beef, is the main cause of dietary inorganic arsenic intake. High levels of arsenic have been observed in the water-logged fields of rice, especially in South Asian countries where rice is a staple food [1,2].
Various natural and anthropogenic activities lead to the release of arsenic in the environment. Human activities like mining, glass manufacturing, pesticide production, and use, production of petroleum products, burning of coal products, are some of the sources of arsenic release in nature. Arsenic can also be released in the environment due to natural phenomenon like weathering of arsenic-containing rocks, geothermal activities, oxidation of arsenic bearing minerals. Arsenic is also found in hydrothermal fluids [1,2].
2. Arsenic Chemistry
Arsenic is known as the ‘King of poisons’ and has also been used to treat leukemias [7]. The unique chemistry of arsenic explains its dual effect as a carcinogen and anticancer drug. Arsenic is a metalloid (atomic number=33), placed in group 15 of the periodic table. Arsenic can easily form covalent bonds with other elements due to its ability to donate electrons. It can also form many biomolecules due to its stable bonding with methyl groups [1]. Arsenic can exist in four redox states, viz., +5 (Arsenate), +3 (Arsenite), 0 (elemental arsenic), and −3 (arsine). Arsenite (As3+) and arsenate (As5+) are the two toxic forms of inorganic arsenic that are predominantly found in water bodies. Arsenite (As3+) is more toxic and mobile compared to arsenate (As5+) [8, 9]. The presence of these two arsenic species in the environment depends upon the pH and the redox potential. Arsenate is found mostly in oxidizing conditions, whereas trivalent arsenite is predominant under reducing conditions in the environment [1].
At physiological pH, arsenite mimics glycerol and hence is taken up by aquaglyceroporin channels in the cells. The toxicity of arsenite is based on its ability to react with thiols that lead to the inactivation of various thiol-containing proteins and enzymes. The phosphate transporters take up arsenate due to its resemblance to phosphate. Arsenate interferes with phosphate transport and leads to competitive inhibition of enzymes that utilize phosphate. It primarily affects the enzymes of metabolic pathways like glycolysis, oxidative phosphorylation that require phosphate [10]. Thus, understanding the chemistry of arsenic is crucial to study its biological effects.
3. Arsenic metabolism
Several epidemiological and case-control studies have established inorganic arsenic as a carcinogen in humans [4]. Arsenic methylation occurs in the liver; however, certain in-vitro studies have suggested that arsenic methylation may also transpire in the cytosol of kidney, lung, and testes. Challenger and his colleagues first illustrated the reductive/oxidative methylation pathway of arsenic in microbes. In this pathway, inorganic pentavalent arsenate (As5+) is reduced to inorganic trivalent arsenite (As3+) in the presence of glutathione (GSH), which can also get conjugated to a protein [11, 12]. Arsenite is further methylated by arsenicmethylatransferases (AS3MT) using S-adenosylmethionine (SAM) as a methyl donor to form monomethylarsonous acid (MMAIII) and dimethylarsinous acid (DMAIII). These protein-bound methylated trivalent species can further be oxidized to pentavalent monomethylarsonic acid (MMAV) and dimethylarsinic acid (DMAV). The methylated pentavalent arsenic species are less toxic compared to inorganic As3+ and its methylated metabolites. The methylated metabolites of arsenite (As3+) have been found to interfere with the enzyme functions in the cells. Also, they are more genotoxic than inorganic As3+ [13] [12].
Metabolism of arsenic has significant consequences in arsenic-carcinogenesis. Glutathione is an antioxidant that protects cells from free radical damage. During arsenic metabolism, GSH donates electrons resulting in reduced GSH levels in cells upon long-term arsenic exposure. Thus, arsenic exposure enhances oxidative stress by antioxidant depletion and impairment of ROS-scavenging enzymes [14].
Arsenic methylation is essential for its excretion from the body. During arsenic metabolism, the use of SAM as a methyl donor by methyltransferases is essential, which leads to a reduction of SAM levels and disruption of DNA/histone methylome (Fig. 1). Epigenetic aberrations are commonly found in cancer which include changes in global methylation patterns, hypermethylation at gene promoter regions, alterations in acetylation/methylation patterns of histone proteins, which can silence tumor suppressor genes or activate the oncogenes reference 1 [15]. Maurizio Mauro and others in recent studies showed that global and gene-specific methylation changes were observed in the V79 cell line, Chinese hamster embryonic lung fibroblasts, human keratinocytes after short term arsenite treatment [16, 17]. Another study by Meredith Eckstein et al. explored the temporary and permanent changes observed in DNA methylation patterns with low-dose inorganic arsenic treatment in HeLa cells. The study linked epithelial-to-mesenchymal transition (EMT) to the arsenic induced changes in methylation patterns in HeLa cells [18]. Although, several studies have investigated the effect of arsenic on the DNA methylome, histone modifications and miRNA for epigenetic targets it is currently at preliminary stage and further in-depth investigations are required. However, it would be apt to state here that arsenic metabolism does influence its role as a carcinogen.
Fig. 1.

Arsenic metabolism and its proposed mechanisms of carcinogenesis. Metabolism of arsenic occurs via oxidation, reduction and methylation reactions. Arsenate (As+5) enters inside the cell via phosphate transporters, where it undergoes a reduction in the presence of glutathione (GSH) to form arsenite (As+3). Arsenite may also directly enter the cells via aquaglyceroporin channels (not shown in the figure). The reduced arsenite undergoes methylation in the presence of arsenic methyltransferases (AS3MT) and S-adenosylmethionine (SAM) to form monomethylarsonous acid (MMAIII) and dimethylarsinous acid (DMAIII) in the following reactions. The dimethylated arsenite may be further trimethylated in some cases. MMAIII and DMAIII may also be oxidized to pentavalent methylated species. The dimethylated arsenic species are excreted from the body. The inorganic arsenite and its methylated species through oxidative stress cause DNA, protein, lipid damage-causing carcinogenic mutations, mitochondrial dysfunction, activation of signaling pathways related to cell proliferation, apoptosis and survival, impaired DNA repair and altered immune mechanisms. Arsenic biotransformation leads to SAM depletion causing epigenetic changes (hypomethylation of oncogenes or hypermethylation of tumor suppressor genes).
4. Mechanisms of arsenic-induced carcinogenesis
Several genotoxic and non-genotoxic mechanisms of arsenic-induced cancer have been proposed at cellular and molecular levels. Oxidative stress induction via reactive oxygen species (ROS) generation, indirect DNA damage due to ROS, epigenetic modifications, and activation of stress and other signaling pathways are some of the proposed mechanisms of arsenic-induced cancers. Inorganic trivalent arsenic is one of the main inducers of ROS in cells. Arsenic metabolism produces different types of ROS like hydroxyl radical, hydrogen peroxide, and superoxide anion radical. Cells convert arsenite to arsenate via oxidation generating two electrons, which are important for hydrogen peroxide production. The hydrogen peroxide further generates hydroxyl radical, a highly active radical by Fenton reaction. Arsenic has also been found to generate reactive nitrogen species (RNS), nitric oxide (NO) in arsenic-treated BAEC, and CHO-K1 cells [19]. Arsenic activates the NADPH oxidase complex responsible for superoxide anion radical generation [2, 20, 21]. Further, it has been noted that arsenic disturbs the mitochondrial membrane integrity leading to the release of ROS into the cytosol. An excessive generation of ROS overcomes the body’s natural antioxidant systems leading to oxidative stress in the cells. The free radicals formed cause oxidative damage to DNA, RNA, lipids, and proteins, which result in mutations in the genetic material [2].
