

Abstract
Ponatinib is a tyrosine kinase inhibitor (TKI) directed against BCR-ABL1 which is successfully used in patients with BCR-ABL1 T315I+ chronic myeloid leukemia (CML). However, BCR-ABL1 compound mutations may develop during therapy in these patients and may lead to drug resistance. Asciminib is a novel drug capable of targeting most BCR-ABL1 mutant-forms, including BCR-ABL1T315I, but remains ineffective against most BCR-ABL1T315I+ compound mutation-bearing sub-clones. We demonstrate that asciminib synergizes with ponatinib in inducing growth-arrest and apoptosis in patient-derived CML cell lines and murine Ba/F3 cells harboring BCR-ABL1 T315I or T315I-including compound mutations. Asciminib and ponatinib also produced cooperative effects on CRKL phosphorylation in BCR-ABL1-transformed cells. The growth-inhibitory effects of the drug combination ‘asciminib+ponatinib’ was further enhanced by hydroxyurea (HU), a drug which has lately been described to suppresses the proliferation of BCR-ABL1 T315I+ CML cells. Cooperative drug effects were also observed in patient-derived CML cells. Most importantly, we were able to show that the combinations ‘asciminib+ponatinib’ and ‘asciminib+ponatinib+HU’ produce synergistic apoptosis-inducing effects in CD34+/CD38- CML stem cells obtained from patients with chronic phase CML or BCR-ABL1 T315I+ CML blast phase. Together, asciminib, ponatinib and HU synergize in producing anti-leukemic effects in multi-resistant CML cells, including cells harboring T315I+ BCR-ABL1 compound mutations and CML stem cells. The clinical efficacy of this TKI combination needs to be evaluated within the frame of upcoming clinical trials.
Keywords: CML, asciminib, ponatinib, BCR-ABL1T315I, BCR-ABL1 compound mutations, drug combinations, leukemic stem cells, hydroxyurea
Introduction
Tyrosine kinase inhibitors (TKI) targeting BCR-ABL1 are nowadays the standard of care in patients with chronic myeloid leukemia (CML). This treatment usually results in a long-lasting, deep molecular response and thus in normalization of life expectancy [1-5]. However, treatment failure may occur in CML patients due to TKI-resistance [4-10]. This phenomenon can in many cases be explained by BCR-ABL1 mutations [4-10]. In other patients, activation of BCR-ABL1-independent signaling pathways supposedly mediate TKI resistance [4-10]. About 10% of all BCR-ABL1 mutations detected in CML cells are threonine to isoleucine mutations at codon 315, resulting in the expression of BCR-ABL1T315I. This mutation prevents binding of most TKI to BCR-ABL1, and thus leads to multi-drug resistance [3-6]. To date, among all approved and widely available TKI, ponatinib can effectively suppress growth of CML sub-clones expressing BCR-ABL1 T315I [11,12]. Ponatinib has indeed been associated with a high response-rate in heavily pretreated patients with BCR-ABL1 T315I+ CML [12]. However, cardiovascular side effects have been documented in patients after exposure to relatively high doses of ponatinib. This observation implies that ponatinib should not be administered continuously in all patients, especially if cardiovascular risk factors or an overt cardiovascular disease were also reported [12-15]. Furthermore, leukemic cells harboring BCR-ABL1 variants with at least two mutations on the same allele (so called compound mutations) may develop during TKI therapy [16-21]. Indeed, compound mutations were found in a substantial portion of patients with relapsed BCR-ABL1+ leukemia who had been treated subsequently with two or more TKI [20]. Whereas non-T315I compound mutants usually retain sensitivity to at least one TKI (especially ponatinib), T315I-including compound mutations have been demonstrated to confer resistance against all available TKI, including ponatinib [18]. No established therapy is available in these cases, which remains a clinical challenge. Combinations of two or more TKI or of TKI and other drugs, such as hydroxyurea (HU), may be a promising approach to overcome resistance in these patients [22-24].
Asciminib (ABL001) is a novel allosteric inhibitor of BCR-ABL1 which has been successfully applied in clinical trials in advanced CML [25-29]. Indeed, first reports suggest that asciminib is efficient in most patients with heavily pretreated CML and is moreover a safe drug [30]. Unlike other BCR-ABL1 TKI, asciminib does not target the ATP-binding site but the myristoyl-binding site of the oncoprotein [25-27,31]. However, resistance against asciminib may also occur [25,32,33]. With regard to BCR-ABL1 mutations, asciminib has been shown to block BCR-ABL1T315I but fails to inhibit T315I-including compound-mutants, at least when applied as single drug [25,32]. Due to their different binding-sites and different mechanisms leading to drug-resistance, combinations including asciminib and other BCR-ABL1 TKI have recently been considered as a new interesting approach [32-34].
In this study, we examined cooperative effects of asciminib and ponatinib on primary human CML cells, human CML cell lines and cell lines exhibiting T315I-compound-mutant forms of BCR-ABL1. We have recently shown, that HU is efficacious in BCR-ABL T315I-transformed CML [22] and therefore asked, whether anti-leukemic effects of asciminib and ponatinib can be further augmented by HU. In a last step, potential cooperative drug effects on CD34+/CD38- CML stem cells were examined.
Patients and methods
Primary CML cells and cell lines
For this study, 11 CML patients gave peripheral blood (PB): 8 patients were in chronic phase (CP) and 3 were in blast phase (BP) at the time of blood sampling. In 2 patients with BP, BCR-ABL1 T315I was detected, in one patient with BP a BCR-ABL1 V299L/F317L compound mutation was found. Patients’ characteristics are summarized in Table 1. PB samples were prepared as described to isolate mononuclear cells (MNC), which were then used for in vitro experiments [22,23]. Written informed consent was obtained from all patients prior to PB-sampling. An approval from the ethics committee of the Medical University of Vienna was obtained for this study (Nr: 1114/2021). In addition to primary cells, we used the human CML cell lines KU812, KCL22 and K562. Details concerning the origin of these cell lines are depicted in Table 2. Furthermore, BCR-ABL1 T315I+ KCL22 cells (KCL22T315I) were created by culturing cells in the presence of imatinib and dasatinib as described previously [35]. To study drug effects on cells harboring complex BCR-ABL1 mutations, we used Ba/F3 cells expressing BCR-ABL1 T315I or T315I-based compound mutations (BCR-ABL1 T315I/E255K, BCR-ABL1 T315I/F311L, BCR-ABL1 T315I/F359V, BCR-ABL1 T315I/G250E) (Table 2). The generation of these cell lines has been described recently [35,36]. Native Ba/F3 cells (lacking BCR-ABL1) were cultured in 10 ng/ml interleukin-3 (IL-3) as described [23] and served as control. Culture of all cell types was performed using RPMI 1640 medium supplemented with 10% fetal calf serum (FCS) and antibiotics. In case of KCL22T315I, imatinib (5 µM) was added to the medium (Table 2).
Table 1.
