

Abstract
Doxorubicin (DOX) is a highly effective chemotherapy agent that often causes cardiotoxicity. Despite a number of extensive studies, the risk for DOX cardiotoxicity remains unpredictable. The majority of the studies on DOX-induced cardiotoxicity have been focused on the effects on cardiomyocytes that lead to contractile dysfunction. The roles of systemic inflammation, endothelial injury and neutrophil recruitment, all induced by the DOX, are increasingly recognized as the mechanisms that trigger the development and progression of DOX-induced cardiomyopathy. This review explores recent data regarding the possible mechanisms and biomarkers of early subclinical DOX-associated cardiotoxicity.
Keywords: Doxorubicin, cardiotoxicity, mechanisms, neutrophils, coronary vasculature, biomarkers
Introduction
Anthracyclines, such as doxorubicin (DOX) are very potent chemotherapeutic drugs that have significantly improved cancer survival [1]. DOX and other anthracyclines (i.e. epirubicin, daunorubicin, idarubicin) are used for treatment of many cancers of the breast, endometrial and gastric tissues, childhood solid tumors, soft tissue sarcomas, and pediatric leukemia, as well as post-heart and bone marrow transplantation [2]. Although many anti-neoplastic therapies are cardiotoxic, anthracycline-induced cardiomyopathy and heart failure (HF) are the most thoroughly studied [3]. Cardiotoxicity of anthracyclines may not be detected until several years after the treatment and may significantly impact a patient’s survival and quality of life independently of the oncological prognosis [4]. Survivors of childhood cancer treated with DOX are at the greatest risk of cardiovascular morbidity and mortality. About 60% of pediatric cancer patients are treated with DOX-based chemotherapy [5] and 10% of these patients develop cardiomyopathy up to 15 years after the end of chemotherapy [6]. The majority of the studies on DOX-induced cardiotoxicity are focused on the effects on cardiomyocytes that lead to contractile dysfunction. The role of the systemic inflammation associated with neutrophil recruitment and vascular endothelial injury has been recently recognized as a mechanism that triggers the development and progression of DOX-induced cardiomyopathy. This review discusses the available data regarding the possible mechanisms and biomarkers of the early subclinical DOX-associated cardiotoxicity.
Clinical aspects
Clinically recognized DOX-induced cardiotoxicity can occur at any point during and after treatment with anthracyclines. Acute/subacute cardiovascular complications can arise from the initiation of therapy to several weeks after treatment termination [7] and manifest themselves as chest pain, palpitation, dysplasia, and/or tachycardia arrhythmias, as well as a decline in the left ventricle ejection fraction (LVEF) from more than 10% to 50% [8]. Electrocardiograms reveal nonspecific ST-T changes, left axis deviation, and decreased amplitude of QRS complexes [9]. The mechanism of the acute cardiotoxicity is not quite clear, but may be associated with DOX-induced myocardial edema or inflammatory response, which is reversible and can be controlled with appropriate treatment [10]. DOX-induced cardiotoxicity may become clinically evident after one-year chemotherapy completion (late-onset chronic cardiotoxicity), which is characterized by dilated cardiomyopathy, including dilation of ventricles, in some cases of atria, reduced LVEF and contractile function, diastolic dysfunction, and mural thrombi in some patients [11]. DOX-induced pathological alterations indicative of restrictive cardiomyopathy following DOX chemotherapy have been reported in survivors of childhood cancer (i.e. reduced LV mass and diffuse interstitial fibrosis) [12] and in experimental animals (i.e. decreased end-diastolic LV volume, increased posterior wall thickness and a ratio between the E-wave and A wave velocities of the mitral valve >2) [13].
Monitoring
Several different methods exist for the assessment of chemotherapy-induced cardiotoxicity, including electrocardiography, echocardiography, biopsy, scintigraphy, serum analysis, and some genomic markers [14]. Each of these has certain limitations, such as low sensitivity, high invasiveness, elevated costs, and/or relatively late detection of heart dysfunction. The assessment of a decreased LVEF decrease radionuclide scintigraphy or angiocardiography or 12-lead electrocardiogram have been the most common non-invasive methods in clinical practice to monitor cardiotoxicity. However, LVEF is considered a fairly late manifestation of cancer chemotherapy-associated cardiotoxicity, reflecting the presence of irreversible myocardial damage and is usually insensitive to early stages of subclinical injury [15]. Endomyocardial biopsy, traditionally considered as the “gold standard” test for the evaluation of DOX cardiomyopathy is invasive and does not correlate with the subsequent risk of congestive HF [16]. Traditional blood-based cardiac biomarkers, such as cardiac troponins and B-type natriuretic peptide (BNP), have been suggested in the diagnostics of HF, but several studies failed to detect any correlation between their blood results and DOX-induced cardiotoxicity [17]. High-sensitivity troponin assays have been shown to have better accuracy in the diagnostics of acute coronary syndrome and cancer patients treated with anthracyclines [18], but they have failed to predict DOX cardiotoxicity in breast cancer patients in a recently clinical study [19].
Risk factors
A major risk for DOX-induced cardiotoxicity is the total dose of the drug administered. DOX-induced cardiotoxicity is usually cumulative dose-dependent, which begins with the first dose. There has not been established a “safe” dose of DOX which does not result in cardiotoxicity [20]. Clinical HF incidences of 3%, 7%, 18% and 40% have been shown with cumulative doses of 400, 550, 600 or >650 mg/m2 respectively, therefore doses below 450 mg/m2 are recommended [21]. The risk of DOX-induced cardiotoxicity increases with other added chemotherapy drugs, such as trastuzumab or cyclophosphamide, or with added chest or mediastinal radiation [22]. Other suspected risk factors are the age at diagnosis [23], follow-up time, and female sex [24]. Prepubescent girls are at higher risk of DOX-induced cardiotoxicity in comparison with boys [25], as are women older than 65 [26]. It has been suggested that women with higher circulating estrogen are more resistant than an age-matched man to DOX-induced cardiomyopathy [27]. The increased risk of cardiomyopathy induced by DOX in the young and older patients has also been attributed in part to the immature liver function in young children and declining liver activity among older adults, both of which slow DOX clearance and prolong exposure to circulating DOX [28]. Because the liver is a major site of DOX clearance, any alteration in DOX metabolism caused by liver disease or concurrent medications would be expected to result in elevated levels of DOX and increased exposure to toxic concentrations of the drug [29]. Additionally, hypertension, diabetes, dyslipidemia, obesity also increase the risk of DOX-induced cardiotoxicity [30].
Susceptibility to DOX cardiotoxicity is largely individual with some patients developing cardiomyopathy at doses of 200-400 mg/m2, while others tolerating well >1000 mg/m2 [31], suggesting the presence of a genetic predisposition. Several recent studies have addressed the existence of gene variants predisposing to DOX-induced cardiotoxicity. Candidate SNPs or gene panels have previously been associated with the DOX metabolism and transport, oxidative stress, and DNA damage [32], some of which are shown in Table 1. The overall reproducibility of the published studies has been limited, due to small cohorts, failure to assess population ancestry, and lack of replication, including our study on breast cancer patients with or without DOX-induced cardiotoxicity [33]. We have identified 15 SNPs in nine genes in the human leukocyte antigen (HLA) region (NFKBIL1, TNF-alpha, ATP6V1G2, MSH5, MICA, LTA, BAT1, NOTCH4), and three SNPs in the psoriasis susceptibility region of HLA-C as potential candidates for association with DOX-cardiotoxicity in breast cancer patients (Table 1). The function of most of these molecules remains insufficiently characterized, although evidence suggests a role in immune and inflammatory responses [34]. All of the candidate genes in our study are located on chromosomes 6p32 and 6p33. Six of the candidate SNPs in the BAT1-NFKBIL1-LTA region in our dataset have previously been reported in association with myocardial infarction [35] and autoimmune disorders [36]. SNPs within TNF-α, NOTCH4, and C6orf10 have been associated with coronary artery disease, and myocardial infarction, while SNPs within MSH5, MICA, and HLA-C have been reported in autoimmune inflammatory disorders (Table 1). The telomeric class III region of HLA bordering the class I region is particularly gene-dense containing at least 10 genes in addition to TNF alpha within an 82 kb interval, including BAT1, ATP6V1G2, NFkBIL1, LTA, TNF, LTB, LST1, NCR3, AIF-1, BAT3 and BAT2 [37]. Our findings are consistent with reports showing the presence of susceptibility loci within the HLA-gene region for coronary artery disease (CAD) [38] and inflammatory/autoimmune disorders [39]. Autoimmune features and rheumatic manifestations have been reported in cancer patients after chemotherapy [40] including rheumatism, and systemic lupus erythematosus (SLE) in those with breast cancer [41].