Free radicals generated by arsenic affect the expression of genes involved in the DNA damage repair systems like the mismatch repair, nucleotide excision repair (NER) and base-excision repair systems. A study by Wu et al. found that arsenic-induced mdig (mineral dust induced gene) expression in human lung adenocarcinoma A549 cells and human bronchial epithelial BEAS-2B cells. Mdig gene expression has been linked to lung cancers. Proteomic and functional analysis revealed that mdig directly interacted with XRCC6, XRCC5, and DNAPK, the non-homologous end joining (NHEJ) repair complex and this interaction impaired the DNA double-strand repair function of the NHEJ complex 46 [22-24]. The oxidative stress caused by arsenic also affects the signal transduction processes, including mitogen-activated protein kinases (MAPK), NF-κB, Wnt/β-catenin, c-myc, p53, JNK/AP-1, HIF-1, Ras, and Src signaling [2]. Exposure to As3+ has been found to activate the PI3K/AKT signaling pathway, which results in the activation of downstream target proteins that play an important role in cell growth, metabolism, and survival. Human bronchial epithelial cells treated with As3+ activated AKT, which induced expression of vascular endothelial growth factor (VEGF), important in angiogenesis and tumor survival [23]. Taken together, arsenic-induced ROS/RNS overproduction leads to antioxidant depletion, impairment of ROS scavenging enzymes, and mitochondrial dysfunction, which further enhances oxidative stress. The oxidative stress causes DNA damage, post-translational modifications (PTMs) of DNA repair enzymes, disruption of protein tyrosine phosphorylations impairing DNA repair systems, disrupting DNA damage signaling, and promoting uncontrolled cell proliferation, which may be responsible for arsenic carcinogenesis [14].
Genetic mutations have primarily been the focus of research on cancers. However, recent studies have also highlighted the importance of epigenetics, endoplasmic reticulum stress responses, and metabolism in carcinogenesis. Epigenetics refers to the changes in gene expression without altering the DNA sequence. Epigenetic modifications include DNA methylation in gene promoter regions that regulate gene expression, histone tail acetylation, methylation, phosphorylation, ubiquitination, and modifications by non-coding RNAs like miRNA [13, 25, 26]. Arsenic biotransformation utilizes SAM as a methyl donor. The utilization of SAM leads to lower SAM levels and disturbance of the methylome. Several studies have demonstrated the impact of arsenic exposure on DNA/histone methylation. Exposure of a high concentration of inorganic arsenic has been reported to cause hypermethylation of death-associated protein kinase (DAPK) promoter and low expression of the DAPK gene in urothelial carcinoma patients. DAPK is a serine-threonine kinase involved in apoptosis signaling downstream of Tumor Necrosis Factor-α (TNFα), interferon-gamma, and Fas signaling. DAPK promoter hypermethylation is known to be an essential epigenetic modification in esophageal, lung, head and neck, and gastric malignancies. Several studies established that As3+ led to promoter hypermethylation of tumor suppressor genes, induced global DNA hypomethylation, alterations in histone methylation/acetylation that contributes to abnormal gene expression, and arsenic malignancies [27].
Micro-RNAs (miRNAs) are small non-coding RNAs that mediate the silencing of gene expression. miRNAs bind to complementary mRNA sequences that lead to mRNA degradation, deadenylation or decreased translation. Dysregulation of miRNA can lead to the development of cancers, immune diseases, and neurodegenerative disorders. miRNAs can act as oncogenes by suppressing tumor suppressors [28, 29]. In human bronchial epithelial BEAS-2B cells treated with 2.5 μM arsenite for 13 weeks, malignant transformation was observed. Increased expression of miR-155 levels and decreased NRF2 expression was observed in these arsenic-transformed cells. Inhibition of miR-155 led to increased NRF2 levels, where NRF2 is vital in the regulation of antioxidant protein expression. It decreased colony formation suggesting the role of miR-155 in NRF2 signaling in arsenic-induced malignant transformation [30].
Cancer stem cells (CSCs) comprise a small population of the tumor bulk that possess the properties of self-renewal, differentiation, and resistance to chemotherapy. They play a key role in tumor initiation, progression, and metastasis. The CSCs are known to be responsible for the heterogeneous nature of cancer. The CSCs can develop from somatic cells due to de-differentiation, loss of differentiation from terminally differentiated cells or arise from mutations in the normal stem cells [2, 31, 32]. Several studies had shown that arsenic is able to induce transformation of normal stem cells to CSCs or generation of CSCs from non-cancerous cells [4]. One of the most notable features of arsenic-induced CSCs is the metabolic shift from mitochondrial TCA cycle to glycolysis, a process depending on arsenic-induced activation of NRF2 and HIF1α [33]. Both NRF2 and HIF1α are also key regulators of ER-stress UPR and autophagy.
In summary, arsenic elicits its effect as a carcinogen by causing indirect DNA damage, inhibition of the DNA repair systems, epigenetic modifications affecting gene expressions, alterations/activation of signaling pathways involved in cell proliferation, apoptosis, differentiation, and immune surveillance failure, all of which play critical roles on the malignant transformation and the generation of the CSCs.
5. Endoplasmic reticulum (ER) stress-associated unfolded protein response (UPR) in cells
Recent studies have demonstrated that there is an association of ER stress with arsenic-related malignancies, diabetes, inflammation, autoimmune and neurodegenerative diseases. Physiological and pathological processes induce ER stress like iron imbalance, oxidative stress, nutrient deprivation, infection, and cancer, leading to the accumulation of misfolded/unfolded proteins in the ER lumen. Unfolded protein response (UPR) is an adaptive response to resolve the intracellular ER stress (Fig. 2). The ER is a membrane-bound organelle of a network of flattened sacs and tubules connected via the ER space. It plays an important role in the maturation and folding of more than one-third of proteins made in the cell apart from regulating carbohydrate metabolism, calcium homeostasis, and lipid biogenesis [34, 35]. A variety of enzymes like chaperones, oxidoreductases, and foldases are involved in the folding of polypeptides, which are translocated in the ER lumen after synthesis. Under normal physiological conditions, proteins are appropriately folded before their transportation to other organelles in the cells for further post-translational modifications. However, pathological, or physiological stresses like oxidative stress, nutrient deprivation, disturbances in calcium homeostasis, viral infections, increased protein secretions, and hypoxia can disturb the normal protein folding processes in the ER. This leads to the accumulation of unfolded/misfolded proteins in the ER lumen, which causes ER stress. The UPR is triggered in response to ER stress to reinstate normal metabolic and protein folding processes in the cells. The UPR aims to ameliorate ER stress by reducing the protein folding load on the ER by inhibiting translation, increasing the synthesis of chaperones, and stimulating the ER-associated protein degradation (ERAD) to degrade misfolded proteins (Fig. 2). When the ER stress crosses the threshold level, then UPR induces apoptosis [36].
Fig. 2.