Patients’ characteristics: CML samples used for in vitro studies and response to ‘asciminib+ponatinib’
| Patient no. | Age (years) | Gender | Source | CML phase | BCR-ABL1 mutations | Therapy before cell sampling | ‘asciminib+ponatinib’ |
|---|---|---|---|---|---|---|---|
| #1 | 54 | f | PB | CP | n.t. | none | Synergistic (p); Cooperative CD34+/CD38- (a-fc) |
| #2 | 27 | m | PB | CP | n.t. | none | Synergistic (p) Cooperative CD34+/CD38- (a-fc) |
| #3 | 19 | m | PB | CP | n.t. | none | Synergistic (p); Cooperative CD34+/CD38- (a-fc) |
| #4 | 34 | f | PB | CP | n.t. | none | Synergistic (p) |
| #5 | 36 | m | PB | CP | n.t. | none | Synergistic (p) |
| #6 | 25 | f | PB | CP | n.t. | Imatinib (dis.) | Synergistic (p) |
| #7 | 46 | f | PB | CP | n.t. | none | Synergistic (p) |
| #8 | 52 | f | PB | CP | n.t. | none | Cooperative (p) |
| #9 | 31 | m | BM | BP | T315I | Imatinib (res.) | Cooperative CD34+/CD38- (a-fc) |
| Dasatinib (res.) | |||||||
| #10 | 48 | f | BM | BP | T315I | Dasatinib (res.) | Cooperative CD34+/CD38- (a-fc) |
| #11 | 43 | m | PB | BP | V299L/F317L | Imatinib (res.) | Synergistic CD34+/CD38- (a-fc) |
| Dasatinib (res.) | |||||||
| CHT+GO HSCT |
No., number; f, female; m, male; PB, peripheral blood; BM, bone marrow; CP, chronic phase; BP, blast phase; n.t.: not tested; dis: discontinued by the patient; res, resistant; CHT: polychemotherapy; GO: gemtuzumab ozogamycin; HSCT: hematopoietic stem cell transplantation. The in vitro response of leukemic cells to the combination ‘asciminib+ponatinib’ was assessed by 3H-thymidine uptake experiments (cell proliferation: p) and/or by determination of apoptotic CD34+/CD38- stem-cells by Annexin V-FITC/DAPI staining and flow cytometry (a-fc).
Table 2.
Cell lines and culture conditions
| Cell line (Name) | Provider/Origin | Comments |
|---|---|---|
| K562 | Provided by Dr. M. Deininger (University of Utah, Salt Lake City, UT, USA) | - |
| KU812 | Provided by Dr. K. Kishi (Niigata University, Niigata, Japan) | Basophil-committed |
| KCL22 | Purchased from the German Collection of Microorganism and Cell Culture (DSMZ, Braunschweig, Germany) | Complex karyotype; 2 Philadelphia Chromosomes |
| KCL22T315I | Produced in our lab: Dr. K. Byrgazov and Dr. T. Lion (Children’s Cancer Research Institute (CCRI), Vienna, Austria) | Complex karyotype; 2 Philadelphia Chromosomes Imatinib-resistant; Kept in 5 µM imatinib. |
| Ba/F3 native | Purchased from the German Collection of Microorganism and Cell Culture (DSMZ, Braunschweig, Germany) | Kept in 10 ng/ml IL-3 |
| Ba/F3p210T315I | Provided by Dr. M. Deininger (University of Utah, Salt Lake City, UT, USA) | Imatinib-resistant; the generation of this cell line is described in reference 35 |
| Ba/F3p210T315I/E255V | Produced in our lab: Dr. K. Byrgazov and Dr. T. Lion (Children’s Cancer Research Institute (CCRI), Vienna, Austria) | Ponatinib-resistant; the generation of these cell lines is described in reference 36 |
| Ba/F3p210T315I/F311L | ||
| Ba/F3p210T315I/F359V | ||
| Ba/F3p210T315I/G250E |
Human and murine cell lines expressing BCR-ABL1 were purchased or provided by cooperation partners as listed in the table. All cell lines were maintained in RPMI 1640 medium with 10% FCS and antibiotics.
Reagents
Asciminib was ordered from Active Biochem (Kowloon, Hong Kong), ponatinib and imatinib from Chemietek (Indianapolis, IN, USA), and HU from Sigma-Aldrich (St. Louis, MO, USA). Stock solutions of asciminib, ponatinib and imatinib were generated by dissolving in dimethyl sulfoxide (DMSO) (Merck, Darmstadt, Germany) (stock concentration: 10 mM) and HU by dissolving in distilled water (stock concentration: 100 mM). 3H-thymidine was purchased from Perkin Elmer (Boston, MA, USA). Annexin V/FITC and Propidium Iodide (PI) were obtained from eBioscience (San Diego, CA, USA) and 4’,6-diamidino-2-phenylindole (DAPI) from Sigma-Aldrich. RPMI 1640 medium and penicillin+streptomycin were from Lonza (Verviers, Belgium), fetal calf serum (FCS) from Gibco life technologies (Gaithersburg, MD, USA), amphotericin from PAN Biotech (Aidenbach, Germany) and murine IL-3 from PeproTech (Rocky Hill, NJ, USA).
Measurement of 3H-thymidine uptake
To study cell proliferation, cell lines and primary cells were kept in control medium or in medium supplemented with various concentrations of drugs (asciminib, ponatinib, HU) alone or in 2-drug or 3-drug combinations at fixed ratios of drug-concentrations at 37°C for 48 hours. Thereafter, 3H-thymidine-uptake was measured as described [22,23].
Evaluation of apoptosis
Do determined the percentage of apoptotic cells following drug-exposure, BCR-ABL1+ cell lines were cultured in standard medium or in medium supplemented with asciminib, ponatinib or a combination of both drugs for 48 hours. Next, cells were stained by Annexin V-FITC and 4’,6-diamidino-2-phenylindole (DAPI) before apoptosis was quantified by flow cytometry as described previously [22,23]. In 6 patients (CP, n=3; BP, n=3), apoptosis was measured in drug-exposed CD34+/CD38- stem cells by multi-color flow cytometry as reported [23] using FlowJo software (BD Biosciences, San José, CA, USA). Analysis was performed on FACSCanto II (BD Biosciences).
Western blot analysis
To study whether cooperative anti-proliferative effects of ‘asciminib+ponatinib’ can be related to direct effects on BCR-ABL1, we determined expression of the phosphorylated CRK Like Proto-Oncogene (p-CRKL), a substrate of the BCR-ABL1 tyrosine kinase. For this purpose, proteins isolated from drug-exposed CML cell lines was analyzed by Western blotting. Cells were cultured in control medium or in medium supplemented with suboptimal concentrations (which per se failed to completely block p-CRKL) of asciminib and/or ponatinib. After 4 hours, cells were harvested, lysed in lysing buffer, and subjected to Western blot analysis as previously published [22,23] using antibodies directed against p-CRKL or β-tubulin as loading control (Table 3).
Table 3.
Antibodies used for Western blotting
| Protein/Epitope | Clone | Source | Dilution |
|---|---|---|---|
| p-CrkL (Tyr207) | polyclonal | rabbit | 1:1000 |
| β-Tubulin | D2N5G | mouse | 1:1000 |
Both antibodies were purchased from Cell Signaling Technology (Berverly, MA, USA). p, phosphorylated; Tyr, tyrosine.
Statistical analysis
Drug-interactions (additive versus synergistic) were assessed by calculating CI values employing Calcusyn software [38]. Whereas a CI of 1 stands for an additive drug-effect, CI values <1 are indicating synergistic drug effects.
Results
Asciminib and ponatinib synergize in inhibiting growth and viability of BCR-ABL1+ cell lines, including cells harboring BCR-ABL1T315I or T315I-including compound mutations
It has recently been shown that asciminib augments the growth-inhibitory effects of ponatinib in Ba/F3 cells exhibiting BCR-ABL1T315I [32]. We confirmed these drug-effects and extended our studies to human CML cell lines (KU812, K562, KCL22, KCL22T315I). These cells were incubated with ponatinib and asciminib at suboptimal concentrations alone or in combination. We found that asciminib and ponatinib synergize in blocking growth of all human CML cell lines when applied at low concentrations. Synergism was also observed in KCL22T315I cells when using clinically relevant drug concentrations (Figure 1A). In these cells, asciminib and ponatinib concentrations required to induce growth inhibition were higher when compared to KCL22 or BCR-ABL1T315I+ Ba/F3 cells (Figure 1A, 1B). This phenomenon may be best explained by the fact that KCL22T315I (a cell line generated from BP CML) harbor a complex karyotype and carry two Philadelphia chromosomes. These results suggest that the combination ‘asciminib+ponatinib’ may also have the potential to suppress sub-clones harboring complex genetic abnormalities in advanced CML.
