

Abstract
Mitochondria are organelles that provide energy to cells through ATP production. Mitochondrial dysfunction has long been postulated to mediate cellular declines that drive biological aging. Many well-characterized hallmarks of aging may involve underlying energetic defects that stem from loss of mitochondrial function with age. Why and how mitochondrial function declines with age is an open question and one that has been difficult to answer. Mitochondria are powered by an electrochemical gradient across the inner mitochondrial membrane known as the protonmotive force (PMF). This gradient decreases with age in several experimental models. However, it is unclear if a diminished PMF is a cause or a consequence of aging. Herein, we briefly review and define mitochondrial function, we summarize how PMF changes with age in several models, and we highlight recent studies that implicate PMF in aging biology. We also identify barriers that must be addressed for the field to progress. Emerging technology permits more precise in vivo study of mitochondria that will allow better understanding of cause and effect in metabolic models of aging. Once cause and effect can be discerned more precisely, energetics approaches to combat aging may be developed to prevent or reverse functional decline.
Keywords: Membrane potential, Metabolism, AMPK, mTOR, Autophagy
Introduction
Mitochondria have long been thought to play a central role in biological aging. The mitochondrial free radical theory of aging, developed as a variation on Harman’s free radical theory of aging [1], posits that biological aging is driven largely by reactive oxygen species produced in mitochondria as a by-product of oxidative metabolism [2]. Although it now seems clear that the mitochondrial free radical theory of aging is not sufficient to explain many aspects of aging, there is growing consensus that mitochondria can also impact aging through multiple non-free radical mechanisms.
Mitochondrial dysfunction is recognized as one of the nine “hallmarks of aging” [3], and a case can be made that mitochondrial dysfunction may causally contribute to each of the other aging hallmarks [4, 5] (Fig. 1). Mitochondrial reactive oxygen species (ROS) produced as a by-product of oxidative metabolism likely represent an important source of damage within cells, both to mitochondria themselves and to other macromolecules and cellular components [6–8]. Damage to proteins arising from ROS likely contributes to loss of proteostasis and damage to nuclear DNA, which can give rise to genome instability, telomere dysfunction, and cell senescence [9–12]. Dysregulation of mitochondrial metabolism has been associated with declining stem cell function, epigenetic alterations, and dysregulated nutrient sensing during aging [13–15]. Mitochondria also play a central role in apoptotic cell death, which when perturbed can exacerbate many aging phenotypes [16]. More recently, mitochondrial DNA has been shown to induce a potent inflammatory response during aging which likely contributes to the well described phenomenon of “inflammaging” [17–19]. These interactions are by no means a one-way street, with several other aging hallmarks also contributing to age-associated mitochondrial dysregulation, but they do illustrate the importance of mitochondrial function for preserving youthful cellular, tissue, and organismal health.
Fig. 1.
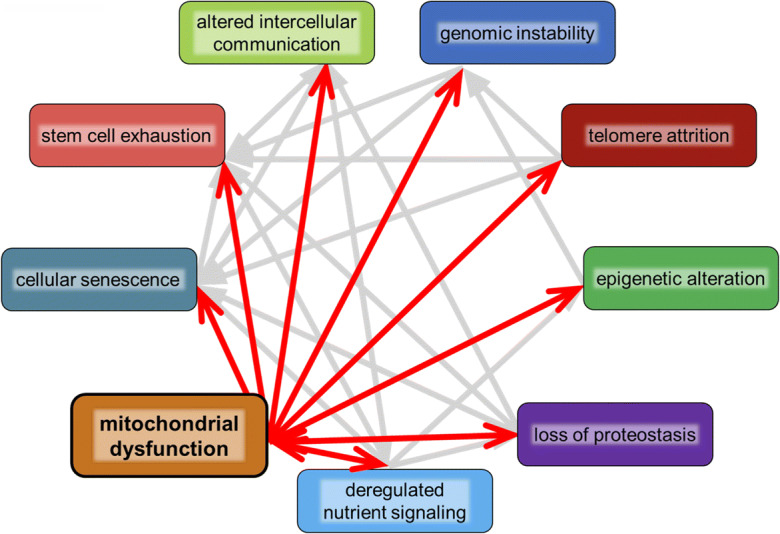
Mitochondrial contribution to hallmarks of aging. Mitochondria interact with each of the “hallmarks of aging,” and underlying mitochondrial dysfunction likely contributes to cellular dysfunctions associated with each hallmark. These complicated interactions suggest that targeting mitochondrial function directly and specifically may improve health as organisms age
One challenge the field has faced in attempting to define the multitude of ways that mitochondria are important during aging has been the limitations of available technologies for studying mitochondrial structure and function in living systems. Recently developed methods have allowed more precise observation and control over mitochondria in their natural state in living tissue or in whole organisms [20–25]. One important step forward has been the development of in vivo imaging technology. Microscopic advances have allowed interrogation of mitochondrial structure within tissues [26]. This has revealed that mitochondria are more diverse than the stereotypical bean-shaped organelles found in textbooks. Mitochondria can form networks or reticula that take on strikingly different morphologies in different cell types [27–30]. In addition, mitochondria and associated metabolic parameters can now be imaged and experimentally perturbed in living mammalian tissues [21, 31]. New understanding of how mitochondria differ between tissues has also expanded metabolic research to consider context when studying metabolism. For example, isolation of tissue-specific mitochondria is now possible and has revealed that mitochondrial genomes are passed on in different amounts across tissues [32]. It is now clear that metabolic study of one tissue or cell type cannot necessarily be applied to a different tissue or cell type. Recent years have seen a paradigm shift away from considering mitochondria as simple powerhouses as researchers in many fields have begun to recognize mitochondria as signaling organelles important for diverse aspects of cellular physiology [33].
Here we approach geroscience from an energetics perspective where we consider the mitochondrial protonmotive force (PMF) as a potential driver of biological aging and a potential target for intervention. We describe the function of the PMF, evidence for its role in aging and disease, and new technologies that can be applied to experimentally manipulate PMF in cells and animals.
Mitochondrial PMF
Mitochondria are powered by an electrochemical gradient termed protonmotive force (PMF). The PMF is a proton gradient across the mitochondrial inner membrane (IM). This proton gradient is made up of two connected potential energies: an electrical membrane potential (∆ψm) and a pH differential (∆pH). Mitochondria remove electrons from cellular metabolites and shuttle them through an electron transport chain (ETC) located in the IM. As electrons are passed through oxidation-reduction reactions, some energy from each transfer is used to pump protons across the IM from the matrix to the intermembrane space (Fig. 2). Proton translocation by the ETC establishes PMF that is then used by ATP synthase, also located in the IM, to make ATP for cells to use. In addition to this energetic function, mitochondria also regulate calcium signaling, apoptosis, redox homeostasis, and stress resistance signaling [34–36]. Here, we define mitochondrial function as respiration (electron flux and oxygen consumption), energetics (ATP production and availability), and the resultant signaling outputs dependent on respiration and energetics (Fig. 2). Each of these aspects of mitochondrial function is independent, but often works together. Importantly, each aspect is ultimately governed by the PMF. The collapse of PMF will eventually halt all mitochondrial function and cause cell death.
Fig. 2.
