

Abstract
Anti-estrogens as hormone therapy are the mainstay treatment for estrogen receptor (ER)-positive breast cancer. ER inhibitors through modulating the transcriptional function of ER have been the frontline anti-estrogens to which refractory phenotype often developed in advanced cancer. The anti-estrogen fulvestrant is currently the only clinically approved pure anti-estrogen which causes ER degradation. However, resistance to fulvestrant still occurs and unfortunately it leaves few choices other than chemotherapy as the later-line treatments to fulvestrant-resistant tumors. Here we show that fulvestrant resistance was accompanied by increased expression of a number of innate immune response genes including the natural killer (NK) cell ligand B7-H6 on the cell surface. In an attempt to overcome the drug resistance phenotype, a NK-based molecular approach taking advantage of a chimeric antigen receptor (CAR) system targeting B7-H6 was established and tested in cells with acquired resistance to fulvestrant. The results demonstrate that the cell therapy approach as a single agent can effectively induce cell death of the resistant cancer cells which is enhanced by the increased expression of cell surface B7-H6. This approach departs from the traditional strategies of conquering anti-estrogen resistant breast cancer and offers a new avenue to eradicate hormone-refractory malignant solid tumors.
Keywords: Chimeric antigen receptor, B7-H6, NKp30/NCR3, natural killer cell, anti-estrogen, fulvestrant, estrogen receptor, cell therapy, breast cancer
Introduction
More than 70% of metastatic breast cancer is of luminal origin and expresses estrogen receptor (ER) [1]. Anti-estrogens of specific ER modulators (SERM) and the aromatase inhibitors (AI) such as tamoxifen and letrozole, respectively, are the mainstay treatments for ER-positive patients [2,3]. Although most early-stage ER-positive breast cancer responds to these conventional anti-estrogens, including tamoxifen and the aromatase inhibitors [3], resistant tumors eventually develop following prolonged treatment [4-6].
Started as a second-line anti-estrogen therapy in 2002 [7], fulvestrant (ICI182,780; Faslodex) has become the mainstay in latest development of frontline anti-estrogen therapy [8-11]. Fulvestrant as a specific ER degrader (SERD) suppresses ER transcriptional activity by directly binding to ER [5,12-14]. Interaction between fulvestrant and ER also induces degradation of ER through a proteasome-dependent manner [12]. At present and for the foreseeable future, fulvestrant is the only pure anti-estrogen approved by the FDA to treat breast cancer refractory to other hormonal therapy. However, numerous laboratory studies [15-17] and clinical observations [5,6,18,19] have shown that breast cancer cells retain the ability to progress on fulvestrant treatment, often accompanied with deadly metastasis. Options of clinical intervention for these patients are limited and efficient regimen has yet to be developed.
Multiple mechanisms underlying the development of resistance to fulvestrant have been demonstrated, such as mutation of the ER coding gene ESR1 [20], aberrant activation of the PI3K/AKT pathway [21], and deregulated insulin-like growth factor 1 receptor (IGF1R) signaling [22]. Given that tumor tissues have high degree of inherent heterogeneity, developing molecular targeted intervention for fulvestrant-refractory tumors has been a challenging task. The strategy of chimeric antigen receptors (CAR) entails an extracellular receptor recognizing cell surface antigens of the target cells which is linked with designated transmembrane and intracellular domains with signaling functions [23]. The intracellular domain of the chimeric receptor is adopted from natural cell surface proteins such as CD3ζ which mediates activation of immune response when the extracellular receptor domain recognizes the ligand and the induced conformation changes consequently stimulate signaling through the immune-receptor tyrosine-based activation motifs (ITAM) [24].
Innate immunity is a major cellular mechanism in the heterogeneous cancer microenvironment playing important role in tumor suppression [25]. Natural killer (NK) cells represent the major anti-cancer taskforce in innate immunity and play an important function in regulating the adaptive immune activities [26]. The function of NK cells are regulated through an array of receptors on the cell surface. For example, engagement of the membrane receptor NKp30 (NCR3) of NK cells with its ligand B7-H6 results in activation of NK cells for secretion of cytokines, interferons, and cytolytic factors [27]. This activation is mediated by coupling NKp30, which has no major intracellular domain for activation signaling, with CD3ξ and FcεRIγ in trans through an opposing charge contacts within the corresponding transmembrane domains of these proteins [28]. In this study, a chimeric molecule is constructed in which the NKp30 including its extracellular ligand-binding domain, the transmembrane domain, together with the intracellular stub is linked to the intracellular domain of CD3ξ which contains four immunoreceptor tyrosine-based activation motifs (ITAM) for immune activation of NK cells. This chimeric antigen receptor (CAR) construct is transduced into the NK92 cell line as a CAR-NK system. We show a proof-of-concept demonstrating the efficacy of targeting breast cancer cells with acquired resistance to fulvestrant. Our results demonstrate that CAR-based cell therapy is a promising clinical armamentarium to overcome endocrine-refractory breast cancer.