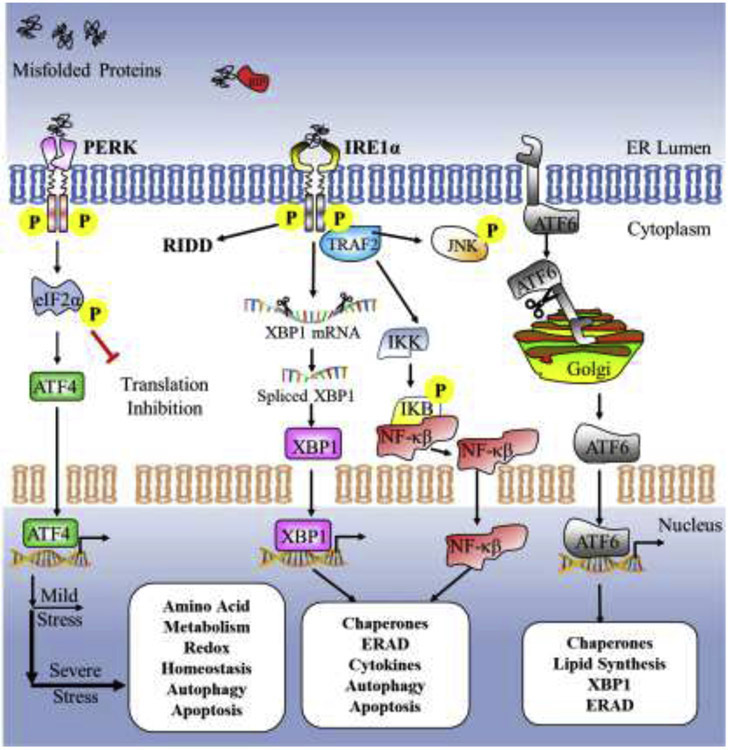
ER stress-activated UPR. Accumulation of misfolded or unfolded proteins in the ER lumen leads to ER stress, which activates the adaptive unfolded protein response (UPR). Various stresses, such as calcium disturbances, increase in protein secretion, oxidative stress, nutrient deprivation, hypoxia, oncogene activation, and infection, disturb the normal folding process in the ER leading to the build-up of misfolded/unfolded proteins in the ER. Under non-stress conditions, chaperone BiP/Grp78 is attached to three ER sensors viz., the protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), inositol requiring kinase 1α (IRE1α) and activating transcription factor 6 (ATF6), and maintains them in a dormant state. During ER stress, BiP binds to the hydrophobic exposed regions of misfolded/unfolded proteins activating the ER stress sensors PERK, IRE1α and ATF6. These sensors primarily activate downstream UPR genes involved in protein folding, ER-associated protein degradation (ERAD), lipid biosynthesis, autophagy. Prolonged ER stress, however, leads to activation of pro-death UPR signaling pathways to restore proteostasis in the ER.
As an evolutionarily conserved stress response, the UPR is orchestrated by three ER transmembrane proteins, viz., protein kinase R (PKR)-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α) and activating transcription factor 6 (ATF6). The ER chaperone glucose-regulated protein78 (Grp78)/BiP binds to the luminal domains of these three ER stress sensors and maintains them in an inactive state. However, in the presence of misfolded/unfolded proteins in the ER lumen, Grp78 dissociates from PERK, IRE1α, and ATF6, and binds to the exposed regions of the misfolded proteins. This results in the activation of PERK, IRE1α and ATF6 [34, 37]. PERK is a kinase activated by ER stress that phosphorylates eukaryotic translation initiation factor 2α (eIF2α) to inhibit global protein translation. In contrast, PERK activation increases the translation of transcription factor ATF4 with an aim to restore ER homeostasis by inducing pro-survival genes like Xbp1. PERK also phosphorylates NRF2 to promote expression of antioxidant genes. PERK-ATF4 signaling upregulates the transcription factor C/EBP-homologous protein (CHOP) on prolonged ER stress. Sustained PERK-ATF4 signaling can lead to high CHOP levels that inhibit anti-apoptotic BCL-2 and activates pro-apoptotic BIM causing mitochondrial apoptosis [34, 36, 38, 39]. IRE1α is an ER stress sensor having a cytoplasmic tail with two enzymatic activities, including serine/threonine kinase and an endoribonuclease (RNase) domain. Under moderate ER stress conditions, IRE1α RNase splices a 26-nucleotide intron from X-box binding protein (XBP1), which translocates to the nucleus to activate genes associated with ERAD, chaperones and lipid biogenesis. However, conditions of severe ER stress hyperactivates the IRE1α RNase, cleaving microRNAs that under normal circumstances suppress pro-apoptotic proteins. IRE1α hyperactivation upregulates pro-oxidant protein TXNIP (thioredoxin-interacting protein), activating the caspase-1 dependent apoptotic pathway. IRE1α also activates ASK1 (apoptosis signal-regulating kinase) and downstream JNK (c-Jun NH2-terminal kinase) kinase leading to caspase-3 mitochondrial apoptotic cell death [34, 40]. ATF6 is a 90 kDa type II transmembrane ER protein that is activated on ER stress and translocates to the Golgi apparatus, where it is cleaved by Site-1 and Site-2 proteases. The cleaved 50 kDa ATF6(N) transcription factor moves to the nucleus to increase transcription of UPR target genes such as XBP1, GRP78, CRYAB, and VEGF [40, 41].
UPR elicits both survival and apoptosis outcomes in cells. The nature and duration of the stressors decide the balance in UPR-dependent cell fate [42]. It remains to be fully elucidated what roles of UPR plays in various stages of cancer and how UPR is involved in arsenic-associated malignancies. Emerging evidence revealed abnormal activation of UPR signaling in tumor development and progression in breast, prostate, brain, and colon cancers [43, 44]. Tumor development begins with cellular transformation, followed by uncontrolled cell proliferation, metastasis, angiogenesis, metabolic reprogramming, cancer stem cell generation and development of resistance to chemotherapy [43, 45]. During growth, tumors experience various stresses such as hypoxia, oxidative stress, glucose deficiency, and inadequate amino acid supplies that affect the protein-folding capacity of the ER. Glucose-derived ATP is required for chaperone-mediated protein folding. Also, glycosylation, which is a post-translational modification that occurs in the ER, requires glucose as a substrate. Hence, glucose deprivation activates the ER stress related UPR [46, 47] Similarly, upregulated protein translation due to oncogene activation, uncontrolled cell proliferation and increased glycolysis puts an additional folding load on the ER. Genomic instability can further impair the folding processes and cause ER stress [48].
Arsenic impairs the innate and cell-mediated immunity through UPR activation. In mouse macrophage cell line RAW 264.7, arsenic trioxide (ATO) was found to disrupt macrophage function via PERK-ATF4 signaling. The ATO-mediated macrophage function disruption was diminished by antioxidant N-acetyl cysteine (NAC) or chemical chaperone PBA treatment suggesting the role of ROS and UPR [49, 50]. Chronic exposure of sodium arsenite (10 and 100 ppb) via drinking water/food caused significant alterations in gene expression related to the innate immune responses in mouse lungs. IL-1/TLR pathway components like Myd88 and Traf6 proteins were decreased by arsenite treatment. IκBα protein levels, which is an inhibitory protein for NF-κB were significantly increased at both arsenite concentrations. Transcriptome and protein expression studies confirmed significant alterations for many cytokines and cytokine receptor genes in the arsenite treated mouse lungs [51]. One of the limitations of this study is that the arsenic concentration used is 100ppb (~13.35 uM), which is significantly higher than the WHO or US EPA permissible level, 10ppb, of arsenic in drinking water. However, there are certain parts of the world like China, India, Chile, Mexico, Argentina, etc. where arsenic concentration in drinking water reached to more than 100ppb [4, 5], which may justify the high concentration of arsenic, 100ppb, is used in cellular experiments [1].