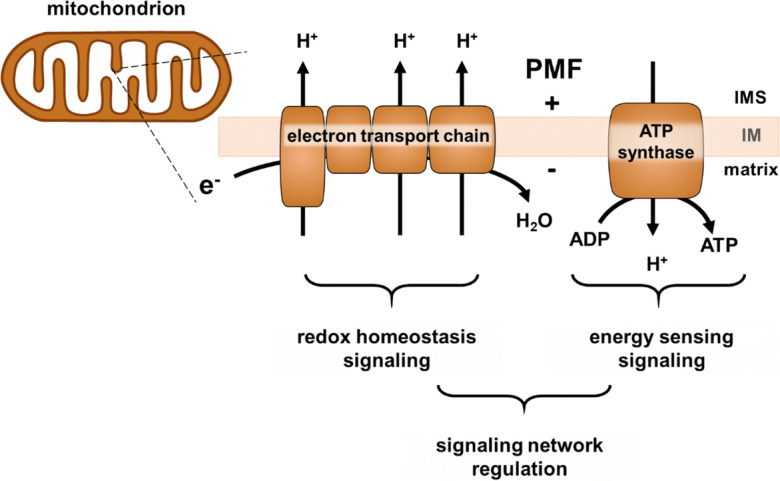
Mitochondrial function schematic. A mitochondrion is pictured with a magnified inner membrane (IM). The electron transport chain (ETC) in the IM passes electrons, consumes oxygen, and pumps protons (H+) from the mitochondrial matrix to the intermembrane space (IMS). This is the respiration aspect of mitochondrial function. Proton pumping by the ETC establishes protonmotive force (PMF), an electrochemical gradient across the IM (represented by + and -). ATP synthase uses the PMF to make ATP by allowing proton reentry into the matrix. ATP production provides energy for the cell and will be referred to as energetics. Downstream of the ETC, PMF, and ATP production are diverse signaling outputs that coordinate cellular physiology
A popular analogy for mitochondrial function in the bioenergetics field is the “proton circuit” [37, 38]. Electron flux through the ETC is analogous to amperage in an electrical circuit; electron flow through the ETC is like electron flow through the wires of a circuit. The PMF is analogous to voltage, a potential energy that powers the circuit. Both components are intrinsic to the circuit and can be observed separately. This analogy holds true for mitochondria as well; respiration and PMF work together, and they can be studied either together or independently, depending on the methods used to study them. This not only is a useful analogy, but it also serves as a warning for experimental interpretation of mitochondrial experiments. If only one parameter (either respiration or PMF) is measured or perturbed in a study, interpretation cannot be applied to the unmeasured component. Similarly, studying mitochondria in isolation from the in vivo, physiologic context may produce confounding results, just like isolated components of a circuit cannot fully describe a city’s power grid. This nuance can cause confusion when trying to interpret the literature from different model systems or under different metabolic conditions. Like the circuit analogy, we will consider mitochondrial PMF like batteries in that respiration charges up a voltage to provide energy.
A nuanced principle in metabolic research is the experimental separation of respiration and PMF. For example, one study genetically inhibited mitochondria in cells which resulted in decreased PMF, respiration, and mitochondrial signaling and in increased cell death [39]. This study then independently rescued either respiration or PMF through genetic regulators. The authors found that PMF rescue led to different outcomes than respiratory rescue. Rescued PMF preserved cell viability, redox homeostasis, and restored normal mitochondrial signaling through AMP-activated protein kinase (AMPK), a cellular energy-sensor [40]. Rescued respiration also preserved cell viability but did not restore AMPK signaling [39]. This concisely demonstrates that PMF and respiration are separable in a cellular context and that mitochondrial signaling outputs can be different depending on which mitochondrial function is perturbed. However, it remains difficult to specifically manipulate PMF acutely and reversibly in vivo, which limits progress in understanding physiologic mitochondrial functions. Regardless, this principle of separate but related mitochondrial functions may be particularly important in aging systems where multiple processes become dysregulated in cells and tissues and the charge of the mitochondrial battery declines with age.
PMF and aging
Many lifespan-extending interventions seem to implicate mitochondrial function. Mutations in, or RNAi knockdown of, electron transport chain components, for example, have long been known to extend lifespan in Caenorhabditis elegans [41–43]. This counter-intuitive example illustrates the complexity of mitochondrial biology, as reduced mitochondrial function can, in some cases, induce a protective response through a process referred to as mitohormesis [44]. Although the precise mechanisms underlying this protective response remain unclear and somewhat controversial [45, 46], examples of mitohormesis-associated lifespan extension have been observed in budding yeast [47, 48], nematode worms [49, 50], fruit flies [51], and mice [51–53], with suggestive evidence for such a mechanism in humans as well [54]. It is worth also noting, however, that a mouse model with deficient respiration was neither long-lived nor short-lived and had no obvious deleterious phenotypes [55]. Thus, the relationship between mitochondrial respiration and lifespan is complex, and the role of mitohormesis in mammalian aging remains to be fully elucidated.
In contrast to mitohormesis, most longevity- and health span–promoting interventions are associated with preservation of mitochondrial function during aging. Exercise is a classic example in humans where enhanced mitochondrial biogenesis and function are thought to promote preservation of muscle function [56, 57], reduced age-associated disease incidence [58, 59], and increased life expectancy [60, 61]. Another example from pre-clinical studies of aging is reduced signaling through the mechanistic target of rapamycin (mTOR) pathway. Genetic or pharmacologic inhibition of mTOR increases lifespan and functional measures of health during aging in yeast, worms, fruit flies, and mice [62, 63]. Experimental evidence suggests that inhibition of mTOR enhances mitochondrial function during aging through multiple mechanisms, including maintaining mitochondrial protein quality control, increasing degradation of damaged mitochondria via autophagy, and regulation of expression of nuclear encoded mitochondrial proteins both transcriptionally and translationally [64, 65]. Intriguingly, inhibition of mTOR with the drug rapamycin can also rescue severe mitochondrial dysfunction in animal models and cell culture [66–70] with initial indications of efficacy in human patients [71, 72].
Several lines of evidence converge to suggest that regulation of PMF specifically may play an important role in linking mitochondrial function to health during aging. In budding yeast, for example, an early indicator of biological age is decreased PMF [73]. Decreased PMF is also a well-known feature of cells from old animals [74], cells used as models for diseases of aging [75], and replicatively aged cells in culture [38, 75]. Further, PMF and respiration decreases in mammalian tissues in different models of aging and diseases of aging [75, 76]. Importantly, measures of both PMF and respiration decline in mitochondria isolated from old mice, supporting a model of declining PMF with age [77]. Causation between declining PMF and biological aging has yet to be confirmed, however. Thus far, the literature supports a model of mitochondrial decline as an early, potentially causative factor in functional decline with age (Fig. 3a). Therefore, developing methods to target PMF specifically may be a preventive strategy to combat aged pathophysiology.
Fig. 3.