Table 1.
Candidate SNPs associated with sensitivity to DOX-induced cardiotoxicity
Cellular and molecular mechanisms and biomarkers oxidative stress
The in vitro metabolism of DOX in cardiac and liver microsomal membranes has been described in several earlier studies. The initial reduction of DOX to a semiquinone free radical is catalyzed by NADPH-dependent cytochrome P450 reductase and reconversion to the quinone, a process involving one-electron reduction, which leads to the persistent production of superoxide anion radical and secondary ROS (e.g., O2∙, H2O2, OH·) [42,43]. H2O2 and O2∙- may also generate highly reactive and toxic hydroxyl radicals (OH) during the iron-catalyzed Haber-Weiss reaction, resulting in an iron cycling between Fe3+ and Fe2+, thus altering iron homeostasis [44]. The increased production of ROS induced by DOX leads to excessive oxidative stress, strongly linked to cell damage involving reduced protein synthesis and redox modifications of proteins, lipids, and DNA [45].
DOX accumulates primarily in the mitochondria [46], which accounts for DOX cardio-selective toxicity, combined with a less active antioxidant network in the heart compared with other tissues such as the liver [47]. DOX, being a cationic drug, binds to the negatively charged phospholipid cardiolipin located on the inner mitochondrial membrane, leading to disruption of the activity of complexes I-IV of the electron transport chain, peroxidation of lipids, oxidative damage of proteins and mitochondrial DNA, loss of ATP levels and mitochondrial permeability transition 3 one integrity [48]. The Keap1-Nrf2 pathway is the major regulator of cytoprotective responses to oxidative and xenobiotic stress by activating antioxidants and anti-electrophiles [49]. The key signaling protein within the pathway is the transcription factor Nrf2, which under the regulation of Keap-1 can protect the cells and tissues from oxidative stress by increasing the expression of several downstream cytoprotective genes, including antioxidants and phase II and phase III detoxification enzymes [50]. It has been demonstrated that deficiency of Nrf2 amplified DOX-induced cardiotoxicity and cardiac dysfunction [51]. We have examined the effect of DOX on the early (48 hours post-injection) gene expression of rat hearts and found that at this time-point there was significant downregulation of NRF2 gene (NFE2L2), cardiolipin gene (CRLS1), along with a widespread reduction in the expression of multiple genes encoding for proteins of complexes I-IV, and ATP synthase [52]. We observed significant downregulation of mitochondrial oxidative phosphorylation (OXPHOS) complexes I-IV and ATP synthase, including, 25 transcripts coding for NADH dehydrogenase in complex I, all four transcripts coding for succinate dehydrogenases in complex II, four transcripts of ubiquinone-cytochrome c reductase in complex III, 13 genes coding for several subunits of cytochrome c oxidase in complex IV, and 8 ATP synthases from complex V. Downregulation of OXPHOS complexes and the associated increased superoxide production and 4-HNE protein adducts tend to predispose to hypertension, coronary artery disease and HF [53].
Along with the increased oxidative stress, DOX also reduces the antioxidant defense of the cells, for example, reducing SOD, and catalase content or activity, thus contributing to enhance and prolong mitochondrial damage [54]. Various antioxidant supplements have shown some protection when combined with DOX, including vitamins C and E [55], glutathione and metallothionein [56], and glutamine [57]. Chandran et al. [58] demonstrated that co-administration of MitoQ, a triphenylphosphonium-conjugated analog of coenzyme Q, to rats treated with DOX resulted in improved LV function. Because MitoQ is a mitochondria-targeted antioxidant, enrichment of mitochondrial membranes with the active antioxidant is beneficial against DOX toxicity.
Calcium homeostasis dysregulation
Intracellular ionized Ca concentration ([Ca2+]i) regulates cardiomyocytes’ contractility through excitation-contraction coupling, a process that links the electric excitation of the sarcolemma surface membrane (action potential) to the mechanical contraction. During the cardiac action potential, Ca2+ enters the cell through the L-type calcium channel, which triggers additional Ca2+ release from the sarcoplasmic reticulum (SR). The elevated [Ca2+]i concentration allows Ca2+ to bind to the myofilament protein troponin C (Tn-C), which then switches on the contractile machinery [59]. For relaxation to occur [Ca2+]i must decline and allow Ca2+ to dissociate from troponin, which involves SR Ca2+-ATPase (SERCA2a), Na+/Ca2+-exchanger (NCX), and plasmalemmal Ca2+-ATPase (PMCA) [60]. The elevated [Ca2+]I levels resulting in mitochondrial Ca2+ overload, combined with unregulated ROS production, causes opening of the mitochondrial permeability transition pore (MPTP), causing permeabilization of the mitochondrial inner membrane to molecules of less than 1.5 kDa in molecular weight [61]. MPTP opening results in inner membrane potential reduction and collapse, respiratory chain uncoupling, halt of mitochondrial ATP synthesis, and eventually, mitochondrial swelling, rupture, and cell death, as reported in DOX cardiotoxicity in the human heart [62].
DOX binds and activates sarcoplasmic reticulum (SR) ryanodine receptors (RYRs) to increase cytosolic ionized Ca2+, while at the same time downregulating calcium transport ATPase (SERCA), which pumps Ca2+ back to SR, leading to an abnormal cytoplasmic Ca2+ and increased generation of ROS [63,64]. It has been shown that DOX directly affects RYR2 activity by rapid reversible activation of the channel, followed later by irreversible inhibition [65]. We have tested the effects of RYR antagonist dantrolene (DNT) on DOX-induced cardiotoxicity in a rat model of breast cancer [66]. We found that DNT improved DOX-induced alterations in the echocardiographic and histopathological parameters, without affecting the anti-tumor efficacy of DOX. Rats treated with DNT lost less body weight, had higher blood GSH levels and lower troponin I level than DOX-treated rats. These data indicate that DNT can provide protection against DOX cardiotoxicity without reducing its antitumor activity.
Topoisomerases
Topoisomerases (Tops) catalyze the relaxation of DNA supercoils and unknotting of DNA helices and strands [67]. Top2 enzymes introduce double-strand breaks in the DNA molecule, passes another unbroken DNA helix through it, and then re-ligates the cut strands. DOX binds to both DNA and Top2, forming the DOX-Top-DNA complex, which inhibits Top2 activity, resulting in DNA double-strand breaks, activating DNA damage response, and apoptosis [68]. Top2 isoforms, Top2α and Top2β, are differentially regulated during the cell cycle in normal and neoplastic tissues, as Top2α is overexpressed in highly proliferative tumor cells and undetectable in quiescent cardiomyocytes [69], while Top2β is overexpressed in terminally differentiated quiescent cells, such as cardiomyocytes [70,71]. Top2β is a mechanism thought to be responsible for DOX-induced cardiotoxicity through enhancement of oxidative stress and impairment of mitochondrial biogenesis [72]. Cardiomyocyte-specific deletion of Top2β protected the cardiomyocytes from DOX-induced DNA double-strand breaks and mice from the development of DOX-induced HF [73]. Currently, dexrazoxane, a Top2 poison, an iron chelator, and free radical scavenger, is the only approved drug for the treatment of DOX-induced cardiotoxicity in clinical settings [74].
