

Abstract
Our previous studies found that deletion of nuclear receptor interacting protein 1 (Nrip1) extended longevity in female mice and delayed cell senescence. The current study investigates the role of NRIP1 in regulating functions of adipose-derived mesenchymal stem cells (ADMSCs) and explores the mechanisms of NRIP1 in skin aging. We first verified the skin aging phenotypes in young (6 months) and old (20 months) C57BL/6J (B6) mice and found deletion of Nrip1 can delay skin aging phenotypes, including reduced thickness of dermis and subcutaneous white adipose tissue (sWAT), as well as the accumulation of senescent cells in sWAT. In ADMSCs isolated from sWAT, we found that deletion of Nrip1 could decrease cell proliferation, prevent cell apoptosis, and suppress adipogenesis. Interestingly, deletion of Nrip1 also reduced cell senescence and maintain cell quiescence of ADMSCs. Moreover, the expressions of genes associated with senescence (p21, and p53), inflammation (p65, IL6, and IL1a), and growth factor (mTOR, Igf1) were reduced in Nrip1 knockout ADMSCs, as well as in siNrip1-treated ADMSCs. Suppression of Nrip1 by siNrip1 also decreased the expressions of mTOR, p-mTOR, p65, and p-p65 in ADMSCs. Reduced expressions of p65 and p-p65 were also confirmed in the skin of Nrip1 knockout mice. These findings suggest that NRIP1 plays an important role in delaying skin aging by reducing ADMSCs senescence and maintaining ADMSCs quiescence.
Keywords: NRIP1, Skin aging, ADMSC, Senescence, Quiescence
Introduction
Skin aging is a continuous dynamic process. Clinically, skin aging is mainly manifested as wrinkle formation, loss of elasticity, and pigmentation. Histologically, skin aging is characterized by thinning of epidermis, atrophy of dermis, flattening of the dermoepidermal junction, accumulation of abnormal elastic tissue in the dermis, and reduced amounts of subcutaneous white adipose tissue (sWAT) [43]. Although the pathological mechanism of skin aging is complex, previous studies found that the pathogenesis mainly included mutations of mitochondrial DNA, accumulated oxidative stress, overexpressed matrix metalloproteinases (MMPs), and increased inflammatory reactions [20]. Recently, increasing evidence has shown that sWAT is correlated with intrinsic aging, as its structure and volume are significantly altered during the process of aging. The alterations of sWAT structure include reduction of the sWAT thickness, reduction of the number and the proliferative ability of preadipocytes, change of the intercellular matrix, and modification of the size of adipocytes [41]. Notably, adipocytes have an important impact on immune reaction and inflammation. Accumulating evidence suggests that the alteration of function in adipocytes plays an important role in aging-related pathological changes.
Nuclear receptor interacting protein 1 (NRIP1), also known as RIP140 (receptor interacting protein of 140 kDa), regulates gene expression by interacting with nuclear receptors, such as estrogen receptor (ER) and androgen receptor (AR). NRIP1 is highly expressed in adipose tissue, liver, and skeletal muscle, and is involved in metabolism, aging, and other physiological process. Compared to wild-type mice, Nrip1 knockout mice are lean, have lower body fat content, and are resistant to obesity induced by a high-fat diet [40]. Our previous study found that deletion of Nrip1 extends longevity of female mice and delays senescence in white adipose tissue (WAT) [40]. We also found that NRIP1 is overexpressed in lesions and peripheral blood mononuclear cells (PBMCs) of psoriasis patients. Suppression of NRIP1 in both CD4+ T cells of psoriasis patients and the IMQ-induced psoriasis-like mouse model can downregulate the expression of NFκB. Additionally, in macrophages, NRIP1 functions as a co-activator for NFκB through direct interaction with RelA/p65 and upregulates the downstream inflammatory gene expression such as TNF-α and interleukin-6 [44].
To investigate the potential role of NRIP1 in skin aging, we first tested the skin aging phenotypes in young (6 months) and old (20 months) C57BL/6J (B6) mice, as well as in Nrip1+/+ and Nrip1-/- mice at old age (20 months). We also isolated adipose-derived mesenchymal stem cells (ADMSCs) from sWAT of both Nrip1+/+ and Nrip1-/- mice and tested the cell phenotypes, including cell senescence, proliferation, apoptosis, and adipogenic differentiation ability. Accordingly, we examined cell cycle and quiescence of ADMSCs to understand the reason why depletion of Nrip1 can reduce cell senescence. Moreover, we tested the genes associated with senescence (p16, p21, and p53), growth factors (mTOR and Igf1), and inflammation-related genes (p65, IL6, and IL1a) in ADMSCs and skin to further delineate the mechanism of how NRIP1 regulates ADMSCs in skin aging.
Materials and methods
Mice
The C57BL/6J (B6, strain ID 000664) mice were purchased from The Jackson Laboratory. The Nrip1 global knockout strain (RY-Nrip1 KO) was created by using the Cre/LoxP strategy described previously [40]. Mice were housed in the Division of Laboratory Animal Medicine at Southern Illinois University School of Medicine (SIU-SOM). All animal experiments were conducted in accordance with NIH guidelines and protocols approved by the SIU-SOM Laboratory Animal Care and Use Committee.
Tissue collection
Mice were euthanized by cervical dislocation for tissue collection. Skin tissue was collected. Tissues for RNA assay were stored in RNAlater (Invitrogen, Cat. #: AM7020) at −20°C. Tissues for protein assays and senescence-associated β-Gal (SA-β-Gal) staining were flash-frozen in dry ice with alcohol and stored at −80°C. Tissues for histology were fixed in 10% formalin.
SA-β-gal staining and histological analysis
Senescent cells were detected by using SA-β-gal staining kit (Cell Signaling Technology, Cat. #: 9860) according to the manufacturer’s instructions. After SA-β-gal staining, tissues were embedded in paraffin blocks and sectioned (4 μm thick). Then, slides were stained with hematoxylin (Thermo Scientific, Cat. #: 6765001) and eosin (Acros Organics, Cat. #: 17372-87-1). To analyze SA-β-gal positive cells, we randomly selected four vision fields under microscope (×40, Olympus IX71) for each slide and calculated the percentage of positive cells. For measuring thickness of dermis and subcutaneous, as well as the diameter of subcutaneous adipocyte, four pictures were taken randomly for each H&E-stained slide under microscope (×40, Olympus IX71).
Immunohistochemistry
Immunohistochemistry staining was performed by using Pierce Peroxidase Detection Kit (Thermo Scientific, Cat. #: 36000). In short, antigen retrieval was performed using 10 μM citrate buffer (pH=6.0) for 15 min at 90–95°C. After antigen retrieval, sections were treated with peroxidase suppressor to block endogenous peroxidase. Incubation of blocking buffer provided by the kit for 1 h was followed by treatment with primary antibodies at 4°C overnight. Sections were then incubated with secondary antibody HRP polymer-Rabbit IgG (1:100, R&D, Cat. #: VC003-025), followed by the incubation with secondary antibody, DAB/Metal Substrate Working Solution, for 5–15 min until desired stain. Finally, sections were counterstained with hematoxylin, dehydrated, and mounted. Primary antibody used in this study includes p-NFκB p65 (1:200, Santa Cruz, Cat. #: sc-33039). Since the primary antibody was raised in rabbit, rabbit IgG (1:200, Invitrogen, Cat. #:10500C) was used as false positive control.
