

Abstract
Tenascin-C is upregulated during inflammation and tumorigenesis, and its expression level is correlated with a poor prognosis in several malignancies. Nevertheless, the substantial role of tenascin-C in cancer progression is poorly understood. Previously, we found that a peptide derived from tenascin-C, termed TNIIIA2, acts directly on tumor cells to activate β1-integrin and induce malignant progression. Here, we show that β1-integrin activation by TNIIIA2 in human fibroblasts indirectly contributes to cancer progression through the induction of cellular senescence. Prolonged treatment of fibroblasts with TNIIIA2 induced cellular senescence, as characterized by the suppression of cell growth and the induction of senescence-associated-β-galactosidase and p16INK4a expression. The production of reactive oxygen species and subsequent DNA damage were responsible for the TNIIIA2-induced senescence of fibroblasts. Interestingly, peptide FNIII14, which inactivates β1-integrin, inhibited fibroblast senescence induced not only by TNIIIA2 but also by H2O2, suggesting that β1-integrin activation plays a critical role in the induction of senescence in fibroblasts. Moreover, TNIIIA2-induced senescent fibroblasts secreted heparin-binding epidermal growth factor-like growth factor (HB-EGF), which caused preneoplastic epithelial HaCaT cells to acquire malignant properties, including colony-forming and focus-forming abilities. Thus, our study demonstrates that tenascin-C-derived peptide TNIIIA2 induces cellular senescence in fibroblasts through β1-integrin activation, causing cancer progression via the secretion of humoral factors such as HB-EGF.
Keywords: Tenascin-C, β1-integrin, cellular senescence, SASP, cancer-associated fibroblast, heparin-binding epidermal growth factor-like growth factor
Introduction
Matricellular proteins, including the CCN (CYR61/CTGF/NOV) family of proteins, fibulins, osteopontin, tenascins, and thrombospondins, which are present in the extracellular matrix (ECM), are characterized by their regulated expression and cell adhesion-modulatory function [1]. They are highly expressed in pathological states, including inflammation and tumorigenesis [1]. Therefore, matricellular proteins have been considered to play important roles in the pathogenesis of inflammatory diseases and cancers by affecting the adhesive interactions of cells with the ECM [1]. Tenascin-C (TNC), a typical matricellular protein, is expressed at low levels in normal adult tissues but is highly expressed in several malignancies, including colorectal cancer and glioma, and its expression levels are correlated with a poor prognosis [2,3]. Therefore, TNC is presumed to be involved in cancer development and malignant progression. A number of reports have demonstrated that TNC acts directly on cancer cells to influence cellular properties integral to cancer aggressiveness, such as dysregulated proliferation and invasive migration [4-7]. On the other hand, TNC is also associated with cancer progression through an indirect mechanism via non-cancerous cells [8-10]. In particular, TNC is highly expressed in cancer-associated fibroblasts, which are major components of the tumor microenvironment [11], and activates these cells to develop a stiffer stroma in tumor tissues, including breast cancer [12,13]. However, the substantial role of this protein in cancer progression in the tumor microenvironment is unknown.
At least some of the diverse biological functions of matricellular proteins, including TNC, are known to be derived from their proteolytic fragments released by inflammatory proteinases including as matrix metalloproteinases and a disintegrin and metalloproteinase with thrombospondin motifs, where tissue remodeling occurs actively in injured tissues [14-16]. Previously, we reported that the cancer-associated alternative splicing domain of the fibronectin type III repeat A2 in the TNC molecule has a cryptic functional site that induces β1-integrin activation [17]. The activation of β1-integrin by a peptide containing this functional site, termed TNIIIA2, is characterized by extremely potent and persistent effects [18]. Based on these effects, TNIIIA2 acts directly on glioma cells and colon cancer cells to enhance cancer-associated malignant properties, such as hyper-proliferation, disseminative migration, and metastatic potential [19-22]. Recently, we found that TNIIIA2 stimulates normal fibroblasts to promote the secretion of humoral factors, resulting in the hyper-proliferation of preneoplastic cells [23]. Thus, aberrant β1-integrin activation by the cryptic functional TNIIIA2 site within TNC, which is highly expressed in the tumor microenvironment, might cause malignant transformation through an indirect mechanism via non-cancerous cells stimulated by TNIIIA2 as well as the direct stimulation of cancer cells.
Senescence is a physiological and pathological cellular program triggered by various types of cellular stress, such as telomere erosion, oncogene activation, and DNA damage [24-26]. Senescent cells are found not only in aged tissues but also in embryonic development, wound healing, and tumor onset [27,28]. Senescent cells in preneoplastic lesions or the tumor microenvironment can secrete a number of soluble factors, such as growth factors, inflammatory cytokines, and proteases, collectively referred to as the senescence-associated secretory phenotype (SASP) [29]. Recent compelling evidence indicates that SASP factors in the tumor microenvironment act through cell non-autonomous (paracrine) and autonomous (autocrine) signaling pathways to play crucial roles in cancer progression [30]. It is widely accepted that senescent cells usually exhibit a flattened and enlarged morphology, regardless of the stimuli that cause senescence [31]. This suggests that the state of integrin-mediated cell adhesion to the ECM substratum may play a role in the induction of cellular senescence. Indeed, cysteine-rich angiogenic inducer 61 (CCN1/Cyr61) and connective tissue growth factor (CCN2/CTGF), typical matricellular proteins present in the ECM, have been reported to induce cellular senescence in fibroblasts by binding to integrin α6β1 [32,33], although the association of integrin-mediated cell adhesion with the induction of senescence was not necessarily proven in that study. Subsequent reports provided the important suggestion that integrin-mediated cell adhesion to the ECM is involved in the induction of cellular senescence [34]. However, the biochemical role of integrin in the induction of cellular senescence and its role in cancer progression are insufficiently understood.
Here, we show that TNIIIA2 derived from TNC, which is highly expressed in the tumor microenvironment, induces cellular senescence in human fibroblasts by activating β1-integrin. In addition, TNIIIA2-induced senescent fibroblasts confer malignant properties on preneoplastic epithelial cells via the secretion of SASP factors. Our results suggest that β1-integrin activation may serve as a common driving force in the induction of cellular senescence, at least in human fibroblasts.
Materials and methods
Reagents
Human plasma fibronectin was purified as described previously [35]. The peptides TNIIIA2 and FNIII14 have been described previously [19,36]. N-acetyl cysteine, sodium butyrate (NaB), rhodanile blue, and an anti-β-actin antibody were purchased from Sigma-Aldrich (Tokyo, Japan). Recombinant human heparin-binding epidermal growth factor-like growth factor (HB-EGF) and an anti-HB-EGF neutralizing antibody (AF-259-NA) were obtained from R&D Systems (Minneapolis, MN). An antibody against the COOH-terminally phosphorylated form of the histone variant H2AX (γH2AX) was purchased from Cell Signaling Technology (Danvers, MA). An anti-talin antibody was purchased from Biomol (Hamburg, Germany), an anti-β1-integrin neutralizing antibody (BV7) was purchased from Abcam (Cambridge, UK).
Cell culture
The human diploid lung embryonic fibroblast WI-38 and TIG-1 cell lines, which were obtained from RIKEN Bio Resource Center (Tsukuba, Japan), were maintained in Eagle’s minimal essential medium (Nissui Pharmaceutical, Tokyo, Japan) supplemented with 10% fetal bovine serum (FBS; SAFC Biosciences, St. Louis, MO). The human diploid lung embryonic fibroblast HFDF cell line, which was kindly provided by Dr. Motoyoshi Nomizu (Tokyo University of Pharmacy and Life Sciences), and human preneoplastic epidermal keratinocyte HaCaT cell line, which was kindly provided by Dr. Yasuhiko Masuho (Tokyo University of Science), were maintained in Dulbecco’s modified Eagle’s medium (Nissui Pharmaceutical) supplemented with 10% FBS. These cell lines were passaged soon after receipt, divided, and stored in liquid nitrogen. Each experiment was carried out using thawed cells without further authentication. These cell lines were also authenticated by routine monitoring of cell morphology and proliferation, kept in a humidified incubator at 37°C with 5% CO2, and cultured for up to 15 passages.