Heat shock proteins
DOX-induced oxidative stress and ATP depletion induce high expression of heat shock proteins (HSPs), which regulate the activity of multiple signaling intermediates involved in the execution of apoptotic signaling pathways [75]. For example, HSP10 and HSP60 overexpression led to an increase in the post-translational modification of Bcl-2 proteins induced by DOX [129], HSP20 reduced DOX-associated oxidative stress, and cardiotoxicity via interacting with AKT phosphorylation [76]. A significant 3,9-fold upregulation of the HSP90 gene (HSPCB) was detected in the rat hearts at 48 hours post-DOX administration [85], a finding that correlated with the 16-fold HSP90 induction by cardiac ischemia [77], due to ROS accumulation [78] and reduction of ATP concentration [79].
Cellular senescence
Many chemotherapeutic drugs, such as anthracyclines, cyclophosphamide, cisplatin, mitoxantrone, and gamma irradiation are known to alter cellular states and induce senescence in cancer cells and the tumor microenvironment [80]. Cellular senescence is a potent tumor-suppressive mechanism that arrests the growth of cells at risk for malignant transformation [81]. Senescence-associated secretory phenotype (SASP) is characterized by arrested cell growth, resistance to apoptosis, high metabolism and secretion of proinflammatory cytokines (e.g. IL-6, IL-1α-6, -8, 10), growth factors (e.g. IGF/IGFBP, FGF, TGF-β, IFN-γ), proteases (e.g. MMP1, MMP-3). It has been demonstrated that cell cycle inhibitors p16 and p21 are overexpressed by senescent cells, making them the most well-established senescence markers [82]. The contribution of senescent cells to coronary heart diseases [83] and atherosclerosis [84] have been shown in several studies. Studies in cancer showed that therapy-induced senescence can stimulate immunosurveillance to eliminate tumor cells, but it can also be a source of chronic inflammation and drug resistance [85]. For example, treatment of breast cancer patients with DOX and alkylating agents induced cellular senescence in a p16INK4a-dependent, telomere-independent fashion [86]. Demaria et al. [87] showed that DOX-induced senescence could persist and contributed to local and systemic inflammation in mice, and elimination of the senescent cells reduced several short- and long-term effects of the drugs, including bone marrow suppression, cardiac dysfunction, cancer recurrence, and physical activity and strength.
Inflammation, endothelial dysfunction, and hypercoagulability
Several inflammatory markers may be able to predict future cardiovascular events [88]. For example, IL-6 is associated with an increased risk of myocardial infarction and cardiovascular death [89]. Elevated pro-inflammatory cytokines, such as TNF, IL-6, monocyte chemotactic protein 1 (MCP-1), and Interferon-γ (INF-γ) were found in the serum of mice after administration of DOX [90]. Wang et al. [91] demonstrated that DOX-induced upregulation of the proinflammatory Toll-like receptor TLR4 in macrophages was associated with DOX-triggered leakage of endotoxin into the circulation of rats. Elevated levels of CRP were associated with decreased LVEF in patients with varying cardiovascular diseases ranging from myocardial infarction to HF [92]. We used multiplex assays for chemokines to examine plasma samples collected before and after the first cycle of DOX-based chemotherapy of breast cancer patients [19]. The results showed that the initial DOX dose-induced chemokine “immune/inflammatory signature” including CCL23, CCL27 and MIF was able to predict the abnormal decrease of LVEF after chemotherapy. Previous studies showed the association of CCL23 and MIF with coronary atherosclerosis, myocardial infarction, and CCL27 was reported in autoimmune diseases (Table 2), but no report correlated these chemokines with cardiotoxicity.
Table 2.
Candidate circulating biomarkers for prediction of DOX-induced cardiotoxicity
| Biomarker | Function |
|---|---|
| CCL23 | Elevated in coronary atherosclerosis [151] |
| Promising biomarker of injury in patients with ischemic stroke through upregulation of TNF-α [152] | |
| Elevated in autoimmune diseases (psoriasis and eczema) [153] | |
| CCL27 | Associated with diminished kidney transplant and autoimmune disease (psoriasis and eczema) [154] |
| MIF | Elevated in myocardial ischemia/reperfusion injury [155] |
| Upregulated during the progression of atherosclerosis [156] | |
| Elevated in myocardial infarction and inflammatory diseases [157] | |
| PGLYRP1 (Tag7, PGRP1) | Increased gene expression and protein levels in patients with myocardial infarction [158] |
| Present in atherosclerotic plaques [159] | |
| Circulating levels were independently associated with coronary and peripheral atherosclerosis [160,161] | |
| Elevated circulating levels predicted poor outcome in patients with HF [162] | |
| Marker of coronary artery disease and HF [163] | |
| CAMP (hCAP-18/LL-37) (Cramp in mouse) | Associated with acute HF rodents [164] |
| Presence in human atherosclerotic plaques [165,166] | |
| Associated with platelet activation and induction of thrombosis [167] | |
| Produced in atherosclerotic lesions, where it may function as an immune modulator [168] | |
| MMP9 | Involved in plaque destabilization resulting in acute coronary syndrome and stroke [169,170] |
| Elevated circulating levels predicted poor outcome in acute coronary syndrome [171] | |
| Role in remodeling atherosclerotic plaques in myocardial infarction [172] | |
| Higher levels associated with acute coronary syndrome [173-176] | |
| MMP8 | Circulating levels correlate with acute coronary syndrome [177] |
| Baseline serum is a significant predictor of LV remodeling and cardiovascular outcome after myocardial infarction [178] | |
| Serum levels associated with subclinical atherosclerosis [179] | |
| MPO | Circulating levels correlate with acute coronary syndrome [180,181] |
| Marker of ischemic heart disease and acute coronary syndrome [182] | |
| Associated with a risk of coronary artery disease [183] | |
| Serum levels predict acute coronary syndrome [184] | |
| Circulating levels appeared as a strong independent marker of coronary artery disease [185] | |
| Potential biomarker of the risk for a subsequent cardiac dysfunction in cancer patients [186] | |
| DEFA 1-4, human neutrophil peptidases (HNPs) | Exert pro-atherosclerotic properties by promoting monocyte adhesion, platelet activation, and foam cell formation [187] |
| Significantly increased in acute coronary syndrome and sepsis [187] | |
| CEACAM8 (CD66b) (Ly6G in mice) | Upregulation in granulocytes polymorphonuclear leukocytes was a risk factor for atherothrombosis [188] |
| Mice treated with anti-LY6G to deplete neutrophils were protected against CD8+ T-cell-dependent myocarditis [189] | |
| TM | Facilitates the thrombin-mediated activation of protein C and plays role in coagulation, fibrinolysis and inflammation [190] |
| Marker of generalized endothelial injury [191] | |
| Elevated circulating levels in intravascular coagulation and venous thrombosis [192] | |
| Circulating levels predict the risk of coronary heart disease [193,194] | |
| OLR1 (LOX-1) | Marker of atherosclerosis and vasculopathy [195] |
| Marker of coronary artery disease, stroke, and acute aortic dissection [196] | |
| Correlate with acute coronary syndrome [197] | |
| Elevated in hypertensive rats [198] | |
| Upregulated during myocardial ischemia-reperfusion [199] | |
| Expressed in atherosclerotic lesions [200] | |
| Markers of NETs | Elevated circulating levels independently associated with severe coronary atherosclerosis and a prothrombotic state [201] |
| Presence in human coronary and ischemic stroke thrombi [202] | |
| Biomarker of STEMI myocardial infarction [203,204] | |
| Biomarker of acute ischemic stroke, coronary and peripheral artery disease [205] | |
| Markers of acute myocardial infarction [206] | |
| Activate ECs and platelets, resulting in endothelial dysfunction, proinflammatory immune response, and thrombotic lesions [207,208] | |
| Presence in atherosclerotic lesions [209] | |