An energetics perspective on aging. Mitochondrial PMF is represented as battery charge level. a The maximum potential energy or battery charge of mitochondria decreases with age in many models. b A hypothetical age prevention therapy may target and preserve mitochondrial PMF to promote cellular health and longevity. c A hypothetical rejuvenation intervention may target lost PMF therapeutically to restore lost battery charge to reverse cellular dysfunction associated with age
PMF and energy sensing
Organisms can sense and communicate nutrient availability throughout cells and tissues. Perturbing nutrient or energy sensing has powerful effects on aging. Caloric restriction, which can be defined as reducing calories without malnutrition, is by far the most well-studied intervention for slowing aging across many different species [78, 79]. Some beneficial effects of calorie restriction may be mediated through changes in PMF. In rodents, fasting was able to slightly ameliorate metabolic decline that occurs in middle aged brains, indicating that modulation of nutrient signaling involves mitochondrial function directly [76]. Further, mitochondria from calorie-restricted old rats had preserved PMF and respiration levels [80, 81]. In C. elegans, increased lifespan triggered by fasting requires functional mitochondrial dynamics (fission and fusion) and respiration [82]. Also in C. elegans, lifespan extension from mTOR inhibition requires the energy sensor, AMPK [83]. How PMF regulates the interplay between mTOR, AMPK, mitochondrial dynamics, and fasting is still unclear.
The lifespan-extending drugs metformin and rapamycin are thought to mimic conditions of calorie restriction, possibly through mitochondrial signals [65, 84–86]. Metformin targets both AMPK and mitochondrial respiration, and rapamycin specifically targets mTOR. In a study using both metformin and rapamycin, toxic effects of metformin in old C. elegans were ameliorated by rapamycin [87]. Metformin toxicity was likely ameliorated by preserved PMF via rapamycin, as demonstrated in cell models [87, 88]. This shows that rapamycin may have some of its beneficial effects through stabilization of PMF. Overall, these studies indicate that the effects of true or mimicked calorie restriction may function through mitochondrial PMF regulation of energy sensing. Testing the effects of calorie restriction and mimetic drugs on the PMF will be important to better understand the molecular mechanisms of fasting and longevity.
PMF and proteostasis
Protein homeostasis, or proteostasis, is a balance between protein production and clearance of damaged or misfolded proteins [89, 90]. One link between mitochondrial PMF and proteostasis is through lysosomal function. Lysosomes maintain an acidic internal environment for proteolytic activities to support proteostasis [91, 92]. An early indicator of biological aging is decreased PMF that is associated with decreased lysosomal acidity [73]. Pharmacologic or genetic rescue of lysosomal acidity rescued mitochondrial PMF in yeast [73]. PMF maintenance is thought to depend on availability of iron sulfur clusters, essential components of the mitochondrial ETC [93]. Lysosomes regulate metal ion availability, including iron for iron sulfur cluster assembly in mitochondria [94, 95]. This linked regulation could explain the mechanism of rescue between lysosomal and mitochondrial functions; functional lysosomes provide iron sulfur clusters for functional ETC activity and PMF generation. This link makes it a likely assumption that dysfunction in one organelle will translate to dysfunction in the other. Moreover, functional interaction between lysosomes and mitochondria may be mediated through signaling outputs of the PMF.
Mitochondria are also recycled like other organelles and proteins in cells. Mitochondria continually undergo fission and fusion in vivo for various purposes [96, 97]. It is thought that mitochondrial fission acts to separate dysfunctional mitochondria, or mitochondria with low PMF, away from healthy mitochondria. The dysfunctional mitochondria can then be degraded and recycled by lysosomes through mitochondrial autophagy (mitophagy) [98–100]. Mitophagy may become dysfunctional with age, allowing damaged mitochondria to accumulate and cause cellular dysfunction. Causation has been difficult to establish between dysfunctional mitophagy and persistence of mitochondria with low PMF. However, recent evidence suggested that direct and specific PMF dissipation in cells caused initiation of mitophagy [25]. In addition, in a study that linked beneficial autophagy to longevity, permeabilizing the mitochondrial IM switched autophagy to a damaging force and shortened lifespan in C. elegans [101]. Permeabilizing the IM in vivo would result in dissipated PMF. This study showed that beneficial effects of autophagy on lifespan are dependent on sufficient PMF. This again supports the hypothesis that maintained PMF is beneficial for healthy aging and connected to proteostasis. It will be important to test precisely how changes in PMF are linked to proteostasis, mitophagy, and lysosomal acidity in vivo.
PMF and stem cell function
Stem cells have regenerative potential that decline with age in many tissues, and restoration of stem cell function has been associated with reversal of age-related functional declines [102]. Mitochondria are directly involved in stem cell function through their role in providing the raw material and energy for growth. In Drosophila, old stem cell mitochondria had decreased ATP levels and decreased PMF. Glycolytic metabolism was concurrently upregulated in these old stem cells with low PMF [103]. It remains unclear whether low PMF occurred as a signal for the metabolic switch to glycolysis or if the metabolic switch itself resulted in the low PMF. This uncertainty is representative of many metabolic studies that are limited by the experimental tools available.
In aging mouse or human hematopoietic stem cells, PMF can vary across cell populations [104]. In such variable stem cell populations, cells with relatively higher mitochondrial PMF had transcriptional profiles similar to younger cells [105]. Stem cells with lower PMF had lower transcriptional rates and reduced DNA repair responses, indicating an older biological phenotype. When PMF was preserved by feeding animals the mitochondrial targeted antioxidant, mitoQ [106], youthful stem cell phenotypes were reestablished [105]. This work highlights how low PMF is associated with aged biology, and that intervening to increase PMF in vivo can reverse aged phenotypes. While the mechanism of mitoQ PMF preservation is unclear, this study provided some evidence for the causal nature of PMF changes in stem cell metabolism and aging. Testing how PMF affects stem cell functions will require precise control over mitochondrial function in vivo.
An energetics perspective on aging
The battery power of mitochondrial function declines with age in many models across species and tissues (Fig. 3a). Here, we outline an emerging geroscience hypothesis based on literature suggesting that preserving or boosting mitochondrial PMF may act to prevent or reverse age-associated dysfunction. This idea builds upon previous goals of broadly targeting mitochondria for anti-aging therapy [107]. Intervening before mitochondrial function significantly declines could prevent the loss of mitochondrial function and PMF that may drive cellular aging (Fig. 3b). Similarly, rejuvenating mitochondrial PMF in aged organisms may act to restore mitochondrial functions and reverse aging phenotypes (Fig. 3c). To test these hypotheses, precise control of PMF and mitochondrial functions is necessary in vivo to produce meaningful physiologic results. The work described herein began to test these hypotheses, and the results thus far have informed our proposed concept of an energetics perspective on aging.
Testing how PMF affects aging
It seems clear that determining cause and effect between PMF changes and physiology will drive progress in understanding disease. It is also clear, however, that determination of cause and effect is difficult and sometimes impossible given limitations of experimental methods. A recent study addressed this difficulty and coined the phrase “causal metabolism.” The study described a method to fundamentally alter metabolism in living mice and asked what the effects were [108]. Expressing an enzyme to artificially decrease the NADH/NAD+ ratio in vivo revealed changes in metabolism as a direct result of the changed redox ratio [108]. The NADH/NAD+ ratio governs electron metabolism and metabolic signaling through many signaling networks in cells and subcellular compartments [109]. Oxidizing NADH to NAD+ directly in live animals changed metabolism through affecting one variable. The resultant physiologic observations could then be attributed directly to the changed NADH/NAD+ ratio upstream. This intervention improves upon classical approaches of observing many aspects of metabolism that are associated with each other in different disease models. Therefore, this study is a great model for causal metabolism studies in aging. Manipulation of single metabolic variables with proper experimental controls should be a standard of mitochondrial research in aging models. First characterizing which aspects of mitochondrial function (ETC flux, ATP/ADP levels, PMF) are affected is required for clear interpretations. Once these effects are characterized for a specific intervention, in vivo application can be used to test what aspects of mitochondrial function cause physiologic changes that may be associated with aging.