It has been known that long-term low dose arsenite (0.125-0.25 μM) exposure in BEAS-2B cells leads to the generation of CSCs [17, 33]. In the arsenic-induced CSCs, a significant downregulation was observed in genes involved in oxidative phosphorylation, TCA cycle, ubiquitination, and DNA repair pathway. In contrast, genes in the glycolytic pathway were upregulated. Glycolysis and correlated pathways like pentose phosphate, serine-glycine and hexosamine pathways are essential for the maintenance of cancer cell stemness [52]. Hexosamine biosynthetic pathway (HBP), which produces uridine diphosphate N-acetyl glucosamine (O-GlcNAc), a substrate for O-GlcNAc transferase (OGT), is found to be upregulated in many cancers. O-GlcNAc is required for protein post-translational modifications, which play crucial roles in cancer-associated processes such as cell division and signaling, metabolism and cytoskeletal regulation [53]. ER stress-induced UPR has been reported to be an upstream activator of HBP in recent studies. XBP1 and ATF4 knockdown was demonstrated to hamper glucose starvation-activated O-GlcNAcylation, suggesting that O-GlcNAc post-translation modification (PTM) was modulated by UPR and may assist metabolic reprogramming of cancer cells [43]. These studies demonstrate that there may be a relationship between the UPR and metabolism in arsenic-induced carcinogenesis and CSC maintenance. The roles played by UPR in tumors were summarized in Figure 3.
Fig. 3.

Interactions among ER, mitochondria and autophagy in ER stress. The ER and mitochondria are connected at multiple sites to form mitochondria-ER associated membranes (MAMs). Mitofusin proteins (Mfn-1/2) binds the mitochondrion and ER in the MAM. MAMs play essential roles in autophagy, ROS-mediated signals, immune signaling, inflammation, and calcium transfer due to the presence of various proteins and transporters. ATG14 is present in the MAMs, an essential protein for phagosome nucleation for mitophagy or degradation of misfolded proteins. Protein folding occurs in the ER and generates ROS, which under normal conditions, is taken care of by antioxidants like glutathione (not shown in the figure). However, arsenite metabolism depletes GSH antioxidants, which may disturb the redox environment and thus the ER protein folding process. Similarly, NOX activation also generates hydrogen peroxide in the ER. Oxidative stress from ROS generation along with nutrient deficiency, increase in protein secretion, infection, hypoxia, oncogene activation disturbs the ER protein folding and quality control systems, causing ER stress. ER stress activates the unfolded protein response (UPR), which stimulates the transcription of autophagy genes, chaperones, ERAD, apoptosis. Calcium overload in the ER results in calcium transfer from the ER to the mitochondria through calcium transporters like VDAC, SERCA. Calcium ions transferred from ER to mitochondria stimulates the electron-transport chain (ETC) generation of ROS, disturbs mitochondrial membrane potential and leads to cytochrome c release.
6. Autophagy and arsenic carcinogenesis
Autophagy is an evolutionarily conserved cell-eating process that plays an important housekeeping role during physiological and pathological conditions. It clears defective/damaged organelles, misfolded or aggregates of proteins, via the formation of unique structures that engulf the cellular cargo and fuse with the lysosomes leading to degradation. Macroautophagy is the major autophagy pathway to clear cellular debris, and henceforward will be designated as “autophagy” [54, 55]. Autophagy is essential for cellular differentiation, development, immunity, stress response, and maintaining normal cellular homeostasis. Defective autophagy is involved in many diseases like cancer, diabetes mellitus, Parkinson's disease and pathogen infections. The autophagy process can have both cytoprotective as well as pro-death outcomes in tumors. In K-ras-driven lung cancer cells, autophagy promoted tumorigenicity by providing metabolic substrates to maintain ATP levels and nucleotide pools whereas, autophagy can prevent cancers by clearing cellular debris and misfolded proteins in the early tumor stage. Hence, a context-dependent role for autophagy has been suggested in cancer [56-58].
Autophagy levels are low under normal physiological conditions. However, nutrition starvation and other stresses stimulate autophagy, leading to high basal autophagy [59]. Stress signals such as hypoxia, low ATP levels, nutrient deprivation, and ER stress, are some of the inducers of autophagy. Autophagy occurs in several steps, namely the formation of autophagosome, cargo selection, the fusion of autophagosome with lysosome, and cellular debris degradation. Under stress conditions like starvation, unc-51-like kinase (ULK1) dissociates from the mammalian target of rapamycin complex1 (mTORC1), leading to ULK1 activation and autophosphorylation. ULK1 also requires direct 5’ AMP protein kinase (AMPK) activation to initiate the autophagy process. ULK1 further phosphorylates mAtg13 and FIP200. The activated ULK1-Atg13-FIP200-Atg101 complex leads to the initiation of the formation of the isolated membrane/phagophore. ULK1 also phosphorylates Beclin1 to advance autophagy nucleation. Beclin1 forms a part of the PI3K complex along with AMBRA, VPS34 and p150, which drive the nucleation step in autophagy. The phagophore almost digests the cellular debris/cargo at this stage, followed by the phagophore's elongation. The elongation step of autophagy is implemented by the ubiquitin-conjugating systems Atg5-Atg12-Atg16L and LC3II-PE conjugates. Thus, the three-step initiation, nucleation and elongation of phagophore encapsulating the cellular debris lead to the autophagosome formation. The autophagosome fuses with lysosomes leading to the formation of autophagolysosome and degradation by the lysosomal enzymes [55, 60].
6.1. Activation of autophagy in ER stress conditions
Autophagy is activated by the ER stress associated UPR pathway to restore homeostasis. Apoptosis is activated in certain conditions where the UPR or autophagy is unable to relieve ER stress. The three ER stress sensors, ATF6, IRE1 and PERK, are involved in the activation of the autophagy pathway. In the PERK branch, activation of the transcription factor ATF4 leads to the upregulation of the autophagy genes LC3 and Atg12. The transcription factor CHOP activated via the PERK- eIF2α-ATF4 or the transcription factor ATF6 can transcriptionally upregulate Atg5 and p62 levels. In the ATF6 UPR pathway, cleaved ATF6 transcription factor induces DAPK1 kinase expression, which further, via Atg9 activation, affects the autophagy process. ATF6 activates TSC2 via the Rheb-mTOR pathway, which inhibits mTORC1, the negative regulator of the ULK1-mATG13-FIP200-ATG101 complex to induce autophagy [61-63]. In the IRE1 branch, both IRE1/XBP1 and IRE1/JNK pathways are involved in activating the autophagy. IRE1 splices XBP1 mRNA, affecting acetylated forkhead O family protein, FoxO1 binding to Atg7, mediating the autophagic process. IRE1 via the TRAF2/ASK1/JNK activation phosphorylates Bcl2 protein causing its dissociation from Beclin1, promoting autophagy [61, 64, 65]. Thus, all three ER stress-mediated UPR pathways are involved in autophagy induction.
6.2. Autophagy and UPR in cancers
Autophagy is known to have either an oncogenic or tumor suppressor role in cancers. Cheng and others demonstrated that tunicamycin (TM), an ER stress inducer, induced autophagy in the breast cancer cell lines MCF-7 and MDA-MB-231. Increased expression of LC3-II, beclin1, p62, GRP78, IRE1, pJNK, JNK, ERK1/2, and pERK1/2 was observed in the TM-treated breast cancer cells. Cells treated with 3-methyl adenine, an autophagy inhibitor, led to apoptosis. To conclude, ER stress (via TM treatment) induced autophagy, which was regulated by IRE1/JNK/Beclin1 pathway. Utilization of autophagy inhibitors or ER stress promoters can improve chemotherapy's efficiency in breast cancers [66]. A study showed that ER stress inducers thapsigargin, tunicamycin and brefeldin induced autophagy in HCT116 colon cancer cells and DU145 prostate cancer cells. The ER stress-induced autophagy was crucial in mitigating ER stress and preventing cancer cell death by clearing ubiquitinated misfolded/unfolded proteins promoting tumor survival. However, treatment with the same chemicals in normal human colon cell lines and non-transformed murine embryonic fibroblasts did not provide protection against apoptosis [67].