Methods and materials
Cell lines, chemicals and antibodies
MCF7 and T47D cell lines were maintained in DMEM/F-12 with FBS (10%) and PSA (1%). NK92 cells were maintained in alpha-MEM with FBS (12.5%), horse serum (12.5%), β-mecaptoethanol (0.1 mM), IL-2 (100 U/ml), folic acid (0.02 mM), inositol (0.2 mM) and PSA (1%). All materials of cell culture were purchased from Invitrogen, Thermo Fisher Scientific Inc. The following antibodies were purchased: primary antibody pHER2, HER2, ERα (Cell Signaling Technology), pHER2 (Santa Cruz) and β-actin (Genetex). PE-conjugated anti-B7-H6 (Biolegend) and annexin V-conjugated antibodies (Biolegend) and all secondary antibodies for immunohistochemistry staining (Jackson ImmunoResearch). The following shRNA clones were purchased from the RNA Technology Platform and Gene Manipulation Core of Academia Sinica in Taiwan: B7-H6 (TRCN0000139616 and TRCN0000138996), Luciferase (TRCN0000076978 and TRCN0000076980).
Cell viability assay
Cell viability was detected by MTT (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) assay. Briefly, cells were incubated with MTT reagent (final concentration 0.5 mg/ml) for 4 h until purple precipitate is visible which was dissolved by 100 μL of dimethyl sulfoxide (DMSO) at room temperature in the dark for 2 h. Absorbance at 570 nm was measured by a microplate reader (Synergy™ HTX, BioTek).
Western blotting
Cell lysates were isolated by incubation with RIPA buffer (150 mM NaCl, 50 mM Tris [pH 7.5], 1% NP-40, 0.5% DOC, 10% SDS, phosphatase inhibitors consisting of 25 mM NaF and 2 mM Na3VO4, and the protease inhibitors 20 ul/ml aprotinin and 0.1 M PMSF). Cell lysates were separated on an acrylamide gel, transferred to a PVDF membrane (Bio-Rad), and probed with the antibodies of interest. Bands were visualized by a chemiluminescence-based detection method (Fisher/Pierce) that used a horseradish peroxidase-conjugated secondary antibody, and images were acquired by the ChemiDoc™ MP Imaging System (Bio-Rad).
Quantitative real time polymerase chain reaction (qRT-PCR)
Total RNA was extracted with Trizol (Invitrogen), and purified by Direct-zol RNA Miniprep Plus kit (Zymo Research). Reverse-transcription for cDNA was performed with M-MLV transcriptase (Applied Biosystems). For quantitative real-time PCR, cDNA targets were amplified with gene-specific primers using SYBR Green Master Mix (Bio-Rad) on the QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific). In a typical condition, the PCR reaction was initiated by heat activation of the DNA polymerase at 95°C for 10 min, followed by 35 cycles of 95°C for 15 sec, 60°C for 30 sec, and 72°C for 60 sec. Relative levels were calculated using the comparative CT method. Data were normalized to GAPDH. The following primers were used: Human B7-H6-forward, 5’-CTTTTATTCCCAACCCCTCAACA-3’; Human B7-H6-reverse, 5’-CACATCGGTACTCTCCTGCTT-3’; Human ULBP1-forward, 5’-TAAGTCCAGACCTGAACCACA-3’; Human ULBP1-reverse, 5’-TCCACCACGTCTCTTAGTGTT-3’; Human ULBP3-forward, 5’-TCTATGGGTCACCTAGAAGAGC-3’; Human ULBP3-reverse, 5’-TCCACTGGGTGTGAAATCCTC-3’; Human PVR-forward, 5’-TGGAGGTGACGCATGTGTC-3’; Human PVR-reverse, 5’-GTTTGGACTCCGAATAGCTGG-3’; Human ICAM1-forward, 5’-ATGCCCAGACATCTGTGTCC-3’; Human ICAM1-reverse, 5’-GGGGTCTCTATGCCCAACAA-3’; Human ICAM2-forward, 5’-CGGATGAGAAGGTATTCGAGGT-3’; Human ICAM2-reverse, 5’-CACCCACTTCAGGCTGGTTAC-3’; Human PVRL2-forward, 5’-GGATGTGCGAGTTCAAGTGCT-3’; Human PVRL2-reverse, 5’-TGGGACCCATCTTAGGGTGG-3’; Human CD58-forward, 5’-AGAGCATTACAACAGCCATCG-3’; Human CD58-reverse, 5’-ATCTGTGTCTTGAATGACCGC-3’; Human GAPDH-forward, 5’-ACAACTTTGGTATCGTGGAAGG-3’; Human GAPDH-reverse, 5’-GCCATCACGCCACAGTTTC-3’.