Causal metabolic studies that achieve some or all of these standards are emerging in invertebrate models. Traditional genetic interventions to change metabolism were applied in C. elegans, and both respiration and PMF were monitored to show how an unfolded protein response coordinated mitochondrial biogenesis [110]. This showed that mitochondrial function, protein expression and stress resistance signaling programs depend on one another for proper function. The reverse situation—changing the PMF and monitoring physiologic responses—is now possible in C. elegans as well, through optogenetics. Using light-sensitive proton pumps targeted to mitochondria allowed precise control of PMF in isolation from other metabolic processes [111]. Optogenetic tools like these were developed to both increase or decrease mitochondrial PMF in live animals using light [22, 23]. Elevating PMF in vivo revealed a causal connection between PMF and energy sensing though AMPK. When PMF was artificially increased in living C. elegans, starvation-induced AMPK activation was reversed [22]. Similarly, optogenetic dissipation of PMF resulted in activation of AMPK and could modulate starvation behavior [23]. Boosting PMF simulated a fed state and deactivated AMPK signaling, and dissipated PMF activated AMPK and signaled a starved state. These results confirmed a causal link between PMF and energy sensing though AMPK. Therefore, it is likely that changing levels of PMF experimentally would affect aging physiology through the effects on AMPK and nutrient sensing signaling. Determining cause and effect between PMF changes that happen with age and cellular dysfunctions will provide much needed clarity and unity between models of metabolism in aging. Controlling PMF in vivo could be used to reverse decline of PMF that happens in (at least some) tissues with age. Increasing PMF in old animals and monitoring dysfunction associated with specific hallmarks of aging will directly test whether lost PMF causes physiologic dysfunction for that hallmark. Ultimately, artificially increasing PMF in animals and monitoring lifespan would test if the observed decline in PMF with age is causing functional decline and mortality.
In addition to PMF intervention, control over oxidative stress and ROS production is now also amenable to aging research questions. ROS can be produced or scavenged specifically in mitochondria and physiologic responses can be easily monitored [112–114]. One study has already causally implicated mitochondrial ROS with telomere dysfunction in the nucleus [113]. Using these new reagents allows control over mitochondrial redox signaling, and lifespan can be monitored to determine the causal effects of mitochondrial function on aging in a more precise way than has been possible previously. This experimental system can begin to test what levels and types of mitochondrial ROS production are beneficial versus detrimental.
These causal metabolism approaches could be used to answer questions brought up in earlier examples reviewed here, like “does decreased PMF in stem cells cause a switch to glycolytic metabolism?”, and “does increased PMF prevent initiation of mitophagy?” Answering these questions will provide directionality in models that will streamline future investigations and focus hypotheses. Eventually, these approaches and knowledge generated in invertebrates could be applied to mammalian models. Many rodent studies use observational techniques like genomics, proteomics, and metabolomics [65, 115, 116] to observe metabolic changes that impact physiology. These observations could be paired with causal metabolism techniques and optogenetics to enhance the interpretation of these studies.
Further, these techniques can be used tissue specifically. Drosophila and C. elegans could be used to identify causal effects of PMF changes or ROS production in specific tissues to inform more targeted mammalian approaches. Some of this work has already begun in C. elegans, where neuronal AMPK was shown to respond to PMF changes [23]. Moreover, neuronal mTOR inhibition resulted in AMPK-mediated lifespan extension that involved preservation of healthy mitochondrial structure [83]. Overall, using invertebrate models will aid in identifying targets for adapting new interventions in mammals. This will advance our understanding of how mitochondrial PMF causes physiologic change that can be precisely targeted to prevent or reverse the decline that occurs with age in humans.
Integrating an energetics perspective on aging with other models
While we propose that PMF maintenance may support healthy longevity, this model must be reconciled with previous findings suggesting a seemingly opposite interpretation. In studies using isolated mitochondria from long-lived animals such as the naked mole rat, for example, decreased PMF and intrinsically increased basal respiration have been observed [117, 118]. The same is true for bats [119], pigeons [120], and some other animals that are the same size as mice but longer lived. Further, pharmacologic PMF dissipation using protonophores to collapse electrochemical gradients has also been reported to extend lifespan in C. elegans and in female Swiss Webster outbred albino mice [121, 122]. However, these observations are still compatible with the model that preserved PMF during aging may support enhanced longevity and health span. First, it is important to recognize that correlations between decreased PMF and longevity come almost exclusively from laboratory-isolated mitochondria, cells, or tissues. Naked mole rat cells, in particular, require substantially different culture conditions compared to mouse cells, which will likely have an impact on measurements of mitochondrial structure and function. Also, correlations between in vitro PMF measurements and any physiologic output (such as lifespan) should be cautiously interpreted due to methodological considerations [38]. Therefore, whether differences in PMF are actually present in vivo in these long-lived animals remains unknown. The use of protonophores to extend lifespan is particularly interesting but could conceivably act through mitohormesis, as described above, and even potentially result in an overall preservation of PMF into old age. Consistent with this, treatment of mice with protonophores enhanced mitochondrial biogenesis [123], which could lead to more functional mitochondria and preserved PMF.
Another mechanism by which a protonophore could promote longevity is through reduced oxidative stress [117]. Decreased PMF reduces mitochondrial oxidant overproduction in isolated mitochondria [124–126], and the longer-lived mice treated with a protonophore showed some markers of decreased oxidative damage [121]. The relationship between oxidative stress and longevity has been mixed [127], however, and direct in vivo evidence for decreased oxidative stress causing increased longevity is still lacking. Long-lived naked mole rats, for example, show signs of high levels of systemic oxidative damage [128]. Moreover, the free radical theory of aging has not been universally supported experimentally, with studies showing that redox status remains unchanged across mammalian tissues with age [115], and that in some cases increased oxidation is associated with longevity [129]. This underlines a central theme of mitochondrial research—outcomes can seem opposite depending on experimental methods and models. To address these disparate results, experiments that test cause versus effect between PMF and lifespan are necessary to bring clarity and mechanistic understanding.
One recent study illustrates how the seemingly inconsistent evidence for increased or decreased PMF promoting aging can be reconciled. In a naturally aged rodent model, mitochondrial matrix pH in cardiomyocytes was measured under different conditions as a proxy for PMF levels. Old cardiomyocytes had decreased PMF and dysfunctional metabolism [130]. One known mediator of PMF and respiration is the adenine nucleotide transporter (ANT) [131, 132]. The ANT transports ADP into mitochondria for conversion to ATP, and it transports ATP out of mitochondria to be used to do work in the cell. In addition, the ANT can allow protons to reenter the mitochondrial matrix resulting in decreased PMF [132]. In this study, the ANT was pharmacologically inhibited to preserve PMF in the old cells. Upon ANT inhibition, respiratory dysfunction was reversed, PMF was stabilized, and youthful physiology was restored in the old cardiomyocytes [130]. Part of the proposed mechanism involved stabilized ETC flux and redox state. The authors reasoned that if ETC flux is uninhibited and oxidative stress is low, then increasing PMF with age is beneficial to cell physiology and is still compatible with models of decreasing PMF to decrease oxidative stress.