Isolation of ADMSCs and cell culture
Inguinal adipose tissue was obtained from B6, RY-Nrip1+/+, and RY-Nrip1-/- female mice at 6 to 8 months of age. After being minced, tissue was digested by collagenase/dispase (1 mg/ml, Sigma, Cat. #: 10269638001) with 10 mM CaCl2 with constant agitation for 45 min at 37°C. The digestion medium was filtered through a 100-μm cell strainer, washed with PBS, and centrifuged at 500g for 10 min. After centrifugation, the cells were filtered through and a 70-μm strainer and then resuspended in α-MEM (Corning, Cat. #: 10-022-CV) supplemented with 10% FBS, 1% penicillin and streptomycin, and 250 ng/ml amphotericin B (Sigma, Cat. #: A2942). Culture medium was changed 2 h after plating the cells, followed by another medium change after 12 h, and then every 2 days. Cells of the first four generations were used in the following studies.
Flow cytometry
The surface phenotypic characterization of ADMSCs was determined by flow cytometric analysis. Briefly, 3×106 cells were suspended in 1 ml PBS containing 10% FBS and aliquots of 100 μl were incubated with labeled antibodies (purified anti-mouse CD29, CD31, and CD44, Cat. #: MUXMX-09011, Cyagen) for 30 min at 4°C and were washed twice with PBS. Then, cells were incubated with fluorescein isothiocyanate (FITC) or phycoerythrin (PE)-conjugated goat anti-rat IgG secondary antibodies for 30 min at 4°C. Fluorescence of 15,000 viable cells was analyzed using a flow cytometer (FACSCalibur, BD Biosciences, USA).
siRNA transfection
siCON (Santa Cruz, Cat. #: sc-37007) and siRIP140 (Santa Cruz, Cat. #: sc-36428) were mixed with Lipofectamine 3000 (Life Technologies, Cat. #: L3000-015) and incubated for 45 min at room temperature. Cells were incubated in the transfection medium for 6 h, and then, an equal volume of complete medium was added without removing the transfection medium. Culture medium was changed after 24 h. At 48, 72, and 96 h post-transfection, cells were stained with SA-β-Gal staining or collected for qPCR and western blot analysis
Cell proliferation assay
MTT (MTT, Sigma, Cat. #: M2003) assay was applied to evaluate cell proliferation. Cells were seeded at the concentration of 4 × 104 cells/well in 6-well plates. 100 μl of 5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide and 1.9 ml serum-free culture medium were added to each well at 24, 48, 72, 96, and 120h after being seeded. Following 4-h incubation, wells were aspirated and 1 ml of MTT solvent (4 mM HCl, 0.1% Nondet P-40 (NP40) all in isopropanol) was added to each well. After being resolved completely, solution of each well was aliquoted to four wells of a 96-well plate (250μl/well), and the concentration of cells was calculated by the absorptions at 570 nm and 690nm measured by spectrophotometry (BioTek PowerWave XS, Winooski).
Examination of cumulative population doubling level
ADMSCs were isolated from female Nrip1+/+ and Nrip1-/- mice at middle age (13months). The ADMSCs were seeded at a density of 3 × 104 cells per well in a 6-well cell culture plate and maintained for 5 days. Cells were passaged every 5 days with trypsinization and re-plated at the same density (3× 104 cell/well) for the cumulative population doubling level (cPDL). During continuous passages, the numbers of ADMSCs at both the seeding and harvesting times were determined to calculate the cPDL. We calculated cPDL using the formula: cPDL = log (nf/n0)/log2 + X, where n0 is the initial number of cells at the beginning of the subculture, and nf the final number of cells at the end of that subculture. X is the doubling level of the cells at the initiation of the subculture.
Cell apoptosis assay
Annexin-V staining was applied to evaluate cell apoptosis. The cells were stained with PE Annexin-V and 7-Amino-Actinomycin (7-AAD) following the manufacturer’s instructions (BD Pharmingen, Franklin Lakes). The percentage of live (PE Annexin-V-/7-AAD-), apoptotic (PE Annexin-V+/7-AAD-), and necrotic (PE Annexin-V+/7-AAD+) cells was analyzed with flow cytometry (BD Accuri C6).
Cell cycle assays
Propidium iodide (PI; BD Pharmingen, Franklin Lakes) staining was applied to determine cell cycle. Cells were fixed in 75% ethanol overnight at 4°C, then stained with PI (50 μg/ml) containing RNase A (16 μg/ml), and incubated for 45 min at 37°C. For each sample, at least 30,000 events were collected and examined by flow cytometry (BD Accuri C6).
Adipogenic differentiation of ADMSCs
The potential of cultured ADMSCs to differentiate into adipocytes was determined. Cells were cultured in 35-mm dishes until 100% confluent, and then, the differentiation was induced by completed StemXVivo adipogenic differentiation media (1:100 dilution of StemXVivo® Adipogenic Supplement in α-MEM; R&D, Cat. #: CCM011). Oil Red O (Sigma, Cat. #: O0625) staining was used to detect intracellular lipid droplets after 4-, 8-, and 12-day induction.
Cell senescence
For detecting senescence, ADMSCs were seeded in 6-well plates at the concentration of 5 × 103 cells/well. At days 5, 7, 9, and 11, cells were stained by SA-β-gal. To quantitate the SA-β-gal positive cells, after staining with DAPI, 4 random vision fields were selected under microscope (×40) for each well and the percentage of positive cells was calculated.
Immunocytochemistry
Cells were stained with 4% paraformaldehyde in PBS (pH 7.5) at room temperature for 15 min. Then, cells were permeabilized with 0.3% Triton X-100 in PBS for 5 min at room temperature. After blocking with 5% BSA for 1 h, cells were incubated with primary antibody against Ki67 (1:100; Novus Biologicals, Cat. #: NB600-1209) at 4°C overnight. After washing, cells were reacted with chicken anti-rabbit IgG (H+L) secondary antibodies (FITC; Novex life technologies, Invitrogen, Cat. #: A15988). Nuclei were stained with NucBlue™ Live Cell Stain (life technologies, Invitrogen, Cat. #: R37605). Images were captured using a Zeiss LSM800 scanning confocal microscope (Carl Zeiss, Jena, Germany). The total numbers of cells and Ki67-positive cells were counted and the percentage of Ki67-positive cells was calculated.
RNA extraction and qRT-PCR
Total RNA was isolated by TRIzol Reagent (Sigma, Cat. #: T9424). Complementary DNA was synthesized using the All-in-One cDNA Synthesis SuperMix (Biotool, Cat. #: B24403). One micrograms of total RNA was reverse transcribed in 10μl reaction following the manufacturer’s instructions. qRT-PCR was performed using the SYBR Green One Step qPCR Kit (Biotool, Cat. #: B21202) on the ABI 7500 Real-Time PCR machine (Applied Biosystems). Primers used for qPT-PCR are listed in Table 1.
Table 1.