Induction of cellular senescence
The cells were treated with TNIIIA2 (in medium supplemented with 1% FBS for the indicated periods), 125 μM H2O2 (in medium supplemented with 10% FBS for 30 min and then washed and incubated with fresh medium for the indicated periods), and 4 mM NaB (in medium supplemented with 10% FBS for the indicated periods). The induction of cellular senescence was confirmed by staining for senescence-associated β-galactosidase (SA-β-gal) as described previously [37].
Cell survival and proliferation
The cells (6.0 × 103 cells/well) were seeded on 96-well culture plates coated with fibronectin (2.0 μg/mL) or type I collagen (12.5 μg/mL) in serum-free medium. The number of viable cells was evaluated using a WST-8 assay (Dojindo, Kumamoto, Japan), as described previously [18].
Measurement of oxidative stress
Reactive oxygen species (ROS) levels were measured by carboxy-H2DCFDA (Molecular Probes, Eugene, OR) as per the manufacturer’s instructions. The cells were incubated with carboxy-H2DCFDA for 30 min. Images were captured with a BIOREVO BZ-9000 (Keyence, Osaka, Japan).
Semi-quantitative PCR
Semi-quantitative PCR analysis was performed as described previously [19]. mRNA levels were examined using the following primers: p16, forward 5’-CGGAACCTCCCTCAGACATC-3’ and reverse 5’-TCATGAAGTCGACAGCTTCCG-3’; β-actin, forward 5’-TGAAGTACCCCATTGAACACG-3’ and reverse 5’-GTGCTAGGAGCCAGGGCAGT-3’; HB-EGF, forward 5’-GTGGTGCTGAAGCTCTTTC-3’ and reverse 5’-CCCCTTGCCTTTCTTCTTTC-3’; and GAPDH, forward 5’-TTCACCACCATGGAGAAGGC-3’ and reverse 5’-GGCATGGACTGTGGTCATGA-3’. Primers were obtained from Eurofins Genomics (Tokyo, Japan).
Western blot analysis
Western blot analysis was conducted as described previously [18].
Talin knockdown using small interfering RNA (siRNA)
WI-38 cells (6.0 × 104 cells/well) were transfected with 20 nM talin siRNA or 20 nM siPerfect negative control siRNA (Sigma-Aldrich) using the Lipofectamine RNAiMAX reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions and seeded on 12-well culture plates in growth medium without antibiotics. At 48 h after transfection, the cells were subjected to western blot or cellular senescence analysis. The talin-1 siRNA sequence was 5’-AAUCGUGAGGGUACUGAAACU-3’ (Sigma-Aldrich), corresponding to positions 6043-6063 relative to the mRNA start codon.
Preparation of culture supernatant from senescent fibroblasts
Fibroblasts (2.0 × 105 cells/well) were seeded on 12-well culture plates coated with fibronectin (2.0 μg/mL) or type I collagen (12.5 μg/mL) and treated with TNIIIA2 or NaB and/or peptide FNIII14 for 5 days. The cells were washed and incubated with 5 mg/mL bovine serum albumin for 3 days. The collected media were centrifuged to remove cell debris and the supernatants (conditioned media; CM) were collected and stored at -80°C until further use.
2D coculture experiments
WI-38 cells (2.5 × 104 cells/well) were seeded on 24-well culture plates for 24 h. HaCaT cells (1.0 × 104 cells/well) in a 1:1 mixture of Dulbecco’s modified Eagle’s medium-Eagle’s minimal essential medium in the presence or absence of TNIIIA2 and/or peptide FNIII14 were overlaid on the WI-38 cells. After 48 h, the cells were fixed with 4% paraformaldehyde and stained with 1% rhodanile blue for epithelial cell staining [38]. Images were captured and analyzed by Motic Image Plus 2.2S (Shimadzu Rika, Kyoto, Japan).
Focus formation assay
HaCaT cells (2.0 × 105 cells/well) were seeded on 24-well culture plates coated with fibronectin (2.0 μg/mL) or type I collagen (12.5 μg/mL) in the presence of CM. After 8-10 days, the cells were fixed with 4% paraformaldehyde and stained with crystal violet. Images were captured and analyzed by Motic Image Plus 2.2S.
Colony formation assay
A colony formation assay was performed as described previously [19].
Statistical analysis
Data are expressed as the mean ± standard deviation (SD). A two-tailed Student’s t-test or one-way analysis of variance was used to determine statistical differences. Values of P < 0.05 were considered significant.
Results
Induction of cellular senescence in normal human fibroblasts through the activation of β1-integrin by a TNC-derived peptide
To clarify the indirect role of TNC in cancer progression via non-cancerous cells, we investigated the effects of TNIIIA2 on human fibroblasts, the major component of the cancer stroma. When the human diploid fibroblast WI-38 cell line was cultured in the presence of TNIIIA2 for several days, the cells exhibited an enlarged and flattened morphology (Figure 1A), which is morphologically characteristic of senescent cells [31]. In fact, TNIIIA2-treated WI-38 cells were positive for SA-β-gal-staining, a lysosomal enzyme used widely as a marker of senescence (Figure 1A). Although the number of SA-β-gal-positive cells increased in a TNIIIA2 concentration-dependent manner (Figure 1A), the obvious appearance of SA-β-gal-positive cells required more than 3 days of culture in medium containing TNIIIA2 at concentrations higher than 12.5 μg/mL (Figure 1B and 1C), suggesting that the appearance of SA-β-gal-positive cells requires the strong and continuous stimulation of fibroblasts with TNIIIA2. The induction of SA-β-gal-positive cells by TNIIIA2 treatment was also reproduced with other normal human diploid fibroblast cell lines (TIG-1 and HFDF cells) (Figure 1D and 1E). Moreover, these changes in response to TNIIIA2 treatment were accompanied by the upregulation of p16INK4a, a marker of senescence growth arrest, concomitant with a reduction in cell proliferation (Figure 1F and 1G). Furthermore, the growth arrest caused by culture with TNIIIA2 was not recovered even when cells were re-cultured without TNIIIA2 (Figure 1H). In addition, no significant change in the appearance of SA-β-gal-positive cells was observed after removal of TNIIIA2 and further culture without TNIIIA2 (Figure 1I), confirming that the cellular changes induced by TNIIIA2 were irreversible. From these results, we concluded that strong stimulation of cells with TNIIIA2 over several days caused cellular senescence in the WI-38 fibroblasts.
Figure 1.