These data again demonstrate how the PMF can signal independently from other mitochondrial functions such as respiration efficiency and redox signaling. Importantly, the data show that if redox status is maintained, preserved PMF is beneficial, as long as it is opposing a natural decline. On the other hand, if metabolism is pathologically increased and oxidative stress occurs, then decreasing PMF would be beneficial to decrease mitochondrial oxidant production [133]. Ultimately, when both electron flux and PMF are maintained, aging phenotypes can be delayed [130]. These phenomena demonstrate the importance of tissue, cell, organ, and time specific studies to understand precisely when, where, and how mitochondrial function is impaired. These seemingly opposed models of PMF regulation and longevity can be reconciled through precise methods, experimental design, and interpretation. Applying general metabolic results from isolated systems to broad whole-organism models may not be as informative because of this dichotomy in results from PMF regulation and aging.
Conclusions
In much the same way that portable batteries must be replaced after repeated charge/drain cycles, mitochondria decline in function with age. Restoring mitochondrial function, like replacing a battery, can restore cells’ ability to do work and may have a positive impact on human health. Some ways to “replace the battery” of mitochondrial function may include stimulating mitophagy and mitochondrial dynamics, and modulating energy-sensing signaling, possibly through interventions such as calorie restriction. To learn how to effectively use these approaches to maintain mitochondrial health, it will be important to precisely answer when, where, and how PMF is regulated and if boosting PMF in vivo will affect aging biology and lifespan. Therefore, testing whether PMF changes are a cause or a consequence of physiologic dysfunction is a priority for metabolic models of aging. Further, it will be exciting to test whether PMF levels can be an early indicator of aging dysfunction. This analysis will depend on development of reliable measures of PMF and mitochondrial function in whole organisms, and eventually in humans [134, 135]. This may include identifying appropriate tissues to monitor and non-invasive methods to assess in situ PMF. Considering the impact of bioenergetics on hallmarks of aging may also reveal novel mechanisms of regulating hallmarks of health [136]. These newly described hallmarks also likely depend on cellular energetics, and deeper understanding of mitochondria will help to integrate hallmarks of aging and health with molecular underpinnings that can be targeted therapeutically.
Acknowledgements
The authors thank Dr. Gavin Pharoah for critically reviewing the manuscript.
Author contribution
BJB wrote and revised the manuscript; MK wrote and revised the manuscript.
Funding
BJB is supported by the Biological Mechanisms for Healthy Aging (BMHA) Training Grant NIH T32AG066574. MK is the director of the BMHA Training Program and the Nathan Shock Center of Excellence in the Basic Biology of Aging (NIH P30AG013280) at the University of Washington. This work was supported by NIH grant R01NS98329 to MK.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Brandon J. Berry, Email: bberry5@uw.edu
Matt Kaeberlein, Email: kaeber@uw.edu.
References
- 1.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 2.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Rijt S, Molenaars M, McIntyre RL, Janssens GE, Houtkooper RH. Integrating the hallmarks of aging throughout the tree of life: a focus on mitochondrial dysfunction. Front Cell Dev Biol. 2020;8:594416. doi: 10.3389/fcell.2020.594416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bornstein R, Gonzalez B, Johnson SC. Mitochondrial pathways in human health and aging. Mitochondrion. 2020;54:72–84. doi: 10.1016/j.mito.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longo VD, Liou LL, Valentine JS, Gralla EB. Mitochondrial superoxide decreases yeast survival in stationary phase. Arch Biochem Biophys. 1999;365:131–142. doi: 10.1006/abbi.1999.1158. [DOI] [PubMed] [Google Scholar]
- 7.Papa S, Skulachev VP. Reactive oxygen species, mitochondria, apoptosis and aging. Mol Cell Biochem. 1997;174:305–319. doi: 10.1023/A:1006873518427. [DOI] [PubMed] [Google Scholar]
- 8.Auten RL, Davis JM. Oxygen toxicity and reactive oxygen species: the devil is in the details. Pediatr Res. 2009;66:121–127. doi: 10.1203/PDR.0b013e3181a9eafb. [DOI] [PubMed] [Google Scholar]
- 9.Stefanatos R, Sanz A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018;592:743–758. doi: 10.1002/1873-3468.12902. [DOI] [PubMed] [Google Scholar]
- 10.Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. 2010;12:503–535. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Q, Huang J, Wang G. Mitochondria, telomeres and telomerase subunits. Front Cell Dev Biol. 2019;7:274. doi: 10.3389/fcell.2019.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saberi M, Zhang X, Mobasheri A (2021) Targeting mitochondrial dysfunction with small molecules in intervertebral disc aging and degeneration. Geroscience doi:10.1007/s11357-021-00341-1 [DOI] [PMC free article] [PubMed]
- 13.Zhang H, Menzies KJ, Auwerx J (2018) The role of mitochondria in stem cell fate and aging. Development 145 doi:10.1242/dev.143420 [DOI] [PMC free article] [PubMed]
- 14.Peleg S, Feller C, Ladurner AG, Imhof A. The metabolic impact on histone acetylation and transcription in ageing trends. Biochem Sci. 2016;41:700–711. doi: 10.1016/j.tibs.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 15.Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11:230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Pollack M, Leeuwenburgh C. Apoptosis and aging: role of the mitochondria. J Gerontol A Biol Sci Med Sci. 2001;56:B475–B482. doi: 10.1093/gerona/56.11.b475. [DOI] [PubMed] [Google Scholar]
- 17.Picca A et al. (2017) Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. International Journal of Molecular Sciences 18 doi:10.3390/ijms18050933 [DOI] [PMC free article] [PubMed]
- 18.Zampino M, Brennan NA, Kuo PL, Spencer RG, Fishbein KW, Simonsick EM, Ferrucci L. Poor mitochondrial health and systemic inflammation? Test of a classic hypothesis in the Baltimore Longitudinal Study of Aging Geroscience. 2020;42:1175–1182. doi: 10.1007/s11357-020-00208-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vizioli MG, et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020;34:428–445. doi: 10.1101/gad.331272.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolf DM, et al. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 2019;38:e101056. doi: 10.15252/embj.2018101056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glancy B, et al. Power grid protection of the muscle mitochondrial reticulum. Cell Rep. 2017;19:487–496. doi: 10.1016/j.celrep.2017.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berry BJ, et al. Optogenetic control of mitochondrial protonmotive force to impact cellular stress resistance. EMBO Rep. 2020;21:e49113. doi: 10.15252/embr.201949113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berry BJ, Baldzizhar A, Nieves TO, Wojtovich AP. Neuronal AMPK coordinates mitochondrial energy sensing and hypoxia resistance in C. elegans. FASEB J. 2020;34:16333–16347. doi: 10.1096/fj.202001150RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tkatch T, et al. Optogenetic control of mitochondrial metabolism and Ca(2+) signaling by mitochondria-targeted opsins. Proc Natl Acad Sci U S A. 2017;114:E5167–E5176. doi: 10.1073/pnas.1703623114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ernst P, et al. Precisely control mitochondria with light to manipulate cell fate decision. Biophys J. 2019;117:631–645. doi: 10.1016/j.bpj.2019.