Primer sequences used for qRT-PCR
| Gene | Forward primer sequence (5′-3′) | Reverse primer sequence (5′-3′) |
|---|---|---|
| Adiponection | TATCGCTCAGCGTTCAGTGT | AGAGTCCCGGAATGTTGCAG |
| Fabp4 | CGATGAAATCACCGCAGACG | CCAGCTTGTCACCATCTCGT |
| Gapdh | ATGTGTCCGTCGTGGATCTGAC | AGACAACCTGGTCCTCAGTGTAG |
| Igf1 | TGCTCTTCAGTTCGTGTG | ACATCTCCAGTCTCCTCAG |
| IL1a | TGCAGTCCATAACCCATGATC | ACAAACTTCTGCCTGACGAG |
| IL6 | CAAAGCCAGAGTCCTTCAGAGAG | GCCACTCCTTCTGTGACTCC |
| mTOR | TGCTTGTGGTTGGCATAACA | TTCAGAGCCACAAACAGAGC |
| Nrip1 | AGCAGGACAAGAGTCACAGAAAC | TGTGATGATTGGCAGTATCTACG |
| p16 | GTGTGCATGACGTGCGGG | GCAGTTCGAATCTGCACCGTAG |
| p21 | AACATCTCAGGGCCGAAA | TGCGCTTGGAGTGATAGAAA |
| p53 | CTAGCATTCAGGCCCTCATCC | TCCGACTGTGACTCCTCCAT |
| p65 | CTTGGCAACAGCACAGACC | GAGAAGTCCATGTCCGCAAT |
| Pparγ | GTGAGACCAACAGCCTGAC | TATCAGTGGTTCACCGCTTC |
ELISA
The level of IL-6 in culture medium was evaluated using ELISA kit (Thermo Scientific, Cat. #: 88-7064-22), according to the protocol provided by the manufacturer.
Protein extraction and western blot
Skin tissues were homogenized in ice-cold homogenization buffer (50mM Tris HCL pH 8.0, 0.5mM DTT, 0.1% NP-40) in the presence of protease/phosphatase inhibitor using a handheld pestle. After homogenization, tissues were lysed in RIPA buffer (20mM Tris HCL pH 8.0, 300 mM NaCl, 1 mM DTT, 2% NP-40, 1% sodium deoxycholate, 0.2% SDS) containing Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific, Cat. #: 78440) for 1 h on ice. Protein was obtained after centrifugation for 20 min at 12,000×g at 4°C. Protein concentration was measured by Pierce BCA Concentration Test Kit (Thermo Scientific, Cat. #: 23225). Protein samples were heat-denatured in Laemmli Loading Buffer (Alfa Aesar, J60660-AC) with 1% β-ME. 20–40 μg protein was separated on sodium dodecylsulfate-polyacrylamide gel (SDS-PAGE) and transferred to a 0.22-μm polyvinylidene difluoride membrane (NEN LifeScience Products). Non-specific binding sites were blocked with 5% BSA in TBST. After blocking, the membranes were incubated overnight at 4°C with primary antibody against NFκB (1:200, Santa Cruz, Cat. #: sc-8008), p-NFκB (1:500, Santa Cruz, Cat. #: sc-33039), and GAPDH (1:2000, CST, Cat. #: 2118S). Immune complexes were detected with RDye 800CW goat anti-rabbit IgG (LiCor, Cat. #: C30521-01) and the intensity of protein bands was quantified using an Odyssey Infrared Imaging System (Li-COR).
Statistical analysis
All data are shown as means ± standard errors (SE). Data were analyzed by two-tailed Student’s t test. P<0.05 was considered as a statistically significant difference.
Results
Skin aging phenotypes in young and old B6 mice
To verify skin aging phenotypes in B6 mice, dorsal skin sections of young (6-month-old, n=4) and old (20-month-old, n=4) female B6 mice were subjected to histopathological analysis. The thickness of dermis and subcutaneous white adipose tissue (sWAT) from old mice was reduced significantly compared to young mice (168.50±13.89μm vs. 229.56±14.80μm, P=0.040, 257.55±54.43μm vs. 436.42±31.59μm, P=0.046, Fig. 1a, b). Moreover, the diameter of subcutaneous adipocyte from old mice was significantly reduced compared to young mice (33.06±1.12μm vs. 43.09±1.55μm, P=0.006, Fig. 1c). Importantly, the number of senescent cells, detected by SA-β-gal staining, in sWAT of old mice was over five times more than that of young mice (71.51±1.71% vs. 14.65±1.29%, P<0.001, Fig. 1a, d)
Fig. 1.

Skin aging phenotypes in young and old B6 mice. a Dermal and subcutaneous white adipose tissue (sWAT) thickness decreases and senescence increases with age (upper scale bar 100μm, down scale bar 50μm). b Quantification of the dermal and sWAT thickness in young and old mice. c Quantification of the subcutaneous adipocyte diameter in young and old mice. d The percentage of senescent cells was calculated by SA-β-gal staining. *P<0.05, **P<0.01
Depletion of Nrip1 alters skin aging phenotypes
To investigate the role of NRIP1 in skin aging, we collected skin tissues from female Nrip1+/+ (n=3) and Nrip1-/- (n=4) mice at the age of 20 months. We measured the thickness of dermis and sWAT, as well as the percentage of SA-β-gal positive adipocytes. The results showed that the thickness of dermis and sWAT is significantly increased in Nrip1-/- mice compared with Nrip1+/+ mice (214.68.50±15.38μm vs. 152.82±5.46μm, P=0.036, 427.72±22.27μm vs. 230.30±9.34μm, P=0.0028, Fig. 2a, b). The diameter of subcutaneous adipocyte from Nrip1-/- mice and Nrip1+/+ mice showed no difference (37.50±0.40μm vs. 39.24±2.57μm, P=0.66, Fig. 2c). The percentage of SA-β-gal positive cells was significantly decreased in Nrip1-/- mice (41.86±6.04% vs. 71.26±2.38%, P=0.02, Fig. 2a, d).
Fig. 2.

Skin aging phenotypes in Nrip1+/+ and Nrip1-/- mice. a Dermal and sWAT thickness decreases and senescence increases in Nrip1-/- mice (upper scale bar 100μm, down scale bar 50μm). b Quantification of the dermal and sWAT thickness in Nrip1+/+ and Nrip1-/- mice. c Quantification of the subcutaneous adipocyte diameter in Nrip1+/+ and Nrip1-/- mice. d The percentage of senescent cells was calculated by SA-β-gal staining. *P<0.05, **P<0.01
Phenotypic characterization of ADMSCs
As previously described, we isolated ADMSCs from inguinal adipose tissue in B6 mice. The cells were cultured to passage 3 and exhibited a fibroblast-like morphology when reaching full confluence (Fig. 3a). After maintaining in the complete StemXVivo adipogenic differentiation media for 14 days, cells differentiated into adipocytes with lipid vesicle (Fig. 3b). Oil Red O staining also confirmed the adipogenic differentiation of ADMSCs (Fig. 3c). The immunophenotypic characteristics of ADMSCs were analyzed by flow cytometry. The FACS results showed that ADMSCs were positive for CD29 and CD44 and negative for CD31 (Fig. 3d).
Fig. 3.

Phenotypic characterization of ADMSCs. a Morphological feature of ADMSCs at full confluence (scale bar 200μm). b Adipogenic differentiation of ADMSCs after 14 days (scale bar 200μm). c Adipogenesis was confirmed in ADMSCs by Oil Red O staining (scale bar 100μm). d Detection of MSC-specific marker (CD29+, CD44+, and CD31-) of ADMSCs by flow cytometric analysis
Depletion of Nrip1 reduces ADMSCs senescence
After isolating ADMSCs from sWAT of Nrip1+/+ and Nrip1-/- mice at young age (6 months), cell senescence was performed to evaluate the senescent property. Cell senescence data showed that the number of SA-β-gal positive cells accumulated with culture time in both Nrip1+/+ and Nrip1-/- ADMSCs. Importantly, the percentage of SA-β-gal positive cells from Nrip1-/- ADMSCs was significantly lower than that from Nrip1+/+ ADMSCs in day 7, day 9, and day 11 (P=0.028, P=0.0005, and P=0.009, Fig. 4a, b). To further confirm the role of NRIP1 in regulating senescence in ADMSCs and explore a potential therapeutic method to delay stem cell senescence, ADMSCs isolated from B6 mice were treated with either siRNA targeting Nrip1 (siNrip1) or siRNA scramble control (siCon) on day 4 post-isolation. SA-β-gal staining was performed at 72 h after treatment. The data showed that siNrip1-treated group had a significantly lower percentage of senescent cells compared to the siCon group (15.3% vs. 23.7%, P=0.034, Fig. 4c, d).