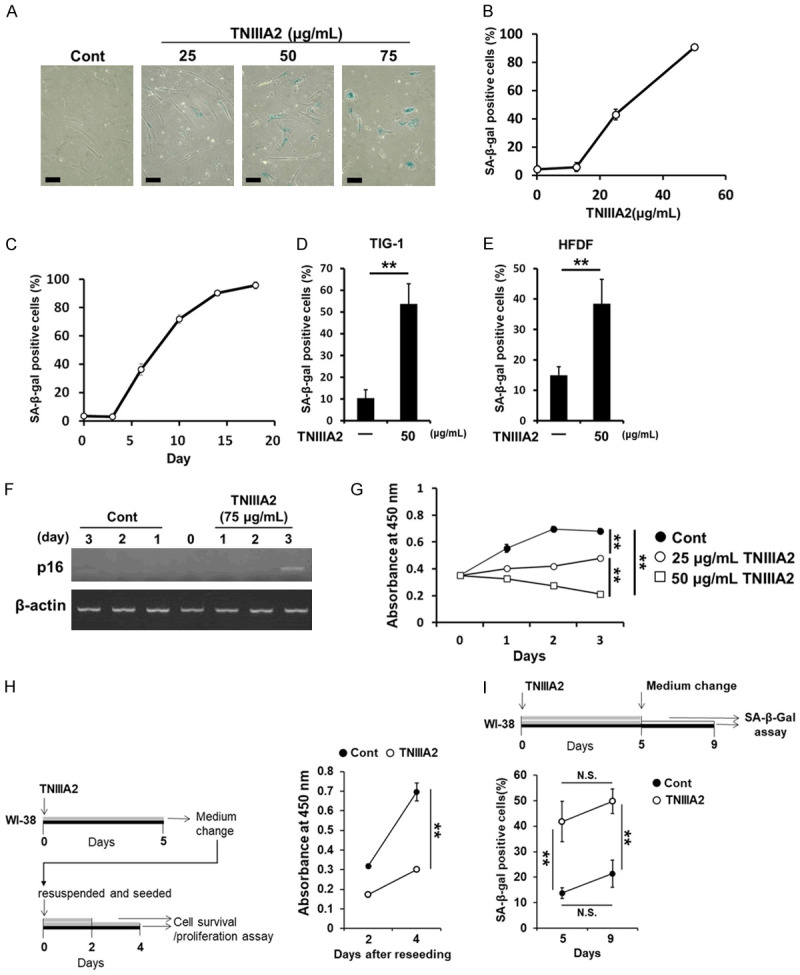
TNIIIA2 induces cellular senescence in normal human fibroblasts. WI-38 cells adhered to the fibronectin substrate were stimulated with TNIIIA2. (A and B) Cells were treated with TNIIIA2 for 5 days at the indicated concentrations and then stained for SA-β-gal activity. SA-β-gal-positive cells were counted and expressed as a percentage of the total number of cells. Scale bar, 200 μm. (C) Cells were treated with TNIIIA2 (25 μg/mL) for the indicated number of days and then subjected to an SA-β-gal assay. TIG-1 cells (D) and HFDF cells (E) were treated with TNIIIA2 for 5 days. The cells were subjected to an SA-β-gal assay. (F) After TNIIIA2 treatment for the indicated number of days after which p16 and β-actin mRNA levels were measured by semi-quantitative PCR. (G) The effect of TNIIIA2 on the proliferation of WI-38 cells as shown by a WST-8 assay. (H) WI-38 cells were treated with or without TNIIIA2 (50 μg/mL) for 5 days and then washed, resuspended, and plated. The cells were then incubated for 2 or 4 days, and subjected to a WST-8 assay. (I) WI-38 cells were treated with or without TNIIIA2 (50 μg/mL) for 5 days. Then the cells were washed, the medium was replaced, and the cells were incubated for 4 days, after which the cells were subjected to an SA-β-gal assay. Data are presented as means ± SD, **P < 0.01, N.S. not significant.
Many recent studies have shown that ROS generated in cells are the main cause of stress-induced cellular senescence [39]. As can be seen from Figure 2A, TNIIIA2 treatment caused the production of ROS. Addition with N-acetyl cysteine, a ROS scavenger, prevented the TNIIIA2-induced production of ROS (Figure 2A) and the subsequent increase in the number of SA-β-gal-positive cells (Figure 2B). Furthermore, TNIIIA2 treatment also caused significant DNA damage, as judged by the phosphorylation of γH2AX, a marker of DNA damage (Figure 2C). These results suggest that TNIIIA2-induced cellular senescence in fibroblasts is due to ROS production and subsequent DNA damage.
Figure 2.
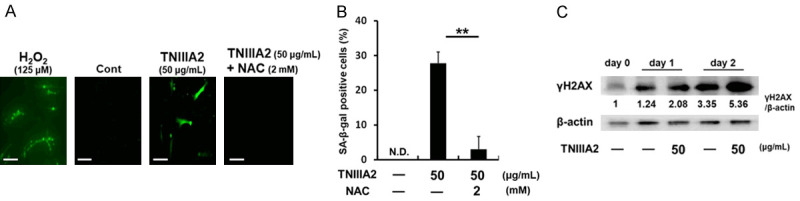
TNIIIA2-induced senescence of fibroblasts is attributed to the DNA damage response. (A) WI-38 cells were treated with TNIIIA2 in the presence or absence of N-acetyl cysteine (NAC) for 6 h, and ROS production was visualized by carboxy-H2DCFDA. Scale bar, 200 μm. (B) Cells treated with TNIIIA2 in the presence of absence of NAC were subjected to an SA-β-gal assay. Data are presented as means ± SD, **P < 0.01. N.D., not detected. (C) Cells treated with TNIIIA2 were subjected to western blot analysis of γH2AX and β-actin.
Because TNIIIA2 was previously found to be a peptide factor capable of activating β1-integrin [36], β1-integrin activation may play a role in TNIIIA2-induced cellular senescence. To clarify this, we performed the following examinations. First, flow cytometric analysis using a monoclonal antibody that recognizes a conformation-specific epitope of active β1-integrin showed that TNIIIA2 activated β1-integrin in WI-38 cells (Figure 3A), followed by accelerated adhesion to the fibronectin substrate (Figure 3B). Second, pretreatment of WI-38 cells with the anti-β1-integrin neutralizing BV7 antibody prevented the appearance of SA-β-gal-positive cells induced by TNIIIA2 (Figure 3C). Third, siRNA silencing of talin, an integrin-binding cytoplasmic adaptor protein that is required for the activation and retention of β1-integrin [40], inhibited the appearance of SA-β-gal-positive cells induced by TNIIIA2 (Figure 3D and 3E). Fourth, peptide FNIII14, which was previously found as a β1-integrin inactivator [36], actually inactivated β1-integrin in WI-38 cells (Figure 3A and 3B) and efficiently inhibited the appearance of SA-β-gal-positive cells induced by TNIIIA2 (Figure 3F). Taken together, these results demonstrate that β1-integrin activation is the main cause of TNIIIA2-induced cellular senescence in WI-38 fibroblasts.
Figure 3.

TNIIIA2-induced cellular senescence is based on β1-integrin activation. (A) WI-38 cells were incubated with or without TNIIIA2 (50 μg/mL) or Mn2+ (4 mM) in the presence or absence of peptide FNIII14 (50 μg/mL) for 30 min. The activation of β1-integrin was measured by flow cytometry using antibody AG89, which recognizes a conformation-specific epitope of active β1-integrin. Dashed lines in each panel show the mean fluorescence intensity of the control. (B) WI-38 cells suspended in serum-free medium were seeded with or without TNIIIA2 in the presence or absence of peptide FNIII14 on 96-well plates coated with fibronectin (2.0 μg/mL). Adhered cells were stained with crystal violet and counted under a microscope. Statistical analysis was conducted to compare the total percentages of spread and adhered cells. (C) WI-38 cells were pre-incubated with function-blocking antibodies against β1-integrin (BV7, 10 μg/mL), and then treated with TNIIIA2 for 5 days. The cells were subjected to an SA-β-gal assay. (D) At 48 h after talin siRNA transfection, talin levels were measured by western blot analysis. (E) Effect of siRNA-mediated talin depletion on TNIIIA2-induced SA-β-gal activity. At 48 h after siRNA transfection, the cells were treated with TNIIIA2 for 5 days. The cells were subjected to an SA-β-gal assay. (F) WI-38 cells were treated with TNIIIA2 in the presence or absence of peptide FNIII14 (50 μg/mL) for 5 days and then subjected to an SA-β-gal assay.