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glancy B, et al. Mitochondrial reticulum for cellular energy distribution in muscle. Nature. 2015;523:617–620. doi: 10.1038/nature14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glancy B, Kim Y, Katti P, Willingham TB. The functional impact of mitochondrial structure across subcellular scales. Front Physiol. 2020;11:541040. doi: 10.3389/fphys.2020.541040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bleck CKE, Kim Y, Willingham TB, Glancy B. Subcellular connectomic analyses of energy networks in striated muscle. Nat Commun. 2018;9:5111. doi: 10.1038/s41467-018-07676-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shami GJ, Cheng D, Verhaegh P, Koek G, Wisse E, Braet F. Three-dimensional ultrastructure of giant mitochondria in human non-alcoholic fatty liver disease. Sci Rep. 2021;11:3319. doi: 10.1038/s41598-021-82884-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kowaltowski AJ, et al. Mitochondrial morphology regulates organellar. Ca FASEB J. 2019;33:13176–13188. doi: 10.1096/fj.201901136R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willingham TB, Zhang Y, Andreoni A, Knutson JR, Lee DY, Glancy B. MitoRACE: evaluating mitochondrial function in vivo and in single cells with subcellular resolution using multiphoton NADH autofluorescence. J Physiol. 2019;597:5411–5428. doi: 10.1113/JP278611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahier A, Dai CY, Tweedie A, Bezawork-Geleta A, Kirmes I, Zuryn S. Affinity purification of cell-specific mitochondria from whole animals resolves patterns of genetic mosaicism. Nat Cell Biol. 2018;20:352–360. doi: 10.1038/s41556-017-0023-x. [DOI] [PubMed] [Google Scholar]
- 33.Chandel NS. Evolution of mitochondria as signaling organelles. Cell Metab. 2015;22:204–206. doi: 10.1016/j.cmet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 34.Nicholls DG. Mitochondria and calcium signaling. Cell Calcium. 2005;38:311–317. doi: 10.1016/j.ceca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 35.Fox TD. Mitochondrial protein synthesis, import, and assembly. Genetics. 2012;192:1203–1234. doi: 10.1534/genetics.112.141267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olsen RK, Cornelius N, Gregersen N. Redox signalling and mitochondrial stress responses; lessons from inborn errors of metabolism. J Inherit Metab Dis. 2015;38:703–719. doi: 10.1007/s10545-015-9861-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicholls DG. Bioenergetics 4. 4th ed: Elsevier; 2013.
- 38.Nicholls DG. Mitochondrial membrane potential and aging. Aging Cell. 2004;3:35–40. doi: 10.1111/j.1474-9728.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 39.Martinez-Reyes I, et al. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol Cell. 2016;61:199–209. doi: 10.1016/j.molcel.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burkewitz K, Zhang Y, Mair WB. AMPK at the nexus of energetics and aging. Cell Metab. 2014;20:10–25. doi: 10.1016/j.cmet.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Felkai S, Ewbank JJ, Lemieux J, Labbe JC, Brown GG, Hekimi S. CLK-1 controls respiration, behavior and aging in the nematode Caenorhabditis elegans. EMBO J. 1999;18:1783–1792. doi: 10.1093/emboj/18.7.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng J, Bussiere F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell. 2001;1:633–644. doi: 10.1016/S1534-5807(01)00071-5. [DOI] [PubMed] [Google Scholar]
- 43.Dillin A, et al. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 44.Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis) Exp Gerontol. 2010;45:410–418. doi: 10.1016/j.exger.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 45.Bennett CF, Kaeberlein M. The mitochondrial unfolded protein response and increased longevity: Cause, consequence, or correlation? Exp Gerontol. 2014;56C:142–146. doi: 10.1016/j.exger.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bennett CF, Vander Wende H, Simko M, Klum S, Barfield S, Choi H, Pineda VV, Kaeberlein M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat Commun. 2014;5:3483. doi: 10.1038/ncomms4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Longo VD, Shadel GS, Kaeberlein M, Kennedy B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 2012;16:18–31. doi: 10.1016/j.cmet.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pan Y. Mitochondria, reactive oxygen species, and chronological aging: a message from yeast. Exp Gerontol. 2011;46:847–852. doi: 10.1016/j.exger.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 49.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 50.Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Owusu-Ansah E, Song W, Perrimon N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 2013;155:699–712. doi: 10.1016/j.cell.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cox CS, et al. Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell Metab. 2018;28(776-786):e775. doi: 10.1016/j.cmet.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dell'agnello C, et al. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- 54.Sparks LM, Redman LM, Conley KE, Harper ME, Hodges A, Eroshkin A, Costford SR, Gabriel ME, Yi F, Shook C, Cornnell HH, Ravussin E, Smith SR. Differences in mitochondrial coupling reveal a novel signature of mitohormesis in muscle of healthy individuals. J Clin Endocrinol Metab. 2016;101:4994–5003. doi: 10.1210/jc.2016-2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deepa SS, Pharaoh G, Kinter M, Diaz V, Fok WC, Riddle K, Pulliam D, Hill S, Fischer KE, Soto V, Georgescu C, Wren JD, Viscomi C, Richardson A, van Remmen H. Lifelong reduction in complex IV induces tissue-specific metabolic effects but does not reduce lifespan or healthspan in mice. Aging Cell. 2018;17:e12769. doi: 10.1111/acel.12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saito Y, Chikenji TS, Matsumura T, Nakano M, Fujimiya M. Exercise enhances skeletal muscle regeneration by promoting senescence in fibro-adipogenic progenitors. Nat Commun. 2020;11:889. doi: 10.1038/s41467-020-14734-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reddy A et al. (2020) pH-gated succinate secretion regulates muscle remodeling in response to exercise Cell 183:62-75.e17 doi:10.1016/j.cell.2020.08.039 [DOI] [PMC free article] [PubMed]
- 58.Trewin AJ, et al. Acute exercise alters skeletal muscle mitochondrial respiration and H2O2 emission in response to hyperinsulinemic-euglycemic clamp in middle-aged obese men. PLoS One. 2017;12:e0188421. doi: 10.1371/journal.pone.0188421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lear SA, et al. The effect of physical activity on mortality and cardiovascular disease in 130 000 people from 17 high-income, middle-income, and low-income countries: the PURE study. Lancet. 2017;390:2643–2654. doi: 10.1016/S0140-6736(17)31634-3. [DOI] [PubMed] [Google Scholar]