Figure 1.
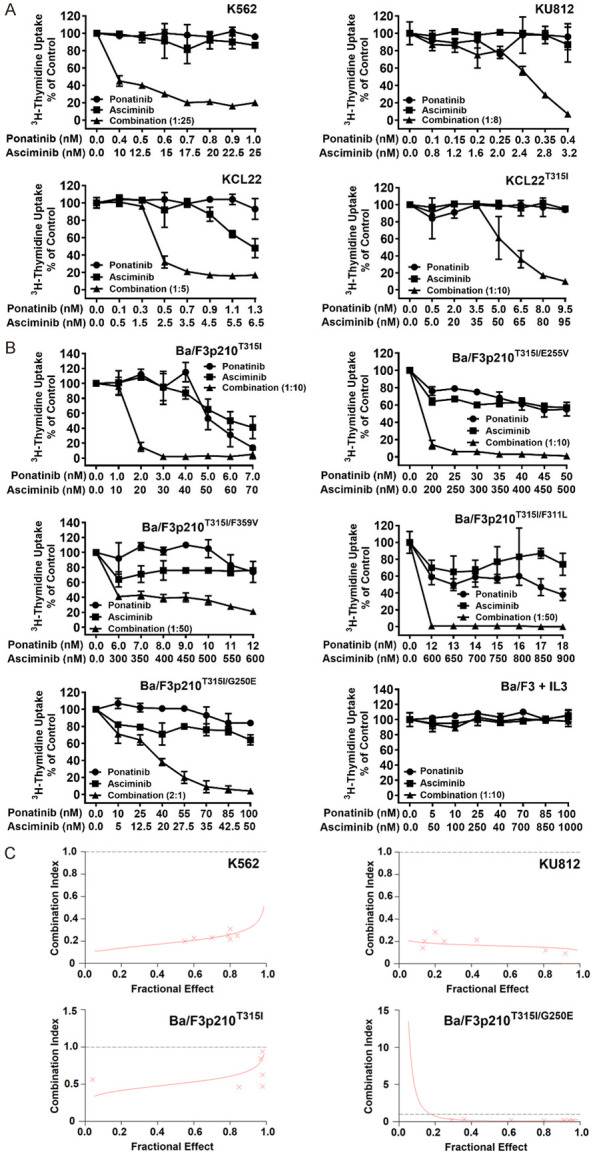
Asciminib and ponatinib synergize in producing growth inhibition in BCR-ABL1+ cell lines. The human CML cell lines KU812, K562, KCL22 and KCL22T315I (A) and Ba/F3 cells expressing various BCR-ABL1-mutations (Ba/F3p210T315I, Ba/F3p210T315I/E255V, Ba/F3p210T315I/F359V, Ba/F3p210T315I/F311L, Ba/F3p210T315I/G250E) or untransfected Ba/F3 cells supplemented with 10 ng/ml IL-3 (B) were kept in control medium or in the presence of asciminib (■-■), ponatinib (●-●), or a combination of both drugs at a fixed ratio (▲-▲) for 48 hours before 3H-thymidine-uptake was evaluated. Results are calculated as percent of control and represent the mean ± S.D. of triplicates. (C) The nature of drug interaction (additive versus synergistic) shown in (A) and (B) was determined for each experiment by calculating combination index (CI) values using Calcusyn software. A CI value of 1 indicates an additive effect, whereas CI values below 1 indicate synergistic drug effects. Examples are shown for K562 and KU812 as well as Ba/F3p210T315I and Ba/F3p210T315I/G250E cells as indicated.
Next, Ba/F3 cells harboring BCR-ABL1 T315I, BCR-ABL1 T315I/E255V, BCR-ABL1 T315I/F311L, BCR-ABL1 T315I/F359V or BCR-ABL1 T315I/G250E were subjected to the drug-combination ‘asciminib+ponatinib’. We found that asciminib synergizes with ponatinib in blocking growth of all BCR-ABL1-transformed Ba/F3 clones, whereas no effect was seen in untransfected (BCR-ABL1-negative) Ba/F3 cells (Figure 1B). Synergistic drug-interactions were confirmed by determining combination index (CI) values. Examples of synergistic drug effects are shown in Figure 1C. Asciminib and ponatinib also produced strong synergistic apoptosis-inducing effects in KU812, K562 and KCL22T315I cells (Figure 2A) as well as in BCR-ABL1-transformed Ba/F3 cells (Figure 2B). To investigate whether drug-synergism is related to direct effects on BCR-ABL1, we determined expression of phosphorylated CRKL (p-CRKL), a downstream-target of BCR-ABL1. Whereas suboptimal concentrations of asciminib or ponatinib failed to counteract p-CRKL expression, the combination of both drugs led to complete suppression of p-CRKL, suggesting that synergistic anti-leukemic effects of ‘asciminib+ponatinib’ resulted from dual targeting of BCR-ABL1 (Figure 3).
Figure 2.

Asciminib and ponatinib synergize in producing apoptosis in BCR-ABL1+ cell lines. KU812, K562 and KCL22T315I (A) and Ba/F3 cells expressing various BCR-ABL1-mutations (Ba/F3p210T315I, Ba/F3p210T315I/E255V, Ba/F3p210T315I/F359V, Ba/F3p210T315I/F311L, Ba/F3p210T315I/G250E) (B) were kept in control medium or in the presence of asciminib, ponatinib or a combination of both drugs as indicated for 48 hours before apoptosis was determined by Annexin V-FITC/DAPI staining and flow cytometry. One typical experiment (left panels) or the mean ± S.D. of three independent experiments. (right panels) are shown. Asterisk (right panels): P<0.05 compared to control.
Figure 3.
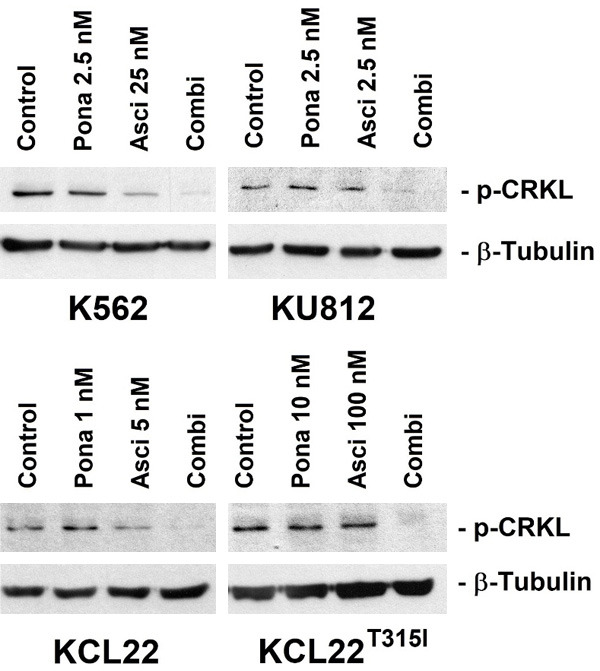
Asciminib and ponatinib synergize in inhibiting CRKL phosphorylation in human CML cell lines. KU812, K562, KCL22 and KCL22T315I were kept in control medium (Control) or in the presence of asciminib (Asci), ponatinib (Pona) or a combination (Combi) of both drugs as indicated for 4 hours before expression of p-CRKL and β-tubulin (loading control) were analyzed by Western blotting.