It has been reported that cellular senescence can be induced by exogenous addition of H2O2 [31]. Actually, the addition of H2O2 to the culture system resulted in cellular senescence in the WI-38 fibroblasts, as judged by SA-β-gal staining (Figure 4A). Interestingly, this H2O2-induced cellular senescence was also prevented by further addition of either an anti-β1-integrin neutralizing antibody (Figure 4A) or peptide FNIII14 (Figure 4B). These results suggest that β1-integrin activation functions as the common driving force of cellular senescence, at least in human fibroblasts. Supporting this assumption, Mn2+ [41], a well-known activator of β1-integrin (Figure 3A), induced cellular senescence in WI-38 fibroblasts, as determined by SA-β-gal-staining (Figure 4C). As observed with the induction of cellular senescence by TNIIIA2 (see Figure 1C), Mn2+ treatment also took a few days to induce senescence (Figure 4C). Taken together with the results of Figure 1B and 1C, these findings suggest that the induction of cellular senescence in human fibroblasts might require the sustained activation of β1-integrin. On the other hand, peptide FNIII14 prevented induction of cellular senescence caused by β1-integrin activation as mentioned above, but could not revert the state of SA-β-gal-staining of cells induced by TNIIIA2 (Figure 4D), further supporting that cellular senescence induced by β1-integrin activation is irreversible.
Figure 4.
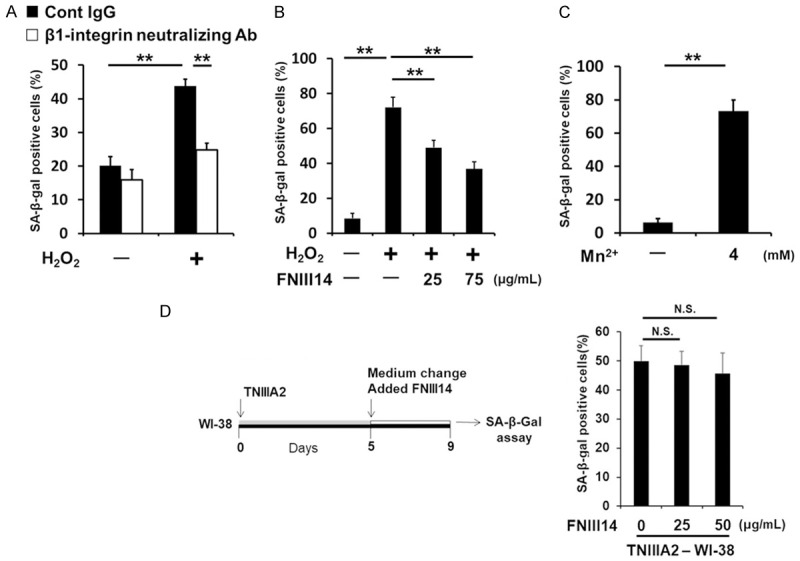
β1-integrin activation is a common driving force for cellular senescence in human fibroblasts. WI-38 cells were treated with or without an anti-β1-integrin-blocking antibody (BV7, 10 μg/mL) (A) or with or without peptide FNIII14 (B), and the cells were exposed to 125 μM H2O2 for 30 min, and then washed out and cultured in the presence or absence of BV7 (A) or peptide FNIII14 (B), and subjected to an SA-β-gal assay. (C) WI-38 cells were treated with Mn2+ for 5 days, and subjected to an SA-β-gal assay. (D) WI-38 cells were treated with TNIIIA2 for 5 days and then washed and treated with or without peptide FNIII14 for 4 days before being subjected to an SA-β-gal assay. Data are presented as means ± SD, **P < 0.01, N.S. not significant.
Malignant transformation of preneoplastic epithelial cells by TNIIIA2-induced senescent fibroblasts
Next, we examined whether TNIIIA2-induced senescent fibroblasts are associated with malignant transformation. For this purpose, preneoplastic epithelial HaCaT cells, which exhibit aneuploidy, were used. HaCaT cells were cocultured with non-senescent WI-38 cells or TNIIIA2-induced senescent WI-38 cells and then stained with rhodanile blue, which selectively stains epithelial cells [38]. As a result, HaCaT cell growth was found to be promoted by coculturing with TNIIIA2-induced senescent WI-38 cells (represented as “TNIIIA2-WI-38” in Figure 5A) compared with non-senescent WI-38 cells (“Cont-WI-38” in Figure 5A). The promotion of cell growth by TNIIIA2-induced senescent cells was comparable to that induced by WI-38 cells treated with NaB, a histone deacetylase inhibitor, which has been reported to induce cellular senescence, accompanied by induction of SASP factors [42,43] (“NaB-WI-38” in Figure 5A). These findings suggest that TNIIIA2-induced senescent WI-38 cells secrete SASP factors that promote HaCaT cell growth. Indeed, CM collected from TNIIIA2-induced senescent WI-38 cells promoted HaCaT cell growth (Figure 5B and 5C). Similar results were obtained with TNIIIA2-induced senescent TIG-1 cells (Figure 5D). Importantly, CM from TNIIIA2-induced senescent WI-38 cells strongly enhanced the anchorage-independent colony-forming ability of HaCaT cells in soft agarose (Figure 5E). Furthermore, HaCaT cells treated with CM from TNIIIA2-induced senescent fibroblasts (WI-38 and TIG-1) grew in multiple layers in 2D coculture, forming foci consisting of multilayered HaCaT cells (Figure 5F and 5G). These results suggest that TNIIIA2-induced senescent fibroblasts secrete SASP factors that have the ability to induce the malignant transformation of preneoplastic epithelial HaCaT cells.
Figure 5.
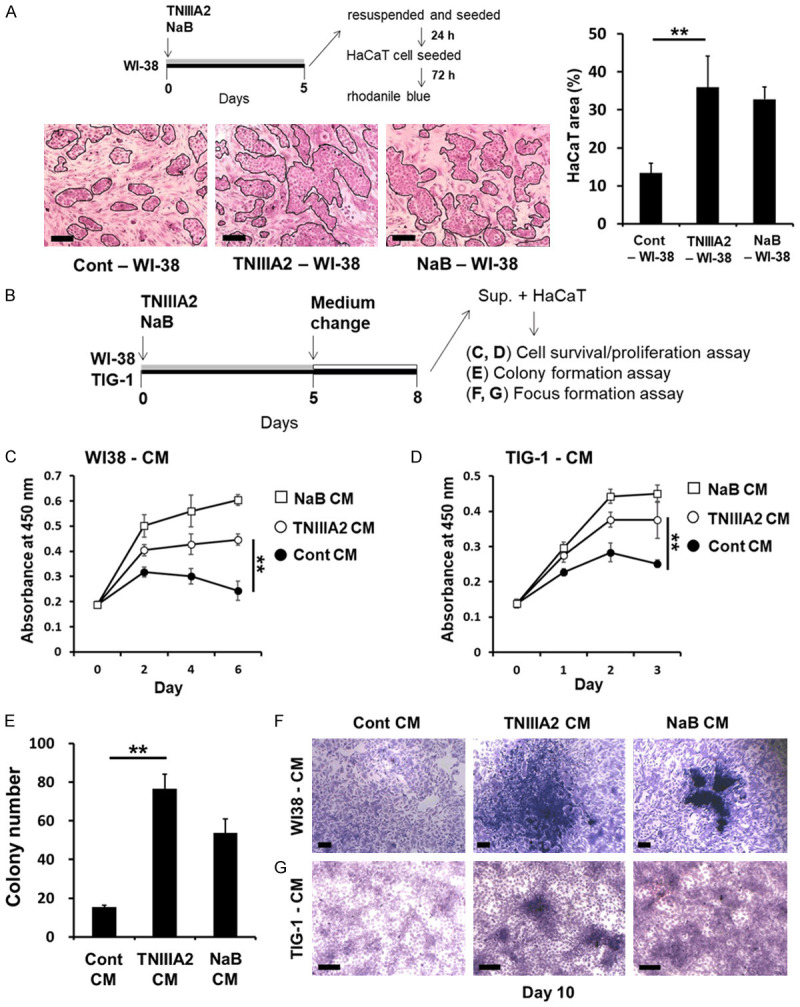
Secreted factors from TNIIIA2-induced senescent fibroblasts induce the malignant transformation of HaCaT cells. (A) WI-38 cells were treated with either TNIIIA2 (50 μg/mL) or NaB (4 mM) for 5 days, and the cells were resuspended and plated. After incubation for 24 h, WI-38 cells were cocultured with HaCaT cells in 2D coculture experiments for 3 days. HaCaT area was measured by rhodanile blue staining, as described in the Materials and Methods. Scale bar, 200 μm. (B) WI-38 or TIG-1 cells were treated with either TNIIIA2 (50 μg/mL) or NaB (4 mM) for 5 days, and the media were removed and replaced, and then the cells were incubated for 3 days. CM were collected (Cont CM: control; TNIIIA2 CM: cultured with TNIIIA2; NaB CM: cultured with NaB). In (C and D), HaCaT cells were cultured in CM from WI-38 cells (C) or TIG-1 cells (D), and cell proliferation was assayed by a WST-8 assay. (E) HaCaT cells were cultured in CM from WI-38 cells, and HaCaT cells suspended in CM were subjected to a colony formation assay, as described in the Materials and Methods. HaCaT cells were cultured in CM from WI-38 cells (F) or TIG-1 cells (G), and the cells were subjected to a focus formation assay. Data are presented as means ± SD, **P < 0.01. Scale bar, 200 μm.