- 60.Warburton DE, Nicol CW, Bredin SS. Health benefits of physical activity: the evidence. CMAJ. 2006;174:801–809. doi: 10.1503/cmaj.051351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mandsager K, Harb S, Cremer P, Phelan D, Nissen SE, Jaber W. Association of cardiorespiratory fitness with long-term mortality among adults undergoing exercise treadmill testing. JAMA Netw Open. 2018;1:e183605. doi: 10.1001/jamanetworkopen.2018.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson SC, Sangesland M, Kaeberlein M, Rabinovitch PS (2015) Modulating mTOR in aging and health Interdisciplinary topics in gerontology 40:107-127 doi:10.1159/000364974 [DOI] [PubMed]
- 64.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 65.Martin-Perez M, et al. PKC downregulation upon rapamycin treatment attenuates mitochondrial disease. Nat Metab. 2020;2:1472–1481. doi: 10.1038/s42255-020-00319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson SC, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013;342:1524–1528. doi: 10.1126/science.1244360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson SC, Yanos ME, Bitto A, Castanza A, Gagnidze A, Gonzalez B, Gupta K, Hui J, Jarvie C, Johnson BM, Letexier N, McCanta L, Sangesland M, Tamis O, Uhde L, van den Ende A, Rabinovitch PS, Suh Y, Kaeberlein M. Dose-dependent effects of mTOR inhibition on weight and mitochondrial disease in mice. Front Genet. 2015;6:247. doi: 10.3389/fgene.2015.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ito TK, et al. Hepatic S6K1 partially regulates lifespan of mice with mitochondrial complex I deficiency. Front Genet. 2017;8:113. doi: 10.3389/fgene.2017.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang A, Mouser J, Pitt J, Promislow D, Kaeberlein M. Rapamycin enhances survival in a Drosophila model of mitochondrial disease. Oncotarget. 2016;7:80131–80139. doi: 10.18632/oncotarget.12560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng X, Boyer L, Jin M, Kim Y, Fan W, Bardy C, Berggren T, Evans RM, Gage FH, Hunter T (2016) Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. eLife 5 doi:10.7554/eLife.13378 [DOI] [PMC free article] [PubMed]
- 71.Johnson SC, et al. mTOR inhibitors may benefit kidney transplant recipients with mitochondrial diseases. Kidney Int. 2019;95:455–466. doi: 10.1016/j.kint.2018.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sage-Schwaede A, Engelstad K, Salazar R, Curcio A, Khandji A, Garvin JH, Jr, De Vivo DC. Exploring mTOR inhibition as treatment for mitochondrial disease. Ann Clin Transl Neurol. 2019;6:1877–1881. doi: 10.1002/acn3.50846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hughes AL, Gottschling DE. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 2012;492:261–265. doi: 10.1038/nature11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ames BN, Shigenaga MK, Hagen TM. Mitochondrial decay in aging. Biochim Biophys Acta. 1995;1271:165–170. doi: 10.1016/0925-4439(95)00024-x. [DOI] [PubMed] [Google Scholar]
- 75.Sugrue MM, Tatton WG. Mitochondrial membrane potential in aging cells. Biol Signals Recept. 2001;10:176–188. doi: 10.1159/000046886. [DOI] [PubMed] [Google Scholar]
- 76.Bayliak MM, Sorochynska OM, Kuzniak OV, Gospodaryov DV, Demianchuk OI, Vasylyk YV, Mosiichuk NM, Storey KB, Garaschuk O, Lushchak VI. Middle age as a turning point in mouse cerebral cortex energy and redox metabolism: Modulation by every-other-day fasting. Exp Gerontol. 2020;145:111182. doi: 10.1016/j.exger.2020.111182. [DOI] [PubMed] [Google Scholar]
- 77.Kokoszka JE, Coskun P, Esposito LA, Wallace DC. Increased mitochondrial oxidative stress in the Sod2 (+/-) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci U S A. 2001;98:2278–2283. doi: 10.1073/pnas.051627098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anderson RM, Weindruch R. The caloric restriction paradigm: implications for healthy human aging. American journal of human biology: the official journal of the Human Biology Council. 2012;24:101–106. doi: 10.1002/ajhb.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hagopian K, Harper ME, Ram JJ, Humble SJ, Weindruch R, Ramsey JJ. Long-term calorie restriction reduces proton leak and hydrogen peroxide production in liver mitochondria. Am J Physiol Endocrinol Metab. 2005;288:E674–E684. doi: 10.1152/ajpendo.00382.2004. [DOI] [PubMed] [Google Scholar]
- 81.Bevilacqua L, Ramsey JJ, Hagopian K, Weindruch R, Harper ME (2005) Long-term caloric restriction increases UCP3 content but decreases proton leak and reactive oxygen species production in rat skeletal muscle mitochondria Am J Physiol Endocrinol Metab 289:E429-438 doi:10.1152/ajpendo.00435.2004 [DOI] [PubMed]
- 82.Weir HJ et al. (2017) Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab 26:884-896.e885 doi:10.1016/j.cmet.2017.09.024 [DOI] [PMC free article] [PubMed]
- 83.Zhang Y et al. (2019) Neuronal TORC1 modulates longevity via AMPK and cell nonautonomous regulation of mitochondrial dynamics in Elife 8 doi:10.7554/eLife.49158 [DOI] [PMC free article] [PubMed]
- 84.Vial G, Detaille D, Guigas B. Role of mitochondria in the mechanism(s) of action of metformin. Front Endocrinol (Lausanne) 2019;10:294. doi: 10.3389/fendo.2019.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M, Pollak M, Zhang Y, Yu Y, Becker KG, Bohr VA, Ingram DK, Sinclair DA, Wolf NS, Spindler SR, Bernier M, de Cabo R. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mohsin AA, Chen Q, Quan N, Rousselle T, Maceyka MW, Samidurai A, Thompson J, Hu Y, Li J, Lesnefsky EJ. Mitochondrial complex I inhibition by metformin limits reperfusion injury. J Pharmacol Exp Ther. 2019;369:282–290. doi: 10.1124/jpet.118.254300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Espada L, et al. Loss of metabolic plasticity underlies metformin toxicity in aged Caenorhabditis elegans. Nat Metab. 2020;2:1316–1331. doi: 10.1038/s42255-020-00307-1. [DOI] [PubMed] [Google Scholar]
- 88.Lerner C, et al. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell. 2013;12:966–977. doi: 10.1111/acel.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019;20:421–435. doi: 10.1038/s41580-019-0101-y. [DOI] [PubMed] [Google Scholar]
- 90.Klaips CL, Jayaraj GG, Hartl FU. Pathways of cellular proteostasis in aging and disease. J Cell Biol. 2018;217:51–63. doi: 10.1083/jcb.201709072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mauvezin C, Nagy P, Juhasz G, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015;6:7007. doi: 10.1038/ncomms8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Settembre C, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hughes CE, Coody TK, Jeong MY, Berg JA, Winge DR, Hughes AL (2020) Cysteine Toxicity Drives Age-Related Mitochondrial Decline by Altering Iron Homeostasis. Cell 180:296-310. e218 doi:10.1016/j.cell.2019.12.035 [DOI] [PMC free article] [PubMed]
- 94.Weber RA et al. (2020) Maintaining iron homeostasis is the key role of lysosomal acidity for cell proliferation. Mol Cell 77:645-655 e647 doi:10.1016/j.molcel.2020.01.003 [DOI] [PMC free article] [PubMed]
- 95.Chen KL, et al. Loss of vacuolar acidity results in iron-sulfur cluster defects and divergent homeostatic responses during aging in Saccharomyces cerevisiae. Geroscience. 2020;42:749–764. doi: 10.1007/s11357-020-00159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21:204–224. doi: 10.1038/s41580-020-0210-7. [DOI] [PubMed] [Google Scholar]