| Increase with age [210] | |
| TAT complex | Diagnosis of hypercoagulability [211] |
| Diagnosis and assessment of treatment-induced intravascular coagulation, deep vein thrombosis, and pulmonary thromboembolism [212,213] | |
| Elevated circulating levels associated with coronary artery disease [214] | |
| Increased plasma levels associated with atrial fibrillation [215] | |
| CRP | A well-known marker of inflammation [216] and endothelial dysfunction [217,218] |
| Marker of coronary artery disease [219] | |
| Serum levels correlate with severity of coronary artery lesions [220] | |
| Associated with atherothrombosis [221] | |
| Predict cardiovascular disease [222] | |
| Marker of early vascular aging [223] | |
| vWF | Elevated plasma levels associated with myocardial infarction [224], coronary artery disease [225] and ischemic stroke [226] |
| P-selectin | Promotes acute myocardial infarction [227] |
| Associated with atherothrombosis [228] | |
| hsa-mir-1 | Downregulated in cardiac hypertrophy [229] |
| Has-mir-16-5p | Aggravates myocardial infarction injury by targeting IRS1 [230] |
| hsa-mir133a | Downregulated in cardiac hypertrophy [231] |
| hsa-miR-92 | Upregulated in dilated cardiomyopathy [232] |
| hsa-mir-34a | Upregulation in myocardial infarction [233] |
| Has-mir-15a | Upregulated in myocardial ischemia [234] |
| hsa-mir-30 | Downregulated in dilated cardiomyopathy [235] |
DOX-induced inflammation is associated with endothelial dysfunction, which is a complex process involving ROS production, pro-inflammatory cytokines secretion and inactivation of NO production, resulting in disruption of vascular contractility [93]. Dysfunctional endothelial cells (ECs) secrete TNF-α, IL-1β, IL-6, IL-8, which along with granulocyte colony-stimulating factor (G-CSF) promote mobilization of neutrophils from the bone marrow and their recruitment to the vascular endothelium [94]. Neutrophils adhere to the endothelium and release neutrophil granular peptides and proteins, such as myeloperoxidase (MPO), elastase, matrix metalloproteases (MMPs) disrupting inner endothelial junction. Further damage can be induced by the production of neutrophil extracellular traps (NETs), which enhance inflammation and endothelial permeability. Neutrophils activate platelets, which bind to the extracellular matrix (ECM) beneath the endothelial layer and create platelet plug to maintain hemostasis within an injured vessel [95]. The interactions between ECs, neutrophils, and platelets are influenced by several factors released from ECs, such as thrombomodulin (TM), von Willebrandt factor (vWF), P-selectin, which modulate platelet activity, coagulation, and vascular contractility, all of which contribute to the thrombotic formation, myocardial infarction, coronary artery disease and ischemic stroke [93]. It is known that cancer chemotherapy increases the risk of cancer-related thrombosis, which is a major risk factor for cardiovascular diseases [96]. We have found, that circulating biomarkers of inflammation, hypercoagulability and endothelial injury before or after the initial infusion of DOX-based chemotherapy were able to predict the risk of early subclinical DOX-induced cardiotoxicity in breast cancer patients [97]. Patients with an abnormal decline of LVEF had significantly elevated levels of MPO and TM both at baseline, and after the first dose of DOX-based chemotherapy relative to patients with normal LVEF. The first dose of DOX also induced higher circulating levels of thrombin-anti-thrombin complex (TAT) complex, C-reactive protein (CRP), markers of NETs, vWF, and P-selectin in patients with cardiotoxicity in comparison with patients without. These findings indicate that the risk of DOX-induced cardiotoxicity in breast cancer is associated with endothelial dysfunction, inflammation, and prothrombotic state before and after the first dose of chemotherapy. Furthermore, the increased circulating levels of MPO and TM before and after the first DOX infusion in cancer patients with DOX-induced low LVEF suggest their potential to be used as predictive biomarkers and the need for future validation studies. In addition, the circulating levels of TAT, NETs, CRP, and vWF might also be potentially predictive for the risk of DOX-induced cardiotoxicity after the first chemotherapy dose.
Cardiac microvascular ECs, being the most abundant cell type in adult myocardium are in direct contact with the adjacent cardiomyocytes and fibroblasts, and actively secret many proteins, which can modulate cardiac contractility and remodeling [98,99]. DOX-induced endothelial damage has been associated with the development of severe chronic vascular diseases, such as the atherosclerosis [100]. A prospective study of 7289 childhood survivors showed that 10% developed coronary artery disease 10 years after diagnosis [101]. A recent review by Luu et al. [102] indicates that the initial endothelial damage could be asymptomatic with a long delay between the end of DOX treatment and the onset of vascular disorder, but with time, the declining health of the endothelium progressively renders ECs more vulnerable to chronic inflammatory stressors. The ability of DOX to affect cardiac microvascular endothelial cell permeability in vivo has been reported in rat studies where cardiac permeability changes correlated with decreased LV function [103]. Wilkinson et al. [104] showed that DOX could increase cardiac microvascular endothelial cell permeability, which could potentially lead to cardiomyopathy. It has been shown that DOX reduces the expression of phosphorylated eNOS (Ser1177), which leads to a decreased bioavailability of NO and endothelial dysfunction [105]. Urschel et al. [106] demonstrated that DOX administration enhanced proinflammatory TNF-α signaling by activating NF-κB, which led to endothelial dysfunction characterized by increased permeability, enhanced oxidative stress, followed by adhesion of leukocytes to the activated endothelium. Clayton et al. [107] showed that DOX-induced aortic stiffness through TNFα-mediated endothelial inflammation, which was associated with adverse structural changes, including collagen deposition (fibrosis), elastin fragmentation, and formation of AGEs (advanced glycation end-products). AGEs, also known as glycotoxins are highly oxidant compounds with pathogenic significance in many degenerative diseases, such as atherosclerosis, Alzheimer’s disease, diabetes, and chronic kidney disease [108,109]. Chow et al. [110] assessed endothelial-dependent vasodilatation in anthracycline-treated patients and impairment of endothelial-dependent arterial vasodilatation, which was sustained for months to years, suggesting its important role in the progression of coronary disease. These findings correlate with several clinical studies showing that chemotherapy with DOX induces damage to the coronary microcirculation, which might contribute to the adverse cardiovascular outcomes in cancer survivors, including those who did not develop symptomatic cardiotoxicity [111].
Gene expression of circulating blood cells
The effect of DOX on gene expression of easily obtainable tissue such as the blood is an opportunity to identify non-invasive biomarkers for the prediction and identification of DOX-induced cardiotoxicity. McCaffrey et al. [112] demonstrated that breast cancer patients with low LVEF after DOX chemotherapy had significantly altered transcripts in comparison with patients with normal LVEF, including genes associated with apoptosis, immunity, detoxification, and drug transport, in comparison with patients who maintained normal LVEF. Two of the decreased genes were suggested as potential biomarkers of DOX-induced cardiotoxicity, T Cell Leukemia/Lymphoma protein 1A (TCL1A), major pro-survival factor for cardiomyocytes, and ABCB1, which codes for the multidrug resistance protein 1 (MDR1), an efflux pump for DOX, potentially leading to higher cardiac levels of the drug. Doroshow et al. [113] showed that patients exposed to continuous DOX infusions have increased DNA-base oxidation within their blood cells compared to pre-treatment.