Notably, function-blocking experiments revealed that TNIIIA2-induced senescent cells secreted HB-EGF as a detrimental SASP factor; a neutralizing antibody against HB-EGF impeded the increased proliferation (Figure 6A-D) and enhanced the colony-forming ability of the HaCaT cells (Figure 6C and 6E), which were conferred by TNIIIA2-induced senescent WI-38 cells. Supporting these results, the induction of HB-EGF mRNA was observed in TNIIIA2-induced senescent fibroblasts (Figure 6F). Meanwhile, exogenously application of recombinant human HB-EGF enhanced HaCaT cell proliferation on the 2D culture plates (Figure 6G) as well as colony formation in soft agarose (Figure 6H). In conclusion, TNIIIA2 peptide derived from TNC, which is highly expressed in the tumor stroma, induces cellular senescence in stromal fibroblasts by activating β1-integrin, resulting in the malignant progression of preneoplastic epithelial cells though the secretion of SASP factors such as HB-EGF.
Figure 6.
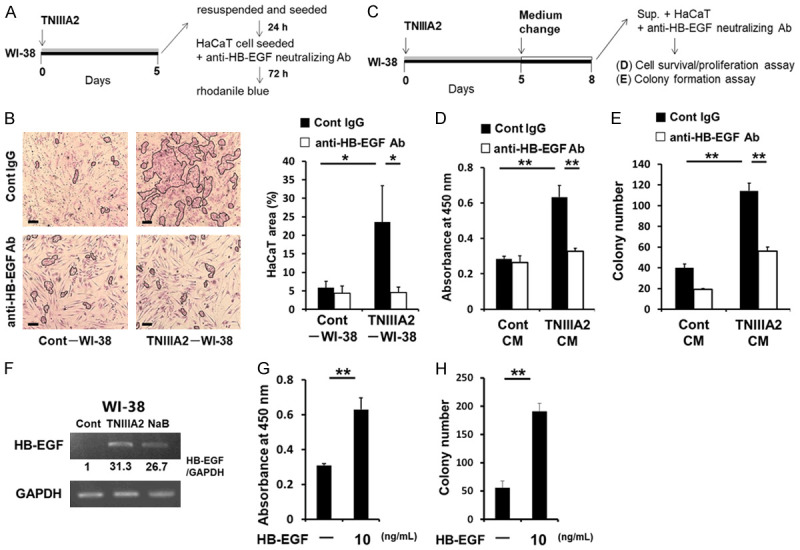
TNIIIA2 secreted SASP factors, including HB-EGF, induce the malignant transformation of HaCaT cells. In (A and B), HaCaT cells cocultured with TNIIIA2-induced senescent WI-38 cells were incubated with either an anti-HB-EGF neutralizing antibody or isotype control. After incubation for 72 h, the HaCaT area was measured by rhodanile blue staining. Scale bar, 200 μm. (C) HaCaT cells treated with CM from TNIIIA2-induced senescent WI-38 cells were incubated with either an anti-HB-EGF neutralizing antibody or isotype control and then subjected to a WST-8 (D) or colony formation (E) assay. (F) WI-38 cells were treated with either TNIIIA2 (50 μg/mL) or NaB (4 mM) to induce cellular senescence, as described in the Materials and Methods. HB-EGF and GAPDH mRNA levels were then measured by semi-quantitative PCR. In (G and H), HaCaT cells were treated with recombinant human HB-EGF, and the cells were subjected to a WST-8 assay on day 2 (G) or colony formation assay on day 10 (H). Data are presented as means ± SD, *P < 0.05, **P < 0.01.
Discussion
TNC is considered to be closely involved in cancer development and progression [2], but the substantial role of this protein is controversial. In the present study, we showed that the TNC-derived peptide TNIIIA2 has the ability to induce cellular senescence in fibroblasts, and the soluble factors secreted from TNIIIA2-induced senescent fibroblasts contribute to the malignant transformation of preneoplastic epithelial cells (Figure 7). The induction of cellular senescence by TNIIIA2 was attributed to β1-integrin activation and the resulting production of ROS, but it was necessary to activate β1-integrin for an extended period of time (Figure 1B and 1C). TNC, the parent molecule of TNIIIA2, is upregulated in inflammatory lesions, including inflammatory bowel disease and fibrosis, and its expression is further upregulated with increased disease activity [44,45]. Moreover, TNC positively regulates its own expression [13], resulting in a spiraling increase in TNC expression [46]. Indeed, it was reported that tissue concentrations of TNC can reach approximately single-digit milligram per milliliter levels [47], although they are dependent on tissue location and pathological state. Thus, TNIIIA2 may be highly released from continually increasing levels of TNC in precancerous lesions and confer malignant properties on preneoplastic epithelial cells through the induction of senescence in stromal fibroblasts based on β1-integrin activation, serving as an important determinant of cancer development and progression.
Figure 7.
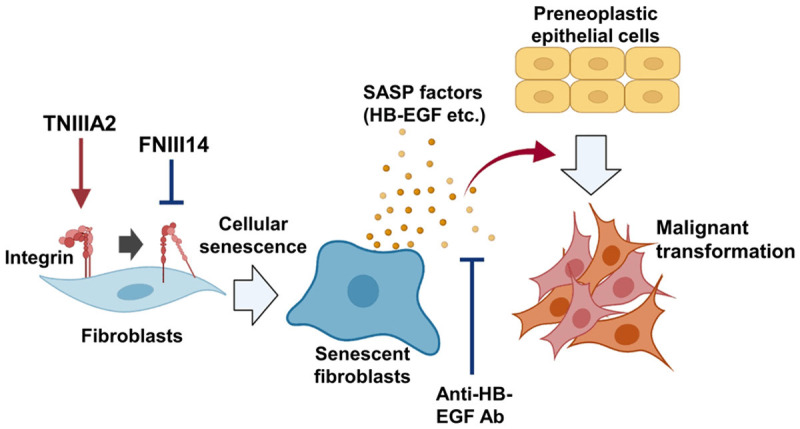
A model of malignant transformation of preneoplastic cells via factors secreted from TNIIIA2-induced senescent fibroblasts. Activation of β1-integrin by TNIIIA2 induces fibroblast senescence. Factors secreted by TNIIIA2-induced senescent fibroblasts, including HB-EGF, are involved in the malignant transformation of preneoplastic cells. Inactivation of β1-integrin by peptide FNIII14 inhibits TNIIIA2-induced cellular senescence. Created using BioRender.com.