Fig. 4.

Depletion of Nrip1 reduces ADMSCs senescence. a Cell senescence was examined in ADMSCs isolated from Nrip1+/+ and Nrip1-/- mice at days 5, 7, 9, and 11 (scale bar 100μm). b Quantification of SA-β-gal positive cells in Nrip1+/+ and Nrip1-/- ADMSCs. c Quantification of SA-β-gal positive cells in siCon and siNrip1 ADMSCs isolated from B6 mice at 72 h after treatment. d Cell senescence was examined by SA-β-gal staining in siCon and siNrip1 ADMSCs at 72 h after treatment. *P<0.05, **P<0.01
Depletion of Nrip1 alters cell proliferation and cell apoptosis of AMDSCs
To verify whether depletion of Nrip1 alters cell proliferation, cell apoptosis, and colony formation of ADMSCs, we performed MTT assay and Annexin-V staining. As the MTT assay showed, the proliferation rate of Nrip1-/- ADMSCs was significantly lower than that of Nrip1+/+ ADMSCs at day 2 and day 4 after being seeded (P=0.029 and P= 0.012, Fig. 5a). The Annexin-V staining results showed that the percentage of apoptotic cells decreased significantly in Nrip1-/- ADMSCs compared with Nrip1+/+ ADMSCs at 24, 48, and 72 h after the cells being seeded (P<0.001, Fig. 5b). Taken together, these data indicated that depletion of Nrip1 could decrease cell proliferation and prevent cell apoptosis.
Fig. 5.

Depletion of Nrip1 alters cell proliferation and cell apoptosis of ADMSCs. a MTT assay showed the proliferation rate decreased significantly in Nrip1-/- ADMSCs compared to Nrip1+/+ ADMSCs at day 2 and day 4 after being seeded. b Annexin V showed that depletion of Nrip1 inhibited the apoptosis of AMDSCs at 24, 48, and 72 h after being seeded. *P<0.05, **P<0.01
Depletion of Nrip1 delay adipogenic differentiation of ADMSCs
To assess the adipogenic differentiation ability of ADMSCs from Nrip1-/- and Nrip1+/+ mice, differentiation was induced by culturing with adipogenic differentiation media. Lipid dye Oil Red O was performed to confirm adipogenesis. Starting from day 4, lipid droplets could be observed under a microscope and gradually increased with culture time (Fig. 6a). The result of semi-quantitative detection showed that the absorbance values in Nrip1-/- ADMSCs were significantly lower compared to Nrip1+/+ ADMSCs at days 8 and 12 (P=0.013 and P=0.001, Fig. 6b). To quantify the adipogenic differentiation of ADMSCs, genes related to adipogenic differentiation (Fabp4, Pparγ, and Adiponectin) were examined at days 4, 8, and 12 after induction (Fig. 6c). Consistent with Oil Red O staining, the expressions of Fabp4, Pparγ, and Adiponectin were significantly lower in Nrip1-/- ADMSCs compared to Nrip1+/+ ADMSCs at days 8 and 12. These results suggested that Nrip1-/- ADMSCs exhibited lower response to induction factors, indicating that depletion of Nrip1 could suppress the differentiation of ADMSCs.
Fig. 6.

Adipogenic differentiation in Nrip1+/+ and Nrip1-/- ADMSCs. a Adipogenesis was detected by Oil Red O staining in Nrip1+/+ and Nrip1-/- ADMSCs at 4, 8, and 12 days after induction. b Semi-quantitative detection of adipogenesis showed that the absorbance values in Nrip1-/- ADMSCs were significantly lower compared to Nrip1+/+ ADMSCs at days 8 and 12. c Genes related to adipogenic differentiation (Fabp4, Pparγ, and Adiponectin) were analyzed by qRT-PCR. *P<0.05, **P<0.01
Depletion of Nrip1 enhances cellular quiescence of ADMSCs
PI staining and flow cytometry assay showed that the percentage of G0/G1 cells was significantly increased in Nrip1-/- ADMSCs (P<0.01, Fig. 7a, b). Moreover, compared to Nrip1+/+ ADMSCs, the percentages of both S and G2/M cells were significantly reduced in Nrip1-/- ADMSCs (P<0.05, Fig. 7a, b). To further identify the quiescent cell population, immunocytochemistry was performed to stain cells with Ki67. Cells without Ki67 staining were identified as quiescent cells (Fig. 7c). As expected, the percentage of Ki67-positive cells in Nrip1-/- ADMSCs was significantly lower compared to Nrip1+/+ ADMSCs, indicating that depletion of NRIP1 sustains quiescence of ADMSC (P<0.001, Fig. 7d).
Fig. 7.

Depletion of Nrip1 enhances cellular quiescence of ADMSCs. a, b Cell cycle distribution of ADMSCs in Nrip1+/+ and Nrip1-/- ADMSCs. c Ki67-positive cells were presented by immunocytochemistry in Nrip1+/+ and Nrip1-/- ADMSCs. d Quantification of Ki67-positive cells in Nrip1+/+ and Nrip1-/- ADMSCs. *P<0.05, **P<0.01
Cell phenotypes of ADMSCs from middle age Nrip1+/+ and Nrip1-/- mice
The cPDL showed that the proliferation rate of Nrip1-/- ADMSCs at middle age was significantly higher than that of Nrip1+/+ ADMSCs (Fig. 8a). The expression of p21 and p53 was significantly lower in Nrip1-/- ADMSCs compared to Nrip1+/+ ADMSCs at passage 3 (P<0.05, Fig. 8b). ELISA result showed that the level of IL-6 was also significantly lower in Nrip1-/- ADMSCs compared to Nrip1+/+ ADMSCs at passage 3 (P<0.05, Fig. 8c). These cell phenotypes results suggested that Nrip1+/+ ADMSCs at middle age began senescing earlier than Nrip1-/- ADMSCs.
Fig. 8.