In human tumor cell lines HT29 colorectal adenocarcinoma, MCF7 mammary adenocarcinoma, DU145 prostate carcinoma, HCT116 colorectal adenocarcinoma and U373 MG glioblastoma, hypoxia was found to induce transcription of autophagy genes microtubule-associated protein1 light chain 3β and autophagy-related gene 5 (ATG5) through the transcription factors ATF4 and CHOP, respectively. PERK, an UPR transmembrane ER stress sensor, regulates ATF4 and CHOP genes' transcription. These tumor cell lines became more sensitive to hypoxia and irradiation on autophagy inhibition, suggesting the pro-tumorigenic role of ER stress-induced autophagy in the hypoxic environment [68]. Thus, ER stress and the associated UPR play a vital role in autophagy and apoptosis.
Fewer studies have focused mainly on the direct connections between ER stress and autophagy in arsenic-induced carcinogenesis. A study conducted in human pancreatic cell lines where arsenic trioxide (ATO) and BET bromodomain inhibitor JQ1 combination therapy was investigated as an anticancer treatment. ATO-sensitive cells were found to stimulate ER stress leading to autophagosome formation and cell death in the pancreatic cell lines [69].
6.3. Interaction among UPR, mitochondria and autophagy in arsenic carcinogenesis
Various in vitro and in vivo studies have shown that arsenic induces malignant transformation by ROS generation/oxidative stress that damage DNA, lipids, and proteins, impairment of the DNA repair systems, alterations in epigenetics and genetics, activation of cell signaling pathways such as NF-κB, JNK, and influences on immune surveillance [2]. The ER, mitochondria and autophagy are closely related in response to cellular stress signals. During stress conditions such as nutrient deficiency, autophagy and ER stress, UPR is essential in the recycling of improperly folded proteins and organelles, which are used for energy production [4]. Similarly, ER and autophagy play an essential role in maintaining mitochondrial function and dynamics (Fig. 3).
The ER and mitochondrion are connected via mitochondrial-associated ER membranes (MAMs), which are specialized subcellular compartments shaped by ER subdomains collocated to mitochondria. Calcium transport channels, enzymes engaged in lipid synthesis and transport, and proteins with oncogenic/oncosuppressive functions that regulate cell signaling pathways are present in MAMs. MAMs play a crucial role in ER stress, calcium transport and signaling, mitochondrial bioenergetics, morphology and motility, lipid transport, lipid synthesis, and inflammation signaling. Accumulation of calcium in mitochondria affects apoptosis, autophagy, and metabolic processes. MAMs play an essential role in cell signaling pathways that affect cancer cell functions [70]. Autophagy is a catabolic process that recycles damaged organelles, misfolded proteins using the lysosomal machinery. It also protects against oxidative stress by clearing damaged mitochondria and peroxisomes, which are important sources of ROS generation. Defects in autophagy have been associated with genomic instability, chromosomal aberrations, DNA mutations, and increased cancer risk [71]. Autophagy can have dual impacts on tumorigenesis. “Mitophagy” or autophagy of defective/damaged mitochondria can protect cells against ROS-generated oxidative damage. Contrarily, under stress conditions like nutrient deprivation, recycling of defective organelles and misfolded proteins can provide nutrition for tumor growth. A study by Zhang et al. reported that BEAS-2B cells treated with low-dose arsenic for short-term elevated ROS levels, but due to autophagy protection, this treatment did not cause cellular transformation. However, higher intracellular ROS levels were observed and caused cell transformation on impairment of autophagy process [72].
Similar to BEAS-2B cells, HBE and HEK293 cells exposed to environmentally relevant concentrations of arsenite (500nm-4μM) was found to deregulate autophagy, which caused prolonged NRF2 activation and p62 accumulation. Prolonged NRF2 activation plays a role in human diseases like cancers [73]. In another study by Dodson et al., low dose arsenite treatment (< 5μM) in HEK293, NIH 3T3, and HeLa cells caused transient ER stress response via autophagy inhibition [74]. Arsenic trioxide (ATO) was noted to cause ROS-mediated oxidative stress in GC-1 spermatogonial cells, which correlated with mitochondrial dysfunction and autophagy. Metabolomics studies also identified that ATO exposure in these cells altered ten metabolic pathways involved in the metabolism of amino acids, lipids, glycans, and vitamins [75].
Various animal and cellular studies have demonstrated that inorganic arsenic causes mitochondrial dysfunction via excessive ROS production, mitochondrial DNA damage, elevated proton leak from depolarization, and metabolic reprogramming due to mitochondrial respiration uncoupling and mitochondrial membrane damage. Oxidative stress also affects proteostasis in the ER. In cancer cells, proteotoxic stress/imbalance in proteostasis is associated with mitochondrial dysfunction. Proteostasis is maintained by cellular processes that regulate protein biosynthesis, folding, trafficking, and degradation. Molecular chaperones are regulators of proteostasis which enable proper folding of proteins and prevent aggregation of misfolded proteins. Trivalent arsenic has been shown to influence the functions of chaperones by impeding the binding of chaperones proteins and their substrates and affecting functions of ATP-dependent chaperones through inhibition of ATP generation in mitochondria. Inorganic trivalent arsenic exposure in drinking water has been found to disturb autophagy and ubiquitin-proteasome (UPS) systems affecting protein clearance machineries in the cells [76, 77]. Inorganic arsenic and its methylated metabolites are believed to cause inflammation via oxidative stress induction. Arsenic induced oxidative stress, and prolonged inflammation causes protein misfolding, triggering ER stress [76].
Protein folding and formation of disulfide bonds occur in the ER in a highly oxidizing environment where glutathione (GSH) is the primary redox buffer that protects cells from oxidative stress by neutralizing ROS species. Two ROS generating mechanisms have been suggested during disulfide bond formation in ER. The first is during the transfer of electrons from protein thiol to oxygen and the second is during protein misfolding caused by GSH depletion. Inorganic trivalent arsenic has been reported to deplete intracellular reduced glutathione by binding to it and suppressing glutathione interaction with sulfhydryl groups in proteins [76]. Similarly, the accumulation of misfolded proteins increases ROS production in the mitochondria due to Ca2+ leakage from the ER. Thus, both mitochondrial respiration and ER protein oxidation give rise to ROS production. Thus, there is an intricate connection between oxidative stress, ER and mitochondria in arsenic-induced carcinogenesis [78].
Several other studies have also associated arsenic with the induction of ER stress, mitochondrial dysfunction, and autophagy. In human lung adenocarcinoma A549 cells, arsenic trioxide (ATO)-induced ER stress was observed by an increase in the protein expression of GRP78, CHOP and caspase-12. Treatment of A549 cells with NAC, a ROS scavenger, reduced ER stress suggesting that ATO induces ER stress via ROS generation. Mitochondrial dysfunction was also observed in the ATO treated cells [79]. Low doses of sodium arsenite (< 1μM) upregulated ROS levels triggering ER stress in human lung epithelial cells BEAS-2B and skin keratinocytes. ER stress-activated the UPR, as observed by an increase in BiP and phosphorylated PERK levels. Accordingly, low dose arsenite treatment increased ROS levels, activating ER stress-related UPR and masked p53 function, leading to malignant transformation of cells [80]. The crosstalk and interaction among ER stress-mediated UPR, mitochondria and autophagy have been illustrated in Fig.3.