Increasing evidence suggests that senescent fibroblasts promote the phenotypic transformation of preneoplastic cells and their malignant progression [38,48,49]. TNC expression, which is upregulated in the tumor stroma as a marker of cancer-associated fibroblasts, is correlated with a poor prognosis in several malignancies [46,50]. In particular, high levels of large TNC variants are independently associated with disease progression [51,52] and a poor prognosis, and high degradation levels of TNC variants result in the increased incidence of recurrence in patients with early stage non-small cell lung cancer [53], indicating that the cryptic functional TNIIIA2 site within large TNC variants can become functional in the tumor microenvironment and acts on fibroblasts to confer malignant properties on preneoplastic cells. To verify whether the findings of the present study actually occur in vivo, further investigations are needed using more complex experimental systems such as in vivo cancer models and clinical samples.
Interestingly, in addition to the effect of β1-integrin activation by TNIIIA2 on the induction of cellular senescence in fibroblasts, we showed that Mn2+, a β1-integrin activator, also induces cellular senescence, and an anti-β1-integrin neutralizing antibody and peptide FNIII14, both of which inactivate β1-integrin, suppressed H2O2-induced cellular senescence. CCN1 and CCN2, which are typical matricellular proteins, have been reported to induce cellular senescence in fibroblasts by binding to β1-integrin [32,33]. More recently, Shin and colleagues demonstrated that the depletion of βPAK-interacting exchange factor in fibroblasts induces cellular senescence via the phosphorylation of focal adhesion kinase and paxillin, and the induced cellular senescence was suppressed by treatment with arginyl-glycyl-aspartic acid peptide or focal adhesion kinase inhibitor [34]. These studies support the legitimacy of our conclusion that the activation of β1-integrin plays a pivotal role as a driving force in the induction of cellular senescence, at least in human fibroblasts. This implies that drugs capable of inactivating β1-integrin can provide a novel strategy, so-called “senotherapy”, for chemotherapy/chemoprevention of senescence-related diseases including cancer.
Many recent studies have indicated that the ECM stiffness of tumors increases with cancer progression [54,55]. Moreover, the interplay between cancer cells and cancer-associated fibroblasts has been implicated in increased ECM stiffness in the tumor microenvironment, leading to cancer aggression [56,57]. In studies on mammary tumorigenesis in the MMTV/PyMT mouse model, preneoplastic lesions show increased ECM stiffness compared with normal mammary gland, and ECM stiffness also correlates with malignant stage [58]. In an analysis of human samples, ductal in situ carcinoma, which is the preneoplastic stage of breast carcinoma, shows increased β1-integrin activation compared with normal breast tissue, and this activation also correlates with malignant stage [59]. More recently, glioblastoma aggression was shown to correlate with the stiffness of a TNC-enriched ECM, and ECM stiffness is increased in glioblastoma cells expressing an auto-clustering β1-integrin mutant, thereby promoting tumor burden [60,61]. In addition, cells cultured on stiff substrata promote the production of ROS compared with those cultured on soft substrata, and ROS production is increased in cells expressing the auto-clustering β1-integrin mutant [62]. These observations support the suggestion that the TNIIIA2 matricryptic site derived from TNC may create a tumor microenvironment with pro-adhesive activity based on the aberrant activation of β1-integrin, leading to increased ECM stiffness and the induction of senescence in fibroblasts via the β1-integrin-ROS pathway.
HB-EGF is highly expressed in multiple types of cancer and its expression level is correlated with a poor prognosis in several malignancies [63,64]. Murata and colleagues reported that HB-EGF expression is not detected in normal cervical stroma, but is expressed at high levels in stromal fibroblasts in the tumor microenvironment, and its levels correlate with disease stage in uterine cervical cancers, indicating that HB-EGF may play a critical role in cancer progression via the interaction of cancer cells with cancer-associated fibroblasts [65]. Moreover, Sasaki and colleagues reported that HB-EGF derived from colonic fibroblasts promotes colon carcinogenesis in colitis-associated carcinogenesis in mice [66]. Our observations indicate that the procancer effect of TNIIIA2-induced senescent fibroblasts might result from, at least in part, HB-EGF secretion. Additional experiments to examine whether TNIIIA2-induced senescent fibroblasts secrete other SASP factors with procancer effects on preneoplastic epithelial cells are now in progress.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (Grant# 23590090) from the Japan Science and Technology Agency. This work was also supported by Center for Clinical and Translational Research of Kyushu University.
Disclosure of conflict of interest
None.
References
- 1.Murphy-Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix Biol. 2014;37:1–14. doi: 10.1016/j.matbio.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Midwood KS, Hussenet T, Langlois B, Orend G. Advances in tenascin-C biology. Cell Mol Life Sci. 2011;68:3175–3199. doi: 10.1007/s00018-011-0783-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Z, Zhang C, Feng Y, Quan M, Cui Y, Xuan Y. Tenascin-C predicts poor outcomes for patients with colorectal cancer and drives cancer stemness via hedgehog signaling pathway. Cancer Cell Int. 2020;20:122. doi: 10.1186/s12935-020-01188-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angel I, Pilo Kerman O, Rousso-Noori L, Friedmann-Morvinski D. Tenascin C promotes cancer cell plasticity in mesenchymal glioblastoma. Oncogene. 2020;39:6990–7004. doi: 10.1038/s41388-020-01506-6. [DOI] [PubMed] [Google Scholar]
- 5.Sarkar S, Mirzaei R, Zemp FJ, Wei W, Senger DL, Robbins SM, Yong VW. Activation of NOTCH signaling by tenascin-C promotes growth of human brain tumor-initiating cells. Cancer Res. 2017;77:3231–3243. doi: 10.1158/0008-5472.CAN-16-2171. [DOI] [PubMed] [Google Scholar]