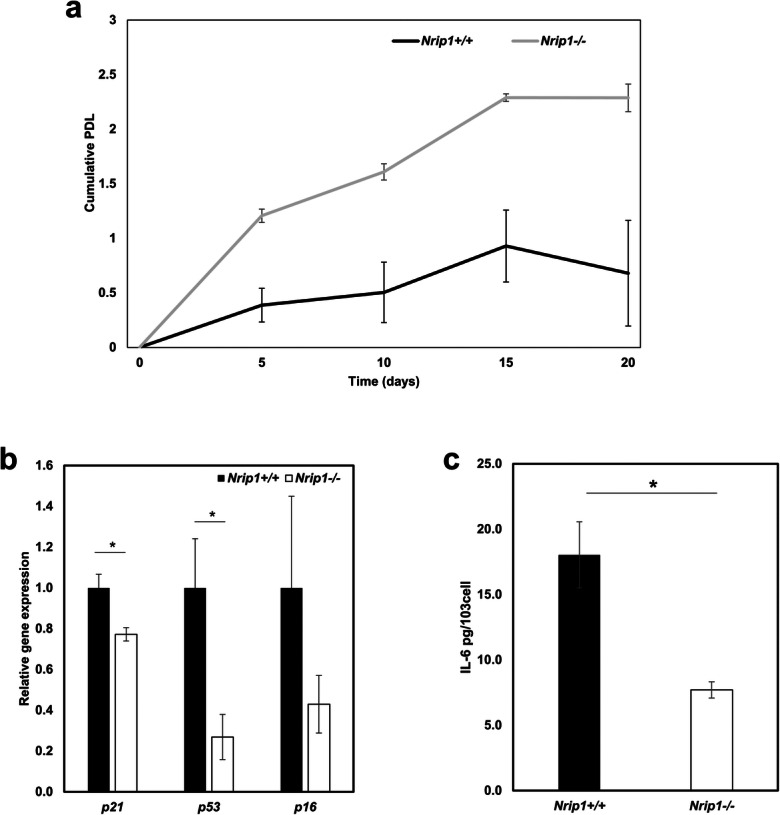
Cell phenotypes of ADMSCs from middle age Nrip1+/+ and Nrip1-/- mice. a cPDL of Nrip1+/+ and Nrip1-/- ADMSCs passaged every 5 days. b qRT-PCR examined the expression of p16, p21, and p53 in Nrip1+/+ and Nrip1-/- ADMSCs at passage 3. c The level of IL-6 in Nrip1+/+ and Nrip1-/- ADMSCs at passage 3. *P<0.05
Depletion of Nrip1 alters expression of senescence- and inflammation-associated genes
To investigate the molecular role of NRIP1 in cell senescence, we tested the expression of genes associated with senescence (p16, p21, and p53), growth factors (mTOR and Igf1), and senescence-associated secretory phenotype (SASP) (IL6 and IL1a). First, the efficiency of knocking out and knocking down Nrip1 was confirmed by testing the expression of Nrip1 (P<0.05, Fig. 9a, b). The qRT-PCR results showed a 95% depletion of Nrip1 expression in Nrip1-/- ADMSCs (1.00±0.28 vs. 0.05±0.03, Nrip1+/+ vs. Nrip1-/-, P=0.015). Comparing to siCon, siNrip1 suppressed 60% of Nrip1 expression in ADMSCs (1.00±0.09 vs. 0.40±0.04, siCon vs. siNrip1, P=0.026). The expression of senescence-associated genes (p21 and p53) and SASP (IL6 and IL1a) decreased in both Nrip1-/- and siNrip1 ADMSCs compared to Nrip1+/+ and siCon ADMSCs. However, the expression of p16 increased in Nrip1-/- ADMSCs compared to Nrip1+/+ ADMSCs. Moreover, gene expression results showed that Igf1, mTOR, and p65 were all significantly reduced in Nrip1-/- and siNrip1 ADMSCs (Fig. 9a, b). Consistently, the western blot results showed that at protein level, mTOR, p65, and p-p65 were significantly lower in siNrip1 ADMSCs than in siCon ADMSCs, while p-mTOR showed suggestively lower in siNRIP1 ADMSCs (P=0.08, Fig. 9c, d).
Fig. 9.

Depletion of Nrip1 alters expression of senescence- and inflammation-associated genes. a qRT-PCR verified the deletion of Nrip1 and examined the expression of senescence-associated genes (p16, p21, and p53), growth factors (mTOR and Igf1), and inflammation-related genes (p65, IL6, and IL1a) in Nrip1+/+ and Nrip1-/- ADMSCs. b qRT-PCR verified the deletion of Nrip1 and examined the expression of senescence-associated genes (p16, p21, and p53), growth factors (mTOR and Igf1), and inflammation-related genes (p65, IL6, and IL1a) in siCon and siNrip1 ADMSCs isolated from B6 mice. c, d Western blot quantification of mTOR, p-mTOR, p65, and p-p65 in siCon and siNrip1 ADMSCs isolated from B6 mice. *P<0.05, **P<0.01
Depletion of Nrip1 reduces p-p65 level in skin
In order to confirm whether depletion of Nrip1 would regulate the expression of p65 in skin, expressions of Nrip1, p65, mTOR, and Igf1 in mRNA levels were measured by qRT-PCR in the skin of Nrip1-/- and Nrip1+/+ mice. The reduced expression of Nrip1 confirmed the successful knockout of Nrip1 in skin. Consistent with the results of ADMSCs, expressions of mTOR and Igf1 were significantly reduced in the skin of Nrip1-/- mice (P<0.001 and P<0.032, Fig. 10a). The results showed that p65 expression in the skin was suggestively reduced in Nrip1-/- mice compared to Nrip1+/+ mice (P=0.12, Fig. 10a). In western blot results, p-p65 was significantly reduced in the skin of Nrip1-/- mice (P=0.048, Fig. 10b), while p65 was suggestively reduced. We also performed immunohistochemistry to verify the level of p-p65 in young (6-month-old) and old (20-month-old) B6 mice, as well as Nrip1+/+ and Nrip1-/- mice at old age (20-month-old). Increased p-p65 levels were seen in the sWAT at old age (Fig. 10c). Moreover, p-p65 levels were reduced in Nrip1-/- mice compared with Nrip1+/+ mice (Fig. 10c).
Fig. 10.

Depletion of Nrip1 reduces p-p65 level in skin. a qRT-PCR verified the deletion of Nrip1 and examined the expression of p65, mTOR, and Igf1 in skin of Nrip1+/+ and Nrip1-/- mice. b Western blot quantification of p65 and p-p65 in skin of Nrip1-/- and Nrip1+/+ mice. c Immunohistochemical detection of p-p65 expression in young and old B6 mice, as well as in Nrip1-/- and Nrip1+/+ mice at old age (upper scale bar 100μm, down scale bar 50μm). *P<0.05, **P<0.01
Discussion
NRIP1 acts as a co-regulator by interacting with other nuclear receptors and transcription factors [2]. Using bioinformatics and genetic strategies, a previous study suggested that NRIP1 might be a strong candidate aging-related gene [7]. It is reported that NRIP1 might be involved in the aging process by suppressing mitochondrial biogenesis via regulating peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1 α) [25]. Moreover, the expression of NRIP1 in liver, kidney, and adipose tissue showed age-specific changes [10]. Most importantly, our group found deletion of Nrip1 extended longevity in female mice and delayed senescence of mouse embryonic fibroblasts (MEFs), indicating that NRIP1 plays an important role in aging [40].
To our knowledge, this is the first study to evaluate the role of NRIP1 in skin aging. Previous studies in mice found some common histologic characteristics of skin aging that is also found in the human, including atrophy of dermis, loss of subcutaneous fat, graying of hair, and alopecia [12, 18]. In our study, we verified that the thickness of dermis and sWAT and the diameter of subcutaneous adipocyte in old B6 mice are significantly reduced compared to young B6 mice, which is in accordance with other previous studies [12, 35, 41]. Also, the current study found that the deletion of Nrip1 could significantly improve some skin aging phenotypes, including the increased thickness of dermis and sWAT, as well as reduced accumulation of senescent adipocytes. Nevertheless, the diameter of subcutaneous adipocyte showed no difference in old Nrip1-/- and Nrip1+/+ mice, which suggests that the increased thickness of sWAT in Nrip1-/- results from the increased number of cells. This is consistent with the reduced senescence and apoptosis detected in vitro and in vivo. Interestingly, we found that the diameter data of old B6 mice and old Nrip1+/+ mice showed significant difference, which emphasizes the importance of using littermate controls in our study. Importantly, the aging-related inflammatory cytokines p65, IL1a, and IL6 are also significantly reduced. These results indicate that suppression of Nrip1 may delay the skin aging in female mice.