HU augments anti-neoplastic effects of ‘asciminib+ponatinib’ in TKI-resistant CML cells
HU is prescribed for palliative cytoreduction in advanced CML, often in combination with a TKI. We have recently shown that HU exerts strong antineoplastic effects on BCR-ABL1 T315I-mutated CML cells, presumably by interfering with CDK4/CDK6 activity and thus cell cycle progression [22]. We therefore asked, whether addition of HU would augment the effects of the drug-combination ‘asciminib+ponatinib’. To address this question, we exposed BCR-ABL1+ cells to low concentrations of asciminib, ponatinib, or HU either alone or in 2- or 3-drug combinations. In these experiments, HU further enhanced the effects of a suboptimal ‘asciminib+ponatinib’ combination in all four CML cell lines tested, including BCR-ABL1T315I-bearing KCL22 cells (Figure 4A). Drug synergism was also confirmed in Ba/F3 cells expressing compound mutations involving BCR-ABL1T315I (Figure 4B).
Figure 4.
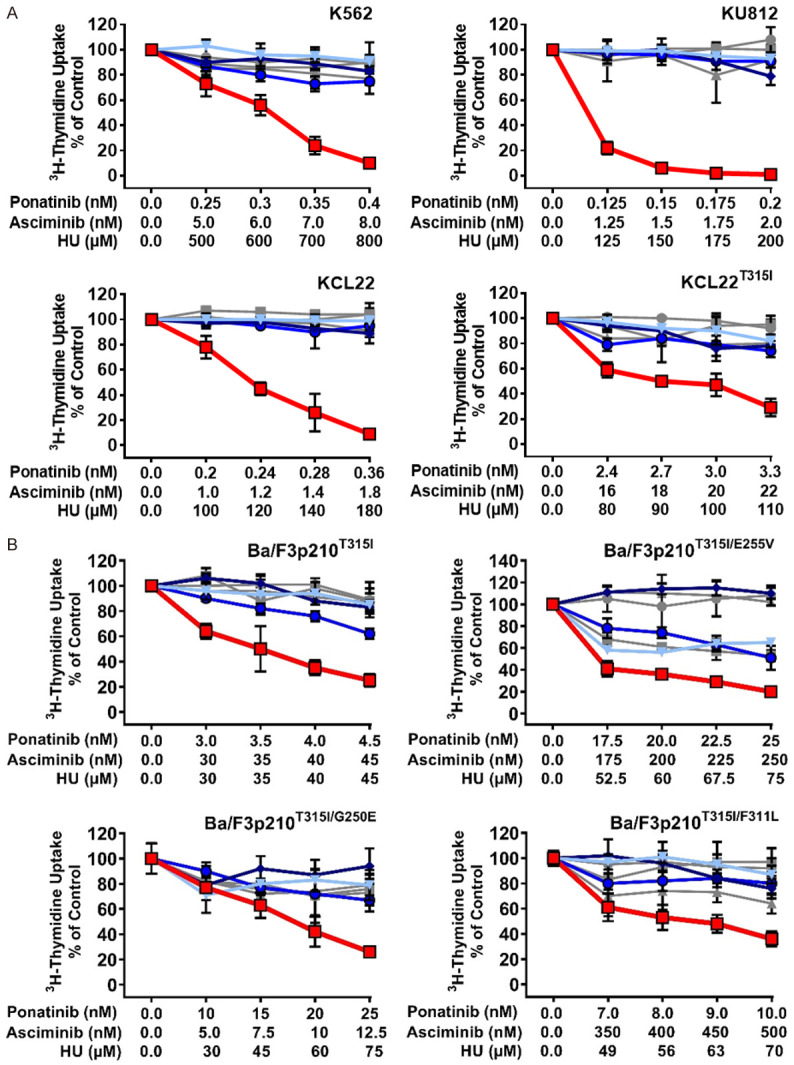
Hydroxyurea (HU) augments anti-neoplastic effects of ‘asciminib+ponatinib’ in TKI-resistant CML cells. Human CML cell lines (A) and Ba/F3 cells expressing various BCR-ABL1-mutations (B) were kept in control medium or in the presence of a single drug (gray lines: asciminib (■-■); ponatinib (●-●); hydroxyurea (HU; ▲-▲), a two-drug combination (blue lines: ‘asciminib+ponatinib’ (▼-▼); ‘ponatinib+HU’ (♦-♦); ‘asciminib+HU’ (○-○) or the three-drug combination ‘asciminib+ponatinib+HU’ (red line; □-□) for 48 hours before 3H-thymidine-uptake was evaluated. Results are calculated as percent of control and represent the mean ± S.D. of triplicates.
Synergistic drug effects are also seen in primary TKI-resistant CML cells
Next, we asked whether synergistic drug effects can also be demonstrated in primary patient-derived CML cells. To address this question, leukemic cells from 8 patients with CP CML were exposed to asciminib and/or ponatinib and/or HU. Asciminib and ponatinib induced cooperative growth-inhibitory effects in primary CML cells in all CML samples tested (Figure 5A). Moreover, HU was found to enhance the growth-inhibitory effects of a suboptimal combination of ‘asciminib+ponatinib’ (Figure 5B).
Figure 5.
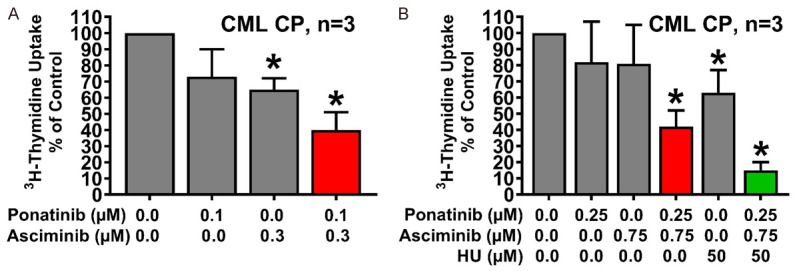
Asciminib, ponatinib and HU synergize in counteracting the growth of primary CML cells. Primary neoplastic cells isolated from 3 patients with CP CML (A: Patients #1, #3 and #4; B: Patients #2, #6 and #7) were kept in control medium or in the presence of asciminib, ponatinib, HU, or drug-combinations as indicated for 48 hours before 3H-thymidine-uptake was evaluated. Results are expressed as percent of control and represent the mean ± S.D. of three patients. Asterisk: P<0.05 compared to control medium. Patients’ numbers refer to Table 1.
Asciminib and ponatinib synergize in producing apoptosis in CD34+/CD38- CML stem cells
Intrinsic and acquired resistance of leukemic stem cells (LSC) against TKI represents a major clinical challenge in CML [4,6]. We asked whether the combination ‘asciminib+ponatinib’ induces apoptosis in primary patient-derived CD34+/CD38- CML stem cells. The gating strategy applied to detect CML LSC is shown in Figure 6A. We found that asciminib promotes the apoptosis-inducing effects of ponatinib on CD34+/CD38- stem cells in all donors with CP CML (Figure 6B) and in all samples from patients with BCR-ABL1 T315I+ or BCR-ABL1 V299L/F317L+ BP CML (Figure 6C). In one patient with BCR-ABL1 T315I+ BP CML and one patient with BCR-ABL1 V299L/F317L+ BP CML, we also added HU and found that the combination ‘asciminib+ponatinib+HU’ exerts a strong cooperative effect on survival of CD34+/CD38- LSC (Figure 6D).
Figure 6.