- 6.Cai J, Du S, Wang H, Xin B, Wang J, Shen W, Wei W, Guo Z, Shen X. Tenascin-C induces migration and invasion through JNK/c-Jun signalling in pancreatic cancer. Oncotarget. 2017;8:74406–74422. doi: 10.18632/oncotarget.20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Z, Schwenzer A, Rupp T, Murdamoothoo D, Vegliante R, Lefebvre O, Klein A, Hussenet T, Orend G. Tenascin-C promotes tumor cell migration and metastasis through integrin α9β1-Mediated YAP inhibition. Cancer Res. 2018;78:950–961. doi: 10.1158/0008-5472.CAN-17-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirzaei R, Sarkar S, Dzikowski L, Rawji KS, Khan L, Faissner A, Bose P, Yong VW. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oncoimmunology. 2018;7:e1478647. doi: 10.1080/2162402X.2018.1478647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steitz AM, Steffes A, Finkernagel F, Unger A, Sommerfeld L, Jansen JM, Wagner U, Graumann J, Müller R, Reinartz S. Tumor-associated macrophages promote ovarian cancer cell migration by secreting transforming growth factor beta induced (TGFBI) and tenascin C. Cell Death Dis. 2020;11:249. doi: 10.1038/s41419-020-2438-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murdamoothoo D, Sun Z, Yilmaz A, Riegel G, Abou-Faycal C, Deligne C, Velazquez-Quesada I, Erne W, Nascimento M, Mörgelin M, Cremel G, Paul N, Carapito R, Veber R, Dumortier H, Yuan J, Midwood KS, Loustau T, Orend G. Tenascin-C immobilizes infiltrating T lymphocytes through CXCL12 promoting breast cancer progression. EMBO Mol Med. 2021;13:e13270. doi: 10.15252/emmm.202013270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, Hynes RO, Jain RK, Janowitz T, Jorgensen C, Kimmelman AC, Kolonin MG, Maki RG, Powers RS, Puré E, Ramirez DC, Scherz-Shouval R, Sherman MH, Stewart S, Tlsty TD, Tuveson DA, Watt FM, Weaver V, Weeraratna AT, Werb Z. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20:174–186. doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brechbuhl HM, Barrett AS, Kopin E, Hagen JC, Han AL, Gillen AE, Finlay-Schultz J, Cittelly DM, Owens P, Horwitz KB, Sartorius CA, Hansen K, Kabos P. Fibroblast subtypes define a metastatic matrisome in breast cancer. JCI Insight. 2020;5:e130751. doi: 10.1172/jci.insight.130751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katoh D, Kozuka Y, Noro A, Ogawa T, Imanaka-Yoshida K, Yoshida T. Tenascin-C induces phenotypic changes in fibroblasts to myofibroblasts with high contractility through the integrin αvβ1/transforming growth factor β/SMAD signaling axis in human breast cancer. Am J Pathol. 2020;190:2123–2135. doi: 10.1016/j.ajpath.2020.06.008. [DOI] [PubMed] [Google Scholar]
- 14.Midwood KS, Chiquet M, Tucker RP, Orend G. Tenascin-C at a glance. J Cell Sci. 2016;129:4321–4327. doi: 10.1242/jcs.190546. [DOI] [PubMed] [Google Scholar]
- 15.Siri A, Knäuper V, Veirana N, Caocci F, Murphy G, Zardi L. Different susceptibility of small and large human tenascin-C isoforms to degradation by matrix metalloproteinases. J Biol Chem. 1995;270:8650–8654. doi: 10.1074/jbc.270.15.8650. [DOI] [PubMed] [Google Scholar]
- 16.Davis GE, Bayless KJ, Davis MJ, Meininger GA. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol. 2000;156:1489–1498. doi: 10.1016/S0002-9440(10)65020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saito Y, Imazeki H, Miura S, Yoshimura T, Okutsu H, Harada Y, Ohwaki T, Nagao O, Kamiya S, Hayashi R, Kodama H, Handa H, Yoshida T, Fukai F. A peptide derived from tenascin-C induces beta1 integrin activation through syndecan-4. J Biol Chem. 2007;282:34929–34937. doi: 10.1074/jbc.M705608200. [DOI] [PubMed] [Google Scholar]
- 18.Tanaka R, Seki Y, Saito Y, Kamiya S, Fujita M, Okutsu H, Iyoda T, Takai T, Owaki T, Yajima H, Taira J, Hayashi R, Kodama H, Matsunaga T, Fukai F. Tenascin-C-derived peptide TNIIIA2 highly enhances cell survival and platelet-derived growth factor (PDGF)-dependent cell proliferation through potentiated and sustained activation of integrin α5β1. J Biol Chem. 2014;289:17699–17708. doi: 10.1074/jbc.M113.546622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujita M, Yamamoto T, Iyoda T, Fujisawa T, Sasada M, Nagai R, Kudo C, Otsuka K, Kamiya S, Kodama H, Fukai F. Aggressive progression in glioblastoma cells through potentiated activation of integrin α5β1 by the tenascin-C-derived peptide TNIIIA2. Mol Cancer Ther. 2019;18:1649–1658. doi: 10.1158/1535-7163.MCT-18-1251. [DOI] [PubMed] [Google Scholar]
- 20.Fujita M, Yamamoto T, Iyoda T, Fujisawa T, Nagai R, Kudo C, Sasada M, Kodama H, Fukai F. Autocrine production of PDGF stimulated by the tenascin-C-derived peptide TNIIIA2 induces hyper-proliferation in glioblastoma cells. Int J Mol Sci. 2019;20:3183. doi: 10.3390/ijms20133183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujita M, Sasada M, Iyoda T, Nagai R, Kudo C, Yamamoto T, Osada S, Kodama H, Fukai F. Anoikis resistance conferred by tenascin-C-derived peptide TNIIIA2 and its disruption by integrin inactivation. Biochem Biophys Res Commun. 2021;536:14–19. doi: 10.1016/j.bbrc.2020.12.050. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki H, Sasada M, Kamiya S, Ito Y, Watanabe H, Okada Y, Ishibashi K, Iyoda T, Yanaka A, Fukai F. The promoting effect of the extracellular matrix peptide TNIIIA2 derived from tenascin-c in colon cancer cell infiltration. Int J Mol Sci. 2017;18:181. doi: 10.3390/ijms18010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujita M, Ito-Fujita Y, Iyoda T, Sasada M, Okada Y, Ishibashi K, Osawa T, Kodama H, Fukai F, Suzuki H. Peptide TNIIIA2 derived from tenascin-C contributes to malignant progression in colitis-associated colorectal cancer via β1-Integrin activation in fibroblasts. Int J Mol Sci. 2019;20:2752. doi: 10.3390/ijms20112752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlseder J, Smogorzewska A, de Lange T. Senescence induced by altered telomere state, not telomere loss. Science. 2002;295:2446–2449. doi: 10.1126/science.1069523. [DOI] [PubMed] [Google Scholar]
- 25.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 26.Chen Q, Fischer A, Reagan JD, Yan LJ, Ames BN. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci U S A. 1995;92:4337–4341. doi: 10.1073/pnas.92.10.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhinn M, Ritschka B, Keyes WM. Cellular senescence in development, regeneration and disease. Development. 2019;146:dev151837. doi: 10.1242/dev.151837. [DOI] [PubMed] [Google Scholar]
- 28.Schosserer M, Grillari J, Breitenbach M. The dual role of cellular senescence in developing tumors and their response to cancer therapy. Front Oncol. 2017;7:278. doi: 10.3389/fonc.2017.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malaquin N, Martinez A, Rodier F. Keeping the senescence secretome under control: molecular reins on the senescence-associated secretory phenotype. Exp Gerontol. 2016;82:39–49. doi: 10.1016/j.exger.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 30.Wyld L, Bellantuono I, Tchkonia T, Morgan J, Turner O, Foss F, George J, Danson S, Kirkland JL. Senescence and cancer: a review of clinical implications of senescence and senotherapies. Cancers (Basel) 2020;12:2134. doi: 10.3390/cancers12082134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–453. doi: 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jun JI, Lau LF. CCN2 induces cellular senescence in fibroblasts. J Cell Commun Signal. 2017;11:15–23. doi: 10.1007/s12079-016-0359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shin EY, Park JH, You ST, Lee CS, Won SY, Park JJ, Kim HB, Shim J, Soung NK, Lee OJ, Schwartz MA, Kim EG. Integrin-mediated adhesions in regulation of cellular senescence. Sci Adv. 2020;6:eaay3909. doi: 10.1126/sciadv.aay3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miekka SI, Ingham KC, Menache D. Rapid methods for isolation of human plasma fibronectin. Thromb Res. 1982;27:1–14. doi: 10.1016/0049-3848(82)90272-9. [DOI] [PubMed] [Google Scholar]