In vitro co-culture cytotoxicity assay
3×104 target cells were seeded and cultured in 12-well plate overnight. NK92 cells were then added to the wells at an E:T (Effector-to-Target) ratio of 1:1 and co-cultured in a humidified incubator at 37°C with 5% CO2 for 24 h. After incubation, cells were stained with an annexin V-conjugated antibody and 7-AAD and analyzed by BD FACSVerse™ flow cytometer (BD Biosciences). Alternatively, cell cytotoxicity assay was performed with the IncuCyte imaging system (Essen Biosciences) following the manufacturer’s protocol. 3×103 target cells were seeded in 96-well plate for overnight, then co-cultured with NK92 cells at an E:T ratio of 1:1. Cell apoptosis was detected and assessed real-time by the IncuCyte® Caspase-3/7 Green Apoptosis Assay Reagent substrate (Essen Biosciences).
Flow cytometry
Cells were plated at equal densities and incubated overnight. Resuspended cells were washed with PBS buffer, and incubated on ice for 1 h with the fluorescent dye-conjugated antibody at a concentration of 0.2 μg/well in FACS buffer (1 × PBS, 0.5% bovine serum albumin, 0.05% NaN3). The cells were then analyzed by BD FACSVerse™ Flow Cytometer (BD Biosciences).
The NCR3-NK92 preparation
NK92 cells were maintained in αMEM with FBS (12.5%), horse serum (12.5%), β-mecaptoethanol (0.1 mM), IL-2 (100 U/ml), folic acid (0.02 mM), inositol (0.2 mM) and PSA (1%). The cells were then transduced with a retroviral vector coding for the NCR3 extracellular and transmembrane domain fused to the intracellular domain of CD3ζ, followed by a P2A autocleavage site in frame with a mGFP reporter. One day later, the transduced cells were evaluated for cell number, viability, and transduction efficacy by flow cytometry.
Results
The ER-positive breast epithelial cancer cell line MCF-7 was cultured in escalating concentrations of fulvestrant (from 1×10-7 to 5×10-6 M). Resistant cells of three independent cultures were collected as distinctly pooled fulvestrant-resistant MCF-7 cells (hereafter referred to as MCF7/ICI-R #1, #2, and #3) (Figure 1A). Western blotting analysis showed increased expression of the receptor tyrosine kinases ErbB2/HER2/neu along with its activated phosphorylated forms (pY1222) in two of the lines (#1 and #3) (Figure 1B), while elevated expression of ERα was observed solely in line #2 (Figure 1C). This result implies that both ER-dependent and ER-independent mechanisms were underlying the phenotype of anti-estrogen resistance.
Figure 1.
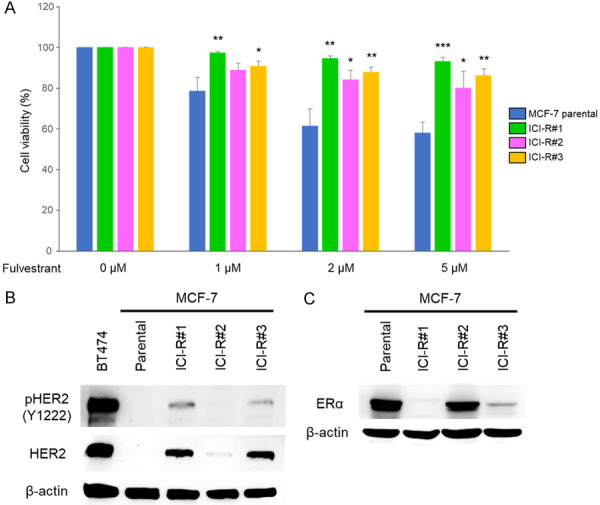
Characterization of MCF-7 derivatives with acquired resistance to fulvestrant. A. The parental MCF-7 cells and its derivatives with acquired fulvestrant resistance (ICI-R) were inoculated in 96-well plates and treated with Fulvestrant of indicated doses or mock treated for 72 h. Cell growth was evaluated by MTT assay. The results are presented as mean ± SD based on three independent experiments. *, P < 0.05, **, P < 0.01, ***, P < 0.001 as determined by t-test. B and C. The levels of indicated endogenous proteins were analyzed by western blotting using the designated antibodies. β-actin was used as the internal controls. Expression of ErbB2/HER2 in BT474 was used as the positive control of HER2 expression.
It has been suggested that the tumor microenvironment including the immune compartment plays important role in the development of resistance to anti-estrogens [29]. Conversely, whether the evolution of the endocrine refractory phenotypes is associated with reprogramming of the response to immune surveillance of cancer cells remains unaddressed. To test this possibility, expression of the cell surface receptors known to play important function in NK immunity, such as CD58 [30], ULBP1 [31], ULBP3 [31], ICAM1 and ICAM2 [32], PVR [33], PVRL2 [33], and B7-H6 [27] were analyzed for the MCF-7 parental cells and the fulvestrant-resistant derivatives.