To further investigate the role of NRIP1 in skin aging by regulating cellular senescence, we performed an in vitro study in ADMSCs isolated from sWAT. It has been reported that mouse ADMSCs could positively expressed CD29, CD44, and CD90 and did not express CD31, CD34, and CD45. Our FACS results showed that ADMSCs were positive for CD29 and CD44 and negative for CD31, which helped to verify the ADMSCs. Due to the differentiation potential, paracrine, and immunoregulation effects, ADMSCs have been reported to be utilized in dermatological applications such as facial atrophy, scars, wound healing, and alopecia [9]. Recent studies also revealed that injection of ADMSCs can prevent skin aging phenotype by stimulating collagen synthesis and increasing angiogenesis [19]. In our study, ADMSCs from Nrip1-/- mice showed reduced senescence in cell senescence assay compared to ADMSCs from Nrip1+/+ mice. Likewise, ADMSCs from B6 mice treated by siNrip1 also showed fewer senescent cells compared to siCon-treated ADMSCs. Cellular senescence is defined as a state of stable cell cycle arrest due to diverse stresses, which is characterized by several molecular markers, including CDKIs (p16, p21) and p53 [21, 33]. It is well-known that senescence relies on two main molecular pathways: p53 and p16 pathways. As a tetrameric transcription factor, p53 is activated by several posttranslational modifications. Active p53 regulates the downstream pro-senescence target p21, which is responsible for G1 cell cycle arrest [32]. Once activated, the cyclin-dependent kinase (CDK) inhibitor p21 suppresses the activity of CDK2 and results in hypo-phosphorylated retinoblastoma protein (pRB), contributing to the onset of cellular senescence [14]. Furthermore, it has been reported that p16-positive cells accumulate during aging in multiple tissues [39]. The p16-mediated senescence also relies on controlling the pRB and leads to G1 cell cycle arrest [30]. Activated p16 prevents CDK4/6 from binding cyclin D and phosphorylating pRB. Otherwise, the phosphorylated pRB dissociates from the transcription factor E2F1, which allows E2F1 to enter the nucleus and to promote the transcription of target genes [23]. Another important phenotype of senescent cells is SASP, including IL1a and IL6. In the process of skin aging, senescent preadipocytes accumulate in adipose tissue and secrete high levels of inflammatory cytokines [36]. Consistent with these findings, our results showed that the expressions of senescence-associated genes (p21 and p53) and SASP (IL1a and IL6) are decreased in both Nrip1-/- and siNrip1 ADMSCs compared to Nrip1+/+ and siCon ADMSCs. Taken together, our results suggest that the deletion of Nrip1 suppresses senescence not only in vivo in sWAT but also in vitro in ADMSCs.
Surprisingly, the expression of p16 increased in Nrip1-/- ADMSCs compared to Nrip1+/+ ADMSCs in the current study. Although p16 is a key gene associated with senescence, it is reported that p16 is also involved in regulating cell quiescence and apoptosis in mammalian cells. It is suggested that the level of p16 is elevated in quiescent smooth muscle cell in rat aorta [16]. Likewise, p16 prevents dentate gyrus stem cells from being activated and contributes to the maintenance of cell quiescence by reserving the self-renewal capacity [28]. RB-E2F signaling plays a vital role in stem cell quiescence as E2F is suppressed by the binding of RB in quiescent cells. Moreover, Rb has been shown to have a specialized role in regulating cell cycle exit [4]. Recent studies have raised a model controlling the conversion of quiescence and proliferation which is called “Rb-E2F bistable switch.” The “E2F off” (quiescence) state means that cells remain in the reversible G0 state when E2F is suppressed. The “E2F on” (proliferation) state means that cells re-enter the G1 of cell cycle when E2F is activated [42]. As we mentioned before, the binding of p16 with CDK4/6 could result in the reduced levels of E2F1 released from the Rb [30]. Together, these studies suggest that p16 functions as an inhibition factor of E2F1 and leads to the “E2F off” state which cells remain quiescent. Additionally, P16 may also act as an inhibitor of DNA damage-induced apoptosis and can protect cells from UV-dependent apoptosis by regulating the expression of Bcl-2 [1]. Therefore, in our study, inhibited apoptosis in ADMSCs by NRIP1 deletion may also be related to the elevated expression of p16. However, the elevated P16 needs to be further verified at the protein level and its effects on stromal cell quiescence and apoptosis needs to be further investigated.
Although ADMSCs are considered to have strong self-renewal and cell proliferation capability, several studies have shown that the function of ADMSCs decreases with aging. The cell morphology, proliferation capability, and differentiation potential of ADMSCs from old donors are significantly different from ADMSCs from young donors, which may limit the therapeutic potential of ADMSCs [6]. Therefore, improving the function of ADMSCs by genetic treatment may provide a new approach for better clinical outcomes. To investigate the effects of NRIP1 on the cellular function of ADMSCs, we tested cell proliferation, apoptosis, and differentiation. Our results showed that ADMSCs from Nrip1-/- mice have impaired proliferation and less apoptosis compared to ADMSCs from Nrip1+/+ mice. Our previous data found that suppressing NRIP1 inhibited cell growth and induced apoptosis in breast cancer cell lines [3]. Indeed, we used the primary ADMSCs as the target cell type in our current study, which have the unique cell quiescent state compared to the cancer cell lines. Interestingly, the quiescent stem cells show decreased differentiation and apoptosis [38]. Regarding the adipogenic differentiation, the lipid droplet accumulation measured by Oil Red assay revealed a significant decrease in Nrip1-/- ADMSCs compared to Nrip1+/+ ADMSCs. Furthermore, the same trend was observed in the quantitative analysis of molecular markers of adipogenic differentiation (Fabp4, Ppparγ, and Adiponectin). Apart from the vital role in senescence, p53 also takes part in the maintenance of stem cells by acting as a differentiation regulator. Studies have shown that activation of p53 can lead to the rapid differentiation of human embryonic stem cells, and knocking out p53 can delay the differentiation [17, 27]. Consistent with these findings, our results indicate that suppressing Nrip1 can decrease the expression of p53 and inhibit adipogenic differentiation. Taken together, these assays show that suppression of Nrip1 leads to reduced proliferation, decreased apoptosis, and adipogenic differentiation.
To understand the reason why depletion of Nrip1 can reduce cell senescence, decrease cell proliferation, prevent cell apoptosis, and decrease adipogenic differentiation potential, we hypothesize cell cycle and quiescence of ADMSC as possible underlying mechanisms. As expected, our results showed that the quiescent cells in ADMSCs from Nrip1-/- mice were significantly increased compared to ADMSCs from Nrip1+/+ mice. Cellular quiescence is the reversible state in which cells exist in the G0 stage of cell cycle and stay as dormant with low-activity [38]. For mammalian stem cells, quiescence can preserve self-renewal, proliferation, and differentiation, which is critical for survival and long-term reconstituting capacity [22]. Based on the unique characteristics of stem cell quiescence, two innovative therapeutic strategies have been established which could provide promising clinical outcomes in the near future. The first is called “lock-out” strategy, which consists of pushing stem cells out of quiescence to induce proliferation and differentiation. The other one is called “lock-in” strategy, which consists of maintaining the quiescent state to prevent aberrant proliferation and differentiation or premature senescence [5]. According to these findings and our results, we believe that depletion of Nrip1 could lead to ADMSCs maintaining a quiescent cell cycle state to preserve their long-term function and self-renewal capacity.