- 36.Fujita M, Sasada M, Iyoda T, Fukai F. Involvement of integrin-activating peptides derived from Tenascin-C in cancer aggression and new anticancer strategy using the fibronectin-derived integrin-inactivating peptide. Molecules. 2020;25:3239. doi: 10.3390/molecules25143239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu T, Finkel T. Free radicals and senescence. Exp Cell Res. 2008;314:1918–1922. doi: 10.1016/j.yexcr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Z, Costell M, Fässler R. Integrin activation by talin, kindlin and mechanical forces. Nat Cell Biol. 2019;21:25–31. doi: 10.1038/s41556-018-0234-9. [DOI] [PubMed] [Google Scholar]
- 41.Ni H, Li A, Simonsen N, Wilkins JA. Integrin activation by dithiothreitol or Mn2+ induces a ligand-occupied conformation and exposure of a novel NH2-terminal regulatory site on the beta1 integrin chain. J Biol Chem. 1998;273:7981–7987. doi: 10.1074/jbc.273.14.7981. [DOI] [PubMed] [Google Scholar]
- 42.Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106:17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH. Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol Cell Biol. 1996;16:5210–5218. doi: 10.1128/mcb.16.9.5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ning L, Li S, Gao J, Ding L, Wang C, Chen W, Shan G, Zhang F, Yu J, Xu G. Tenascin-C Is increased in inflammatory bowel disease and is associated with response to infliximab therapy. Biomed Res Int. 2019;2019:1475705. doi: 10.1155/2019/1475705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, Lafyatis R, Lee J, Hinchcliff M, Feghali-Bostwick C, Lakota K, Budinger GR, Raparia K, Tamaki Z, Varga J. Tenascin-C drives persistence of organ fibrosis. Nat Commun. 2016;7:11703. doi: 10.1038/ncomms11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yeo SY, Lee KW, Shin D, An S, Cho KH, Kim SH. A positive feedback loop bi-stably activates fibroblasts. Nat Commun. 2018;9:3016. doi: 10.1038/s41467-018-05274-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lightner VA, Slemp CA, Erickson HP. Localization and quantitation of hexabrachion (tenascin) in skin, embryonic brain, tumors, and plasma. Ann N Y Acad Sci. 1990;580:260–275. doi: 10.1111/j.1749-6632.1990.tb17935.x. [DOI] [PubMed] [Google Scholar]
- 48.Pazolli E, Luo X, Brehm S, Carbery K, Chung JJ, Prior JL, Doherty J, Demehri S, Salavaggione L, Piwnica-Worms D, Stewart SA. Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res. 2009;69:1230–1239. doi: 10.1158/0008-5472.CAN-08-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lugo R, Gabasa M, Andriani F, Puig M, Facchinetti F, Ramírez J, Gómez-Caro A, Pastorino U, Fuster G, Almendros I, Gascón P, Davalos A, Reguart N, Roz L, Alcaraz J. Heterotypic paracrine signaling drives fibroblast senescence and tumor progression of large cell carcinoma of the lung. Oncotarget. 2016;7:82324–82337. doi: 10.18632/oncotarget.10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ni WD, Yang ZT, Cui CA, Cui Y, Fang LY, Xuan YH. Tenascin-C is a potential cancer-associated fibroblasts marker and predicts poor prognosis in prostate cancer. Biochem Biophys Res Commun. 2017;486:607–612. doi: 10.1016/j.bbrc.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 51.Takeda A, Otani Y, Iseki H, Takeuchi H, Aikawa K, Tabuchi S, Shinozuka N, Saeki T, Okazaki Y, Koyama I. Clinical significance of large Tenascin-C spliced variant as a potential biomarker for colorectal cancer. World J Surg. 2007;31:388–394. doi: 10.1007/s00268-006-0328-6. [DOI] [PubMed] [Google Scholar]
- 52.Hagiwara K, Harimoto N, Yokobori T, Muranushi R, Hoshino K, Gantumur D, Yamanaka T, Ishii N, Tsukagoshi M, Igarashi T, Tanaka H, Watanabe A, Kubo N, Araki K, Hosouchi Y, Shirabe K. High Co-expression of Large Tenascin C Splice variants in stromal tissue and annexin A2 in cancer cell membranes is associated with poor prognosis in pancreatic cancer. Ann Surg Oncol. 2020;27:924–930. doi: 10.1245/s10434-019-07708-x. [DOI] [PubMed] [Google Scholar]
- 53.Cai M, Onoda K, Takao M, Kyoko IY, Shimpo H, Yoshida T, Yada I. Degradation of tenascin-C and activity of matrix metalloproteinase-2 are associated with tumor recurrence in early stage non-small cell lung cancer. Clin Cancer Res. 2002;8:1152–1156. [PubMed] [Google Scholar]
- 54.Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP, Del Río Hernández A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis. 2017;6:e352. doi: 10.1038/oncsis.2017.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, Hofman P, Bellvert F, Meneguzzi G, Bulavin DV, Estrach S, Feral CC, Chan SY, Bozec A, Gaggioli C. Tumor-stroma mechanics coordinate amino acid availability to sustain tumor growth and malignancy. Cell Metab. 2019;29:124–140.e10. doi: 10.1016/j.cmet.2018.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jang I, Beningo KA. Integrins, CAFs and mechanical forces in the progression of cancer. Cancers (Basel) 2019;11:721. doi: 10.3390/cancers11050721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res. 2012;10:1403–1418. doi: 10.1158/1541-7786.MCR-12-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, Weaver VM. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb) 2015;7:1120–1134. doi: 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miroshnikova YA, Mouw JK, Barnes JM, Pickup MW, Lakins JN, Kim Y, Lobo K, Persson AI, Reis GF, McKnight TR, Holland EC, Phillips JJ, Weaver VM. Tissue mechanics promote IDH1-dependent HIF1α-tenascin C feedback to regulate glioblastoma aggression. Nat Cell Biol. 2016;18:1336–1345. doi: 10.1038/ncb3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barnes JM, Kaushik S, Bainer RO, Sa JK, Woods EC, Kai F, Przybyla L, Lee M, Lee HW, Tung JC, Maller O, Barrett AS, Lu KV, Lakins JN, Hansen KC, Obernier K, Alvarez-Buylla A, Bergers G, Phillips JJ, Nam DH, Bertozzi CR, Weaver VM. A tension-mediated glycocalyx-integrin feedback loop promotes mesenchymal-like glioblastoma. Nat Cell Biol. 2018;20:1203–1214. doi: 10.1038/s41556-018-0183-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee K, Chen QK, Lui C, Cichon MA, Radisky DC, Nelson CM. Matrix compliance regulates Rac1b localization, NADPH oxidase assembly, and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23:4097–4108. doi: 10.1091/mbc.E12-02-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ongusaha PP, Kwak JC, Zwible AJ, Macip S, Higashiyama S, Taniguchi N, Fang L, Lee SW. HB-EGF is a potent inducer of tumor growth and angiogenesis. Cancer Res. 2004;64:5283–5290. doi: 10.1158/0008-5472.CAN-04-0925. [DOI] [PubMed] [Google Scholar]
- 64.Shin CH, Robinson JP, Sonnen JA, Welker AE, Yu DX, VanBrocklin MW, Holmen SL. HBEGF promotes gliomagenesis in the context of Ink4a/Arf and pten loss. Oncogene. 2017;36:4610–4618. doi: 10.1038/onc.2017.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murata T, Mizushima H, Chinen I, Moribe H, Yagi S, Hoffman RM, Kimura T, Yoshino K, Ueda Y, Enomoto T, Mekada E. HB-EGF and PDGF mediate reciprocal interactions of carcinoma cells with cancer-associated fibroblasts to support progression of uterine cervical cancers. Cancer Res. 2011;71:6633–6642. doi: 10.1158/0008-5472.CAN-11-0034. [DOI] [PubMed] [Google Scholar]
- 66.Sasaki S, Baba T, Shinagawa K, Matsushima K, Mukaida N. Crucial involvement of the CCL3-CCR5 axis-mediated fibroblast accumulation in colitis-associated carcinogenesis in mice. Int J Cancer. 2014;135:1297–1306. doi: 10.1002/ijc.28779. [DOI] [PubMed] [Google Scholar]