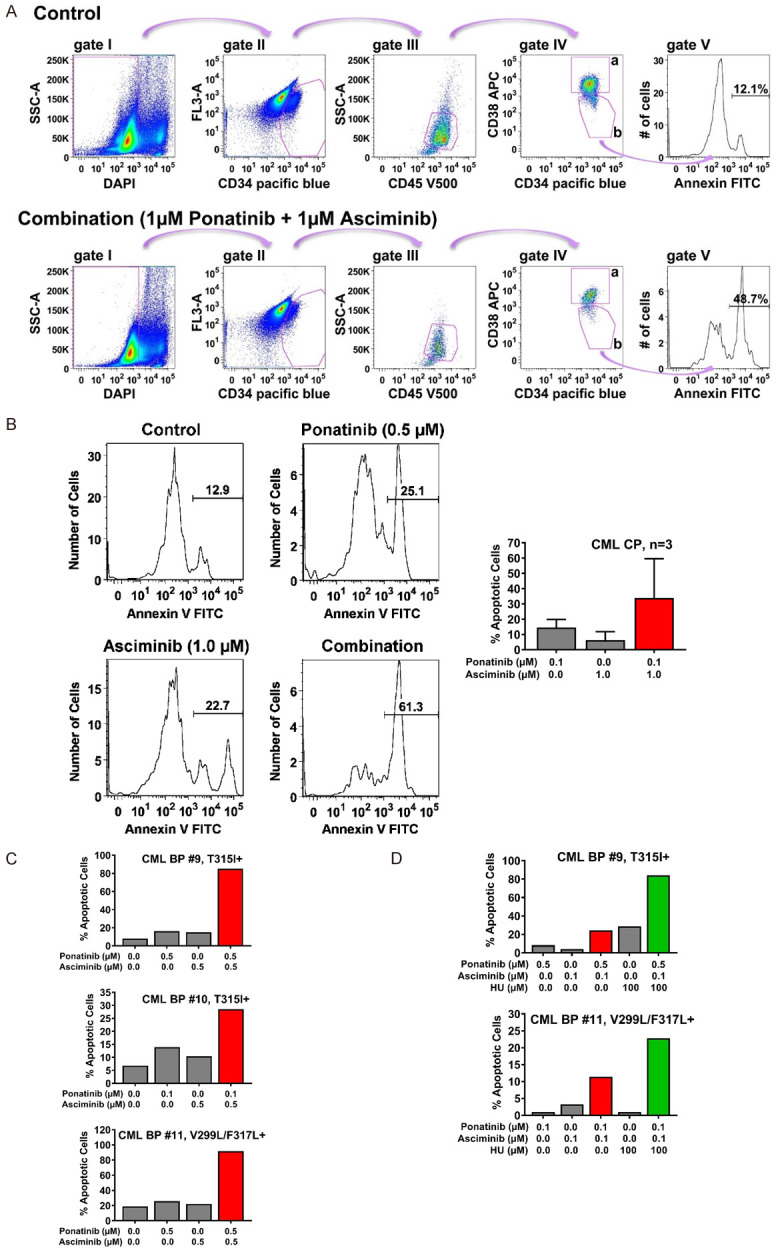
Asciminib, ponatinib and HU synergize in inducing apoptosis in CD34+/CD38- CML stem cells. A. For the detection of Annexin+/DAPI- immature CD34+/CD45dim/CD38- stem cells in peripheral blood samples of CML patients, a step by step gating strategy was applied (steps I-IV) as indicated by pink arrows. After excluding DAPI-negative cells (gate I), immature cells were identified based on their CD34 expression (gate II) and further gated as CD45 dim-positive cells (gate III). Next, cells were separated into progenitor cells (CD34+/CD38+; gate IVa) and stem cells (CD34+/CD38-; gate IVb) by their CD38 expression. Expression of Annexin was determined in stem cells as shown in gate V. The example shows peripheral blood cells from a patient with CML treated with control medium (upper panel) or the combination of ponatinib and asciminib (each 1 µM, lower panel) for 48 hours. B. Primary neoplastic cells isolated from 3 patients with CP CML (patients #1, #2 and #3) were kept in control medium or in the presence of asciminib, ponatinib, or a combination of both drugs as indicated for 48 hours before apoptosis in CD34+/CD38- stem cells was determined by Annexin V-FITC/DAPI staining and flow cytometry. One typical experiment (patient #1) is shown in the left panel; results shown in the right panel represent the mean ± S.D. of the percentage of apoptotic cells (after subtraction of apoptotic cell-counts in control medium) in each condition determined in three patients (patients #1, #2 and #3). C, D. Primary neoplastic cells isolated from 3 patients with BP CML (patients #9, #10 and #11) were kept in control medium or in the presence of asciminib, ponatinib, HU or drug-combinations as indicated for 48 hours before apoptosis within the CD34+/CD38- stem-cell fraction was determined by Annexin V-FITC/DAPI staining and flow cytometry. Results show one typical experiment after subtraction of apoptotic cell-counts in control medium. Patients’ numbers refer to Table 1.
Discussion
The management of TKI-resistant, advanced CML still represents a clinical challenge, especially when BCR-ABL1 T315I is detected [4-6]. Ponatinib is to date the only available and widely used TKI that is directed against BCR-ABL1T315I. However, ponatinib should not be used for long-term treatment in all patients due to potential (vascular) side effects [16-19]. Moreover, T315I-including compound mutations of BCR-ABL1 may occur during ponatinib treatment, resulting in a loss of response [12-15]. In ponatinib-resistant CML or in ponatinib-intolerant patients, other drugs, such as asciminib, which was tested successfully in clinical studies [25-31] and just received breakthrough designation in CML by the FDA, may be considered. Asciminib appears to be a promising drug in heavily pretreated CML patients, which is especially due to the fact, that its binding site differs from binding sites interacting with other approved BCR-ABL1-TKI. However, resistance against asciminib may also occur [25,32,33]. Indeed, asciminib is unable to inhibit the growth of cells harboring T315I-including compound mutations of BCR-ABL1 [25,32]. Furthermore, mutations concerning the myristoyl-binding pocket such as A344P, P465S, and G671R, have been shown to compromise asciminib binding [32]. Finally, resistance against asciminib may be mediated by other resistance mechanisms such as overexpression of the efflux transporters ABCB1 and ABCG2 [33]. All these observations suggest that responses to asciminib may not be durable in all patients with advanced CML.
Combinations of anti-leukemic drugs including BCR-ABL1 TKI may represent an interesting therapeutic option in advanced CML as such drug combinations may suppress drug-resistant subclones [22-24,32-34]. For example, recent data suggest that asciminib cooperates with other BCR-ABL1-targeting TKI in counteracting the survival of BCR-ABL1+ cells [32-34]. In this study, we have confirmed cooperative drug effects and also provide evidence that these drug combinations are effective in human CML cells, including TKI-resistant CML subclones in CML CP and CML PB.
Several mechanisms may contribute to the synergistic anti-leukemic effects produced by combinations of drugs directed against BCR-ABL1. To study these mechanisms, we performed Western blot analysis and found that suboptimal concentrations of asciminib or ponatinib alone (as single agents) are unable to counteract CRKL phosphorylation in leukemic cells, whereas the drug combination resulted in complete suppression of p-CRKL. This observation suggests that synergistic anti-leukemic effects of ‘asciminib+ponatinib’ result primarily from dual targeting of BCR-ABL1 which is most probably due to the different BCR-ABL1 drug-binding sites involved. An alternative explanation could be additional drug targets (apart from BCR-ABL1) involved in synergistic drug interactions. Indeed, whereas asciminib has been postulated to be rather specific for BCR-ABL1 [28], ponatinib reportedly binds to a number of additional target kinases that are critical for growth and survival of CML cells [16,39]. However, so far, it remains unknown what ponatinib-targets may play a role in synergistic drug effects.