Assessment with quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) showed that gene expression of the surface proteins including ULBP1, ICAM1 and B7-H6 were increased in at least two out of the three fulvestrant-resistant lines (Figure 2A). The enhanced expression of these NK-recruiting surface markers predicts sensitization of these cell lines to innate immune cells. Indeed, incubation with the natural killer cell line NK92 provoked more conspicuous cell death in two of the ICI-R derivatives (ICI-R#2 and ICI-R#3) when compared to the parental MCF-7 cells as demonstrated by the increase of apoptotic cells with flow cytometry gated by annexin V and 7-aminoactinomycin D (7-AAD) (Figure 2B and 2C) as well as caspase activation measured by a real-time imaging system (IncuCyte) (Figure 2D and 2E). These results suggest the potential of exploiting NK to target the fulvestrant-refractory cancer cells regardless of the underlying mechanisms of drug resistance.
Figure 2.

Gene expression of cell surface molecules involved in innate immunity and cytotoxicity of NK cells. A. mRNA expression of the indicated cell surface genes was analyzed by qRT-PCR normalized to GAPDH. Results are presented as mean ± SD based on three independent experiments. The statistical significance was assessed by t-test. *, P < 0.05, **, P < 0.01. B. MCF7 and the ICI-R cells were co-cultured with NK-92 cells for 24 h. Apoptotic cells were analyzed by gating with annexin V and 7-AAD running through flow cytometry. C. The quantitative results of cytotoxicity assay by flow cytometry. Results are presented as mean ± SD based on three independent experiments. The statistical significance was assessed by t-test. *, P < 0.05, **, P < 0.01. D. Real time detection of cytotoxicity of NK92 cells co-cultured with MCF-7 and ICI-R cells. Results are presented as mean ± SD based on three independent experiments. E. The quantitative results of cytotoxicity assay by IncuCyte. Results are presented as mean ± SD based on three independent experiments. The statistical significance was assessed by t-test. *, P < 0.05, **, P < 0.01.
Cell surface presentations of B7-H6 in the MCF-7 and MCF-7/ICI-R lines were further confirmed by flow cytometry. Consistent with its increased RNA expression (Figure 2A), protein expression of B7-H6 in the ICI-R lines was enhanced in the MCF-7/ICI-R lines compared to the MCF-7 parental cells (ICI-R#2 and ICI-R#3; Figure 3A and 3B). To exploit the function of surface B7-H6 in NK targeting, a chimeric structure was generated in which the extracellular immunoglobulin-like motif of NCR3 together with its transmembrane domain and part of intracellular region was fused with the intracellular domain of CD3ζ which contains three immunoreceptor tyrosine-based activation motifs (ITAMs) for activation of immune signaling [34] (Figure 3C). The fusion protein was cloned in a lentiviral cassette with co-cistronic expression of the membrane-bound green fluorescence protein (mGFP) gene separated by a P2A self-cleavage site [35]. The construct, termed NCR3-CD3ζ-P2A-GFP, was then packaged in lentivirus and transduced into NK92 cells to produce a chimeric antigen receptor (CAR)-expressing NK designated as NCR3-NK92. Expression of the construct was assessed by flow cytometry for GFP (a transduction rate of 61% in Figure 3D). Depleting B7-H6 by a specific short hairpin RNA (shB7-H6) in the fulvestrant-resistant cells (MCF-7/ICI-R#3) significantly desensitized the cells to NK-mediated cytotoxicity compared to cells transduced with a control shRNA of luciferase (shLuc), suggesting that the enhanced killing by NCR3-NK92 depended at least in part on the corresponding ligand of NCR3 (Figure 3E). Consistently, IFNγ expression in the co-culture of NCR3-NK92 and the B7-H6-depleted cells was lower than in the co-culture of NCR3-NK92 and the control shLuc cells (Figure 3F).
Figure 3.

Increased B7-H6 expression of ICI-R cells serve as a target of NCR3- NK92 cells. A. B7-H6 surface expressions of MCF-7 parental and the ICI-R derivatives were assessed by flow cytometry. B. The mean fluorescent intensity obtained by flow cytometry. Results are presented as mean ± SD based on three independent experiments. Statistical significance was determined by t-test: *, P < 0.05, **, P < 0.01. C. Schematic diagram of the NCR3-CD3ζ-P2A-GFP construct in a retroviral vector. The extracellular, transmembrane (TM), cytoplasmic regions, and immunoreceptor tyrosine-based activation motifs (ITAM) are indicated. D. Left, the transduction efficiency of the CAR construct to NK92 cells was analyzed with the mGFP reporter by flow cytometry. Right, the plots of fluorescence intensities of the mock treated or virus-transduced NK92 cells. E. Cytotoxicity of NCR3-NK92 cells co-cultured with MCF-7 ICI-R#3 harboring shB7-H6 or the control shRNA of luciferase (shLuc). Results are presented as mean ± SD based on three independent experiments. Statistical significance was determined by t-test: **, P < 0.01, ***, P < 0.001. F. IFN-γ production of NCR3-NK92 cells co-cultured with MCF-7 ICI-R#3 harboring shB7-H6 or shLuc. Statistical significance was determined by t-test. *, P < 0.05.
To test whether the NCR3-NK92-mediated cell killing is a general phenomenon to fulvestrant-resistant cells, MCF-7 and the three independent ICI-R derivatives were subjected to killing by parental NK92 or NCR3-NK92 (Figure 4A and 4B). Flow cytometry gated by annexin V and 7-AAD showed that, except for MCF-7/ICI-R#2 which was exceptionally sensitive to NK killing, MCF-7 and the ICI-R derivatives were only moderately sensitive to killing mediated by the parental NK92. In drastic contrast, transduction with the NCR3-CD3ζ-P2A-GFP chimera significantly enhanced the killing activity of NK cells to all fulvestrant-resistant derivatives, in particular ICI-R#2 and ICI-R#3 which expressed higher levels of cell surface B7-H6 than other lines (Figure 4B).
Figure 4.

NCR3-CAR promoted NK killing of fulvestrant-resistant cells. A. MCF7 and the ICI-R derivatives were co-cultured with the B7-H6-directed CAR-NK or the mock-transduced NK cells for 24 h. After incubation, the apoptotic cells were analyzed with annexin V-conjugated antibody and 7-AAD staining and detected by flow cytometry. B. The quantitative results of cytotoxicity assay by flow cytometry. The results are presented by the mean ± SD based on three independent experiments. Statistical significance was determined by t-test. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
Besides MCF-7 cells, acquired fulvestrant resistance also induced increase of B7-H6 in two independently produced fulvestrant-resistant derivatives from the ER+ breast cancer epithelial cells T47D (ICI-R#1 and #2) (Figure 5A and 5B). Furthermore, NK92 cells transduced with the NCR3-CD3ζ-P2A-GFP chimera preferentially killed the two ICI-R derivatives (Figure 5C and 5D).
Figure 5.

Fulvestrant-resistant T47D derivatives expressed higher levels of cell surface B7-H6 and were sensitized to anti-B7-H6 CAR-NK. A. B7-H6 surface expressions of T47D parental and the ICI-R derivatives were assessed by flow cytometry. B. The mean fluorescent intensity obtained by flow cytometry. Results are presented as mean ± SD based on three independent experiments. Statistical significance was determined by t-test: *, P < 0.05, **, P < 0.01. C. T47D and ICI-R derivatives were co-cultured with the B7-H6-directed CAR-NK or mock-transduced NK cells for 12 h. After incubation, the apoptotic cells were analyzed with annexin V-conjugated antibody and 7-AAD staining and detected by flow cytometry. D. The quantitative results of cytotoxicity assay by flow cytometry. The results are presented by the mean ± SD based on three independent experiments. Statistical significance was determined by t-test. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
Discussion
Breast cancer with acquired resistance to hormone therapies poses a daunting challenge. The presented study demonstrates that multiple cellular mechanisms were invoked to acquire fulvestrant resistance. Molecular characterization of the fulvestrant-resistant MCF-7 derivatives suggested that both ER-dependent and ER-independent mechanisms were involved in the development of acquired resistance to fulvestrant. Thus, targeted therapy against a certain cellular target is unlikely to produce a sustainable suppression of the resistant tumors. The development of acquired fulvestrant resistance was associated with enhanced cell surface immunomodulatory genes. This suggests a reprogramming of the crosstalk between tumors and the immune microenvironment which can be exploited for therapeutic opportunities (Figure 6).
Figure 6.
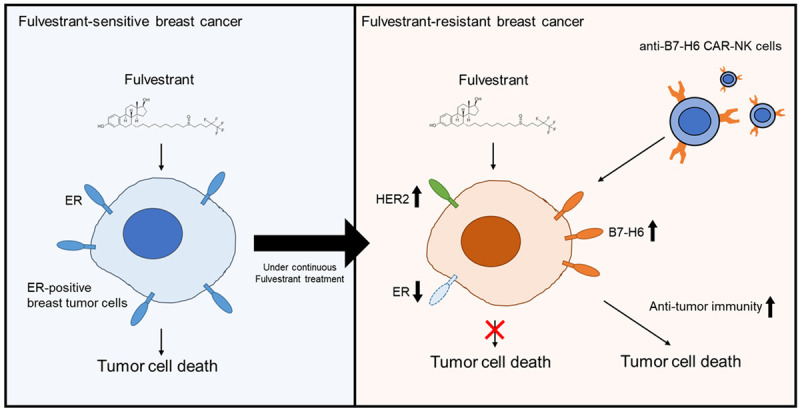
Schematic diagram of targeting fulvestrant resistance by a CAR-NK approach. The model demonstrates that fulvestrant-resistant breast cancer cells have increased B7-H6 expression on their surface, resulting in enhanced vulnerability to the treatment of B7-H6-targeting CAR-NK.
Significant progress has been made in recent years in the establishment of chimeric antigen receptor-based cell therapy in liquid cancers and sophisticated CAR systems have been under development [23,36]. The application of CAR-based cell therapy to advanced solid tumors holds a great promise for advanced cancer [23,37]. The current study demonstrates for the first time that application of CAR-based cell therapy to endocrine-resistant breast cancer is a novel and promising approach to overcome the malignancy. As an effort of proof of concept, the presented study adopted the basic CAR construct with the extracellular ligand-binding motif of NCR3 receptor and replaced its intracellular ITIM with the ITAM motifs of CD3ζ [34]. The efficacy and manipulability can be improved with more sophisticated configurations entailing a plethora of component proteins in immune signaling. It is noteworthy that depleting the expression of B7-H6 did not completely rescue cells from NK killing, suggesting that other surface molecules of fulvestrant-resistant cells are also involved in sensitization to NK-mediated cytotoxicity. It is conceivable that multiple cell surface ligands or receptors are contributing to recruiting or activating NK cells. Further assessment of the interaction between cancer cells with the immune microenvironment can lead to new perspective of overcoming anti-estrogen resistance.
The immune context in ER+ tumor tissues has been suggested to play a role in determining the responsiveness to endocrine therapies [29]. Conversely, whether the evolution of the endocrine refractory phenotypes is associated with reprogramming of the response to immune surveillance of cancer cells remains unaddressed. The current study focuses on the use of NK immunity to target treatment-resistant breast cancer. NK activation is known to be mediated by ligand interaction of the surface receptors of the NK cells. For example, ligation of the CD2 receptor of NK cells by CD58 potentiates NK cell activation [30]. ULBP1 and ULBP3 are the major ligands of the NK-activating pattern recognition receptor NKG2D [31]. Similarly, ICAM1 and ICAM2 are the major ligands of LFA1 to stimulate NK cells [32], while PVR and PVRL2 bind to DNAM1 that can induce NK activation [33].
Among these factors, B7-H6 stands out as the main factor associated with fulvestrant resistance in our cell system. B7-H6 preferentially expresses on the cell surface of tumor cells and is absent in most normal cells, suggesting a tumor-promoting function [27]. Indeed, its functions in tumor growth and metastasis and as a potential therapeutic target have been demonstrated [27,38,39]. B7-H6 is a bona fide ligand of NKp30/NCR3 [27]. Thus, the NCR3-B7-H6 receptor-ligand axis has been exploited for the development of CAR-based strategies for cancer targeting [34]. The current study explores the potential of CAR-based cell therapy to overcome the breast cancer refractory to endocrine therapy. Of the three independent fulvestrant-resistant MCF-7/ICI-R derivatives, the expression of B7-H6 follows the order (low to high) of #1-#2-#3. Among them, however, MCF-7/ICI-R#2 exhibited exceptional sensitivity to killing mediated by the parental NK. It is possible that additional yet to be identified NK-targeted cell surface molecules or regulation of cell surface molecules for NK recognition and activation can be potential mechanisms. It should be noted that all lines including the MCF-7 parental cells were more sensitive to NCR3-NK than the parental NK cells in cell killing, suggesting that the cognate NCR3-ligand signaling can trigger cytotoxicity in different cellular context. The sensitivity to NCR3-NK is in general consistent with the relative expression levels of B7-H6 in the three ICI-R derivatives. Besides the MCF-7 series, association of increased H7-H6 expression with fulvestrant resistance and the enhanced sensitivity to the treatment by B7-H6-targeting CAR-NK was also observed in fulvestrant-resistant derivatives of the T47D cells, suggesting that the mechanism may also be relevant to endocrine resistance in different cellular contexts. In conclusion, our study supports the promising potential of targeted cell therapy in breast cancers which developed resistance to conventional endocrine therapies.
Acknowledgements
This study was supported in part by Ministry of Health and Welfare Cancer Research Center of Excellence grants MOHW109-TDU-B-212-134024 and MOHW109-TDU-B-212-010001 (to S.-C.W.), and the CMUH grant DMR-CELL-17025 (to S.-C.W.). The work was also financially supported in part by the Drug Development Center, China Medical University from the Ministry of Education in Taiwan (S.-C.W.). Experiments and data analysis were performed in part through the use of the Medical Research Core Facilities Center, Office of Research & Development at China medical University, Taichung, Taiwan.
Disclosure of conflict of interest
None.
References
- 1.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soleja M, Raj GV, Unni N. An evaluation of fulvestrant for the treatment of metastatic breast cancer. Expert Opin Pharmacother. 2019;20:1819–1829. doi: 10.1080/14656566.2019.1651293. [DOI] [PubMed] [Google Scholar]
- 3.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 4.Kabos P, Borges VF. Fulvestrant: a unique antiendocrine agent for estrogen-sensitive breast cancer. Expert Opin Pharmacother. 2010;11:807–816. doi: 10.1517/14656561003641982. [DOI] [PubMed] [Google Scholar]
- 5.Howell A, Robertson JF, Quaresma Albano J, Aschermannova A, Mauriac L, Kleeberg UR, Vergote I, Erikstein B, Webster A, Morris C. Fulvestrant, formerly ICI 182, 780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J. Clin. Oncol. 2002;20:3396–3403. doi: 10.1200/JCO.2002.10.057. [DOI] [PubMed] [Google Scholar]
- 6.Osborne CK, Pippen J, Jones SE, Parker LM, Ellis M, Come S, Gertler SZ, May JT, Burton G, Dimery I, Webster A, Morris C, Elledge R, Buzdar A. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J. Clin. Oncol. 2002;20:3386–3395. doi: 10.1200/JCO.2002.10.058. [DOI] [PubMed] [Google Scholar]
- 7.Bross PF, Cohen MH, Williams GA, Pazdur R. FDA Drug approval summaries: fulvestrant. The Oncologist. 2002;7:477–480. doi: 10.1634/theoncologist.7-6-477. [DOI] [PubMed] [Google Scholar]
- 8.Howell A, Osborne CK, Morris C, Wakeling AE. ICI 182,780 (Faslodex™) Cancer. 2000;89:817–825. doi: 10.1002/1097-0142(20000815)89:4<817::aid-cncr14>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 9.DeFriend DJ, Howell A, Nicholson RI, Anderson E, Dowsett M, Mansel RE, Blamey RW, Bundred NJ, Robertson JF, Saunders C, et al. Investigation of a new pure antiestrogen (ICI 182780) in women with primary breast cancer. Cancer Res. 1994;54:408–414. [PubMed] [Google Scholar]
- 10.McClelland RA, Gee JM, Francis AB, Robertson JF, Blamey RW, Wakeling AE, Nicholson RI. Short-term effects of pure anti-oestrogen ICI 182780 treatment on oestrogen receptor, epidermal growth factor receptor and transforming growth factor-alpha protein expression in human breast cancer. Eur J Cancer. 1996;32A:413–416. doi: 10.1016/0959-8049(95)00517-x. [DOI] [PubMed] [Google Scholar]
- 11.McClelland RA, Manning DL, Gee JM, Anderson E, Clarke R, Howell A, Dowsett M, Robertson JF, Blamey RW, Wakeling AE, Nicholson RI. Effects of short-term antiestrogen treatment of primary breast cancer on estrogen receptor mRNA and protein expression and on estrogen-regulated genes. Breast Cancer Res Treat. 1996;41:31–41. doi: 10.1007/BF01807034. [DOI] [PubMed] [Google Scholar]
- 12.Long X, Nephew KP. Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-alpha. Jf Biol Chem. 2006;281:9607–9615. doi: 10.1074/jbc.M510809200. [DOI] [PubMed] [Google Scholar]
- 13.Martin LA, Pancholi S, Chan CM, Farmer I, Kimberley C, Dowsett M, Johnston SR. The anti-oestrogen ICI 182,780, but not tamoxifen, inhibits the growth of MCF-7 breast cancer cells refractory to long-term oestrogen deprivation through down-regulation of oestrogen receptor and IGF signalling. Endocr Relat Cancer. 2005;12:1017–1036. doi: 10.1677/erc.1.00905. [DOI] [PubMed] [Google Scholar]
- 14.Nicholson RI, Gee JM, Manning DL, Wakeling AE, Montano MM, Katzenellenbogen BS. Responses to pure antiestrogens (ICI 164384, ICI 182780) in estrogen-sensitive and -resistant experimental and clinical breast cancer. Ann N Y Acad Sci. 1995;761:148–163. doi: 10.1111/j.1749-6632.1995.tb31376.x. [DOI] [PubMed] [Google Scholar]
- 15.Osborne CK, Coronado-Heinsohn EB, Hilsenbeck SG, McCue BL, Wakeling AE, McClelland RA, Manning DL, Nicholson RI. Comparison of the effects of a pure steroidal antiestrogen with those of tamoxifen in a model of human breast cancer. J Natl Cancer Inst. 1995;87:746–750. doi: 10.1093/jnci/87.10.746. [DOI] [PubMed] [Google Scholar]
- 16.McClelland RA, Barrow D, Madden TA, Dutkowski CM, Pamment J, Knowlden JM, Gee JM, Nicholson RI. Enhanced epidermal growth factor receptor signaling in MCF7 breast cancer cells after long-term culture in the presence of the pure antiestrogen ICI 182,780 (Faslodex) Endocrinology. 2001;142:2776–2788. doi: 10.1210/endo.142.7.8259. [DOI] [PubMed] [Google Scholar]
- 17.Sommer A, Hoffmann J, Lichtner RB, Schneider MR, Parczyk K. Studies on the development of resistance to the pure antiestrogen Faslodex in three human breast cancer cell lines. J Steroid Biochem Mol Biol. 2003;85:33–47. doi: 10.1016/s0960-0760(03)00139-0. [DOI] [PubMed] [Google Scholar]
- 18.Di Leo A, Jerusalem G, Petruzelka L, Torres R, Bondarenko IN, Khasanov R, Verhoeven D, Pedrini JL, Smirnova I, Lichinitser MR, Pendergrass K, Garnett S, Lindemann JPO, Sapunar F, Martin M. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J. Clin. Oncol. 2010;28:4594–4600. doi: 10.1200/JCO.2010.28.8415. [DOI] [PubMed] [Google Scholar]
- 19.Valachis A, Mauri D, Polyzos NP, Mavroudis D, Georgoulias V, Casazza G. Fulvestrant in the treatment of advanced breast cancer: a systematic review and meta-analysis of randomized controlled trials. Crit Rev Oncol/Hematol. 2010;73:220–227. doi: 10.1016/j.critrevonc.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo JA, Cristofanilli M, Gomez H, Arteaga CL, Giltnane J, Balko JM, Cronin MT, Jarosz M, Sun J, Hawryluk M, Lipson D, Otto G, Ross JS, Dvir A, Soussan-Gutman L, Wolf I, Rubinek T, Gilmore L, Schnitt S, Come SE, Pusztai L, Stephens P, Brown M, Miller VA. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757–1767. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beeram M, Tan QT, Tekmal RR, Russell D, Middleton A, DeGraffenried LA. Akt-induced endocrine therapy resistance is reversed by inhibition of mTOR signaling. Ann Oncol. 2007;18:1323–1328. doi: 10.1093/annonc/mdm170. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Moerkens M, Ramaiahgari S, de Bont H, Price L, Meerman J, van de Water B. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res. 2011;13:R52. doi: 10.1186/bcr2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 25.Pierce S, Geanes ES, Bradley T. Targeting natural killer cells for improved immunity and control of the adaptive immune response. Front Cell Infect Microbiol. 2020;10:231. doi: 10.3389/fcimb.2020.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bald T, Krummel MF, Smyth MJ, Barry KC. The NK cell-cancer cycle: advances and new challenges in NK cell-based immunotherapies. Nature Immunol. 2020;21:835–847. doi: 10.1038/s41590-020-0728-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, Haldeman B, Ostrander CD, Kaifu T, Chabannon C, Moretta A, West R, Xu W, Vivier E, Levin SD. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med. 2009;206:1495–1503. doi: 10.1084/jem.20090681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front Immunol. 2019;10:909. doi: 10.3389/fimmu.2019.00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rani A, Stebbing J, Giamas G, Murphy J. Endocrine resistance in hormone receptor positive breast cancer-from mechanism to therapy. Front Endocrinol (Lausanne) 2019;10:245. doi: 10.3389/fendo.2019.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rolle A, Halenius A, Ewen EM, Cerwenka A, Hengel H, Momburg F. CD2-CD58 interactions are pivotal for the activation and function of adaptive natural killer cells in human cytomegalovirus infection. Eur J Immunol. 2016;46:2420–2425. doi: 10.1002/eji.201646492. [DOI] [PubMed] [Google Scholar]
- 31.Vivier E, Tomasello E, Paul P. Lymphocyte activation via NKG2D: towards a new paradigm in immune recognition? Curr Opin Immunol. 2002;14:306–311. doi: 10.1016/s0952-7915(02)00337-0. [DOI] [PubMed] [Google Scholar]
- 32.Tremblay-McLean A, Bruneau J, Lebouché B, Lisovsky I, Song R, Bernard NF. Expression profiles of ligands for activating natural killer cell receptors on HIV infected and uninfected CD4+ T cells. Viruses. 2017;9:295. doi: 10.3390/v9100295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, Levine Z, Beiman M, Dassa L, Achdout H, Stern-Ginossar N, Tsukerman P, Jonjic S, Mandelboim O. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A. 2009;106:17858–17863. doi: 10.1073/pnas.0903474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang T, Wu MR, Sentman CL. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J Immunol. 2012;189:2290–2299. doi: 10.4049/jimmunol.1103495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fowler DK, Stewart S, Seredick S, Eisen JS, Stankunas K, Washbourne P. A multisite gateway toolkit for rapid cloning of vertebrate expression constructs with diverse research applications. PLoS One. 2016;11:e0159277. doi: 10.1371/journal.pone.0159277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, Yin J, You F, Zhu M, Shen W, Chen G, Zhu X, Wu D, Yu J. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8:1083–1089. [PMC free article] [PubMed] [Google Scholar]
- 37.Liu B, Yan L, Zhou M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am J Cancer Res. 2019;9:228–241. [PMC free article] [PubMed] [Google Scholar]
- 38.Ni L, Dong C. New B7 family checkpoints in human cancers. Mol Cancer Ther. 2017;16:1203–1211. doi: 10.1158/1535-7163.MCT-16-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia E, Shen Y, Bhandari A, Zhou X, Wang Y, Yang F, Wang O. Long non-coding RNA LINC00673 promotes breast cancer proliferation and metastasis through regulating B7-H6 and epithelial-mesenchymal transition. Am J Cancer Res. 2018;8:1273–1287. [PMC free article] [PubMed] [Google Scholar]