Perspectives
Arsenic is a well-established human carcinogen that elicits its effects by influencing epigenetics, genetics, DNA repair systems, ER stress-induced UPR, mitochondrial metabolism, autophagy, immune responses and various intracellular signaling pathways. Arsenic is known to induce oxidative stress resulting in mitochondrial dysfunction which can affect the ER homeostasis and its folding ability. Similarly, the protein folding oxido/reduction environment and the depletion of antioxidant GSH during arsenic metabolism can also lead to excessive ROS generation in the ER. The trivalent arsenic species (As3+) have also been shown to inhibit oxidative protein folding in vitro. Taken together, arsenic induced oxidative stress plays an important role in the mitochondrial dysfunction and ER stress contributing to carcinogenesis [2, 77, 78]. As mentioned before, the ER, mitochondrion and autophagy are interlinked to each other and functional disturbances in one of these cellular organelles will affect the functions in the other organelles. In the various in vitro and in vivo studies discussed so far, arsenic has been consistently shown to affect the UPR, mitochondrion and autophagy. The ER stress-mediated UPR was involved in various stages of the cancers such as tumor initiation, progression, migration/invasion, angiogenesis, metabolism, immune responses, and chemoresistance. A context-dependent role was observed of the ER stress-mediated UPR, mitochondrion and autophagy depending upon the cancer type and stage.
UPR is primarily an adaptive survival stress response to maintain proteostasis in cells. However, sustained ER stress results in UPR triggering the apoptotic signaling pathways. Arsenic transformed and the cancer cells in general are able to overcome ER stresses and avoid the UPR-apoptotic pathways in the early stages. These cells can utilize the ER stress-mediated UPR survival pathways, autophagy and metabolic reprogramming process efficiently to ensure cancer cell survival and the stemness of CSCs [81, 82]. Understanding the intricate details in the UPR signaling pathway may provide molecular targets for possible future cancer therapy. Various ER stress-focused clinical trials have been conducted globally for diabetes, ataxia, lung cancer, and obesity [83]. Promoting ER stress-associated apoptotic pathways in the UPR can be implemented to advance cancer cell death. Another course of action could be by using UPR pathway-specific drug inhibitors to impair the use of UPR as a survival pathway for cancers [42]. The challenge for cancer scientists is to choose appropriate UPR targets and to ensure that the drug selectively and maximally targets the UPR leading to effective cancer treatment. Another challenge is to make sure that a proper analysis of the research data is made to overcome limitations associated with varied experimental conditions, understanding the in vitro versus in vivo implications and tumor heterogeneity, etc.
Tumor relapse and chemoresistance have been linked to poor survival in cancer patients. The CSCs are resistant to chemotherapy and play a critical role in tumor relapse. The metabolic reprogramming and autophagy activation determine the cell fate and regulate the stemness properties of the CSCs. Majority of the studies emphasize that the metabolic switch in the CSCs from mitochondrial OXPHOS to glycolysis ensure stemness maintenance, whereas ER stress-UPR activation promotes differentiation of the CSCs. We have observed similar results in the arsenic-induced CSCs. However, a significant downregulation of the genes involved in the ER stress and autophagy was observed in the arsenic-induced CSCs [4]. In summary, arsenic exposure activates ER stress-UPR and autophagy, causes mitochondrial dysfunction which influences arsenic-induced cancers and the generation/maintenance of the CSCs. A better understanding of the upstream and downstream cell signaling pathways involved in the arsenic-induced ER stress-UPR, autophagy and mitochondria and their interconnected roles in arsenic-carcinogenesis and CSCs will lead to identification of key targets for a future anti-cancer therapy.
Acknowledgements
The research work in Chen’s lab is supported by NIH grants R01 ES028263, ES028335, ES031822, and partially supported by NIH grant P30 ES020957.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
No conflict of interest can be declared.
References
- [1].Flora SJS, 1 - Arsenic: Chemistry, Occurrence, and Exposure, in: Flora SJS (Ed.), Handbook of Arsenic Toxicology, Academic Press, Oxford, 2015, pp. 1–49. [Google Scholar]
- [2].Li L, Chen F, Oxidative stress, epigenetics, and cancer stem cells in arsenic carcinogenesis and prevention, Current pharmacology reports 2(2) (2016) 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Farzan SF, Karagas MR, Chen Y, In utero and early life arsenic exposure in relation to long-term health and disease, Toxicology and applied pharmacology 272(2) (2013) 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Li L, Bi Z, Wadgaonkar P, Lu Y, Zhang Q, Fu Y, Thakur C, Wang L, Chen F, Metabolic and epigenetic reprogramming in the arsenic-induced cancer stem cells, Seminars in cancer biology 57 (2019) 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Podgorski J, Berg M, Global threat of arsenic in groundwater, Science 368(6493) (2020) 845–850. [DOI] [PubMed] [Google Scholar]
- [6].Ayotte JD, Medalie L, Qi SL, Backer LC, Nolan BT, Estimating the High-Arsenic Domestic-Well Population in the Conterminous United States, Environ Sci Technol 51(21) (2017) 12443–12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Khairul I, Wang QQ, Jiang YH, Wang C, Naranmandura H, Metabolism, toxicity and anticancer activities of arsenic compounds, Oncotarget 8(14) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kaur S, Kamli MR, Ali A, Role of arsenic and its resistance in nature, Can J Microbiol 57(10) (2011) 769–74. [DOI] [PubMed] [Google Scholar]
- [9].Suhadolnik MLS, Salgado APC, Scholte LLS, Bleicher L, Costa PS, Reis MP, Dias MF, Ávila MP, Barbosa FAR, Chartone-Souza E, Nascimento AMA, Novel arsenic-transforming bacteria and the diversity of their arsenic-related genes and enzymes arising from arsenic-polluted freshwater sediment, Scientific reports 7(1) (2017) 11231–11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang HC, Fu HL, Lin YF, Rosen BP, Pathways of arsenic uptake and efflux, Curr Top Membr 69 (2012) 325–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhou Q, Xi S, A review on arsenic carcinogenesis: Epidemiology, metabolism, genotoxicity and epigenetic changes, Regulatory Toxicology and Pharmacology 99 (2018) 78–88. [DOI] [PubMed] [Google Scholar]
- [12].Khairul I, Wang QQ, Jiang YH, Wang C, Naranmandura H, Metabolism, toxicity and anticancer activities of arsenic compounds, Oncotarget 8(14) (2017) 23905–23926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhou Q, Xi S, A review on arsenic carcinogenesis: Epidemiology, metabolism, genotoxicity and epigenetic changes, Regul Toxicol Pharmacol 99 (2018) 78–88. [DOI] [PubMed] [Google Scholar]
- [14].Tam LM, Price NE, Wang Y, Molecular Mechanisms of Arsenic-Induced Disruption of DNA Repair, Chemical Research in Toxicology 33(3) (2020) 709–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang L, Lu Q, Chang C, Epigenetics in Health and Disease, Adv Exp Med Biol 1253 (2020) 3–55. [DOI] [PubMed] [Google Scholar]