T315I-including compound mutations of BCR-ABL1 represent a clinical challenge in CML, as these mutations cause resistance against all currently available TKI, including ponatinib and asciminib [12-15,25,32]. Therefore, current studies focus on the identification of strategies which lead to the elimination of CML-subclones carrying these mutations. We show that the combination ‘asciminib+ponatinib’ produces synergistic anti-proliferative effects in murine cell lines harboring BCR-ABL1T315I or ponatinib-resistant T315I-including compound mutations, hereby confirming previous observations [32]. Indeed, Eide et al. have recently shown that the combination ‘asciminib+ponatinib’ is able to prevent the outgrowth of drug-resistant Ba/F3p210 cells and can restore efficacy against highly-resistant BCR-ABL1 compound mutations. In our study, we have further investigated this drug combination and found clear synergistic effects between asciminib and ponatinib. In particular, both drugs, when applied together, were demonstrated to cooperate in producing growth inhibition in human CML cell lines, including TKI-resistant BCR/ABL1 T315I+ KCL22 cells, and also Ba/F3p210 cells expressing BCR/ABL1T315I or various T315I-based compound mutations. Moreover, the drug combination ‘asciminib+ponatinib’ was found to cooperate in suppressing the proliferation of primary CML cells, including leukemic cells obtained from patients with CP and BP. Together, this data suggest that ‘asciminib+ponatinib’ may be a powerful drug-combination in advanced CML.
We next asked whether there are additional or even stronger drug combinations through which CML subclones bearing BCR-ABL1 compound mutations (involving T315I) can be suppressed or even can be eradicated. We have recently shown that HU exerts strong anti-neoplastic effects on BCR-ABL1 T315I-mutated CML cells and synergizes with ponatinib in inhibiting cell proliferation [22]. These HU effects on CML cells may be caused by the suppression of CDK4 and CDK6 in leukemic cells. Since the cyclin-dependent kinases CDK4 and CDK6 are not among major targets of ponatinib and asciminib we asked whether additional suppression of CDK4/CDK6 could augment the effects of the drug combination ‘asciminib+ponatinib’ on growth of multi-mutated CML cells. Indeed, we were able to demonstrate that HU augments the anti-leukemic effects of the drug-combination ‘asciminib+ponatinib’ in all human BCR-ABL1+ cell lines evaluated, including BCR-ABL1T315I-bearing KCL22 cells and Ba/F3 cells expressing various BCR-ABL1 compound mutations. Simultaneous application of low doses of these 3 drugs may therefore be sufficient to control cell proliferation in CML cells, even in the presence of compound mutations.
One limitation to the application of ponatinib (at least in a subset of patients) are severe cardiovascular side effects, especially if ponatinib is used at a daily dose of 45 mg [12-15]. Lower concentrations of ponatinib, such as 15 mg or 30 mg/day, were demonstrated to have a more beneficial risk profile [40,41]. We have shown, that substantially lower doses of ponatinib were needed to suppress growth and survival of BCR-ABL1+ cells in vitro when ponatinib was combined with asciminib. Hence, the application of additional or alternative drug combinations could allow the use of lower doses of ponatinib and thus may help avoid side effects. Of note, neither HU nor asciminib have been associated with cardiovascular side effects, arguing for a combination of these drugs with ponatinib. However, drug combinations may also lead to new or unexpected side effects due to simultaneous suppression of multiple ‘off-targets’ that may play a role in non-neoplastic cells, such as endothelial cells or cardiomyocytes. Concerning ‘HU+ponatinib’, a combination which is occasionally used in clinical practice in case of advanced or TKI-resistant CML, no increase of cardiovascular side effects has been reported so far. However, further in vivo experiments and clinical trials are warranted to carefully investigate whether the combinations ‘asciminib+ponatinib’ and ‘asciminib+ponatinib+HU’ would produce unexpected adverse events in patients.
Treatment failure, relapse or progression of CML is not always associated with mutations in BCR-ABL1 [4,7]. Another mechanism that is considered to play a role in this context is intrinsic resistance of CML LSC against TKI [4,7,42-44]. To address this point, we investigated whether the combinations ‘asciminib+ponatinib’ and ‘asciminib+ponatinib+HU’ would be able to induce apoptosis in CD34+/CD38- CML LSC. Indeed, asciminib was found to cooperate or even synergize with ponatinib in inducing apoptosis in CD34+/CD38- CML-LSC, including cells obtained from patients with BCR-ABL1 T315I+ BP CML. Cooperative pro-apoptotic effects could also be confirmed for the triple combination ‘asciminib+ponatinib+HU’. These data implicate that new drug combinations, such as ‘asciminib+ponatinib’ or ‘asciminib+ponatinib+HU’, may be able to overcome intrinsic stem cell resistance and also intrinsic+acquired/mutation-induced resistance of CML LSC.
Together, we provide evidence that the combinations ‘asciminib+ponatinib’ and ‘asciminib+ponatinib+HU’ potently inhibit proliferation and survival of human and murine BCR-ABL1+ cell lines, including leukemic cells exhibiting compound mutations involving BCR-ABL1 T315I. We also provide evidence, that these combinations synergize in inhibiting the survival of primary CML cells, including LSC obtained from patients with CP or BCR-ABL1 T315I+BP. Whether these drug combinations also produce major clinical effects in patients with TKI-refractory, advanced CML remains to be determined within the frame of clinical trials.
Acknowledgements
This study was supported by the Austrian Science Fund (FWF) grants F4701 and F4704. We like to thank Christian Milosits for skillful technical support.
Disclosure of conflict of interest
K.V.G.: honoraria from Novartis, Incyte, BMS, Pfizer and AbbVie. K.B.: employment at Oncopeptides AB. T.L.: honoraria from Incyte, Pfizer, Angelini, Novartis, Bristol-Myers Squibb, and Amgen; research grants from Incyte and Novartis. J.M.: research grant and travel support from Novartis. W.R.S.: honoraria from AbbVie, Amgen, Astellas, Celgene, Daiichi Sankyo, Deciphera, Incyte, Jazz, Lipomed, Novartis, Pfizer und Thermo Fisher. P.V.: research funding and honoraria from Novartis and Incyte, and honoraria from BMS/Celgene, Blueprint, Pfizer, AOP Orphan. The other authors (Y.F., D.B., C.S., I.S., L.D.-S., G.E., M.S.-G.) have not disclosed conflicts of interest.