We have found a high similarity between the gene expression profiles of peripheral blood cells (PBCs) and cardiac tissue in rats with DOX-induced cardiotoxicity [114]. Of the ~4,000 differentially regulated genes in each heart tissue and PBCs of rats, at 48 hours after DOX administration, 2400 genes were similarly differentially regulated. Therefore, in the subsequent clinical study, we have examined the potential of PBMC transcriptome profile after the first dose of DOX chemotherapy to predict DOX-induced cardiomyopathy in breast cancer patients [115]. The results showed that significantly altered transcripts coding for proteins of neutrophils, macrophages, and monocytes were able to predict the risk for DOX-induced cardiotoxicity. The top upregulated DOX-induced transcripts associated with abnormal LVEF decline include neutrophilic anti-microbial proteins such as alpha-defensins (DEFA1-4); cathelicidin (CAMP); MPO; peptidoglycan recognition protein1 (PGLYRP1); matrix metalloproteases (MMP) 8 and 9); carcinoembryonic antigen-related cell adhesion molecule 8 (CEACAM8, known as CD66b), glycosyl-phosphatidylinositol glycoprotein (CD177). These findings are in agreement with the reported role of neutrophils in cardiovascular diseases by Frangogiannis et al. [116]. Some of the neutrophil transcripts (CAMP, MMP9, PGLYRP1, MMP8) in our clinical study were similarly dysregulated in the rat blood cells in our previous study [52]. We have recently found significant elevation of the plasma proteins coded by CAMP, MMP9, PGLYRP1, CEACAM8, ELANE, and MPO, suggesting that these molecules carry the potential for an early prediction of the risk for LVEF decrease. Another significantly upregulated transcript in the group of patients with low LVEF in our study is also OLR1 (oxidized low-density lipoprotein receptor 1), which codes for lectin-type oxidized LDL receptor 1 (LOX-1) protein. LOX-1 is suggested as a marker of atherosclerosis and induces vascular endothelial cell activation, and dysfunction, resulting in pro-inflammatory responses, pro-oxidative conditions, and apoptosis [117]. Recent studies showed that OLR1 was highly expressed in polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC) cells of cancer patients [118] and in low-density granulocytes of patients with lupus who are at high risk of cardiovascular diseases [119], but no data are available on its role in DOX-induced cardiotoxicity. Neutrophil granular proteins are known to be involved in the inflammation and progression of cardiovascular diseases including atherosclerosis, thrombosis, and acute coronary syndrome [120,121]. The elevation of neutrophil granular proteins, including MPO, MMP9, CAMP, PGLYRP1, MMP8, CEACAM8 and DEFA1 has been predictive of atherosclerosis, coronary artery disease, and myocardial infarction (Table 2). We have analyzed the published transcriptional data related to the gene expression of the top upregulated transcripts for cardiovascular diseases through the genetics database for cardiovascular diseases HeartBioPortal (https://www.heartbioportal.com/) [122]. The analysis showed that elevation of several of these genes, including DEFA4, CAMP, PGLYRP1, MMP9, MPO, CEACAM9, ELANE, OLR1 was associated with myocardial infarction (Figure 1). This finding confirms our hypothesis that the early subclinical toxicity of chemotherapy with DOX might in part, be mediated by neutrophil granular proteins through enhancing cardiovascular inflammation, coronary artery disorder, and atherosclerosis, which predispose to myocardial infarction (Figure 2). Therefore, peripheral blood transcriptome and proteome biomarkers, such as MPO, CAMP, PGLYRP1, MMP8, MMP9, CEACAM8, DEFA1-4, OLR-1, ARG-1, ELANE could provide early indications about DOX-induced cardiomyopathy.
Figure 1.
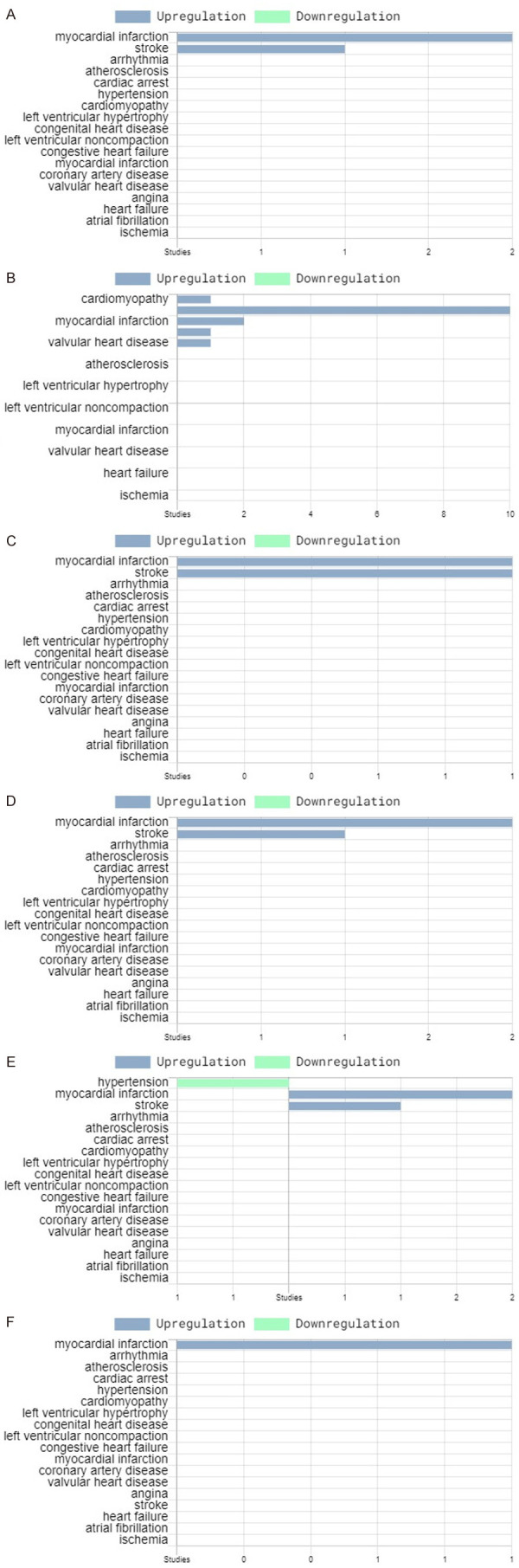
Analysis of six of the top upregulated genes in the blood of breast cancer patients with subclinical DOX-induced cardiotoxicity [115] using HeartBioportal database: (A) MPO; (B) MMP9; (C) CEACAM8; (D) CAMP; (E) PGLYRP1; (F) ELANE. The elevated expression of the six genes after the initial DOX dose in patients with abnormally decline LVEF correlate with upregulation in cardiovascular diseases.
Figure 2.
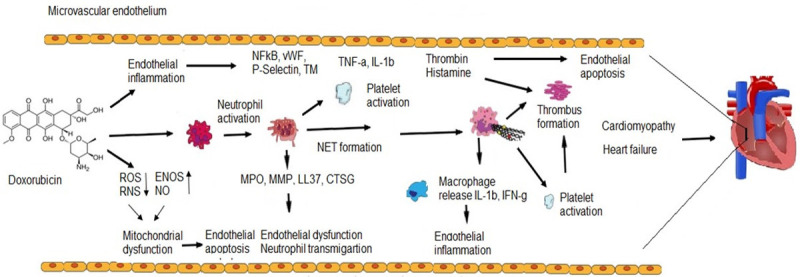
DOX-induced cardiomyocytes damage is preceded by vascular endothelial injury associated with neutrophil degranulation and NET formation. DOX induces ROS generation, inactivation of NO production, endothelial dysfunction and recruitment of neutrophils. Pro-inflammatory cytokines secreted by monocytes, lymphocytes and ECs activate neutrophil degranulation. Neutrophil granule proteins participate in the disruption of EC structure, leading to endothelial inflammation, neutrophil transmigration and apoptosis. NETs provide scaffold for platelet activation and deposition, thus promoting thrombosis formation.