- 97.Wai T, Langer T. Mitochondrial dynamics and metabolic regulation trends. Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 98.Bakula D, Scheibye-Knudsen M. MitophAging: mitophagy in aging and disease. Front Cell Dev Biol. 2020;8:239. doi: 10.3389/fcell.2020.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20:31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, Maccoss MJ, Pallanck LJ. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A. 2013;110:6400–6405. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhou B et al. (2019) Mitochondrial permeability uncouples elevated autophagy and lifespan extension Cell 177:299-314 e216 doi:10.1016/j.cell.2019.02.013 [DOI] [PMC free article] [PubMed]
- 102.Schultz MB, Sinclair DA. When stem cells grow old: phenotypes and mechanisms of stem cell aging. Development. 2016;143:3–14. doi: 10.1242/dev.130633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morris O, Deng H, Tam C, Jasper H. Warburg-like metabolic reprogramming in aging intestinal stem cells contributes to tissue hyperplasia. Cell Rep. 2020;33:108423. doi: 10.1016/j.celrep.2020.108423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sukumar M, et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016;23:63–76. doi: 10.1016/j.cmet.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mansell E et al. (2020) Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy Stem Cell doi:10.1016/j.stem.2020.09.018 [DOI] [PMC free article] [PubMed]
- 106.Tauskela JS. MitoQ--a mitochondria-targeted antioxidant. IDrugs. 2007;10:399–412. [PubMed] [Google Scholar]
- 107.Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Mol Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Goodman RP, et al. Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature. 2020;583:122–126. doi: 10.1038/s41586-020-2337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kulkarni CA, Brookes PS. Cellular compartmentation and the redox/nonredox functions of NAD Antioxid Redox. Signal. 2019;31:623–642. doi: 10.1089/ars.2018.7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shpilka T, du YG, Yang Q, Melber A, Uma Naresh N, Lavelle J, Kim S, Liu P, Weidberg H, Li R, Yu J, Zhu LJ, Strittmatter L, Haynes CM. UPR. Nat Commun. 2021;12:479. doi: 10.1038/s41467-020-20784-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Berry BJ, Wojtovich AP. Mitochondrial light switches: optogenetic approaches to control metabolism. FEBS J. 2020;287:4544–4556. doi: 10.1111/febs.15424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Trewin AJ, Bahr LL, Almast A, Berry BJ, Wei AY, Foster TH, Wojtovich AP. Mitochondrial reactive oxygen species generated at the complex-II matrix or intermembrane space microdomain have distinct effects on redox signaling and stress sensitivity in Caenorhabditis elegans. Antioxid Redox Signal. 2019;31:594–607. doi: 10.1089/ars.2018.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Qian W, et al. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc Natl Acad Sci U S A. 2019;116:18435–18444. doi: 10.1073/pnas.1910574116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xie W, Jiao B, Bai Q, Ilin VA, Sun M, Burton CE, Kolodieznyi D, Calderon MJ, Stolz DB, Opresko PL, St Croix CM, Watkins S, van Houten B, Bruchez MP, Burton EA (2020) Chemoptogenetic ablation of neuronal mitochondria in vivo with spatiotemporal precision and controllable severity Elife 9 doi:10.7554/eLife.51845 [DOI] [PMC free article] [PubMed]
- 115.Xiao H et al. (2020) A quantitative tissue-specific landscape of protein redox regulation during aging Cell 180:968-983.e924 doi:10.1016/j.cell.2020.02.012 [DOI] [PMC free article] [PubMed]
- 116.Mills EL, et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature. 2018;560:102–106. doi: 10.1038/s41586-018-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vyssokikh MY, et al. Mild depolarization of the inner mitochondrial membrane is a crucial component of an anti-aging program. Proc Natl Acad Sci U S A. 2020;117:6491–6501. doi: 10.1073/pnas.1916414117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mookerjee SA, Divakaruni AS, Jastroch M, Brand MD. Mitochondrial uncoupling and lifespan. Mech Ageing Dev. 2010;131:463–472. doi: 10.1016/j.mad.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jürgens KD, Prothero J. Scaling of maximal lifespan in bats. Comp Biochem Physiol A Comp Physiol. 1987;88:361–367. doi: 10.1016/0300-9629(87)90498-1. [DOI] [PubMed] [Google Scholar]
- 120.Barja G. Mitochondrial free radical production and aging in mammals and birds. Ann N Y Acad Sci. 1998;854:224–238. doi: 10.1111/j.1749-6632.1998.tb09905.x. [DOI] [PubMed] [Google Scholar]
- 121.Caldeira da Silva CC, Cerqueira FM, Barbosa LF, Medeiros MH, Kowaltowski AJ. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell. 2008;7:552–560. doi: 10.1111/j.1474-9726.2008.00407.x. [DOI] [PubMed] [Google Scholar]
- 122.Lemire BD, Behrendt M, DeCorby A, Gásková D. C. elegans longevity pathways converge to decrease mitochondrial membrane potential. Mech Ageing Dev. 2009;130:461–465. doi: 10.1016/j.mad.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 123.Cerqueira FM, Laurindo FR, Kowaltowski AJ. Mild mitochondrial uncoupling and calorie restriction increase fasting eNOS, akt and mitochondrial biogenesis. PLoS One. 2011;6:e18433. doi: 10.1371/journal.pone.0018433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Berry BJ, Trewin AJ, Amitrano AM, Kim M, Wojtovich AP. Use the protonmotive force: mitochondrial uncoupling and reactive oxygen species. J Mol Biol. 2018;430:3873–3891. doi: 10.1016/j.jmb.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chouchani ET, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Loschen G, Flohe L, Chance B. Respiratory chain linked H(2)O(2) production in pigeon heart mitochondria. FEBS Lett. 1971;18:261–264. doi: 10.1016/0014-5793(71)80459-3. [DOI] [PubMed] [Google Scholar]
- 127.Perez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y, Richardson A. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790:1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Andziak B, et al. High oxidative damage levels in the longest-living rodent, the naked mole-rat. Aging Cell. 2006;5:463–471. doi: 10.1111/j.1474-9726.2006.00237.x. [DOI] [PubMed] [Google Scholar]
- 129.Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000361. doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang H, Alder NN, Wang W, Szeto H, Marcinek DJ, Rabinovitch PS (2020) Reduction of elevated proton leak rejuvenates mitochondria in the aged cardiomyocyte Elife 9 doi:10.7554/eLife.60827 [DOI] [PMC free article] [PubMed]
- 131.Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, Cornwall EJ. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J. 2005;392:353–362. doi: 10.1042/BJ20050890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bertholet AM, et al. H(+) transport is an integral function of the mitochondrial ADP/ATP carrier. Nature. 2019;571:515–520. doi: 10.1038/s41586-019-1400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chouchani ET, Kazak L, Spiegelman BM. New advances in adaptive thermogenesis: UCP1 and beyond. Cell Metab. 2019;29:27–37. doi: 10.1016/j.cmet.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 134.Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial electron transport chain: oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020;37:101674. doi: 10.1016/j.redox.2020.101674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cardenes N, et al. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood. 2014;123:2864–2872. doi: 10.1182/blood-2013-09-529420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lopez-Otin C, Kroemer G. Hallmarks of health. Cell. 2021;184:33–63. doi: 10.1016/j.cell.2020.11.034. [DOI] [PubMed] [Google Scholar]