References
- 1.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, Cornelissen JJ, Hughes T, Agis H, Fischer T, Verhoef G, Shepherd J, Saglio G, Gratwohl A, Nielsen JL, Radich JP, Simonsson B, Taylor K, Baccarani M, So C, Letvak L, Larson RA IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 2.Castagnetti F, Gugliotta G, Breccia M, Stagno F, Iurlo A, Albano F, Abruzzese E, Martino B, Levato L, Intermesoli T, Pregno P, Rossi G, Gherlinzoni F, Leoni P, Cavazzini F, Venturi C, Soverini S, Testoni N, Alimena G, Cavo M, Martinelli G, Pane F, Saglio G, Rosti G, Baccarani M GIMEMA CML Working Party. Long-term outcome of chronic myeloid leukemia patients treated frontline with imatinib. Leukemia. 2015;29:1823–1831. doi: 10.1038/leu.2015.152. [DOI] [PubMed] [Google Scholar]
- 3.Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, Cervantes F, Deininger M, Gratwohl A, Guilhot F, Hochhaus A, Horowitz M, Hughes T, Kantarjian H, Larson R, Radich J, Simonsson B, Silver RT, Goldman J, Hehlmann R European LeukemiaNet. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J. Clin. Oncol. 2009;27:6041–6051. doi: 10.1200/JCO.2009.25.0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hochhaus A, Erben P, Ernst T, Mueller MC. Resistance to targeted therapy in chronic myelogenous leukemia. Semin Hematol. 2007;44:S15–S24. doi: 10.1053/j.seminhematol.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Hehlmann R, Müller MC, Lauseker M, Hanfstein B, Fabarius A, Schreiber A, Proetel U, Pletsch N, Pfirrmann M, Haferlach C, Schnittger S, Einsele H, Dengler J, Falge C, Kanz L, Neubauer A, Kneba M, Stegelmann F, Pfreundschuh M, Waller CF, Spiekermann K, Baerlocher GM, Ehninger G, Heim D, Heimpel H, Nerl C, Krause SW, Hossfeld DK, Kolb HJ, Hasford J, Saußele S, Hochhaus A. Deep molecular response is reached by the majority of patients treated with imatinib, predicts survival, and is achieved more quickly by optimized high-dose imatinib: results from the randomized CML-study IV. J. Clin. Oncol. 2014;32:415–423. doi: 10.1200/JCO.2013.49.9020. [DOI] [PubMed] [Google Scholar]
- 6.Quintás-Cardama A, Cortes J. Therapeutic options against BCR-ABL1 T315I-positive chronic myelogenous leukemia. Clin Cancer Res. 2008;14:4392–4399. doi: 10.1158/1078-0432.CCR-08-0117. [DOI] [PubMed] [Google Scholar]
- 7.Rinke J, Hochhaus A, Ernst T. CML - Not only BCR-ABL1 matters. Best Pract Res Clin Haematol. 2020;33:101194. doi: 10.1016/j.beha.2020.101194. [DOI] [PubMed] [Google Scholar]
- 8.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 9.Balabanov S, Braig M, Brümmendorf TH. Current aspects in resistance against tyrosine kinase inhibitors in chronic myelogenous leukemia. Drug Discov Today Technol. 2014;11:89–99. doi: 10.1016/j.ddtec.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Jabbour EJ, Cortes JE, Kantarjian HM. Resistance to tyrosine kinase inhibition therapy for chronic myelogenous leukemia: a clinical perspective and emerging treatment options. Clin Lymphoma Myeloma Leuk. 2013;13:515–529. doi: 10.1016/j.clml.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, Metcalf CA 3rd, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, Keats JA, Wang Y, Sundaramoorthi R, Thomas M, Zhou D, Snodgrass J, Commodore L, Sawyer TK, Dalgarno DC, Deininger MW, Druker BJ, Clackson T. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ, Talpaz M, DiPersio J, DeAngelo DJ, Abruzzese E, Rea D, Baccarani M, Müller MC, Gambacorti-Passerini C, Wong S, Lustgarten S, Rivera VM, Clackson T, Turner CD, Haluska FG, Guilhot F, Deininger MW, Hochhaus A, Hughes T, Goldman JM, Shah NP, Kantarjian H PACE Investigators. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369:1783–1796. doi: 10.1056/NEJMoa1306494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valent P, Hadzijusufovic E, Schernthaner GH, Wolf D, Rea D, le Coutre P. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood. 2015;125:901–906. doi: 10.1182/blood-2014-09-594432. [DOI] [PubMed] [Google Scholar]
- 14.Mayer K, Gielen GH, Willinek W, Müller MC, Wolf D. Fatal progressive cerebral ischemia in CML under third-line treatment with ponatinib. Leukemia. 2014;28:976–977. doi: 10.1038/leu.2013.320. [DOI] [PubMed] [Google Scholar]
- 15.Jain P, Kantarjian H, Jabbour E, Gonzalez GN, Borthakur G, Pemmaraju N, Daver N, Gachimova E, Ferrajoli A, Kornblau S, Ravandi F, O’Brien S, Cortes J. Ponatinib as first-line treatment for patients with chronic myeloid leukaemia in chronic phase: a phase 2 study. Lancet Haematol. 2015;2:e376–e383. doi: 10.1016/S2352-3026(15)00127-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khorashad JS, Kelley TW, Szankasi P, Mason CC, Soverini S, Adrian LT, Eide CA, Zabriskie MS, Lange T, Estrada JC, Pomicter AD, Eiring AM, Kraft IL, Anderson DJ, Gu Z, Alikian M, Reid AG, Foroni L, Marin D, Druker BJ, O’Hare T, Deininger MW. BCR-ABL1 compound mutations in tyrosine kinase inhibitor-resistant CML: frequency and clonal relationships. Blood. 2013;121:489–498. doi: 10.1182/blood-2012-05-431379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibbons DL, Pricl S, Posocco P, Laurini E, Fermeglia M, Sun H, Talpaz M, Donato N, Quintás-Cardama A. Molecular dynamics reveal BCR-ABL1 polymutants as a unique mechanism of resistance to PAN-BCR-ABL1 kinase inhibitor therapy. Proc Natl Acad Sci U S A. 2014;111:3550–3555. doi: 10.1073/pnas.1321173111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zabriskie MS, Eide CA, Tantravahi SK, Vellore NA, Estrada J, Nicolini FE, Khoury HJ, Larson RA, Konopleva M, Cortes JE, Kantarjian H, Jabbour EJ, Kornblau SM, Lipton JH, Rea D, Stenke L, Barbany G, Lange T, Hernández-Boluda JC, Ossenkoppele GJ, Press RD, Chuah C, Goldberg SL, Wetzler M, Mahon FX, Etienne G, Baccarani M, Soverini S, Rosti G, Rousselot P, Friedman R, Deininger M, Reynolds KR, Heaton WL, Eiring AM, Pomicter AD, Khorashad JS, Kelley TW, Baron R, Druker BJ, Deininger MW, O’Hare T. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26:428–442. doi: 10.1016/j.ccr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byrgazov K, Lucini CB, Valent P, Hantschel O, Lion T. BCR-ABL1 compound mutants display differential and dose-dependent responses to ponatinib. Haematologica. 2018;103:e10–e12. doi: 10.3324/haematol.2017.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeung DT. Compound mutations in CML-imaginary bogeyman or real arch-nemesis? Leuk Res. 2019;81:102–104. doi: 10.1016/j.leukres.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, Sawyers CL. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117:2562–2569. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneeweiss-Gleixner M, Byrgazov K, Stefanzl G, Berger D, Eisenwort G, Lucini CB, Herndlhofer S, Preuner S, Obrova K, Pusic P, Witzeneder N, Greiner G, Hoermann G, Sperr WR, Lion T, Deininger M, Valent P, Gleixner KV. CDK4/CDK6 inhibition as a novel strategy to suppress the growth and survival of BCR-ABL1T315I+ clones in TKI-resistant CML. EBioMedicine. 2019;50:111–121. doi: 10.1016/j.ebiom.2019.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gleixner KV, Sadovnik I, Schneeweiss M, Eisenwort G, Byrgazov K, Stefanzl G, Berger D, Herrmann H, Hadzijusufovic E, Lion T, Valent P. A kinase profile-adapted drug combination elicits synergistic cooperative effects on leukemic cells carrying BCR-ABL1T315I in Ph+ CML. Leuk Res. 2019;78:36–44. doi: 10.1016/j.leukres.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valent P, Herndlhofer S, Schneeweiß M, Boidol B, Ringler A, Kubicek S, Gleixner KV, Hoermann G, Hadzijusufovic E, Müllauer L, Sperr WR, Superti-Furga G, Mannhalter C. TKI rotation-induced persistent deep molecular response in multi-resistant blast crisis of Ph+ CML. Oncotarget. 2017;8:23061–23072. doi: 10.18632/oncotarget.15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rea D, Lang F, Kim D, Cortes JE, Hughes TP, Minami H, Breccia M, Deangelo DJ, Hochhaus A, Talpaz M, Goh YT, le Coutre P, Deininger MW, Etienne G, Sondhi M, Mishra , Aimone P, Ng-Sikorski J, Mauro MJ. Asciminib, a specific allosteric BCR-ABL1 inhibitor, in patients with chronic myeloid leukemia carrying the T315I mutation in a phase 1 trial. Blood. 2018;132:792. [Google Scholar]