Circulating microRNAs
The involvement of miRNAs in cardiovascular biology and pathology, and their potential as biomarkers has been demonstrated in several studies on cardiovascular diseases [123,124]. In a clinical study, we have examined the potential of circulating miRNAs to predict the risk of DOX-induced cardiotoxicity in breast cancer patients [125]. We have identified 32 differentially regulated microRNAs (DEmiRs) in patients with an abnormal decline of LVEF >10% in comparison with patients who maintained normal LVEF. Several of the DEmiRs in patients with an abnormal decline of LVEF EF have been reported previously in patients with cardiovascular diseases, including downregulation miR-1 and miR-133 in cardiac hypertrophy, upregulation of miR-92 in dilated hypertrophy, upregulation of miR-34a in myocardial infarction, downregulation of miR-15b in myocardial infarction and downregulation of miR-30 in dilated cardiomyopathy (Table 2). Elevated miR-23b suppressed IL-17-, TNF-α- and IL-1β-induced NF-κB activation, which correlated with its downregulation in inflammatory disorders, such as SLE and rheumatoid arthritis [126]. Several of DemiRs have been associated with inflammatory and autoimmune diseases such as elevated miR-16, miR-486, miR 92, miR-532, miR-140 [127-129]. None of these miRNAs have been reported as potential candidate biomarkers for the prediction of DOX-induced cardiotoxicity. The analysis of the targets of DEmiRs in the group of patients with low LVEF showed their association with several of the differentially regulated mRNA in the same patients.
DNA methylation of peripheral blood cells
In our recent study, we have determined the whole-genome DNA methylation of blood cells from breast cancer patients treated with DOX-based chemotherapy [130]. The results showed that 379 differentially methylated CpGs at baseline and 136 CpGs after the first chemotherapy dose significantly correlated with LVEF status. Pathway enrichment analysis using GO, Reactome, KEGG showed that the most significantly positively enriched pathway was “RNA splicing” (included RBM17, DHX9, EIF4A3, CACS3, DHX15, HNRNPH1, HNRNPR, HNRNPU, SF1, SNRPN) and the most significantly negatively enriched pathway was IFN-γ signaling (included IRF6, HLA-DRB1, TRIM14, HLA-A, and HLA-F). Overexpression of DHX15 and EIF4A3 splicing factors have been implicated in the contractile function of cardiomyocytes [131,132] and in cancer [133]. IRF6, a member of the IFN family of transcription factors is one of the significantly hypomethylated genes before the start of chemotherapy that predicted the risk of DOX-induced cardiotoxicity in our study. These findings correlate with previous reports showing that IRF6 has a protective role in the response to endotoxic shock [134] which is one of the suggested mechanisms of DOX-induced inflammation and multiorgan toxicity [135]. Downregulation of IRF6 has been demonstrated in several cancers, including breast cancer [136]. Further analysis with IPA software showed upregulation of estrogen receptor (ER) signaling, ErbB signaling, and Thrombin signaling. ErbB receptor tyrosine kinases, epidermal growth factor receptor (EGFR), and ErbB2 (neu, HER2) are often overexpressed, amplified, or mutated in many forms of cancer, including breast cancer, making them important therapeutic targets and predictors of the therapeutic response to DOX [137]. At the same time, ERB2 overexpression in the heart leads to hypertrophy [138]. It is well known that ER signaling plays an important role in breast cancer progression and the majority of human breast cancers start as estrogen-dependent [139]. The activation of the coagulation cascade in which thrombin plays a key role is closely related to inflammation, development of cardiovascular diseases, and HF prognosis [140]. Accordingly, our previous study demonstrated that elevated markers of inflammation, hypercoagulability, and endothelial function (i.e. thrombomodulin, myeloperoxidase, thrombin-anti-thrombin complex) before and after the first dose of DOX chemotherapy were able to predict the early subclinical DOX-induced cardiotoxicity in patients with breast cancer [97].
Conclusions
DOX is a powerful chemotherapy agent that has improved substantially the cure rate and survivorship of patients with various types of cancer. Unfortunately, the cardiotoxicity of DOX remains an important health concern. Despite the years of substantial research, there is still insufficient understanding of the mechanisms governing cardiac toxicity and no efficient treatment or means for early prediction. DOX-induced cardiotoxicity begins asymptomatically with the initial dose and develops into asymptomatic cardiac dysfunction, and subsequent HF. Our studies have demonstrated that the initial dose of chemotherapy with DOX in cancer patients induced neutrophil activation that could be detrimental to the vascular integrity, as the release of the harmful cargo of their granules could compromise vascular integrity or induce a pro-thrombotic state. We propose that neutrophil degranulation and NETosis during the early stages of vascular inflammation are key mechanisms of DOX-associated cardiovascular toxicity. Neutrophil granular proteins and NETs could potentially act as predictive biomarkers, as well as novel therapeutic targets for DOX-induced cardiotoxicity.
Acknowledgements
This work was supported in part with grants from Susan G Komen for the Cure (grant# BCTR78206), Arkansas Breast Cancer Research Program and the University of Arkansas for Medical Sciences (1UL1RR029884), NIH/NIA Claude Pepper Center (P30AG028718), and funds from the Laura F. Hutchins, M.D. Distinguished Chair for Hematology and Oncology.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Venkatesh P, Kasi A. Anthracyclines. 2021 Mar 10. In: StatPearls [Internet] Treasure Island (FL): StatPearls Publishing; 2021. [Google Scholar]
- 3.Bansal N, Adams MJ, Ganatra S, Colan SD, Aggarwal S, Steiner R, Amdani S, Lipshultz ER, Lipshultz SE. Strategies to prevent anthracycline-induced cardiotoxicity in cancer survivors. Cardiooncology. 2019;5:18. doi: 10.1186/s40959-019-0054-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nebigil CG, Désaubry L. Updates in anthracycline-mediated cardiotoxicity. Front Pharmacol. 2018;9:1262–1271. doi: 10.3389/fphar.2018.01262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kremer LC, van Dalen EC, Offringa M, Voute PA. Frequency and risk factors of anthracycline-induced clinical heart failure in children: a systematic review. Ann Oncol. 2002;13:503–516. doi: 10.1093/annonc/mdf118. [DOI] [PubMed] [Google Scholar]
- 6.Hershman DL, McBride RB, Eisenberger A, Tsai WY, Grann VR, Jacobson JS. Doxorubicin, cardiac risk factors, and cardiac toxicity in elderly patients with diffuse B-cell non-Hodgkin’s lymphoma. J. Clin. Oncol. 2008;26:3159–3165. doi: 10.1200/JCO.2007.14.1242. [DOI] [PubMed] [Google Scholar]
- 7.Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32:302–14. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 8.Tonge CM, Fernandez RC, Harbinson MT. Commentary: current issues in nuclear cardiology. Br J Radiol. 2008;81:270–274. doi: 10.1259/bjr/59260451. [DOI] [PubMed] [Google Scholar]