To further confirm our hypothesis that ADMSCs could preserve their long-term function by maintaining cell quiescence, we investigated the cell phenotypes of ADMSCs from middle age Nrip1+/+ and Nrip1-/- mice. We noticed an interesting phenomenon that the proliferation rate of Nrip1-/- ADMSCs at middle age was higher than that of Nrip1+/+ ADMSCs based on the cPDL result. The expression of cyclin-dependent kinase (p21 and p53) was significantly lower in Nrip1-/- ADMSCs compared to Nrip1+/+ ADMSCs. The level of SASP marker (IL-6) was also significantly lower in Nrip1-/- ADMSCs compared to Nrip1+/+ ADMSCs. According to these results, we found that Nrip1+/+ ADMSCs at middle age began senescing earlier than Nrip1-/- ADMSCs, which suggests that as early as middle age the Nrip1-/- ADMSCs may function better than Nrip1+/+ ADMSCs. Combined with our previous findings, it is reasonable to conclude that deletion of Nrip1 could maintain ADMSCs quiescence and preserve the stem cell ability at young age, thus reducing ADMSCs senescence during aging.
Although the underlying mechanism of how NRIP1 regulates ADMSCs quiescence is unclear, our current results showed that the expression of mTOR is significantly decreased not only in the ADMSCs from Nrip1-/- mice but also in siNrip1-treated ADMSCs. The accumulating data have shown a strong association between mTOR and stem cell quiescence. It has been reported that mTOR activity is necessary for the transition of stem cells from G0 into G(Alert), where G(Alert) stem cells have higher priority to enter cell cycle [31]. Various genetic studies have demonstrated that deletion of several mTOR-negative regulators, including TSC1, PTEN, PML, and Fbxw7, could result in hyper-proliferation and subsequent exhaustion, and defective repopulating potential, which means these mTOR-negative regulators could maintain stem cell quiescence [8]. Furthermore, mTOR signaling has been reported to possess a deleterious role on stem cell maintenance during repeated regeneration cycles [11]. It has been proposed that pharmacological inhibition of mTOR by rapamycin and metformin is sufficient for stem cells to maintain a quiescent state [29]. Based on these findings and our current data, it is suggested that suppressing NRIP1 could maintain ADMSC quiescence by inhibiting mTOR.
As one of the key mediators of aging, increased NFκB activity during skin aging has been observed in several studies [13, 15, 37]. In accordance with this, we found the level of p65 was increased in the sWAT at old age of B6 mice. More importantly, our data firstly demonstrated that the deletion of Nrip1 could reduce the expression of p65 and p-p65 in both skin and ADMSCs. Our previous studies also reported that knocking down Nrip1 can significantly reduce the expression of NFκB in both human keratinocytes and periovarian WAT [26, 40]. Although the underlying mechanism of how NRIP1 regulates NFκB/p65 is unclear, our current results showed that the expression of Igf1 is significantly decreased not only in the skin of Nrip1-/- mice but also in Nrip1-/- and siNrip1 ADMSCs. It has been proposed that IGF1 can activate the downstream PI-3K and Akt, which in turn enhance the NFκB signaling via the IKK complex [34, 37]. In the canonical NFκB signaling pathway, the dimer of p65 and p50 is in an inactive state by the inhibition of IκB in cytoplasm. Upon stimulation, IKKs phosphorylate IκB, which results in their degradation and enables NFκB translocate to the nucleus to induce target gene expression [24]. Given the fact that depletion of NRIP1 leads to the decreased expression of IGF1 and NFκB and reduced level of p65, it is reasonable to conclude that NRIP1 may regulate skin aging through the IGF1/ NFκB pathway.
In conclusion, our results revealed that NRIP1 plays a significant role in skin aging. Deletion of Nrip1 can delay skin aging phenotypes, reduce ADMSCs senescence, and maintain ADMSCs quiescence. This suggests that NRIP1 might be a potential therapeutic target to improve the function of ADMSCs and contribute to delayed skin aging.
Acknowledgments
Lisa Hensley kindly edited the manuscript. Division of Laboratory Animal Medicine of Southern Illinois University School of Medicine provides excellent environment for animal research.
Author contributions
RY conceived and designed the project; YH, YZ, SDG, and JMO performed the experiments; RY, YH, YZ, and SDG analyzed experimental results; YH and RY wrote the manuscript with approval from all authors; RY, HG, and MC revised the manuscript.
Funding
The work is funded by U.S. NIH grant AG046432, internal research seed grants of Southern Illinois University School of Medicine, team science grant of simmons cancer institute at Southern Illinois University School of Medicine, CAMS Innovation Fund for Medical Sciences (CIFMS-2017-I2M-1-017), and China Scholarship Council (No. 201806210434).
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Heng Gu, Email: guheng@pumcderm.cams.cn.
Rong Yuan, Email: ryuan@siumed.edu.
References
- 1.Al-Mohanna MA, Manogaran PS, Al-Mukhalafi Z, Al-Hussein KA, Aboussekhra A. The tumor suppressor p16(INK4a) gene is a regulator of apoptosis induced by ultraviolet light and cisplatin. Oncogene. 2004;23:201–212. doi: 10.1038/sj.onc.1206927. [DOI] [PubMed] [Google Scholar]
- 2.Augereau P, Badia E, Carascossa S, Castet A, Fritsch S, Harmand PO, Jalaguier S, Cavailles V. The nuclear receptor transcriptional coregulator RIP140. Nucl Recept Signal. 2006;4:e024. doi: 10.1621/nrs.04024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aziz MH, Chen X, Zhang Q, DeFrain C, Osland J, Luo Y, Shi X, Yuan R. Suppressing NRIP1 inhibits growth of breast cancer cells in vitro and in vivo. Oncotarget. 2015;6:39714–39724. doi: 10.18632/oncotarget.5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biggar KK, Storey KB. Perspectives in cell cycle regulation: lessons from an anoxic vertebrate. Curr Genomics. 2009;10:573–584. doi: 10.2174/138920209789503905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho IJ, Lui PP, Obajdin J, Riccio F, Stroukov W, Willis TL, Spagnoli F, Watt FM. Mechanisms, Hallmarks, and Implications of Stem Cell Quiescence. Stem Cell Rep. 2019;12:1190–1200. doi: 10.1016/j.stemcr.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fafian-Labora JA, Morente-Lopez M, Arufe MC. Effect of aging on behaviour of mesenchymal stem cells. World J Stem Cells. 2019;11:337–346. doi: 10.4252/wjsc.v11.i6.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flurkey K, Yuan R. How the evolutionary theory of aging can guide us in the search for aging genes. Aging (Albany NY) 2012;4:318–319. doi: 10.18632/aging.100460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gan B, DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle. 2009;8:1003–1006. doi: 10.4161/cc.8.7.8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaur M, Dobke M, Lunyak VV. Mesenchymal Stem Cells from Adipose Tissue in Clinical Applications for Dermatological Indications and Skin Aging. Int J Mol Sci. 2017;18. 10.3390/ijms18010208. [DOI] [PMC free article] [PubMed]