- [16].Mauro M, Caradonna F, Klein CB, Dysregulation of DNA methylation induced by past arsenic treatment causes persistent genomic instability in mammalian cells, Environ Mol Mutagen 57(2) (2016) 137–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chang Q, Chen B, Thakur C, Lu Y, Chen F, Arsenic-induced sub-lethal stress reprograms human bronchial epithelial cells to CD61− cancer stem cells, Oncotarget 5(5) (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Eckstein M, Rea M, Fondufe-Mittendorf YN, Transient and permanent changes in DNA methylation patterns in inorganic arsenic-mediated epithelial-to-mesenchymal transition, Toxicol Appl Pharmacol 331 (2017) 6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shi H, Shi X, Liu KJ, Oxidative mechanism of arsenic toxicity and carcinogenesis, Molecular and Cellular Biochemistry 255(1) (2004) 67–78. [DOI] [PubMed] [Google Scholar]
- [20].Zhang Z, Pratheeshkumar P, Budhraja A, Son Y-O, Kim D, Shi X, Role of reactive oxygen species in arsenic-induced transformation of human lung bronchial epithelial (BEAS-2B) cells, Biochemical and Biophysical Research Communications 456(2) (2015) 643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Maiti S, 9 - Arsenic-Induced Mutagenesis and Carcinogenesis: A Possible Mechanism, in: Flora SJS (Ed.), Handbook of Arsenic Toxicology, Academic Press, Oxford, 2015, pp. 233–279. [Google Scholar]
- [22].Huang HW, Lee CH, Yu HS, Arsenic-Induced Carcinogenesis and Immune Dysregulation, Int J Environ Res Public Health 16(15) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Soza-Ried C, Bustamante E, Caglevic C, Rolfo C, Sirera R, Marsiglia H, Oncogenic role of arsenic exposure in lung cancer: A forgotten risk factor, Critical Reviews in Oncology/Hematology 139 (2019) 128–133. [DOI] [PubMed] [Google Scholar]
- [24].Wu K, Li L, Thakur C, Lu Y, Zhang X, Yi Z, Chen F, Proteomic Characterization of the World Trade Center dust-activated mdig and c-myc signaling circuit linked to multiple myeloma, Sci Rep 6 (2016) 36305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].van Breda SG, Claessen SM, Lo K, van Herwijnen M, Brauers KJ, Lisanti S, Theunissen DH, Jennen DG, Gaj S, de Kok TM, Kleinjans JC, Epigenetic mechanisms underlying arsenic-associated lung carcinogenesis, Arch Toxicol 89(11) (2015) 1959–69. [DOI] [PubMed] [Google Scholar]
- [26].Kim E, Kim M, Woo D-H, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, Lee C, Joo KM, Rich JN, Nam D-H, Lee J, Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells, Cancer cell 23(6) (2013) 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen WT, Hung WC, Kang WY, Huang YC, Chai CY, Urothelial carcinomas arising in arsenic-contaminated areas are associated with hypermethylation of the gene promoter of the death-associated protein kinase, Histopathology 51(6) (2007) 785–92. [DOI] [PubMed] [Google Scholar]
- [28].Pogribny IP, Beland FA, Rusyn I, The role of microRNAs in the development and progression of chemical-associated cancers, Toxicology and Applied Pharmacology 312 (2016) 3–10. [DOI] [PubMed] [Google Scholar]
- [29].Cardoso APF, Al-Eryani L, States JC, Arsenic-Induced Carcinogenesis: The Impact of miRNA Dysregulation, Toxicol Sci 165(2) (2018) 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen C, Jiang X, Gu S, Zhang Z, MicroRNA-155 regulates arsenite-induced malignant transformation by targeting NRF2-mediated oxidative damage in human bronchial epithelial cells, Toxicol Lett 278 (2017) 38–47. [DOI] [PubMed] [Google Scholar]
- [31].Kuşoğlu A, Biray Avci Ç, Cancer stem cells: A brief review of the current status, Gene 681 (2019) 80–85. [DOI] [PubMed] [Google Scholar]
- [32].Daley GQ, Stem cells and the evolving notion of cellular identity, Philosophical transactions of the Royal Society of London. Series B, Biological sciences 370(1680) (2015) 20140376–20140376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bi Z, Zhang Q, Fu Y, Wadgaonkar P, Zhang W, Almutairy B, Xu L, Rice M, Qiu Y, Thakur C, Chen F, NRF2 and HIF1α converge to arsenic-induced metabolic reprogramming and the formation of the cancer stem-like cells, Theranostics 10(9) (2020) 4134–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Oakes SA, Papa FR, The role of endoplasmic reticulum stress in human pathology, Annu Rev Pathol 10 (2015) 173–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Oakes SA, Endoplasmic Reticulum Stress Signaling in Cancer Cells, Am J Pathol 190(5) (2020) 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hetz C, The unfolded protein response: controlling cell fate decisions under ER stress and beyond, Nature Reviews Molecular Cell Biology 13(2) (2012) 89–102. [DOI] [PubMed] [Google Scholar]
- [37].Manalo RVM, Medina PMB, The endoplasmic reticulum stress response in disease pathogenesis and pathophysiology, Egyptian Journal of Medical Human Genetics 19(2) (2018) 59–68. [Google Scholar]
- [38].Papaioannou A, Higa A, Jégou G, Jouan F, Pineau R, Saas L, Avril T, Pluquet O, Chevet E, Alterations of EDEM1 functions enhance ATF6 pro-survival signaling, Febs j 285(22) (2018) 4146–4164. [DOI] [PubMed] [Google Scholar]
- [39].Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA, NRF2 is a direct PERK substrate and effector of PERK-dependent cell survival, Mol Cell Biol 23(20) (2003) 7198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cubillos-Ruiz JR, Bettigole SE, Glimcher LH, Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer, Cell 168(4) (2017) 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang SX, Ma JH, Bhatta M, Fliesler SJ, Wang JJ, The unfolded protein response in retinal vascular diseases: implications and therapeutic potential beyond protein folding, Prog Retin Eye Res 45 (2015) 111–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ojha R, Amaravadi RK, Targeting the unfolded protein response in cancer, Pharmacological Research 120 (2017) 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Madden E, Logue SE, Healy SJ, Manie S, Samali A, The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance, Biol Cell 111(1) (2019) 1–17. [DOI] [PubMed] [Google Scholar]
- [44].Sisinni L, Pietrafesa M, Lepore S, Maddalena F, Condelli V, Esposito F, Landriscina M, Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance between Apoptosis and Autophagy and Its Role in Drug Resistance, Int J Mol Sci 20(4) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Thakur C, Chen F, Connections between metabolism and epigenetics in cancers, Semin Cancer Biol 57 (2019) 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Huber A-L, Lebeau J, Guillaumot P, Pétrilli V, Malek M, Chilloux J, Fauvet F, Payen L, Kfoury A, Renno T, Chevet E, Serge N. Manié, p58IPK-Mediated Attenuation of the Proapoptotic PERK-CHOP Pathway Allows Malignant Progression upon Low Glucose, Molecular Cell 49(6) (2013) 1049–1059. [DOI] [PubMed] [Google Scholar]
- [47].Ding B, Parmigiani A, Divakaruni AS, Archer K, Murphy AN, Budanov AV, Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death, Scientific reports 6 (2016) 22538–22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Oakes SA, Endoplasmic reticulum proteostasis: a key checkpoint in cancer, Am J Physiol Cell Physiol 312(2) (2017) C93–c102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hunt KM, Srivastava RK, Athar M, 11 - Cutaneous Toxicology of Arsenic, in: Flora SJS (Ed.), Handbook of Arsenic Toxicology, Academic Press, Oxford, 2015, pp. 301–314. [Google Scholar]