- 9.Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology. 2010;115:155–162. doi: 10.1159/000265166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. 2007;49:330–352. doi: 10.1016/j.pcad.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Hershman DL, Shao T. Anthracycline cardiotoxicity after breast cancer treatment. Oncology (Williston Park) 2009;23:227–34. [PubMed] [Google Scholar]
- 12.Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD, Colan SD. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2005;23:2629–36. doi: 10.1200/JCO.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 13.Todorova VK, Kaufmann Y, Klimberg VS. Effect of glutamine on doxorubicin-induced cardiotoxicity: an echocardiographic assessment. Cancer Res. 2009;69(Suppl) Abstract nr 6143. [Google Scholar]
- 14.Stone JR, Kanneganti R, Abbasi M, Akhtari M. Monitoring for chemotherapy-related cardiotoxicity in the form of left ventricular systolic dysfunction: a review of current recommendations. JCO Oncol Pract. 2021;17:228–236. doi: 10.1200/OP.20.00924. [DOI] [PubMed] [Google Scholar]
- 15.Gharib MI, Burnett AK. Chemotherapy-induced cardiotoxicity: current practice and prospects of prophylaxis. Eur J Heart Fail. 2002;4:235–242. doi: 10.1016/s1388-9842(01)00201-x. [DOI] [PubMed] [Google Scholar]
- 16.From AM, Maleszewski JJ, Rihal CS. Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86:1095–102. doi: 10.4065/mcp.2011.0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dodos F, Halbsguth T, Erdmann E, Hoppe UC. Usefulness of myocardial performance index and biochemical markers for early detection of anthracycline-induced cardiotoxicity in adults. Clin Res Cardiol. 2008;97:318–26. doi: 10.1007/s00392-007-0633-6. [DOI] [PubMed] [Google Scholar]
- 18.Jones M, O’Gorman P, Kelly C, Mahon N, Fitzgibbon MC. High-sensitive cardiac troponin-I facilitates timely detection of subclinical anthracycline-mediated cardiac injury. Ann Clin Biochem. 2017;54:149–157. doi: 10.1177/0004563216650464. [DOI] [PubMed] [Google Scholar]
- 19.Yu LR, Cao Z, Makhoul I, Daniels JR, Klimberg S, Wei JY, Bai JP, Li J, Lathrop JT, Beger RD, Todorova VK. Immune response proteins as predictive biomarkers of doxorubicin-induced cardiotoxicity in breast cancer patients. Exp Biol Med (Maywood) 2018;243:248–255. doi: 10.1177/1535370217746383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minotti G, Salvatorelli E, Menna P. Pharmacological foundations of Cardio-Oncology. J Pharmacol Exp Ther. 2010;334:2–8. doi: 10.1124/jpet.110.165860. [DOI] [PubMed] [Google Scholar]
- 21.Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97:2869–79. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- 22.Mitry MA, Edwards JG. Doxorubicin induced heart failure: phenotype and molecular mechanisms. Int J Cardiol Heart Vasc. 2016;10:17–24. doi: 10.1016/j.ijcha.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kremer LC, van Dalen EC, Offringa M, Voûte PA. Frequency and risk factors of anthracycline-induced clinical heart failure in children: a systematic review. Ann Oncol. 2002;13:503–512. doi: 10.1093/annonc/mdf118. [DOI] [PubMed] [Google Scholar]
- 24.Lipshultz SE, Lipsitz SR, Mone SM, Goorin AM, Sallan SE, Sanders SP, Orav EJ, Gelber RD, Colan SD. Female sex and drug dose as risk factors for late cardiotoxic effects of doxorubicin therapy for childhood cancer. N Engl J Med. 1995;332:1738–1743. doi: 10.1056/NEJM199506293322602. [DOI] [PubMed] [Google Scholar]
- 25.Lipshultz SE, Sambatakos P, Maguire M. Cardiotoxicity and cardioprotection in childhood cancer. Acta Haematol. 2014;132:391–399. doi: 10.1159/000360238. [DOI] [PubMed] [Google Scholar]
- 26.Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52:1213–1225. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 27.Murphy E, Lagranha C, Deschamps A. Mechanism of cardioprotection: what can we learn from females? Pediatr Cardiol. 2011;32:354–359. doi: 10.1007/s00246-010-9877-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Völler S, Boos J, Krischke M. Age-dependent pharmacokinetics of doxorubicin in children with cancer. Clin Pharmacokinet. 2015;54:1139–1149. doi: 10.1007/s40262-015-0272-4. [DOI] [PubMed] [Google Scholar]
- 29.Koren G, Beatty K, Seto A, Einarson TR, Lishner M. The effects of impaired liver function on the elimination of antineoplastic agents. Ann Pharmacother. 1992;26:363–371. doi: 10.1177/106002809202600311. [DOI] [PubMed] [Google Scholar]
- 30.Armstrong GT, Oeffinger KC, Chen Y, Kawashima T, Yasui Y, Leisenring W, Stovall M, Chow EJ, Sklar CA, Mulrooney DA, Mertens AC, Border W, Durand JB, Robison LL, Meacham LR. Modifiable risk factors and major cardiac events among adult survivors of childhood cancer. J. Clin. Oncol. 2013;31:3673–80. doi: 10.1200/JCO.2013.49.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Von Hoff DD, Layard MW, Basa P, Davis HL Jr, Von Hoff AL, Rozencweig M, Muggia FM. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91:710–717. doi: 10.7326/0003-4819-91-5-710. [DOI] [PubMed] [Google Scholar]
- 32.Leong SL, Chaiyakunapruk N, Lee SW. Candidate gene association studies of anthracycline-induced cardiotoxicity. A systematic review and meta-analysis. Sci Rep. 2017;7:39. doi: 10.1038/s41598-017-00075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Todorova VK, Makhoul I, Dhakal I, Wei J, Stone A, Carter W, Owen A, Klimberg VS. Polymorphic variations associated with doxorubicin-induced cardiotoxicity in breast cancer patients. Oncol Res. 2017;25:1223–1229. doi: 10.3727/096504017X14876245096439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greetham D, Ellis CD, Mewar D, Fearon U, Ultaigh SN, Veale DJ, Guesdon F, Wilson AG. Functional characterization of NF-kappaB inhibitor-like protein 1 (NFkappaBIL1), a candidate susceptibility gene for rheumatoid arthritis. Hum Mol Genet. 2007;16:3027–3031. doi: 10.1093/hmg/ddm261. [DOI] [PubMed] [Google Scholar]
- 35.Koch W, Hoppmann P, Michou E, Jung V, Pfeufer A, Mueller JC, Gieger C, Wichmann HE, Meitinger T, Schömig A, Kastrati A. Association of variants in the BAT1-NFKBIL1-LTA genomic region with protection against myocardial infarction in Europeans. Hum Mol Genet. 2007;16:1821–1827. doi: 10.1093/hmg/ddm130. [DOI] [PubMed] [Google Scholar]
- 36.Okamoto K, Makino S, Yoshikawa Y, Takaki A, Nagatsuka Y, Ota M, Tamiya G, Kimura A, Bahram S, Inoko H. Identification of IkBL as the second major histocompatibility complex-linked susceptibility locus for rheumatoid arthritis. Am J Hum Genet. 2003;72:303–312. doi: 10.1086/346067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neville MJ, Campbell RD. A new member of the Ig superfamily and a V-ATPase G subunit are among the predicted products of novel genes close to the TNF locus in the human MHC. J Immunol. 1999;162:4745–4749. [PubMed] [Google Scholar]
- 38.Davies RW, Wells GA, Stewart AF, Erdmann J, Shah SH, Ferguson JF, Hall AS, Anand SS, Burnett MS, Epstein SE, Dandona S, Chen L, Nahrstaedt J, Loley C, König IR, Kraus WE, Granger CB, Engert JC, Hengstenberg C, Wichmann HE, Schreiber S, Tang WH, Ellis SG, Rader DJ, Hazen SL, Reilly MP, Samani NJ, Schunkert H, Roberts R, McPherson R. A genome-wide association study for coronary artery disease identifies a novel susceptibility locus in the major histocompatibility complex. Circ Cardiovasc Genet. 2012;5:217–25. doi: 10.1161/CIRCGENETICS.111.961243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernando MMA, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abu-Shakra M, Buskila D, Ehrenfeld M, Conrad K, Shoenfeld Y. Cancer and autoimmunity: autoimmune and rheumatic features in patients with malignancies. Ann Rheum Dis. 2001;60:433–436. doi: 10.1136/ard.60.5.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michl I, Zielinski CC. More postchemotherapy rheumatism. J. Clin. Oncol. 1993;11:2051. doi: 10.1200/JCO.1993.11.10.2051. [DOI] [PubMed] [Google Scholar]