- 10.Ghosh S, Thakur MK. Tissue-specific expression of receptor-interacting protein in aging mouse. Age (Dordr) 2008;30:237–243. doi: 10.1007/s11357-008-9062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haller S, Kapuria S, Riley RR, O’Leary MN, Schreiber KH, Andersen JK, Melov S, Que J, Rando TA, Rock J, Kennedy BK, Rodgers JT, Jasper H. mTORC1 Activation during Repeated Regeneration Impairs Somatic Stem Cell Maintenance. Cell Stem Cell. 2017;21:806–818. doi: 10.1016/j.stem.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harkema L, Youssef SA, de Bruin A. Pathology of Mouse Models of Accelerated Aging. Vet Pathol. 2016;53:366–389. doi: 10.1177/0300985815625169. [DOI] [PubMed] [Google Scholar]
- 13.Haustead DJ, Stevenson A, Saxena V, Marriage F, Firth M, Silla R, Martin L, Adcroft KF, Rea S, Day PJ, Melton P, Wood FM, Fear MW. Transcriptome analysis of human ageing in male skin shows mid-life period of variability and central role of NF-kappaB. Sci Rep. 2016;6:26846. doi: 10.1038/srep26846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Invest. 2018;128:1238–1246. doi: 10.1172/JCI95148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Y, Zhu Y, Lian N, Chen M, Bartke A, Yuan R. Metabolic Syndrome and Skin Diseases. Front Endocrinol (Lausanne). 2019;10. 10.3389/fendo.2019.00788. [DOI] [PMC free article] [PubMed]
- 16.Izzard TD, Taylor C, Birkett SD, Jackson CL, Newby AC. Mechanisms underlying maintenance of smooth muscle cell quiescence in rat aorta: role of the cyclin dependent kinases and their inhibitors. Cardiovasc Res. 2002;53:242–252. doi: 10.1016/S0008-6363(01)00444-8. [DOI] [PubMed] [Google Scholar]
- 17.Jain AK, Allton K, Iacovino M, Mahen E, Milczarek RJ, Zwaka TP, Kyba M, Barton MC. p53 regulates cell cycle and microRNAs to promote differentiation of human embryonic stem cells. PLoS Biol. 2012;10:e1001268. doi: 10.1371/journal.pbio.1001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khavkin J, Ellis DA. Aging skin: histology, physiology, and pathology. Facial Plast Surg Clin North Am. 2011;19:229–234. doi: 10.1016/j.fsc.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Kim J-H, Jung M, Kim H-S, Kim Y-M, Choi E-H. Adipose-derived stem cells as a new therapeutic modality for ageing skin. 2011;20:383–7. 10.1111/j.1600-0625.2010.01221.x. [DOI] [PubMed]
- 20.Kohl E, Steinbauer J, Landthaler M, Szeimies RM. Skin ageing. J Eur Acad Dermatol Venereol. 2011;25:873–884. doi: 10.1111/j.1468-3083.2010.03963.x. [DOI] [PubMed] [Google Scholar]
- 21.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li L, Bhatia R. Stem cell quiescence. Clin Cancer Res. 2011;17:4936–4941. doi: 10.1158/1078-0432.CCR-10-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu JY, Souroullas GP, Diekman BO, Krishnamurthy J, Hall BM, Sorrentino JA, Parker JS, Sessions GA, Gudkov AV, Sharpless NE. Cells exhibiting strong p16 (INK4a) promoter activation in vivo display features of senescence. Proc Natl Acad Sci U S A. 2019;116:2603–2611. doi: 10.1073/pnas.1818313116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2. 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed]
- 25.Lopez-Lluch G, Irusta PM, Navas P, de Cabo R. Mitochondrial biogenesis and healthy aging. Exp Gerontol. 2008;43:813–819. doi: 10.1016/j.exger.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luan C, Chen X, Hu Y, Hao Z, Osland JM, Chen X, Gerber SD, Chen M, Gu H, Yuan R. Overexpression and potential roles of NRIP1 in psoriasis. Oncotarget. 2016;7:74236–74246. doi: 10.18632/oncotarget.12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maimets T, Neganova I, Armstrong L, Lako M. Activation of p53 by nutlin leads to rapid differentiation of human embryonic stem cells. Oncogene. 2008;27:5277–5287. doi: 10.1038/onc.2008.166. [DOI] [PubMed] [Google Scholar]
- 28.Micheli L, D’Andrea G, Ceccarelli M, Ferri A, Scardigli R, Tirone F. p16Ink4a Prevents the Activation of Aged Quiescent Dentate Gyrus Stem Cells by Physical Exercise. Front Cell Neurosci. 2019;13:10. doi: 10.3389/fncel.2019.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pavlidou T, Marinkovic M, Rosina M, Fuoco C, Vumbaca S, Gargioli C, Castagnoli L, Cesareni G. Metformin Delays Satellite Cell Activation and Maintains Quiescence. Stem Cells Int. 2019;2019:5980465–5980419. doi: 10.1155/2019/5980465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130:1715–1725. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, Brunson C, Mastey N, Liu L, Tsai CR, Goodell MA, Rando TA. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510:393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129–5143. doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- 33.Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. doi: 10.1101/gad.235184.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salminen A, Kaarniranta K. Insulin/IGF-1 paradox of aging: regulation via AKT/IKK/NF-kappaB signaling. Cell Signal. 2010;22:573–577. doi: 10.1016/j.cellsig.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 35.Schosserer M, Grillari J, Wolfrum C, Scheideler M. Age-Induced Changes in White, Brite, and Brown Adipose Depots: A Mini-Review. Gerontology. 2018;64:229–236. doi: 10.1159/000485183. [DOI] [PubMed] [Google Scholar]
- 36.Starr ME, Saito M, Evers BM, Saito H. Age-Associated Increase in Cytokine Production During Systemic Inflammation-II: The Role of IL-1beta in Age-Dependent IL-6 Upregulation in Adipose Tissue. J Gerontol A Biol Sci Med Sci. 2015;70:1508–1515. doi: 10.1093/gerona/glu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD. NF-kappaB in Aging and Disease. Aging Dis. 2011;2:449–465. [PMC free article] [PubMed] [Google Scholar]
- 38.van Velthoven CTJ, Rando TA. Stem Cell Quiescence: Dynamism, Restraint, and Cellular Idling. Cell Stem Cell. 2019;24:213–225. doi: 10.1016/j.stem.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang AS, Dreesen O. Biomarkers of Cellular Senescence and Skin Aging. Front Genet. 2018;9:247. doi: 10.3389/fgene.2018.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, Chen X, Osland J, Gerber SJ, Luan C, Delfino K, Goodwin L, Yuan R. Deletion of Nrip1 Extends Female Mice Longevity, Increases Autophagy, and Delays Cell Senescence. J Gerontol A Biol Sci Med Sci. 2018;73:882–892. doi: 10.1093/gerona/glx257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wollina U, Wetzker R, Abdel-Naser MB, Kruglikov IL. Role of adipose tissue in facial aging. Clin Interv Aging. 2017;12:2069–2076. doi: 10.2147/CIA.S151599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao G. Modelling mammalian cellular quiescence. Interface Focus. 2014;4:20130074. doi: 10.1098/rsfs.2013.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zouboulis CC, Makrantonaki E. Clinical aspects and molecular diagnostics of skin aging. Clin Dermatol. 2011;29:3–14. doi: 10.1016/j.clindermatol.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 44.Zschiedrich I, Hardeland U, Krones-Herzig A, Berriel Diaz M, Vegiopoulos A, Muggenburg J, Sombroek D, Hofmann TG, Zawatzky R, Yu X, Gretz N, Christian M, White R, Parker MG, Herzig S. Coactivator function of RIP140 for NFkappaB/RelA-dependent cytokine gene expression. Blood. 2008;112:264–276. doi: 10.1182/blood-2007-11-121699. [DOI] [PubMed] [Google Scholar]