- [50].Srivastava RK, Li C, Chaudhary SC, Ballestas ME, Elmets CA, Robbins DJ, Matalon S, Deshane JS, Afaq F, Bickers DR, Athar M, Unfolded protein response (UPR) signaling regulates arsenic trioxide-mediated macrophage innate immune function disruption, Toxicol Appl Pharmacol 272(3) (2013) 879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kozul CD, Hampton TH, Davey JC, Gosse JA, Nomikos AP, Eisenhauer PL, Weiss DJ, Thorpe JE, Ihnat MA, Hamilton JW, Chronic exposure to arsenic in the drinking water alters the expression of immune response genes in mouse lung, Environmental health perspectives 117(7) (2009) 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li L, Bi Z, Wadgaonkar P, Lu Y, Zhang Q, Fu Y, Thakur C, Wang L, Chen F, Metabolic and epigenetic reprogramming in the arsenic-induced cancer stem cells, Semin Cancer Biol 57 (2019) 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Slawson C, Hart GW, O-GlcNAc signalling: implications for cancer cell biology, Nat Rev Cancer 11(9) (2011) 678–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mizushima N, Levine B, Cuervo AM, Klionsky DJ, Autophagy fights disease through cellular self-digestion, Nature 451(7182) (2008) 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ravanan P, Srikumar IF, Talwar P, Autophagy: The spotlight for cellular stress responses, Life Sci 188 (2017) 53–67. [DOI] [PubMed] [Google Scholar]
- [56].Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, Khor S, Chan CS, Rabinowitz JD, White E, Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells, Genes & development 30(15) (2016) 1704–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Guo JY, White E, Autophagy, Metabolism, and Cancer, Cold Spring Harb Symp Quant Biol 81 (2016) 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chen H-Y, White E, Role of autophagy in cancer prevention, Cancer prevention research (Philadelphia, Pa.) 4(7) (2011) 973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Goldsmith J, Levine B, Debnath J, Chapter Two - Autophagy and Cancer Metabolism, in: Galluzzi L, Kroemer G (Eds.), Methods in Enzymology, Academic Press; 2014, pp. 25–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chan EY, mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex, Sci Signal 2(84) (2009) pe51. [DOI] [PubMed] [Google Scholar]
- [61].Lin Y, Jiang M, Chen W, Zhao T, Wei Y, Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response, Biomed Pharmacother 118 (2019) 109249. [DOI] [PubMed] [Google Scholar]
- [62].Yan MM, Ni JD, Song D, Ding M, Huang J, Interplay between unfolded protein response and autophagy promotes tumor drug resistance, Oncol Lett 10(4) (2015) 1959–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhang Z, Zhang L, Zhou L, Lei Y, Zhang Y, Huang C, Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress, Redox Biol 25 (2019) 101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kishino A, Hayashi K, Hidai C, Masuda T, Nomura Y, Oshima T, XBP1-FoxO1 interaction regulates ER stress-induced autophagy in auditory cells, Scientific Reports 7(1) (2017) 4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu W-G, Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity, Nature Cell Biology 12(7) (2010) 665–675. [DOI] [PubMed] [Google Scholar]
- [66].Cheng X, Liu H, Jiang CC, Fang L, Chen C, Zhang XD, Jiang ZW, Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells, Int J Mol Med 34(3) (2014) 772–81. [DOI] [PubMed] [Google Scholar]
- [67].Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM, Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival, J Biol Chem 282(7) (2007) 4702–10. [DOI] [PubMed] [Google Scholar]
- [68].Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P, van der Kogel AJ, Koritzinsky M, Wouters BG, The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5, J Clin Invest 120(1) (2010) 127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Xu C, Wang X, Zhou Y, Chen FX, Wang H, Li K, Fan H, Tang X, Jiang G, Zhang J, Synergy between arsenic trioxide and JQ1 on autophagy in pancreatic cancer, Oncogene 38(47) (2019) 7249–7265. [DOI] [PubMed] [Google Scholar]
- [70].Morciano G, Marchi S, Morganti C, Sbano L, Bittremieux M, Kerkhofs M, Corricelli M, Danese A, Karkucinska-Wieckowska A, Wieckowski MR, Bultynck G, Giorgi C, Pinton P, Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings, Neoplasia 20(5) (2018) 510–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Qi Y, Li H, Zhang M, Zhang T, Frank J, Chen G, Autophagy in arsenic carcinogenesis, Experimental and Toxicologic Pathology 66(4) (2014) 163–168. [DOI] [PubMed] [Google Scholar]
- [72].Zhang T, Qi Y, Liao M, Xu M, Bower KA, Frank JA, Shen H-M, Luo J, Shi X, Chen G, Autophagy Is a Cell Self-Protective Mechanism Against Arsenic-Induced Cell Transformation, Toxicological Sciences 130(2) (2012) 298–308. [DOI] [PubMed] [Google Scholar]
- [73].Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, Zhang DD, Arsenic Inhibits Autophagic Flux, Activating the NRF2-Keap1 Pathway in a p62-Dependent Manner, Molecular and Cellular Biology 33(12) (2013) 2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dodson M, de la Vega MR, Harder B, Castro-Portuguez R, Rodrigues SD, Wong PK, Chapman E, Zhang DD, Low-level arsenic causes proteotoxic stress and not oxidative stress, Toxicology and Applied Pharmacology 341 (2018) 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Chen H, Liu G, Qiao N, Kang Z, Hu L, Liao J, Yang F, Pang C, Liu B, Zeng Q, Li Y, Li Y, Toxic effects of arsenic trioxide on spermatogonia are associated with oxidative stress, mitochondrial dysfunction, autophagy and metabolomic alterations, Ecotoxicology and Environmental Safety 190 (2020) 110063. [DOI] [PubMed] [Google Scholar]
- [76].Tam LM, Wang Y, Arsenic Exposure and Compromised Protein Quality Control, Chemical Research in Toxicology 33(7) (2020) 1594–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ramadan D, Rancy PC, Nagarkar RP, Schneider JP, Thorpe C, Arsenic(III) species inhibit oxidative protein folding in vitro, Biochemistry 48(2) (2009) 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bhandary B, Marahatta A, Kim H-R, Chae H-J, An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases, International journal of molecular sciences 14(1) (2012) 434–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Gu S, Chen C, Jiang X, Zhang Z, ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction underlie apoptosis induced by resveratrol and arsenic trioxide in A549 cells, Chemico-Biological Interactions 245 (2016) 100–109. [DOI] [PubMed] [Google Scholar]
- [80].Ganapathy S, Li P, Fagman J, Yu T, Lafontant J, Zhang G, Chen C, Low doses of arsenic, via perturbing p53, promotes tumorigenesis, Toxicology and Applied Pharmacology 306 (2016) 98–104. [DOI] [PubMed] [Google Scholar]
- [81].Simic MS, Moehle EA, Schinzel RT, Lorbeer FK, Halloran JJ, Heydari K, Sanchez M, Jullié D, Hockemeyer D, Dillin A, Transient activation of the UPR(ER) is an essential step in the acquisition of pluripotency during reprogramming, Sci Adv 5(4) (2019) eaaw0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Peñaranda-Fajardo NM, Meijer C, Liang Y, Dijkstra BM, Aguirre-Gamboa R, den Dunnen WFA, Kruyt FAE, ER stress and UPR activation in glioblastoma: identification of a noncanonical PERK mechanism regulating GBM stem cells through SOX2 modulation, Cell Death & Disease 10(10) (2019) 690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, Luís A, McCarthy N, Montibeller L, More S, Papaioannou A, Püschel F, Sassano ML, Skoko J, Agostinis P, de Belleroche J, Eriksson LA, Fulda S, Gorman AM, Healy S, Kozlov A, Muñoz-Pinedo C, Rehm M, Chevet E, Samali A, Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications, Febs j 286(2) (2019) 241–278. [DOI] [PMC free article] [PubMed] [Google Scholar]