- 26.Hantschel O, Grebien F, Superti-Furga G. The growing arsenal of ATP-competitive and allosteric inhibitors of BCR-ABL. Cancer Res. 2012;72:4890–4895. doi: 10.1158/0008-5472.CAN-12-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, Marzinzik AL, Pelle X, Donovan J, Zhu W, Buonamici S, Hassan AQ, Lombardo F, Iyer V, Palmer M, Berellini G, Dodd S, Thohan S, Bitter H, Branford S, Ross DM, Hughes TP, Petruzzelli L, Vanasse KG, Warmuth M, Hofmann F, Keen NJ, Sellers WR. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 2017;543:733–737. doi: 10.1038/nature21702. [DOI] [PubMed] [Google Scholar]
- 28.Schoepfer J, Jahnke W, Berellini G, Buonamici S, Cotesta S, Cowan-Jacob SW, Dodd S, Drueckes P, Fabbro D, Gabriel T, Groell JM, Grotzfeld RM, Hassan AQ, Henry C, Iyer V, Jones D, Lombardo F, Loo A, Manley PW, Pellé X, Rummel G, Salem B, Warmuth M, Wylie AA, Zoller T, Marzinzik AL, Furet P. Discovery of asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. J Med Chem. 2018;61:8120–8135. doi: 10.1021/acs.jmedchem.8b01040. [DOI] [PubMed] [Google Scholar]
- 29.Hughes TP, Mauro MJ, Cortes JE, Minami H, Rea D, DeAngelo DJ, Breccia M, Goh YT, Talpaz M, Hochhaus A, le Coutre P, Ottmann O, Heinrich MC, Steegmann JL, Deininger MWN, Janssen JJWM, Mahon FX, Minami Y, Yeung D, Ross DM, Tallman MS, Park JH, Druker BJ, Hynds D, Duan Y, Meille C, Hourcade-Potelleret F, Vanasse KG, Lang F, Kim DW. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N Engl J Med. 2019;381:2315–2326. doi: 10.1056/NEJMoa1902328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Gutiérrez V, Luna A, Alonso-Dominguez JM, Estrada N, Boque C, Xicoy B, Giraldo P, Angona A, Alvarez-Larrán A, Sanchez-Guijo F, Ramírez MJ, Mora E, Vélez P, Rosell A, Colorado Araujo M, Cuevas B, Sagüés M, Cortes M, Encinas MP, Casado Montero LF, Moreno Vega M, Serrano L, Gomez V, Garcia-Hernandez C, Lakhwani S, Paz Coll A, de Paz R, Suarez-Varela S, Fernandez-Ruiz A, Perez Lopez R, Ortiz-Fernández A, Jiménez-Velasco A, Steegmann-Olmedillas JL, Hernández-Boluda JC. Safety and efficacy of asciminib treatment in chronic myeloid leukemia patients in real-life clinical practice. Blood Cancer J. 2021;11:16. doi: 10.1038/s41408-021-00420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manley PW, Barys L, Cowan-Jacob SW. The specificity of asciminib, a potential treatment for chronic myeloid leukemia, as a myristate-pocket binding ABL inhibitor and analysis of its interactions with mutant forms of BCR-ABL1 kinase. Leuk Res. 2020;98:106458. doi: 10.1016/j.leukres.2020.106458. [DOI] [PubMed] [Google Scholar]
- 32.Eide CA, Zabriskie MS, Savage Stevens SL, Antelope O, Vellore NA, Than H, Schultz AR, Clair P, Bowler AD, Pomicter AD, Yan D, Senina AV, Qiang W, Kelley TW, Szankasi P, Heinrich MC, Tyner JW, Rea D, Cayuela JM, Kim DW, Tognon CE, O’Hare T, Druker BJ, Deininger MW. Combining the allosteric inhibitor asciminib with ponatinib suppresses emergence of and restores efficacy against highly resistant BCR-ABL1 mutants. Cancer Cell. 2019;36:431–443. doi: 10.1016/j.ccell.2019.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eadie LN, Saunders VA, Branford S, White DL, Hughes TP. The new allosteric inhibitor asciminib is susceptible to resistance mediated by ABCB1 and ABCG2 overexpression in vitro. Oncotarget. 2018;9:13423–13437. doi: 10.18632/oncotarget.24393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elrashedy AA, Ramharack P, Soliman MES. The perplexity of synergistic duality: inter-molecular mechanisms of communication in BCR-ABL1. Anticancer Agents Med Chem. 2019;19:1642–1650. doi: 10.2174/1871520619666190620120144. [DOI] [PubMed] [Google Scholar]
- 35.Yuan H, Wang Z, Gao C, Chen W, Huang Q, Yee JK, Bhatia R, Chen W. BCR-ABL gene expression is required for its mutations in a novel KCL-22 cell culture model for acquired resistance of chronic myelogenous leukemia. J Biol Chem. 2010;285:5085–5096. doi: 10.1074/jbc.M109.039206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.La Rosee P, Corbin AS, Stoffregen EP, Deininger MW, Druker BJ. Activity of the Bcr-Abl kinase inhibitor PD180970 against clinically relevant Bcr-Abl isoforms that cause resistance to imatinib mesylate (Gleevec, STI571) Cancer Res. 2002;62:7149–7153. [PubMed] [Google Scholar]
- 37.Byrgazov K, Lucini CB, Berkowitsch B, Koenig M, Haas OA, Hoermann G, Valent P, Lion T. Transposon-mediated generation of BCR-ABL1-expressing transgenic cell lines for unbiased sensitivity testing of tyrosine kinase inhibitors. Oncotarget. 2016;7:78083–78094. doi: 10.18632/oncotarget.12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 39.Tan FH, Putoczki TL, Stylli SS, Luwor RB. Ponatinib: a novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther. 2019;12:635–645. doi: 10.2147/OTT.S189391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castagnetti F, Pane F, Rosti G, Saglio G, Breccia M. Dosing strategies for improving the risk-benefit profile of ponatinib in patients with chronic myeloid leukemia in chronic phase. Front Oncol. 2021;11:642005. doi: 10.3389/fonc.2021.642005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santoro M, Accurso V, Mancuso S, Contrino AD, Sardo M, Novo G, Di Piazza F, Perez A, Russo A, Siragusa S. Management of ponatinib in patients with chronic myeloid leukemia with cardiovascular risk factors. Chemotherapy. 2019;64:205–209. doi: 10.1159/000504664. [DOI] [PubMed] [Google Scholar]
- 42.Valent P. Emerging stem cell concepts for imatinib-resistant chronic myeloid leukaemia: implications for the biology, management, and therapy of the disease. Br J Haematol. 2008;142:361–378. doi: 10.1111/j.1365-2141.2008.07197.x. [DOI] [PubMed] [Google Scholar]
- 43.Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, Eaves C. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21:926–935. doi: 10.1038/sj.leu.2404609. [DOI] [PubMed] [Google Scholar]
- 44.Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, Barow M, Mountford JC, Holyoake TL. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107:4532–4539. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]