- 42.Pan SS, Bachur NR. Xanthine oxidase cata-lyzed reductive cleavage of anthracycline antibio-tics and free radical formation. Mol Pharmacol. 1980;17:95–99. [PubMed] [Google Scholar]
- 43.Hopkins RZ. Superoxide in biology and medicine: an overview. Reactive Oxygen Species. 2016;1:99–109. [Google Scholar]
- 44.Montaigne D, Hurt C, Neviere R. Mitochondria death/survival signaling pathways in cardiotoxicity induced by anthracyclines and anti-cancer targeted therapies. Biochem Res Int. 2012;2012:951539. doi: 10.1155/2012/951539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem. 1986;261:3060–7. [PubMed] [Google Scholar]
- 46.Wallace KB, Sardão VA, Oliveira PJ. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ Res. 2020;126:926–941. doi: 10.1161/CIRCRESAHA.119.314681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doroshow JH, Locker GY, Myers CE. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: alterations produced by doxorubicin. J Clin Invest. 1980;65:128–13. doi: 10.1172/JCI109642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gorini S, De Angelis A, Berrino L, Malara N, Rosano G, Ferraro E. Chemotherapeutic drugs and mitochondrial dysfunction: focus on doxorubicin, trastuzumab, and sunitinib. Oxid Med Cell Longev. 2018;2018:7582730. doi: 10.1155/2018/7582730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strom J, Xu B, Tian X, Chen QM. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016;30:66–80. doi: 10.1096/fj.14-268904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma Q, He X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol Rev. 2012;64:1055–1081. doi: 10.1124/pr.110.004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li S, Wang W, Niu T, Wang H, Li B, Shao L, Lai Y, Li H, Janicki JS, Wang XL, Tang D, Cui T. Nrf2 deficiency exaggerates doxorubicin-induced cardiotoxicity and cardiac dysfunction. Oxid Med Cell Longev. 2014;2014:748524. doi: 10.1155/2014/748524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Todorova VK, Beggs ML, Delongchamp RR, Dhakal I, Makhoul I, Wei JY, Klimberg VS. Transcriptome profiling of peripheral blood cells identifies potential biomarkers for doxorubicin cardiotoxicity in a rat model. PLoS One. 2012;7:e48398. doi: 10.1371/journal.pone.0048398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emelyanova L, Ashary Z, Cosic M, Negmadjanov U, Ross G, Rizvi F, Olet S, Kress D, Sra J, Tajik AJ, Holmuhamedov EL, Shi Y, Jahangir A. Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation. Am J Physiol Heart Circ Physiol. 2016;311:H54–63. doi: 10.1152/ajpheart.00699.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sayed-Ahmed MM, Khattab MM, Gad MZ, Osman AM. Increased plasma nitric oxide during doxorubicin-induced cardiomyopathy. Pharmacol and Toxicol. 2001;89:140–144. doi: 10.1034/j.1600-0773.2001.d01-148.x. [DOI] [PubMed] [Google Scholar]
- 55.Greenlee H, Kwan ML, Kushi LH. Antioxidant supplement use after breast cancer diagnosis and mortality in the LACE cohort. Cancer. 2012;118:2048–2058. doi: 10.1002/cncr.26526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilliam LA, St Clair DK. Chemotherapy-induced weakness and fatigue in skeletal muscle: the role of oxidative stress. Antioxid Redox Signal. 2011;15:2543–2563. doi: 10.1089/ars.2011.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Todorova VK, Kaufmann Y, Hennings L, Klimberg VS. Oral glutamine protects against acute doxorubicin-induced cardiotoxicity of tumor-bearing rats. J Nutr. 2010;140:44–48. doi: 10.3945/jn.109.113415. [DOI] [PubMed] [Google Scholar]
- 58.Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, Antholine WE, Zielonka J, Srinivasan S, Avadhani NG, Kalyanaraman B. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: cardioprotection by Mito-Q. Biophys J. 2009;96:1388–98. doi: 10.1016/j.bpj.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuo IY, Ehrlich BE. Signaling in muscle contraction. Cold Spring Harb Perspect Biol. 2015;7:a006023. doi: 10.1101/cshperspect.a006023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eisner DA, Caldwell JL, Kistamas K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res. 2017;121:181–195. doi: 10.1161/CIRCRESAHA.117.310230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys. 1979;195:460–7. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 62.Montaigne D, Marechal X, Preau S, Baccouch R, Modine T, Fayad G, Lancel S, Neviere R. Doxorubicin induces mitochondrial permeability transition and contractile dysfunction in the human myocardium. Mitochondrion. 2011;11:22–6. doi: 10.1016/j.mito.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 63.Santulli G, Nakashima R, Yuan Q, Marks AR. Intracellular calcium release channels: an update. J Physiol. 2017;595:3041–3051. doi: 10.1113/JP272781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim SY, Kim SJ, Kim BJ, Rah SY, Chung SM, Im MJ, Kim UH. Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp Mol Med. 2006;38:535–45. doi: 10.1038/emm.2006.63. [DOI] [PubMed] [Google Scholar]
- 65.Hanna AD, Lam A, Tham S, Dulhunty AF, Beard NA. Adverse effects of doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Mol Pharmacol. 2014;86:438–49. doi: 10.1124/mol.114.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Todorova VK, Siegel ER, Kaufmann Y, Kumarapeli A, Owen A, Wei J, Makhoul I, Klimberg VS. Dantrolene attenuates cardiotoxicity of doxorubicin without reducing its antitumor efficacy in a breast cancer model. Transl Oncol. 2020;13:471–480. doi: 10.1016/j.tranon.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang JC. DNA topoisomerases. Ann Rev Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 68.Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67:8839–46. doi: 10.1158/0008-5472.CAN-07-1649. [DOI] [PubMed] [Google Scholar]
- 69.Ali Y, Abd Hamid S. Human topoisomerase II alpha as a prognostic biomarker in cancer chemotherapy. Tumour Biol. 2016;37:47–55. doi: 10.1007/s13277-015-4270-9. [DOI] [PubMed] [Google Scholar]
- 70.McGowan JV, Chung R, Maulik A, Piotrowska I, Walker JM, Yellon DM. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc Drugs Ther. 2017;31:63–75. doi: 10.1007/s10557-016-6711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shandilya M, Sharma S, Das PP, Charak S. Molecular-level understanding of the anticancer action mechanism of anthracyclines. IntechOpen. 2020 [Google Scholar]
- 72.Marinello J, Delcuratolo M, Capranico G. Anthracyclines as topoisomerase ii poisons: from early studies to new perspectives. Int J Mol Sci. 2018;19:3480. doi: 10.3390/ijms19113480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–42. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 74.Langer SW. Dexrazoxane for the treatment of chemotherapy-related side effects. Cancer Manag Res. 2014;6:357–363. doi: 10.2147/CMAR.S47238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shan YX, Liu TJ, Su HF, Samsamshariat A, Mestril R, Wang PH. Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J Mol Cell Cardiol. 2003;35:1135–43. doi: 10.1016/s0022-2828(03)00229-3. [DOI] [PubMed] [Google Scholar]
- 76.Fan GC, Zhou X, Wang X, Song G, