

Abstract
Actin is the most abundant protein in almost all the eukaryotic cells. Actin amino acid sequences are highly conserved and have not changed a lot during the progress of evolution, varying by no more than 20% in the completely different species, such as humans and algae. The network of actin filaments plays a crucial role in regulating cells’ cytoskeleton that needs to undergo dynamic tuning and structural changes in order for various functional processes, such as cell motility, migration, adhesion, polarity establishment, cell growth and cell division, to take place in live cells. Owing to its fundamental role in the cell, actin is a prominent regulator of cell division, a process, whose success directly depends on morphological changes of actin cytoskeleton and correct segregation of duplicated chromosomes. Disorganization of actin framework during the last stage of cell division, known as cytokinesis, can lead to multinucleation and formation of polyploidy in post-mitotic cells, eventually developing into cancer. In this review, we will cover the principles of actin regulation during cell division and discuss how the control of actin dynamics is altered during the state of malignancy.
Keywords: Actin, myosin, mitosis, contractile ring, cleavage furrow, cytokinesis, abscission, cancer
Introduction
The cytoskeleton of eukaryotic cells is made of tubulin and actin polymers that are the most abundant and important components of cellular scaffold. Here actin plays a crucial role in the production of forces required to drive the processes important to sustain cellular functions and to maintain healthy and viable existence of cells. One of the most important events during cell life is cell division. During division, upon separation of duplicated chromosomes and physical cleavage of newly formed daughter cells, this actin- and microtubule-rich cellular cytoskeleton must be reorganized and disassembled, and once the cytokinesis is over, re-assembled to its former state. These tightly regulated morphological changes in cellular framework are guided by the interplay of various intercellular regulators, proteins, and their effectors. This review explains how the success of cell division is dependent on actin-mediated morphological changes that take place during cell division, and how this vital process is affected by the phenotypic characteristics relative to cancer cells.
Actin and its’ isoforms
Vertebrate cells contain three cytoskeleton components: intermediate filaments, microtubules, and actin microfilaments [1]. In eukaryotic cells, actin carries out a huge variety of cellular processes that are made possible by the ability of actin to assemble monomeric globular actin (G-actin) into polymeric filamentous actin (F-actin), and to interchange vice versa. Moreover, the functional diversity is controlled via an actin interaction with actin-binding proteins (ABPs) and other regulatory proteins, which can both induce or inhibit actin polymerization at the designated cellular compartments. This transitional property makes actin a crucial regulator of many cellular functions, varying from the maintenance of cell shape, polarization, cell motility, migration, adhesion, vesicle trafficking, muscle and non-muscle contractility, regulation of transcription, and finally to the regulation of cell division. Reorganization of actin microfilaments results to the morphological modification of cells, transforms cellular proliferative properties, and affects interactions with surrounding environment and cells.
Many eukaryotes, including fission yeast, budding yeast and green alga Chlamydomonas get together by with only one actin gene and protein, which makes up all of the cytoskeletal structures required for living. However, species, such as humans, have several actin genes expressed in different compartments and tissues in the body [2]. In humans, actin is composed of six different cell-type unique isoforms [3,4], which are encoded by separate genes and contribute to the diversity of functions it performs. These isoforms include ACTA1 (α-skeletal, located on chromosome), ACTA2 (α-smooth muscle, located on chromosome 10), ACTB (β-cytoplasmic, located on chromosome 7), ACTC1 (α-cardiac, located on chromosome 15), ACTG1, (γ-cytoplasmic, located on chromosome 17) and ACTG2 (γ-smooth muscle, located on chromosome 2), which mainly differ from one another by their N-terminal amino acid sequence [5-7]. This variation in N-terminus gives rise to different total charge of the molecules, which can be determined by the isoelectric focusing, where the difference in the later (5.40; 5.42, and 5.44) creates the basis for classification of the α-, β-, and γ-actin isoforms. And each of these actin isoforms plays a distinct role in cells, which is also dependent on the tissue type. The expression of actin isoforms in cells is regulated by different modes of action: gene modulation (via promoter elements) [4], translation of messenger RNA (mRNA) (via UTRs) [8], and protein binding (via interactions with ABPs) [9]. As this is not the topic of this review, for more information on this please refer to literature.
The ability of actin to carry out such number of different functions in cells depends on the polymerization and depolymerization capacity of actin fibers. The production of actin starts in cell nucleus, where genes encoding actin are transcribed and get translated into globular monomers (G-actin) once they reach the cytoplasm. Then, these actin monomers adhere to the rapidly growing barbed (plus) end of the extending actin filament in the more stable adenosine triphosphate (ATP)-state at the deep medial cleft that binds ATP molecules, and thus forms fibrous strands (F-actin). These filamentous actin strands perform all the aforementioned functions in cells. As the filament matures, ATP-hydrolysis takes place and adenosine diphosphate (ADP)-monomers start to dissociate from the pointed (minus) end of the filamentous actin strands at a faster pace than the ATP-bound ones are attached (Figure 1). Importantly, released ADP-actin monomers can be converted into ATP-actin monomers via nucleotide exchange and become reusable in another polymerisation reaction [10]. This process of steady-state actin polymerization and depolymerization is called treadmilling [11]. The rates of actin filament assembly and disassembly are regulated by ABPs. Indeed, there are plenty of different ABPs that can modulate the growth and crosslinks of actin filaments. Such ABPs include monomer binders, myosins, bundlers and crosslinkers, cytoskeletal linkers, proteins responsible for capping and severing, branch formation, rulers and stabilisers, side binders and signallers, and lastly anchors to membranes and membrane proteins. The detailed review of ABPs mode of action are thoroughly described by S.J. Winder and K.R. Ayscough [12], and T.D. Pollard [13].
Figure 1.
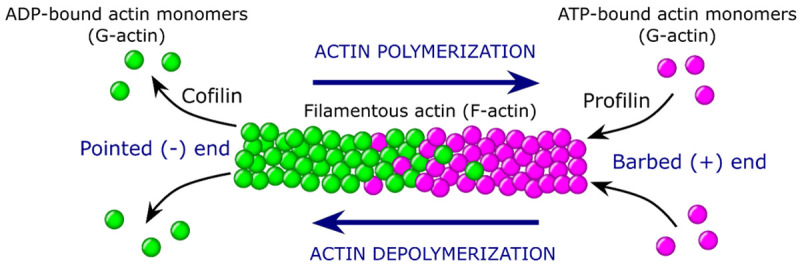
Actin polymerization and steady state treadmilling. The G-actin monomers attach to the continually growing barbed (+) end of the filament in a more stable ATP-state to form F-actin strands, which in cells function to support structural integrity during cell division and migration. Upon ATP-hydrolysis-driven F-actin depolymerization, ADP monomers begin to dissociate from the pointed (-) end of the filament at a faster rate than the ATP monomers are bound. This process of steady-state actin polymerization and depolymerization is known as treadmilling. The dynamic regulation of actin elongation is accelerated/decelerated via an action of profilin and ADF/cofilin.
The broad-spectrum of actin functions in cells
Seven decades of intensive research has led researchers to acquire a creditable understanding of actin structure, its assembly, the role of regulatory proteins, and cellular functions. Actins are fundamental components of the cytoskeleton and play important roles in a variety of cellular processes, including cell growth and re-shaping, motility, migration, invasion, adhesion, polarization, and division. The performance of these functions is made possible due to the ability of actin to form filaments that can swiftly assemble or disassemble based on the cell needs. There are six different highly conserved actin isoforms in vertebrates: four isoforms are expressed in striated (αsk and αca) and smooth (αsm and γsm) muscle cells, and two cytoplasmic β-actin and γ-actin isoforms are expressed ubiquitously [3,5]. To emphasize, vertebrate non-muscle cells express two actin isoforms: cytoplasmic β-actin and γ-actin [14,15]. Even though β- and γ-actins are ubiquitously expressed in all cell types and at the amino acid level are almost identical, they perform very different roles in vivo [16]. Some of the most important actin-mediated cellular functions will be covered in the later topics of this review.
Cell motility: migration, wound-healing and embryonic development
One of the best studied roles of actin is cell motility, which is a fundamental process covering wound healing, migration, and embryonic development. Cell migration is driven by polymerization of actin at the leading edge of the migrating cell, which induces forces to push membrane to foreseen direction. To assist this process, actin-associated proteins co-migrate with actin filaments by interacting with them [17]. There have been several attempts trying to elucidate the β- and γ-actins’ role in cell motility. A study by Peckham et al. demonstrated that overexpression of β-actin increases membrane protrusions and cell migration [18]. In addition, several other studies showed that β-actin is concentrated at the leading edge of migrating cells, whereas γ-actin is more evenly spread throughout the cells [19,20]. Inevitably, studies examining overexpression and silencing of β- and γ-actins provided some contradictory results. For example, Bunnell and Ervasti had studied the roles of both β- and γ-actins’ in cell migration, and showed that γ-actin is not required for migration in vivo and in vitro [19], Meantime, Simiczyjew et al. demonstrated the predominant γ-actin role in controlling the invasiveness of human colon cancer cells [21]. Some of the research covering the regulation of cellular motility did not get around without complications in their experimental flow. However, due to versatility of methodology to be used, switching to alternative experimental models allowed to overcome this. Since the crossed mouse embryonic fibroblasts (MEFs), which were used as a study model, experienced some growth impairment, making it impossible to form confluent cell monolayers for wound-healing assays, the use of cell migration assay instead, established a clear difference in the migratory phenotype between γ-actin- and β-actin-knockout cells [22]. Unlike to γ-actin-knockout cells, β-actin-knockout cells suffered from greatly reduced migration velocity, than compared to controls [22]. To sum up all the data discussed above, one can be said that β-actin is the major actin isoform mediating cell motility.
When considering the regulation of cellular viability, studying of MEFs showed that knocking out γ-actin results in a minor decrease in cell viability [19]. The same group of scientists took an advantage of using a “floxed gene” tool, which allows for spatial and temporal alteration of gene expression [23]. They demonstrated that β-actin-knockout results in a severely impaired growth of primary MEFs, with the characteristic phenotype contributing to this condition, including enhanced apoptosis, increased multinucleation, and an observation that more cells have 4N DNA content [22]. These data suggest that β-actin may be specifically required for cell division. Another study by Shawlot et al. showed that homozygous β-actin knockout leads to an embryonic lethality [24], suggesting that β-actin is an essential gene, responsible for the viability. Interestingly, in this study model embryos survived the gastrulation stage, proposing that compensatory mechanisms of expressing other actin isoforms do exist, which may rescue at the earlier stages of embryonic development [24]. Consistent with this data, Tondeleir et al. observed a switch to γ-actin expression in embryonic stem (ES) cells lacking β-actin, while the β-actin was the only isoform expressed in wild type ES cells [14]. Overall, due to various complications in different studymodels caused by the actin gene knockouts [14], the research of β- and γ-actins’ function in regulating cellular motility has its’ own disadvantages, and thus it will require additional research and effort to answer these fundamental questions.
Cell shape maintenance
The cytoskeleton of animal cells is a very complex, but functionally versatile structure that needs to undergo highly coordinated and dynamic changes during cellular growth or adapt its’ shape in response to the external stimuli. These changes in cytoskeleton are mediated by three major types of cytoskeletal proteins: intermediate filaments, microtubules and actin. The transition of actin cytoskeleton is controlled via a balance of globular G- and filamentous F-actin, and by actin-associated proteins [25]. These, all together form a network of polarized filaments that provide with force generation required for movement, adhesion and cellular re-shaping. Among different actin filament arrays, resides an actin cortex [26], which is thought to play the key roles in cell mechanics. Indeed, the actin cortex enables cells to control its’ shape. A thin network of actin filaments, myosin motors and ABPs lies just below the plasma membrane. Thus, local alterations in mechanical properties of the cortex, especially in cortical tension, carries out cellular shape deformations, such as those that take place during each shape-wise-distinctive phases of mitotic division [27]. The research on actomyosin-dependent mitotic rounding demonstrated that cortical actin thickness is decreased as tension increases, going from prometaphase to metaphase, where the involvement of myosin, rather than actual actin, regulates cortical tension [28]. Surprisingly, some contrary results were published, where modulating actin dynamics without modifying myosin concentration was shown to have an effect on intracellular tension [29]. The actin cortex is a ~300-1000 nm [30] thick layer, made of a mixture of filament bundles and cross-linked filaments. It has a mesh-size of ~50-150 nm [31,32], and thickness of ~50-100 nm [32,33], with a distance to the cell membrane of <20 nm [33]. Despite of the main constituents, the cortex contains a huge variety of cross-linkers (filamin, actinin, and fascin), capping protein, myosin with actin turnover-associated proteins (cofilin and profilin), ERM family proteins (ezrin, radixin, and moesin), nucleating factors (Arp2/3 and formins), and finally, signalling molecules, such as RhoGTPases, RhoGEFs and RhoGAPs [34]. A full description of aforementioned proteins’ roles during actin cytoskeleton and cell cycle progression regulation if not covered in this review, can be readily assessed in Heng and Koh review article [35]. The ERM proteins connect the cortex to the membrane, and thus can pass on forces to the plasma membrane, which determines the shape of the cell [36]. A study by Chugh et al. demonstrated that depletion of cofilin-1 or capping protein enhanced actin cortex thickness and reduced tension in HeLa cells, indicating a function for actin regulating proteins in a cortical stress [27]. The cortical tension is a major feature of the cortex, which makes it possible to regulate dynamics of the cell shape of single cells during cell growth and in response to the external forces. It was shown that the cortex tension depends on actin polymerization and myosin activity, where higher myosin activity and reduced actin polymerization leads to an increased stress of actin cortex [37]. Additionally, a lower cortex tension has been associated with an increased protrusive ability of the cell [38]. So, mechanical properties of the actin cortex determine the rate and extent of cellular deformation during processes important to cells, and in response to other stimuli.
Cytoplasmic actin of mammalian cells is composed of two actin isoforms, β- and γ-actins. The function of these two actin isoforms in cells very much differs. The involvement of β-actin in a control of cell growth was demonstrated in a study by Bunnell et al., where β-actin was shown to be an absolute requirement for sustaining cellular growth potential, while γ-actin was shown to be important for cell survival [22]. In fact, authors observed an increase in the percentage of multinucleated cells in β-actin-knockout MEFs, but not in γ-actin-null MEFs, proposing additional role of β-actin in cell division [19]. This data is consistent with previous reports, showing that β-actin is required for maintaining a proper change in cellular shape during mitosis [39]. In addition, β-actin was shown to accumulate at the ICB in dividing cells [40,41], and due to the better dynamics of the two actin isoforms, was shown to play specific roles in a rapid reorganization of actin cytoskeleton during mitotic division [42]. Thus, the cytoplasmic isoform of β-actin as well as actin cortex itself, provide a base for maintaining and shaping the cell during various spatiotemporally-mediated and vital processes in cells.
Cell division
Actin cytoskeleton experiences a tightly regulated morphological transformation during cell division, which allows for successful DNA content duplication, cellular round-up, condensation and alignment of chromosomes, breakage of chromosomes and movement of sister chromatids to the opposite poles of the cell, and finally physical cleavage of newly formed daughter cells (Figure 2). All of this is possible thanks to the actin-microtubule framework, which depending on physical-structural demand during different phases of mitosis, provides elasticity and rigidity for cellular structure.
Figure 2.
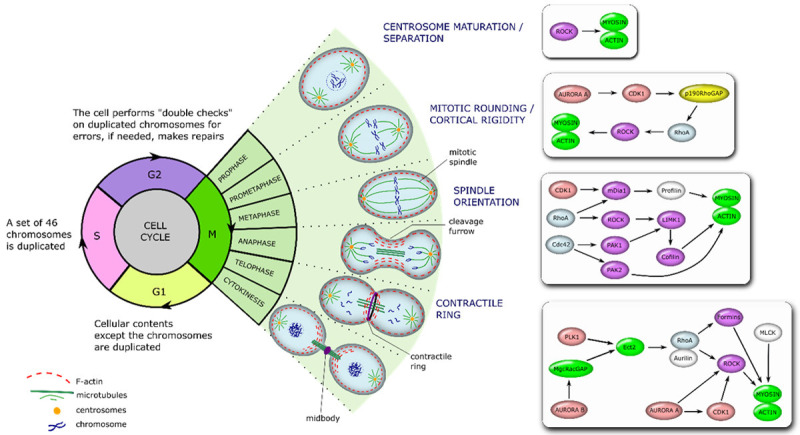
The regulation of actin dynamics during different phases of mitosis. The scheme shows different phases of the cell cycle, depicting the intracellular processes that take place during mitotic (M) phase, and placing a special emphasis on signalling cascades that regulate actin cytoskeleton during the cell cycle progression. During prophase chromosomes condense, spindle fibers emerge from the centrosomes, nuclear envelope breaks down, and centrosomes move towards the opposite poles of the cell. Then, in prometaphase, kinetochores arise at the centrosomes and mitotic spindle microtubules attach to the kinetochores. Next, during metaphase, chromosomes line up at the metaphase plate, while each sister chromatid gets attached to a spindle fiber, outgrowing from the opposite poles. During anaphase, centromere split in two and sister chromatids are pulled towards the opposite poles, while spindle fibers start to elongate the cell. This is followed by telophase, where chromosomes at the opposite poles begin to decondense, and nuclear envelope, surrounding each set of chromosomes, starts to reform. Then, spindle fibers continue to push poles to the opposite directions. Finally, a cleavage furrow, which separates newly formed daughter cells, is formed. The scheme on a right depicts functional relation of proteins, participating in a control of actin dynamics during the M-phase of the cell cycle.
As the name dictates, adherent cells cling onto the substratum during interphase and spread along the surface with a help of adhesion. Mitosis starts with a reduction of cell-matrix adhesions, which in mammalian cells is regulated via integrin and cadherin signalling [43]. The cells’ actin cytoskeleton begins to change during mitosis, where two rounded cells with increased cortical rigidity are formed. After mitosis, the actin cytoskeleton is re-established, enabling cells to regain its’ primary shape and attach to the substratum (Figure 2). This well-organized modulation of such radical changes in cell shape during cell cycle progression proposes a strict regulatory interplay among cytoskeleton signalling, mitotic events, as well as cell-matrix and cell-cell adhesion remodelling [35]. Here we briefly review the regulation of actin cytoskeleton during the cell cycle progression.
Interphase cells are made of an entangled network of actin filaments, which upon cells first entering mitosis, needs to be disassembled and re-organized allowing for mitotic cells to round up. Then, at the end of mitosis, actin rearranges at the cleavage furrow and makes a part of the contractile ring, which mediates the final stage of mitosis, a process known as cytokinesis (Figure 2). The absolute requirement of an intact actin cytoskeleton for the onset of mitosis in a timely manner was demonstrated after cytochalasin D and Latrunculin B had been applied on primary cells and fission yeasts, where it had caused the depolymerisation of actin filaments [44,45]. Additional reports have also demonstrated that stability of the cortical network is vital in ascertaining a correct spindle orientation in mammalian cells [35,46], and that F-actin is prerequisite at the mitotic apparatus during mitotic spindle assembly [47]. Actin network also plays an important mitotic role in the separation of centrosomes, which directly depends on a flow of cortical actin and myosin network. The mitotic cortical actomyosin network, which is cross-attached to the plasma membrane provides elasticity that is prerequisite to counteract the internal pressure inside the metaphase cells [48]. Destruction of this network by actin inhibiting/depolymerising drug Latrunculin or via myosin-II RNA interference (RNAi) leads to a failure of spindle assembly, and subsequent centrosome separation [49,50]. The intracellular forces required for chromosome segregation after spindle formation are generated via actin and myosin interplay as well [51]. The comprehensive summary of drugs’ and chemicals’ effect on actin cytoskeleton can be found in Heng and Koh review article [35].
Finally, for cytokinesis to occur, the constriction of a cleavage furrow must take place. In a typical mitosis, the cleavage furrow forms at the equatorial cortex after anaphase [52], where its’ positioning depends on the mitotic spindle orientation [53]. Here actin gets reorganized and forms a part of the contractile ring (Figure 2), which is essential for the accomplishment of cytokinesis. β-actin is critical for correct change of the cell shape during mitosis [39]. Since β-actin is more dynamic among the two cytoplasmic actin isoforms [6], it may play a specific role during instant reorganization of actin cytoskeleton. Indeed, Dugina et al. showed that β-actin gets concentrated at the contractile ring in dividing cells [40]. Essentially, actin remodelling is major to cortical ingression, where contractile ring transforms into a midbody (MB) ring, and assists in the preparation of future abscission sites [54]. Several proteins have been shown to regulate contractile ring formation via multi-step molecular signalling pathways (Figure 2). Primarily, it was shown that RhoA is responsible for the assembly of the contractile ring that activates the acto-myosin-mediated constriction of the cleavage furrow [55]. Then, the whole signalling cascade with a number of different activating factors has been shown to contribute to the induction of the cleavage furrow formation [56,57]. In addition, several other Rho GTPase regulators were shown to participate in the control of contractile ring formation and cytokinesis, which in more detail will be discussed in the coming paragraphs.
The regulation of actin dynamics during cell division
Cell division is a process that encompasses separation of chromosomes during mitotic phase and physical cleavage of cells during cytokinesis. The reorganization of cellular cytoskeleton during division is coordinated by the actin-microtubule network [58,59]. The first step in mitosis is prophase, during which, the microtubules rearrange and start to polymerize, forming bipolar spindle, while DNA condenses into chromosomes. The transition from G2 to early prophase requires correct spatial cytoskeleton coordination, during which, centrosomes must separate before nuclear envelope breakdown (NEBD). It was shown that centrosome separation before NEBD is slowed down by intracellular forces that push the centrosomes towards the central position below the nucleus [59]. The mechanical force to antagonize centrosome separation requires microtubule and actin polymerization, without which, centrosome separation loses its’ symmetrical position respective to nucleus. Here actin plays a critical role, because inhibition of the linkage between nucleoskeleton and cytoskeleton complex with F-actin leads to mispositioning of centrosomes at the NEBD, which then causes and increased probability of errors during sister chromatid segregation [59]. Next, in prometaphase, nuclear membrane decomposes, making a way for kinetochore microtubules to invade nuclear space and to attach to kinetochores. During the preparation of mitotic entry, centrosomes initiate the formation of mitotic spindle by aligning individual chromosomes at the metaphase plate. Here, during prometaphase, a formation of actin filament and spindle microtubules assembly was identified [60]. Moreover, it was shown that inhibition of actin nucleator in eukaryotic cells Arp2/3 complex, results in a reduced formation of spindle actin. As a result, the mitotic spindle assembly is disturbed, leading to a disorganized chromosome alignment to the metaphase plate, which inevitably leads to mitotic defects [60]. Indeed, a crosstalk between actin and other regulatory proteins was shown to play key roles in maintaining the genomic integrity via a modulation of actin dynamics during mitotic division [61]. Next, in metaphase, chromosomes line up to the metaphase plate, where each chromosome arm is attached to kinetochores, which with a help of extended microtubules reach out to the opposite poles of the spindle. Here, a proper spindle positioning and orientation controlled by the interplay of microtubule and F-actin is an absolute requirement for an accurate mitosis. This would not be possible without a ring-like F-actin structure that is formed around the mitotic spindle, and which was proposed to regulate astral microtubule dynamics and subsequent mitotic spindle orientation [62]. Additionally, it was shown that interrupting either microtubules or F-actin polymerization and inhibiting F-actin-interacting proteins myosin and Plk1, does have a negative effect on a ring-like F-actin structure formation, which consequently modifies spindle assembly and disrupts symmetric division [62]. During the next phase of the cell cycle, which is anaphase, upon checkpoint completion, chromosomes split at centromeres, and resulting sister chromatids are pulled to the opposite sides of the mitotic spindle (Figure 2). As chromosomes separate, the bipolar spindle switches into a collection of interzonal microtubules, which, during telophase, together with actin, condenses into an intercellular bridge (ICB) with a MB in the middle. The nuclear envelope starts to materialize around each set of chromosomes and the mitotic spindle breaks apart. A study by Chen et al. demonstrated that for the completion of cytokinesis, an opposing regulation of the β-(at contractile ring) and γ-actin (at cell cortex) networks is required [63]. What is meant by this is that actin remodelling protein formin DIAPH3 gets activated at the cytokinetic furrow, producing β-actin filaments, while γ-actin gets expended at the cell poles because of the deactivated DIAPH1. Moreover, it was shown that during anaphase, the displacement of actin polymerizing genes inhibits formin’ function, resulting in a reduced cortical stiffness, and subsequent localized membrane blebbing [63]. Finally, cytokinesis takes place, where actin filaments pull the equator of the cell inwards, forming a constriction site (Figure 2). This constriction is also known as a cleavage furrow, deepens as the actin ring retreats, and eventually a physical separation of newly formed daughter cells takes place [64]. In more detail, a cytokinetic cleavage furrow is formed by actomyosin contractile ring contraction, generating the MB, situated in the middle of the ICB. This serves as a platform for the assembly of the abscission machinery that regulates the final separation of newly formed daughter cells. Here, the polymerization of F-actin plays a major role during the assembly, ingression, disassembly, and constriction of the actin contractile ring, and for the cytoskeletal reorganization, leading to a maturation of the MB, which eventually completes the ICB abscission. Therefore, actin filaments must be removed from the sites of abscission for the final cut to take place. Even though many conserved proteins were shown to co-operate in mediating the polymerisation state of actin filaments, we still lack knowledge of molecular mechanisms that drive the final scission machinery during cytokinesis in higher eukaryotes. The research done by Terry et al. reported that actin capping protein and to the barbed end-actin-binding proteins regulate actin polymerization at the very end of cytokinesis [65]. It was shown that even though the depletion of capping proteins results in early MB formation, the cytokinesis still fails. Additionally, the recruitment of endosomal sorting complexes required for transport (ESCRT-III) is disrupted, leading to a failure of abscission [65]. The same group of scientists yet proved another requirement for a successful mitotic division, - the generation of optimal length actin filaments. This was shown to be balanced by formins, which nucleate actin and balance capping proteins’ activity, and if reduced, could lead to an increased accumulation of linear actin filaments, further disrupting cytokinesis [65]. To summarize, during cell division, the dynamics of cellular cytoskeleton made of actin and microtubule filament networks is controlled in a step-by-step manner, where various actin and microtubule regulators join in and leave signalling cascades at different stages, while performing nature-specific functions and adding up to the success and completion of cell division. Importantly, what we know so far are the well-studied class of proteins being involved in a regulation of actin cytoskeletal dynamics during mitotic division, which includes Rho family of small GTPases and their effectors, as well as actin-binding proteins, such as capping protein, profilin and cofilin, and myosin-II, which all share lipid-binding domains [48]. In terms of location, none of these proteins localise at the internal cellular membrane, but rather associate with that membrane through either lipid-binding domains or lipid modifications [48]. Importantly, the reciprocal interaction between the membrane and the cortex assists in cellular division, while performing membrane reorganization in a precise and timely schedule. Rho GTPase and Aurora B kinase, as well as other regulatory proteins of cytokinesis have been studied extensively (Figure 2), and will be discussed in the later paragraphs.
Actin cytoskeleton-associated proteins
The plasma membrane of dividing cell is connected to the cell cortex via a huge diversity of membrane-binding linkers, which connect plasma membrane phospholipids and proteins to the underlying cortical actin cytoskeleton. Such proteins include ezrin, radixin and moesin (ERM) as a regulatory protein complex [66]. It was shown that in normal cells, the removal of ERMs from the cell poles, loosens the rigid cortex during anaphase, which allows for cellular elongation and subsequent chromosome segregation, facilitating the transition to the next phase of the cell cycle. This process in mammalian cells was shown to be driven specifically by phosphatase PPP1R7 [66], which reverses phosphorylation of C-terminal on moesin at the polar cortex. In addition, the cortical ezrin also undergoes dramatic re-localisation during the orientation of centrosome and mitotic spindle [67]. It was demonstrated, that in mammalian colonic epithelial cells, the ezrin- actin-rich cap-like structure forms before the S phase, where it positions the centrosome and one pole of the ensuing mitotic spindle, providing a stable platform for astral microtubule attachment. During the metaphase-anaphase transition, ezrin disappears from the polar cortex and again shows up at the cleavage furrow, forming a lumen around which the dividing cells form a cyst [67]. Contrary to the spatiotemporal ERM regulation during anaphase, upon progression to the cleavage furrow, the ERMs become increasingly restricted where they may serve other mechanical functions during cytokinesis [67]. Equally, a study by Kunda et al. demonstrated that another member of the ERM protein family, moesin, is responsible for mediating cellular rounding and cortical stiffening during mitosis [68]. It contributes to establishing a rigid and uniform cell cortex that functions as a firm base for positioning of the mitotic spindle. Like other ERMs, upon initiation of anaphase, the loss of moesin from cell poles loosens the polar tension, relieving elongation of the mitotic spindle and chromosome separation, which if failed, could contribute to aneuploidy and development of cancer [68]. Finally, last but not least, radixin was reported to accumulate rapidly at the site of furrow formation during anaphase and maintain its’ position at the cleavage furrow until the end of cytokinesis [69]. The relevance to cellular division was proved by overexpression of C-terminal truncated mutant of radixin, which resulted in a multi-nucleation of human cells [70,71]. Another study by Vilmos et al. reaffirmed the mitotic function of ezrin-radixin-moesin protein complex in cross-linking various proteins to the actin filaments of the actin cytoskeleton [72]. In addition, ERMs were shown to aid in the attachment of the mitotic spindle to the cortical actin network and cellular re-shaping during mitosis [73].
Rho GTPases
Being important regulators of actin cytoskeleton, Rho GTPases function as major moderators of many regulatory aspects during mitosis and cytokinesis. Such functions include centrosome duplication and separation, generation of cortical rigidity, microtubule-kinetochore stabilization, cleavage furrow formation, contractile ring formation and subsequent constriction, and finally, abscission [74]. Physical separation of newly formed cells at the end of mitotic phase is mediated by the ingression of actomyosin contractile ring. The GTP-bound and active form of RhoA induces transcriptional changes via F-actin dependent means, and thus, is a key regulator of contractile ring formation [75]. RhoA localizes at the equatorial cell cortex of the emerging cleavage furrow (Figure 3). Once it reaches cytokinesis phase, the primary signal to assemble the contractile ring emanates from Rho-GEF (ECT2), which activates RhoA to promote nucleation, elongation, and movement of actin filaments. Here, coordinated activation of formins (interacts with the barbed end of an actin filament and assembles actin filaments) and myosin-II motors come to an action. Another protein, which is highly concentrated in the cleavage furrow upon anaphase onset and plays an important role during cytokinesis, is anillin (Figure 3). It helps to maintain active myosin in the equatorial plane of the cell, thereby functioning as a scaffold protein and linking RhoA with the contractile ring components actin and myosin [75,76]. It was shown that even though furrows can form and initiate ingression without anillin [76], its’ interaction with RacGAP turns out to promote myosin-II activation via interactions with RhoA [76]. It has been proposed that activated RhoA at the equator of the cell stimulates contractile ring formation via direct formins activation, and indirect myosin-II activation through Rho-kinase and phosphorylation of myosin regulatory light chain (RLC) [77]. Surprisingly, a study by Liu and Weiner demonstrated that RhoA activity can induce furrow formation in all positions of the cell cortex during both, metaphase and anaphase phases [78].
Figure 3.
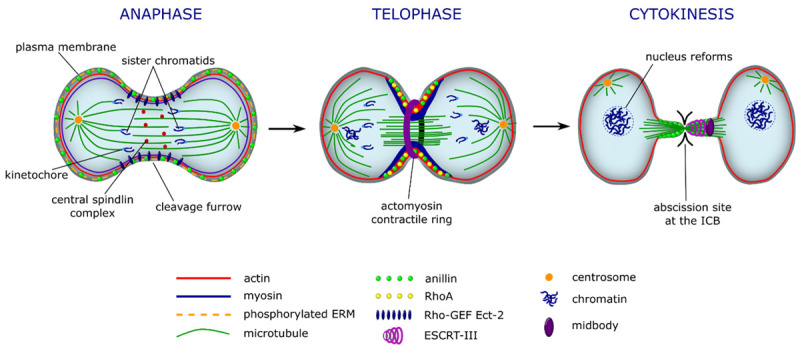
Organization of cellular cytoskeleton during furrow ingression. The picture illustrates the mitotic cleavage furrow formation during anaphase, formation of actomyosin contractile ring in telophase, and final abscission of the ICB during late cytokinesis. In the beginning of anaphase, sister chromatids start to separate upon central spindlin complex entering the spindle midzone. Then, the signalling cascade, which controls reorganization and spatio-temporal dynamics of actin, microtubule, and myosin-II, is activated. This recruits Rho-GEF Ect-2, which activates RhoA and initiates the cleavage furrow formation. In telophase, actomyosin contractile ring and microtubule complex is generated, which produces intracellular forces and tension, required to narrow the furrowing cell at the equatorial actin cortex. In late cytokinetic event, the actin filaments are cleared from the narrowed ICB, where final abscission, mediated by ESCRT-III complex, takes place. Finally, this results in a physical separation of the newly formed daughter cells.
Capping protein
Capping protein is a heterodimer made of structurally similar α- and β-subunits that bind to the barbed ends of actin filaments and is expressed in all eukaryotic cells [79]. Capping protein binds to profilin, and helps to retain actin monomer pool by limiting the number of barbed ends open for growth of actin-based protrusions at the leading edge of the cell, thus stabilizing the barbed ends of actin filaments of striated muscles [13]. Obviously, due its’ ability to interact with barbed ends of the growing actin filaments, capping protein may also play an important role in regulating the length, and thus the growth rate of actin filaments that make up cytoskeleton structure during cytokinesis. Indeed, Terry et al. showed that capping protein participates in the control of actin polymerization towards the end of cytokinesis in human cells [65]. Moreover, cells depleted of capping protein furrow and form premature MBs and fail cytokinesis. Consequently, the proper recruitment of ESCRT-III to the MB fails, impeding further progression into the abscission stage (Figure 3). Importantly, the length of actin filaments optimal for cytokinesis to occur must be strictly balanced. Here actin nucleator formin acts as the major regulator of capping protein activity. Reduction of capping protein activity results in an increased accumulation of formin-based actin filaments. Therefore, depletion of formin FHOD1 leads to a partial rescue of capping-protein-induced cytokinesis defects, proposing that it may antagonize capping protein activity upon midbody formation [65]. Another study by Jo et al. demonstrated that capping protein is crucial for the efficient spindle migration and supervision of the cytoplasmic actin mesh density during asymmetric division of maturing mouse oocytes [80]. A very first study on capping protein function during cell division in fission yeast cells showed that capping protein controls actin dynamics together with actin depolymerization factor (ADF)/cofilin and profiling [79], and is also involved in assembling the F-actin contractile ring [79]. The regulation of subsequent proteins will be discussed in the next paragraph.
Formins, profilin and cofilin
Cellular processes that require morphogenetic alterations in actin cytoskeleton, especially during the formation of contractile ring, foremost, are driven by formins, which are highly conserved initiators of actin assembly. Formins are found in all eukaryotic organisms, where they spatially and temporally control the growth of actin microfilaments (Figure 3). Now, it is widely accepted that formins are required for mammalian cell cytokinesis, where they inhibit or promote elongation of actin filaments [65,81,82]. Most formins are effector proteins of Rho GTPases (Figure 2). Formins are characterised by formin homology domains, that is to say, formin homology domain 1 (FH1) and formin homology domain 2 (FH2) [83]. Specifically FH2 was shown to nucleate and elongate the linear actin filaments by interacting with the barbed end of actin filaments [13,84]. Whereas FH1 domain has multiple binding sites for profilin, -a protein that is enriched during binding to actin monomers and to the barbed end of the actin filament. Profilin is known for spontaneous inhibition of actin nucleation. It binds to the barbed end of actin monomer and suppresses the actin filaments elongation (Figure 1) [13]. It was shown that high concentrations of free profilin can slow down the elongation and even induce dissociation of the terminal subunits [82]. In addition to formins, another well-studied actin filament nucleating protein during cytokinesis is cofilin, which is expressed in high concentrations in most eukaryotic cells [13]. The action of cofilin in cells directly depends on the local concentration of active cofilins: low concentration leads to severing, while high concentration favours nucleation of actin filaments [83]. Wisely, cells use different molecular machinery to regulate cofilin expression. One example is phosphorylation of Ser3 of cofilin, which prevents cofilin binding to actin, and thus inactivates all actin-related cellular functions and stabilizes the actin cytoskeleton [13,85].
Myosin-II
The cell cortex is a fine actin network bound to the plasma membrane and found in most animal cells. It is organized as highly tangled network of hundreds of ABPs, including myosin-II motors. Interaction of myosin-II and actin filaments produce forces required to constrict the contractile ring (~25 nN in the cleavage furrow of echinoderm eggs) [86], and subsequently, to form a cleavage furrow [87]. Myosin-II generates forces strong enough to pull on actin filaments, generating contractile strain in the network, which raises cortical tension of the cell (Figure 3). Variation of such a cortical tension allows for cells to change in shape, -a feature seen during migration and cell division [27,88,89]. Myosin transformation between activated and deactivated states during mitosis is regulated by phosphorylation of RLC of myosin-II at Ser19/Thr18. Two enzymes drive this process, activated myosin phosphatase catalyses the RLC dephosphorylation, causing the disassembly of stress fibers and focal adhesions during prophase, and kinase, which phosphorylates myosin targeting subunit at its’ inhibitory site and reduces the activity of myosin phosphatase during cytokinesis [87]. These modifications in RLC phosphorylation, leading to the activation of myosin-II during cytokinesis are mediated by different kinases present at the cleavage furrow, including Rho-associated kinase (ROCK), citron kinase and MLCK [87]. It was proposed that myosin signalling pathway is foremost triggered by cell cycle kinases cyclin-dependent kinases (CDKs) and polo-like kinases, which coordinate two master regulatory complexes, known as chromosomal passenger complex (CPC) and centralspindlin complex. According to Pollard and O’Shaughnessy, these multi-component complexes determine the site of the cleavage furrow in the cell and activate downstream signalling cascade, including proteins, such as Rho-GEF or ECT2, Rho GTPases and other regulatory proteins of actin contractile ring [87]. Finally, advantages associated to the use of electron micrographs acknowledged the interaction of myosin filaments and actin filaments during a process of contractile ring formation in sea urchin zygotes [90].
Kinases
The cell cycle comprises of consecutive phases mediated by cyclin proteins that function by activating CDKs [91]. In eukaryotes, cellular division starts when CDK1 is activated and carries on its’ function with some help from additional kinases, such as Aurora A, Aurora B, and polo-like kinase 1 (PLK1) (Figure 2). As cell progresses through the cell cycle, unidirectionality of this process is ensured by the targeted proteolysis of the key proteins of the cell cycle. Upon completion of division, phosphatases catalyse the hydrolysis of regulatory molecules, and revert the division state back into interphase phase [92]. It was demonstrated that kinases regulate the timing of the cell cycle; the activation of CDK1/cyclin B pushes the cell into mitosis, but stops division at anaphase by phosphorylating specific sites on proteins that inhibit various cell cycle steps [90]. As an example, phosphorylation of Rho-GEF, inhibits ECT2 activity and further interactions with centralspindlin and other membranes, those concentrate on spindle midzone microtubules at the cleavage furrow [91]. Also, specific inhibition of CDK1 during mitosis can lead to premature cytokinesis [90]. In addition, a study by Basant et al. demonstrated that another kinase Aurora B, can stimulate contractile ring formation by inactivating PAR-5/14-3-3 protein, whose original function is to inhibit oligomerization of centralspindlin [93]. To continue on this topic, formin FHOD1 was identified as a specific Aurora B interactor during cytokinesis, where it localizes to the middle zone of the MB, acting on the barbed ends of actin filaments [94].
Malfunction of actin dynamics during cell division
Remodelling of actin dynamics is necessary for ingression of cellular cortex, transition from a contractile ring to the ICB with a MB ring, and further preparation of the abscission site. The lipid phosphatidylinositol 4,5-biphosphate (PtdIns(4,5)P2) is a major regulator of actin dynamics. It promotes actin polymerization and allocates to the cleavage furrow at a very last step of cell division. The levels of PtdIns(4,5)P2 during cytokinesis is constantly adjusted, where its’ phosphatase OCRL (Lowe oculocerebrorenal syndrome protein) is coordinated locally at the abscission site by Rab35 [95]. Dambournet et al. demonstrated that the correct Rab35 guiding of OCRL, and OCRL-dependent remodelling of F-actin and lipids are of absolute necessity during late stages of cell division. It has been shown that PtdIns(4,5)P2 drives normal cytokinetic-abscission, where OCRL phosphatase is responsible for the hydrolysis of the PtdIns(4,5)P2, and that an improper hydrolysis could lead to the F-actin accumulation in late cytokinetic bridges, thus preventing physical separation of daughter cells [95]. This often leads to aneuploidy [96,97], tetraploidy [96,98], and chromosome instability [99,100], which in worst case scenario can contribute to the progression of tumorigenicity, ultimately developing into cancer [101]. In addition, abnormal cytokinetic abscission due to improper actin clearance in specific cell types during development might explain some of the arising neurological and congenital disorders [102]. Indeed, short F-actin cytoplasmic filaments were frequently encountered in Lowe syndrome patients’ fibroblasts during interphase, suggesting an aberrant F-actin dynamics in these patients’ cells [103]. Luckily, the addition of actin depolymerizing drugs was shown to rescue with actin-accumulation-associated cytokinetic defects. This raises a possibility that to disease-phenotypes-linked OCRL inactivation may be reversed, thus allowing avoiding complications and representing a possible therapeutic option for the disease [95].
Importantly, the PtdIns(4,5)P2 product, phosphatidylinositol 4-phosphate (PtdIns(4)P) coordinates other binding proteins required for cytokinesis, such as the Golgi-associated protein (GOLPH3) [104]. This interaction helps targeting of Rab11 vesicles to the cleavage site. Rab11-endosomes were shown to transport p50RhoGAP that inhibits actin polymerization [105]. Rab11 functions by binding to its’ effector proteins, such as Rab11 family interacting proteins (FIPs) [106]. Specifically, FIP3 is known to play a role in targeting of Rab11 endosomes during mitosis. Knocking down of the FIP3 cargo protein p50RhoGAP, stops actin disassembly at the ICB and inhibits charged multivesicular body protein 4B (CHMP4B), which functions as a component of ESCRT-III complex that mediates membrane remodelling in eukaryotes [107], recruitment to the secondary ingression site, thus halting the cytokinesis [105]. So, one can be said that actin depolymerization is necessary for appropriate ESCRT-III complex localization at the abscission site.
It was demonstrated that by interacting with MICAL1, Rab35 also contributes to actin clearance during cytokinesis [108]. MICAL1 is oxidoreductase and depolymerizes actin filaments, thus promoting actin clearance. Further members of MICAL family also helps to organize other vesicles at the abscission site: MICAL-L1 traffics Rab11 endosomes, while MICAL3 binds Rab8 vesicles [54]. This proposes the importance of these protein roles during cytokinetic events. A study by Fremont et al. demonstrated that MICAL1 depletion triggers accumulation of F-actin at the cytokinetic bridge, and causes several other defects associated with actin clearance from the abscission site [109]. The resultant accumulation of F-actin delays, and in some instances completely inhibits abscission [109]. Nevertheless, around half of the MICAL1-depleted cells could complete abscission in normal timing, proposing that additional compensatory, and yet unknown mechanisms might exist for the F-actin clearance from the abscission site at the ICB. Excessive accumulation of F-actin in cytokinetic bridges inhibits the recruitment of ESCRT-III components to the abscission site, which leads to abscission failure [110]. It is important to mention that MICAL1 was found to play an evolutionarily conserved role in cytokinetic abscission from Drosophila to human cells [108].
There are many other proteins and actin regulators that are plentiful at the site of cytokinetic ring formation, where they change their spatiotemporal dynamics as cell goes through anaphase-telophase. For example, Bai and et al. showed that methionine sulfoxide reductase B2 (MsrB2) is recruited to the MB in response to lagging chromatin and functions within the ICB to stimulate actin polymerization [111]. Upon depletion of MsrB2, F-actin levels drop in the ICB, while dividing cells with lagging chromatin turn to binucleated cells because of unstable cytokinetic bridges. Also, MsrB2 was shown to counteract MICAL1, which is known to depolymerize actin filaments [111]. Another protein that is highly important for the initiation of cytokinetic cleavage furrow is chloride intracellular channel 4 (CLIC4) [112,113]. A study by Kagiali et al. demonstrated that at the early cytokinesis, CLIC4 accumulates at the cleavage furrow, while during late cytokinesis, it localizes to the MB in a RhoA-dependent manner [113]. At the cleavage furrow and MB, CLIC4 interacts with ezrin and anillin, where reciprocally one cannot exist without the other. The knockout of CLIC4 causes abnormal blebbing at the polar cortex and regression of the cleavage furrow. This prolongs cytokinesis, which eventually leads to a formation of multinucleate cells [113]. Thus, CLIC4 functions in concert with ezrin in linking the plasma membrane to the actin cytoskeleton at the polar cortex and cleavage furrow, which helps to maintain cortical stability prerequisite for successful cytokinesis in mammalian cells. To supplement this proposition, a study by Peterman et al. demonstrated that CLIC4 mediates remodelling of the plasma membrane and actomyosin network at the furrow by recruiting MST4 kinase and adjusting phosphorylation of ezrin [112].
Actin dynamics during malignancy
The fundamental property of cancer cells that distinguish them from the benign cells is continues and unregulated proliferation. Instead of responding properly to the growth signals, cancer cells growth and divide in an uncontrolled fashion, invading normal tissues and organs, which eventually leads to tumour disseminating throughout the entire body [114]. One of the most important aspects for tumour cells, allowing them to spread into the distant tissues is the ability to migrate, invade and metastasize. This is possible due to the great plasticity of actin cytoskeleton. In recent years, there have been quite a few studies implicating on different molecular signalling pathways that induce cellular migration and invasion upon progression of different types of cancer [115-117]. A very recent study by Qiao et al. demonstrated that overexpression of yes-associated protein (YAP) causes cytoskeletal rearrangement by changing the dynamics of F-actin/G-actin turnover, and thus promotes migration of cancer cells [115]. Indeed, another study by Yuan et al. has also shown that ADF/cofilin-mediated actin disassembly is regulated via YAP signalling cascade, contributing to non-small cell lung cancer progression [118]. In addition, the most updated information of actin regulators and their structure-related properties that contribute to cancer progression and metastases are all covered in Biber et al. book [119]. In turn, in later chapters, we will discuss the regulation of actin cytoskeleton during epithelial to mesenchymal transition (EMT) and intracellular regulation of actin cytoskeleton-remodelling, - the processes, which add up to creating favourable conditions for metastases. It was estimated that up to 40% of all carcinomas experience the EMT and employ this feature for motility [120].
Actin dynamics during epithelial to mesenchymal transition
The EMT during cancer is generally associated with poor prognosis in patients [121]. Since almost 90% of all cancer-related deaths are due to metastases [122], many studies now concentrate on the repression of EMT. The EMT induces migratory potential and invasiveness of aberrant cells, which increases the potential for disease progression and the risk of metastasis [123]. It was proposed that reorganization of actin dynamics is regulated by the regulatory proteins such as myosin [124], where during cancer, an increased cell contractility and actin stress fibers could be observed [125]. This tight regulation of actin reorganization during the EMT is controlled in response to internal (osmotic pressure) or external stimuli (growth factors). For example, upon signal activation, the ABPs bind to Rho family of small GTPases, which are the main regulators of actin dynamics, and thus control cytoskeletal actin rearrangement during the EMT [126]. The regulation of EMT can also take place at the transcription level. A study by Grzanka et al. showed that sequence-binding protein 1 (SATB1) and F-actin complex may be detected in the border between condensed and decondensed chromatin [127]. The functional value of this complex was supplemented after Wan et al. study, where SATB1 was shown to be overexpressed in bladder transitional cell carcinoma and other bladder cancer cell lines, characterised by high metastasis potential [128]. This suggests that SATB1 participates in a control of the progression of bladder cancer by regulating the genes responsible for the control of the EMT processes.
The plentifulness of research groups in this specific field of cancer research best indicates the importance of the EMT-inducing signalling cascades in cancer cells. Recently, a clinical study by Peng et al. demonstrated that activation of actin cytoskeleton remodelling is crucial for cell density-dependent induction of EMT via hypoxia-inducible factor (HIF) and Notch signalling in cancer cells [126]. Also, it was shown that even though hypoxia is able to induce the HIF signalling, this is not enough to activate the Notch signalling, which then would induce the EMT, unless actin cytoskeleton remodelling would take place at the same time [126]. Most importantly, targeting the signals that activate actin cytoskeleton remodelling via actin polymerization could inhibit the EMT, and thus protect from tumour invasion and further metastasis [126].
The ability of cancer cells to metastasize into other tissues and organs is related to lethality of the disease. The worst case scenario is very often observed in colorectal cancer patients [129]. A recent study, investigating regulatory protein signalling during the EMT suggested that cofilin-1 is required for cancer cells to acquire the EMT morphology, including migratory and invasive phenotype [130]. This is achieved via tightly-controlled actin cytoskeleton organization through the activation of RhoA-LIMK2-cofilin-1 signalling cascade, which upon activation, impacts the cell-cell adhesion organization of colon cancer cells during EMT [130].
The EMT being a crucial process of cancer progression, and thus very often determining the fate of cancer patients, is one of the most widely studied aspects of cancer metastasis. However, the functioning cascade of each individual protein shown to be involved in the regulation of EMT of cancer cells needs to be studied extensively for scientists and clinicians to make a larger image of these tightly controlled processes, and to deal with this misfortune of the humanity.
Actin dynamics during cancer cell adhesion, migration, and invasion
The EMT plays an important role in development, wound healing, and progression of cancer. Especially during metastasis, the primary tumour cells need to invade the surrounding tissue. To do that, tumour cells must terminate cell-cell contacts, remodel cell-extracellular matrix (ECM) adhesion sites and make its’ way through the ECM, following chemo attractants [131].
Upon activation of EMT, epithelial cells experience dramatic changes in their behaviour: cells tend to lose their apical-basal polarity, stable cell-cell adhesions, and most importantly acquire migratory phenotype by reorganizing their actin cytoskeleton [132]. These processes allow for directional migration of disseminating cancer cells. Tumour progression is associated with the loss of E-cadherin-mediated cell-cell adhesions and integrin-mediated adhesions that connect cancer cells to the surrounding ECM [133]. The main locomotion is driven by dynamic protrusions at the leading edge of the migrating epithelial cells [124]. The lamellipodium is one of the main force-generating cellular structure, where actin is polymerized locally at the cell front via Arp2/3, and depolymerized at the back of the lamellipodium by ADF/cofilin [131]. To hold structured lamellipodium in place and prevent withdrawal during migration, which could arise from actin cortex tension, the new cell-ECM contacts are prerequisite. Here, nascent adhesions, requiring myosin II and RhoA to further mature into focal adhesions during lamellipodium extension, are formed [134]. Next, the matured focal adhesions take up a role in functioning as anchorage sites for stress fibers that generate tension. Usually, stress fibers are directly joined to focal adhesions, connecting the cell to the ECM [133]. For efficient migration, the rear end of the cell needs to contract without major obstacles. Thus, for correct and timely contraction, myosin aids in sliding anti-parallel actin fibers along each other, thus creating contractile forces to contract actin filaments anchored to the ECM via focal adhesions. In addition, several studies have revealed an overexpression of RhoA, Rac1 and Cdc42 GTPases to be related to the progression of different cancers [134,135]. Indeed, Cdc42 and Rac were shown to coordinate actin polymerisation at the front of the moving cell [136,137]. The increase in these proteins’ expression is linked to a higher migratory phenotype of cancer cells. For example, the activated Cdc42 leads to a formation of invadopodia and production of matrix metalloproteinases (MMPs) [131], which are absolutely vital enzymes for degradation of the ECM during tumour cells invasion [138]. Thus, the EMT is considered as a teamster of invasion and metastatic spread of cancer cells [139]. However, some cell types do not require lamellipodium and the involvement of Rac or Arp2/3 for the migration and invasion. Instead, they migrate via filopodia, invadopodia or other formin-dependent pseudopodia [137,140].
Concluding remarks and future perspectives
Our knowledge about actin dynamics during cell division and its’ dysregulation, leading to the development of cancer has grown in recent years. It is now clear that actin participates in a huge variety of cellular functions, from the perspective of this review, most importantly, in the regulation of cytoskeleton during a very last step of cell division-cytokinesis. In recent years, there has been increasing evidence, displaying connection between the mis regulation of actin regulatory proteins and prediction of poor clinical prognosis in cancer patients. Even though the increased cytoskeletal activity is frequently associated with cancer progression, the expression of factors responsible for actin polymerization is surprisingly reduced in certain types of tumours, and thus, it is not always completely clear how alterations in the expression of actin regulators contribute to enhancing tumorigenesis and inducing metastases. For this reason, the complex regulation of actin dynamics becomes a huge challenge, when targeting of the specific signalling pathway aimed to inhibit certain cytoskeletal regulators, needs to be chosen. This particularly concerns the discovery and application of new therapeutic strategies, which in a form of drugs, should be appointed as a single agent or a mix of active substances to jointly maintain the cortical stability necessary for successful mitosis, while at the same time ensuring the correct actin levels in late ICBs of dividing cells. Also, therapies to prohibit tumour cells from spreading would be highly significant in preventing cellular motility at the early stage of the disease. Therefore, keeping a constant F-actin/G-actin turnover and inhibiting formation of actin-rich dynamic protrusions as well as exaggerated cytoskeleton plasticity, would add up to reducing the EMT, overall cancer cell motility and further metastasis into distant tissues and organs. Moreover, the actin cytoskeletal machinery in eukaryotic cells is governed by enormous number of proteins, very often transducing signals via multi-step signalling cascades. However, current approaches of cancer therapy very often involve targeting of a single gene, molecule, or molecular signalling pathway alone, where an expected inhibitory feedback is either not specific or insignificant. This is because malignant cells can switch to different modes of signal transduction, which are less reliant on the protein or molecule being inhibited, and thus can thrive without being affected. Therefore, a promising future direction in this field could involve decoding of the whole set of signalling cascades and use a combination of inhibitors, instead of targeting certain key cytoskeletal regulators.
Acknowledgements
This research is/was funded by the European Social Fund under the No 09.3.3-LMT-K-712 “Development of Competences of Scientists, other Researchers and Students through Practical Research Activities” measure.
Disclosure of conflict of interest
None.
References
- 1.Hohmann T, Dehghani F. The cytoskeleton-a complex interacting meshwork. Cells. 2019;8:362. doi: 10.3390/cells8040362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herman IM. Actin isoforms. Curr Opin Cell Biol. 1993;5:48–55. doi: 10.1016/s0955-0674(05)80007-9. [DOI] [PubMed] [Google Scholar]
- 3.Rubenstein PA. The functional importance of multiple actin isoforms. BioEssays. 1990;12:309–315. doi: 10.1002/bies.950120702. [DOI] [PubMed] [Google Scholar]
- 4.Vedula P, Kashina A. The makings of the ‘actin code’: regulation of actin’s biological function at the amino acid and nucleotide level. J Cell Sci. 2018;131:jcs215509. doi: 10.1242/jcs.215509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dugina VB, Shagieva GS, Kopnin PB. Biological role of actin isoforms in mammalian cells. Biochemistry (Mosc) 2019;84:583–592. doi: 10.1134/S0006297919060014. [DOI] [PubMed] [Google Scholar]
- 6.Perrin BJ, Ervasti JM. The actin gene family: function follows isoform. Cytoskeleton (Hoboken) 2010;67:630–634. doi: 10.1002/cm.20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kashina AS. Regulation of actin isoforms in cellular and developmental processes. Semin cell Dev Biol. 2020;102:113–121. doi: 10.1016/j.semcdb.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez A, Kashina A. Posttranscriptional and posttranslational regulation of actin. Anat Rec (Hoboken) 2018;301:1991–1998. doi: 10.1002/ar.23958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajakylä EK, Vartiainen MK. Rho, nuclear actin, and actin-binding proteins in the regulation of transcription and gene expression. Small GTPases. 2014;5:e27539. doi: 10.4161/sgtp.27539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kudryashov DS, Reisler E. ATP and ADP actin states. Biopolymers. 2013;99:245–256. doi: 10.1002/bip.22155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Svitkina T. The actin cytoskeleton and actin-based motility. Cold Spring Harb Perspect Biol. 2018;10:a018267. doi: 10.1101/cshperspect.a018267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winder SJ, Ayscough KR. Actin-binding proteins. J Cell Sci. 2005;118:651–654. doi: 10.1242/jcs.01670. [DOI] [PubMed] [Google Scholar]
- 13.Pollard TD. Actin and actin-binding proteins. Cold Spring Harb Perspect Biol. 2016;8:a018226. doi: 10.1101/cshperspect.a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tondeleir D, Lambrechts A, Müller M, Jonckheere V, Doll T, Vandamme D, Bakkali K, Waterschoot D, Lemaistre M, Debeir O, Decaestecker C, Hinz B, Staes A, Timmerman E, Colaert N, Gevaert K, Vandekerckhove J, Ampe C. Cells lacking β-actin are genetically reprogrammed and maintain conditional migratory capacity. Mol Cell Proteomics. 2012;11:255–271. doi: 10.1074/mcp.M111.015099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simiczyjew A, Pietraszek-Gremplewicz K, Mazur AJ, Nowak D. Are non-muscle actin isoforms functionally equivalent? Histol Histopathol. 2017;32:1125–1139. doi: 10.14670/HH-11-896. [DOI] [PubMed] [Google Scholar]
- 16.Vedula P, Kurosaka S, MacTaggart B, Ni Q, Papoian GA, Jiang Y, Dong D, Kashina A. Different translation dynamics of β- and γ-actin regulates cell migration. Elife. 2021;10:e68712. doi: 10.7554/eLife.68712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katsuno H, Toriyama M, Hosokawa Y, Mizuno K, Ikeda K, Sakumura Y, Inagaki N. Actin migration driven by directional assembly and disassembly of membrane-anchored actin filaments. Cell Rep. 2015;12:648–660. doi: 10.1016/j.celrep.2015.06.048. [DOI] [PubMed] [Google Scholar]
- 18.Peckham M, Miller G, Wells C, Zicha D, Dunn GA. Specific changes to the mechanism of cell locomotion induced by overexpression of beta-actin. J Cell Sci. 2001;114:1367–1377. doi: 10.1242/jcs.114.7.1367. [DOI] [PubMed] [Google Scholar]
- 19.Bunnell TM, Ervasti JM. Delayed embryonic development and impaired cell growth and survival in Actg1 null mice. Cytoskeleton (Hoboken) 2010;67:564–572. doi: 10.1002/cm.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bassell GJ, Zhang H, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, Kosik KS. Sorting of β-Actin mRNA and Protein to Neurites and Growth Cones in Culture. J Neurosci. 1998;18:251–265. doi: 10.1523/JNEUROSCI.18-01-00251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simiczyjew A, Mazur AJ, Popow-Woźniak A, Malicka-Błaszkiewicz M, Nowak D. Effect of overexpression of β- and γ-actin isoforms on actin cytoskeleton organization and migration of human colon cancer cells. Histochem Cell Biol. 2014;142:307–322. doi: 10.1007/s00418-014-1199-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bunnell TM, Burbach BJ, Shimizu Y, Ervasti JM. β-Actin specifically controls cell growth, migration, and the G-actin pool. Mol Biol Cell. 2011;22:4047–4058. doi: 10.1091/mbc.E11-06-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 24.Shawlot W, Deng JM, Fohn LE, Behringer RR. Restricted beta-galactosidase expression of a hygromycin-lacZ gene targeted to the beta-actin locus and embryonic lethality of beta-actin mutant mice. Transgenic Res. 1998;7:95–103. doi: 10.1023/a:1008816308171. [DOI] [PubMed] [Google Scholar]
- 25.Rotty JD, Bear JE. Competition and collaboration between different actin assembly pathways allows for homeostatic control of the actin cytoskeleton. Bioarchitecture. 2015;5:27–34. doi: 10.1080/19490992.2015.1090670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chugh P, Paluch EK. The actin cortex at a glance. J Cell Sci. 2018;131:jcs186254. doi: 10.1242/jcs.186254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chugh P, Clark AG, Smith MB, Cassani DAD, Dierkes K, Ragab A, Roux PP, Charras G, Salbreux G, Paluch EK. Actin cortex architecture regulates cell surface tension. Nat Cell Biol. 2017;19:689–697. doi: 10.1038/ncb3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramanathan SP, Helenius J, Stewart MP, Cattin CJ, Hyman AA, Muller DJ. Cdk1-dependent mitotic enrichment of cortical myosin II promotes cell rounding against confinement. Nat Cell Biol. 2015;17:148–159. doi: 10.1038/ncb3098. [DOI] [PubMed] [Google Scholar]
- 29.Ennomani H, Letort G, Guérin C, Martiel JL, Cao W, Nédélec F, De La Cruz EM, Théry M, Blanchoin L. Architecture and connectivity govern actin network contractility. Curr Biol. 2016;26:616–626. doi: 10.1016/j.cub.2015.12.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clark Andrew G, Dierkes K, Paluch Ewa K. Monitoring actin cortex thickness in live cells. Biophys J. 2013;105:570–580. doi: 10.1016/j.bpj.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eghiaian F, Rigato A, Scheuring S. Structural, mechanical, and dynamical variability of the actin cortex in living cells. Biophys J. 2015;108:1330–1340. doi: 10.1016/j.bpj.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fritzsche M, Li D, Colin-York H, Chang VT, Moeendarbary E, Felce JH, Sezgin E, Charras G, Betzig E, Eggeling C. Self-organizing actin patterns shape membrane architecture but not cell mechanics. Nat Commun. 2017;8:14347. doi: 10.1038/ncomms14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clausen MP, Colin-York H, Schneider F, Eggeling C, Fritzsche M. Dissecting the actin cortex density and membrane-cortex distance in living cells by super-resolution microscopy. J Phys D. 2017;50:064002. doi: 10.1088/1361-6463/aa52a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biro M, Romeo Y, Kroschwald S, Bovellan M, Boden A, Tcherkezian J, Roux PP, Charras G, Paluch EK. Cell cortex composition and homeostasis resolved by integrating proteomics and quantitative imaging. Cytoskeleton (Hoboken) 2013;70:741–754. doi: 10.1002/cm.21142. [DOI] [PubMed] [Google Scholar]
- 35.Heng YW, Koh CG. Actin cytoskeleton dynamics and the cell division cycle. Int J Biochem Cell Biol. 2010;42:1622–1633. doi: 10.1016/j.biocel.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Flores LR, Keeling MC, Sliogeryte K, Gavara N. Ezrin phosphorylation at T567 modulates cell migration, mechanical properties, and cytoskeletal organization. Int J Mol Sci. 2020;21:435. doi: 10.3390/ijms21020435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Charras GT, Coughlin M, Mitchison TJ, Mahadevan L. Life and times of a cellular bleb. Biophys J. 2008;94:1836–1853. doi: 10.1529/biophysj.107.113605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kasza KE, Rowat AC, Liu J, Angelini TE, Brangwynne CP, Koenderink GH, Weitz DA. The cell as a material. Curr Opin Cell Biol. 2007;19:101–107. doi: 10.1016/j.ceb.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 39.Luxenburg C, Pasolli HA, Williams SE, Fuchs E. Developmental roles for Srf, cortical cytoskeleton and cell shape in epidermal spindle orientation. Nat Cell Biol. 2011;13:203–214. doi: 10.1038/ncb2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dugina V, Zwaenepoel I, Gabbiani G, Clément S, Chaponnier C. β- and γ-cytoplasmic actins display distinct distribution and functional diversity. J Cell Sci. 2009;122:2980–2988. doi: 10.1242/jcs.041970. [DOI] [PubMed] [Google Scholar]
- 41.Brockmann C, Huarte J, Dugina V, Challet L, Rey E, Conne B, Swetloff A, Nef S, Chaponnier C, Vassalli JD. Beta- and gamma-cytoplasmic actins are required for meiosis in mouse oocytes. Biol Reprod. 2011;85:1025–1039. doi: 10.1095/biolreprod.111.091736. [DOI] [PubMed] [Google Scholar]
- 42.Bergeron SE, Zhu M, Thiem SM, Friderici KH, Rubenstein PA. Ion-dependent polymerization differences between mammalian β- and γ-nonmuscle actin isoforms. J Biol Chem. 2010;285:16087–16095. doi: 10.1074/jbc.M110.110130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pugacheva EN, Roegiers F, Golemis EA. Interdependence of cell attachment and cell cycle signaling. Curr Opin Cell Biol. 2006;18:507–515. doi: 10.1016/j.ceb.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gachet Y, Reyes C, Goldstone S, Tournier S. The fission yeast spindle orientation checkpoint: a model that generates tension? Yeast. 2006;23:1015–1029. doi: 10.1002/yea.1410. [DOI] [PubMed] [Google Scholar]
- 45.Lee K, Song K. Actin dysfunction activates ERK1/2 and delays entry into mitosis in mammalian cells. Cell Cycle. 2007;6:1487–1495. [PubMed] [Google Scholar]
- 46.Kaji N, Muramoto A, Mizuno K. LIM kinase-mediated cofilin phosphorylation during mitosis is required for precise spindle positioning. J Biol Chem. 2008;283:4983–4992. doi: 10.1074/jbc.M708644200. [DOI] [PubMed] [Google Scholar]
- 47.Woolner S, O’Brien LL, Wiese C, Bement WM. Myosin-10 and actin filaments are essential for mitotic spindle function. J Cell Biol. 2008;182:77–88. doi: 10.1083/jcb.200804062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramkumar N, Baum B. Coupling changes in cell shape to chromosome segregation. Nat Rev Mol Cell Biol. 2016;17:511–521. doi: 10.1038/nrm.2016.75. [DOI] [PubMed] [Google Scholar]
- 49.Rosenblatt J, Cramer LP, Baum B, McGee KM. Myosin II-dependent cortical movement is required for centrosome separation and positioning during mitotic spindle assembly. Cell. 2004;117:361–372. doi: 10.1016/s0092-8674(04)00341-1. [DOI] [PubMed] [Google Scholar]
- 50.Uzbekov R, Kireyev I, Prigent C. Centrosome separation: respective role of microtubules and actin filaments. Biol Cell. 2002;94:275–288. doi: 10.1016/s0248-4900(02)01202-9. [DOI] [PubMed] [Google Scholar]
- 51.Miller AL. The contractile ring. Curr Biol. 2011;21:R976–R978. doi: 10.1016/j.cub.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D’Avino PP, Savoian MS, Glover DM. Cleavage furrow formation and ingression during animal cytokinesis: a microtubule legacy. J Cell Sci. 2005;118:1549–1558. doi: 10.1242/jcs.02335. [DOI] [PubMed] [Google Scholar]
- 53.Glotzer M. Cleavage furrow positioning. J Cell Biol. 2004;164:347–351. doi: 10.1083/jcb.200310112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carlton JG, Jones H, Eggert US. Membrane and organelle dynamics during cell division. Nat Rev Mol Cell Biol. 2020;21:151–166. doi: 10.1038/s41580-019-0208-1. [DOI] [PubMed] [Google Scholar]
- 55.Barr FA, Gruneberg U. Cytokinesis: placing and making the final cut. Cell. 2007;131:847–860. doi: 10.1016/j.cell.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 56.Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev Cell. 2007;12:699–712. doi: 10.1016/j.devcel.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishimura Y, Yonemura S. Centralspindlin regulates ECT2 and RhoA accumulation at the equatorial cortex during cytokinesis. J Cell Sci. 2006;119:104–114. doi: 10.1242/jcs.02737. [DOI] [PubMed] [Google Scholar]
- 58.Dogterom M, Koenderink GH. Actin-microtubule crosstalk in cell biology. Nat Rev Mol Cell Biol. 2019;20:38–54. doi: 10.1038/s41580-018-0067-1. [DOI] [PubMed] [Google Scholar]
- 59.Stiff T, Echegaray-Iturra FR, Pink HJ, Herbert A, Reyes-Aldasoro CC, Hochegger H. prophase-specific perinuclear actin coordinates centrosome separation and positioning to ensure accurate chromosome segregation. Cell Rep. 2020;31:107681. doi: 10.1016/j.celrep.2020.107681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plessner M, Knerr J, Grosse R. Centrosomal actin assembly is required for proper mitotic spindle formation and chromosome congression. iScience. 2019;15:274–281. doi: 10.1016/j.isci.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rizzelli F, Malabarba MG, Sigismund S, Mapelli M. The crosstalk between microtubules, actin and membranes shapes cell division. Open Biol. 2020;10:190314. doi: 10.1098/rsob.190314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu H, Zhao Q, Jiang H, Zhu T, Xia P, Seffens W, Aikhionbare F, Wang D, Dou Z, Yao X. Characterization of ring-like F-actin structure as a mechanical partner for spindle positioning in mitosis. PLoS One. 2014;9:e102547. doi: 10.1371/journal.pone.0102547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen A, Ulloa Severino L, Panagiotou TC, Moraes TF, Yuen DA, Lavoie BD, Wilde A. Inhibition of polar actin assembly by astral microtubules is required for cytokinesis. Nat Comm. 2021;12:2409. doi: 10.1038/s41467-021-22677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mattaini K. Chapter 13. The cell cycle & mitosis. Introduction to Molecular and Cell Biology. 2020 [Google Scholar]
- 65.Terry SJ, Donà F, Osenberg P, Carlton JG, Eggert US. Capping protein regulates actin dynamics during cytokinetic midbody maturation. Proc Natl Acad Sci U S A. 2018;115:2138–2143. doi: 10.1073/pnas.1722281115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McClatchey AI. ERM proteins at a glance. J Cell Sci. 2014;127:3199–3204. doi: 10.1242/jcs.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hebert AM, DuBoff B, Casaletto JB, Gladden AB, McClatchey AI. Merlin/ERM proteins establish cortical asymmetry and centrosome position. Genes Dev. 2012;26:2709–2723. doi: 10.1101/gad.194027.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roubinet C, Decelle B, Chicanne G, Dorn JF, Payrastre B, Payre F, Carreno S. Molecular networks linked by moesin drive remodeling of the cell cortex during mitosis. J Cell Biol. 2011;195:99–112. doi: 10.1083/jcb.201106048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sato N, Yonemura S, Obinata T, Tsukita S, Tsukita S. Radixin, a barbed end-capping actin-modulating protein, is concentrated at the cleavage furrow during cytokinesis. J Cell Biol. 1991;113:321–330. doi: 10.1083/jcb.113.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hiruma S, Kamasaki T, Otomo K, Nemoto T, Uehara R. Dynamics and function of ERM proteins during cytokinesis in human cells. FEBS Lett. 2017;591:3296–3309. doi: 10.1002/1873-3468.12844. [DOI] [PubMed] [Google Scholar]
- 71.Henry MD, Gonzalez Agosti C, Solomon F. Molecular dissection of radixin: distinct and interdependent functions of the amino- and carboxy-terminal domains. J Cell Biol. 1995;129:1007–1022. doi: 10.1083/jcb.129.4.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vilmos P, Kristó I, Szikora S, Jankovics F, Lukácsovich T, Kari B, Erdélyi M. The actin-binding ERM protein moesin directly regulates spindle assembly and function during mitosis. Cell Biol Int. 2016;40:696–707. doi: 10.1002/cbin.10607. [DOI] [PubMed] [Google Scholar]
- 73.Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zuo Y, Oh W, Frost JA. Controlling the switches: Rho GTPase regulation during animal cell mitosis. Cell Signal. 2014;26:2998–3006. doi: 10.1016/j.cellsig.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim JG, Islam R, Cho JY, Jeong H, Cap KC, Park Y, Hossain AJ, Park JB. Regulation of RhoA GTPase and various transcription factors in the RhoA pathway. J Cell Physiol. 2018;233:6381–6392. doi: 10.1002/jcp.26487. [DOI] [PubMed] [Google Scholar]
- 76.Piekny AJ, Glotzer M. Anillin is a scaffold protein that links RhoA, actin, and myosin during cytokinesis. Curr Biol. 2008;18:30–36. doi: 10.1016/j.cub.2007.11.068. [DOI] [PubMed] [Google Scholar]
- 77.Dean SO, Spudich JA. Rho kinase’s role in myosin recruitment to the equatorial cortex of mitotic drosophila S2 cells is for myosin regulatory light chain phosphorylation. PLoS One. 2006;1:e131. doi: 10.1371/journal.pone.0000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wagner E, Glotzer M. Local RhoA activation induces cytokinetic furrows independent of spindle position and cell cycle stage. J Cell Biol. 2016;213:641–649. doi: 10.1083/jcb.201603025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Edwards M, Zwolak A, Schafer DA, Sept D, Dominguez R, Cooper JA. Capping protein regulators fine-tune actin assembly dynamics. Nat Rev Mol Cell Biol. 2014;15:677–689. doi: 10.1038/nrm3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jo YJ, Jang WI, Namgoong S, Kim NH. Actin-capping proteins play essential roles in the asymmetric division of maturing mouse oocytes. J Cell Sci. 2015;128:160–170. doi: 10.1242/jcs.163576. [DOI] [PubMed] [Google Scholar]
- 81.Bohnert KA, Willet AH, Kovar DR, Gould KL. Formin-based control of the actin cytoskeleton during cytokinesis. Biochem Soc Trans. 2013;41:1750–1754. doi: 10.1042/BST20130208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paul AS, Pollard TD. Review of the mechanism of processive actin filament elongation by formins. Cell Motil Cytoskeleton. 2009;66:606–617. doi: 10.1002/cm.20379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Romero S, Clainche CL, Didry D, Egile C, Pantaloni D, Carlier MF. Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell. 2004;119:419–429. doi: 10.1016/j.cell.2004.09.039. [DOI] [PubMed] [Google Scholar]
- 84.Skau CT, Waterman CM. Specification of architecture and function of actin structures by actin nucleation factors. Annu Rev Biophys. 2015;44:285–310. doi: 10.1146/annurev-biophys-060414-034308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 86.Courtemanche N, Pollard TD. Interaction of profilin with the barbed end of actin filaments. Biochem. 2013;52:6456–6466. doi: 10.1021/bi400682n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pollard TD, O’Shaughnessy B. Molecular mechanism of cytokinesis. Annu Rev Biochem. 2019;88:661–689. doi: 10.1146/annurev-biochem-062917-012530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Green RA, Paluch E, Oegema K. Cytokinesis in animal cells. Annu Rev Cell Dev Biol. 2012;28:29–58. doi: 10.1146/annurev-cellbio-101011-155718. [DOI] [PubMed] [Google Scholar]
- 89.Stewart MP, Helenius J, Toyoda Y, Ramanathan SP, Muller DJ, Hyman AA. Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature. 2011;469:226–230. doi: 10.1038/nature09642. [DOI] [PubMed] [Google Scholar]
- 90.Henson JH, Ditzler CE, Germain A, Irwin PM, Vogt ET, Yang S, Wu X, Shuster CB. The ultrastructural organization of actin and myosin II filaments in the contractile ring: new support for an old model of cytokinesis. Mol Biol Cell. 2017;28:613–623. doi: 10.1091/mbc.E16-06-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ding L, Cao J, Lin W, Chen H, Xiong X, Ao H, Yu M, Lin J, Cui Q. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int J Mol Sci. 2020;21:1960. doi: 10.3390/ijms21061960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nasa I, Kettenbach AN. Coordination of protein kinase and phosphoprotein phosphatase activities in mitosis. Front Cell Dev Biol. 2018;6:30. doi: 10.3389/fcell.2018.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Basant A, Lekomtsev S, Tse YC, Zhang D, Longhini KM, Petronczki M, Glotzer M. Aurora b kinase promotes cytokinesis by inducing centralspindlin oligomers that associate with the plasma membrane. Dev Cell. 2015;33:204–215. doi: 10.1016/j.devcel.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Özlü N, Monigatti F, Renard BY, Field CM, Steen H, Mitchison TJ, Steen JJ. Binding partner switching on microtubules and aurora-b in the mitosis to cytokinesis transition. Mol Cell Proteomics. 2010;9:336–350. doi: 10.1074/mcp.M900308-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dambournet D, Machicoane M, Chesneau L, Sachse M, Rocancourt M, El Marjou A, Formstecher E, Salomon R, Goud B, Echard A. Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat Cell Biol. 2011;13:981–988. doi: 10.1038/ncb2279. [DOI] [PubMed] [Google Scholar]
- 96.Potapova T, Gorbsky GJ. The consequences of chromosome segregation errors in mitosis and meiosis. Biology. 2017;6:12. doi: 10.3390/biology6010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Compton DA. Mechanisms of aneuploidy. Curr Opin Cell Biol. 2011;23:109–113. doi: 10.1016/j.ceb.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–162. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 99.Roeles J, Tsiavaliaris G. Actin-microtubule interplay coordinates spindle assembly in human oocytes. Nat Commun. 2019;10:4651. doi: 10.1038/s41467-019-12674-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pfau SJ, Amon A. Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep. 2012;13:515–527. doi: 10.1038/embor.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Godek KM, Venere M, Wu Q, Mills KD, Hickey WF, Rich JN, Compton DA. Chromosomal instability affects the tumorigenicity of glioblastoma tumor-initiating cells. Cancer Discov. 2016;6:532–545. doi: 10.1158/2159-8290.CD-15-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lowe CU, Terrey M, MacLachlan EA. Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am J Dis Child. 1952;83:164–184. doi: 10.1001/archpedi.1952.02040060030004. [DOI] [PubMed] [Google Scholar]
- 103.Suchy SF, Nussbaum RL. The deficiency of PIP2 5-phosphatase in lowe syndrome affects actin polymerization. Am J Hum Genet. 2002;71:1420–1427. doi: 10.1086/344517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sechi S, Colotti G, Belloni G, Mattei V, Frappaolo A, Raffa GD, Fuller MT, Giansanti MG. GOLPH3 is essential for contractile ring formation and Rab11 localization to the cleavage site during cytokinesis in drosophila melanogaster. PLoS Genet. 2014;10:e1004305. doi: 10.1371/journal.pgen.1004305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schiel JA, Simon GC, Zaharris C, Weisz J, Castle D, Wu CC, Prekeris R. FIP3-endosome-dependent formation of the secondary ingression mediates ESCRT-III recruitment during cytokinesis. Nat Cell Biol. 2012;14:1068–1078. doi: 10.1038/ncb2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Prekeris R. Analyzing the functions of Rab11-effector proteins during cell division. Methods Cell Biol. 2015;130:19–34. doi: 10.1016/bs.mcb.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhou Y, Bennett TM, Shiels A. A charged multivesicular body protein (CHMP4B) is required for lens growth and differentiation. Differentiation. 2019;109:16–27. doi: 10.1016/j.diff.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Frémont S, Hammich H, Bai J, Wioland H, Klinkert K, Rocancourt M, Kikuti C, Stroebel D, Romet-Lemonne G, Pylypenko O, Houdusse A, Echard A. Oxidation of F-actin controls the terminal steps of cytokinesis. Nat Commun. 2017;8:14528. doi: 10.1038/ncomms14528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Frémont S, Romet-Lemonne G, Houdusse A, Echard A. Emerging roles of MICAL family proteins - from actin oxidation to membrane trafficking during cytokinesis. J Cell Sci. 2017;130:1509–1517. doi: 10.1242/jcs.202028. [DOI] [PubMed] [Google Scholar]
- 110.Mierzwa B, Gerlich DW. Cytokinetic abscission: molecular mechanisms and temporal control. Dev Cell. 2014;31:525–538. doi: 10.1016/j.devcel.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 111.Bai J, Wioland H, Advedissian T, Cuvelier F, Romet-Lemonne G, Echard A. Actin reduction by MsrB2 is a key component of the cytokinetic abscission checkpoint and prevents tetraploidy. Proc Natl Acad Sci U S A. 2020;117:4169–4179. doi: 10.1073/pnas.1911629117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Peterman E, Valius M, Prekeris R. CLIC4 is a cytokinetic cleavage furrow protein that regulates cortical cytoskeleton stability during cell division. J Cell Sci. 2020;133:jcs241117. doi: 10.1242/jcs.241117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Uretmen Kagiali ZC, Saner N, Akdag M, Sanal E, Degirmenci BS, Mollaoglu G, Ozlu N. CLIC4 and CLIC1 bridge plasma membrane and cortical actin network for a successful cytokinesis. Life Sci Alliance. 2019;3:e201900558. doi: 10.26508/lsa.201900558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cooper GM. The development and causes of cancer. The cell: a molecular approach. 2nd edition. Sunderland (MA): Sinauer Associates; 2000. [Google Scholar]
- 115.Wen S, Hou Y, Fu L, Xi L, Yang D, Zhao M, Qin Y, Sun K, Teng Y, Liu M. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3-p38 MAPK signalling. Cancer Lett. 2019;442:320–332. doi: 10.1016/j.canlet.2018.10.015. [DOI] [PubMed] [Google Scholar]
- 116.Eoh KJ, Kim HJ, Lee JW, Kim LK, Park SA, Kim HS, Kim YT, Koo PJ. E2F8 induces cell proliferation and invasion through the epithelial-mesenchymal transition and notch signaling pathways in ovarian cancer. Int J Mol Sci. 2020;21:5813. doi: 10.3390/ijms21165813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim DK, Kim EK, Jung DW, Kim J. Cytoskeletal alteration modulates cancer cell invasion through RhoA-YAP signaling in stromal fibroblasts. PLoS One. 2019;14:e0214553. doi: 10.1371/journal.pone.0214553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yuan B, Zhang R, Hu J, Liu Z, Yang C, Zhang T, Zhang C. WDR1 promotes cell growth and migration and contributes to malignant phenotypes of non-small cell lung cancer through ADF/cofilin-mediated actin dynamics. Int J Biol. 2018;14:1067–1080. doi: 10.7150/ijbs.23845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Biber G, Ben-Shmuel A, Sabag B, Barda-Saad M. Thomas C, Galluzzi L, editors. Chapter three - actin regulators in cancer progression and metastases: from structure and function to cytoskeletal dynamics. Int Rev Cell Mol Biol. 2020:131–196. doi: 10.1016/bs.ircmb.2020.05.006. [DOI] [PubMed] [Google Scholar]
- 120.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 121.Zhang J, Cai H, Sun L, Zhan P, Chen M, Zhang F, Ran Y, Wan J. LGR5, a novel functional glioma stem cell marker, promotes EMT by activating the Wnt/β-catenin pathway and predicts poor survival of glioma patients. J Exp Clin Cancer Res. 2018;37:225. doi: 10.1186/s13046-018-0864-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 123.Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35:645–654. doi: 10.1007/s10555-016-9648-7. [DOI] [PubMed] [Google Scholar]
- 124.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bade ND, Kamien RD, Assoian RK, Stebe KJ. Curvature and Rho activation differentially control the alignment of cells and stress fibers. Sci Adv. 2017;3:e1700150. doi: 10.1126/sciadv.1700150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Izdebska M, Zielińska W, Grzanka D, Gagat M. The role of actin dynamics and actin-binding proteins expression in epithelial-to-mesenchymal transition and its association with cancer progression and evaluation of possible therapeutic targets. Biomed Res Int. 2018;2018:4578373. doi: 10.1155/2018/4578373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Grzanka D, Gagat M, Izdebska M. Involvement of the SATB1/F-actin complex in chromatin reorganization during active cell death. Int J Mol Med. 2014;33:1441–1450. doi: 10.3892/ijmm.2014.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wan F, Cheng C, Wang Z, Xiao X, Zeng H, Xing S, Chen X, Wang J, Li S, Zhang Y, Xiang W, Zhu Z, Johnson C, Zhu Z. SATB1 overexpression regulates the development and progression in bladder cancer through EMT. PLoS One. 2015;10:e0117518. doi: 10.1371/journal.pone.0117518. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 129.Riihimäki M, Hemminki A, Sundquist J, Hemminki K. Patterns of metastasis in colon and rectal cancer. Sci Rep. 2016;6:29765. doi: 10.1038/srep29765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sousa-Squiavinato ACM, Rocha MR, Barcellos-de-Souza P, de Souza WF, Morgado-Diaz JA. Cofilin-1 signaling mediates epithelial-mesenchymal transition by promoting actin cytoskeleton reorganization and cell-cell adhesion regulation in colorectal cancer cells. Biochim Biophys Acta Mol Cell Res. 2019;1866:418–429. doi: 10.1016/j.bbamcr.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 131.Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, Condeelis J. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–452. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhitnyak IY, Rubtsova SN, Litovka NI, Gloushankova NA. Early events in actin cytoskeleton dynamics and E-cadherin-mediated cell-cell adhesion during epithelial-mesenchymal transition. Cells. 2020;9:578. doi: 10.3390/cells9030578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Canel M, Serrels A, Frame MC, Brunton VG. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci. 2013;126:393–401. doi: 10.1242/jcs.100115. [DOI] [PubMed] [Google Scholar]
- 134.Choi CK, Vicente-Manzanares M, Zareno J, Whitmore LA, Mogilner A, Horwitz AR. Actin and α-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat Cell Biol. 2008;10:1039–1050. doi: 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 136.Mohammadi H, Sahai E. Mechanisms and impact of altered tumour mechanics. Nat Cell Biol. 2018;20:766–774. doi: 10.1038/s41556-018-0131-2. [DOI] [PubMed] [Google Scholar]
- 137.Sahai E. Mechanisms of cancer cell invasion. Curr Opin Genet Dev. 2005;15:87–96. doi: 10.1016/j.gde.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 138.Ye H, Zhang Y, Geng L, Li Z. Cdc42 expression in cervical cancer and its effects on cervical tumor invasion and migration. Int J Oncol. 2015;46:757–763. doi: 10.3892/ijo.2014.2748. [DOI] [PubMed] [Google Scholar]
- 139.Diepenbruck M, Christofori G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr Opin Cell Biol. 2016;43:7–13. doi: 10.1016/j.ceb.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 140.Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278:16–27. doi: 10.1111/j.1742-4658.2010.07919.x. [DOI] [PubMed] [Google Scholar]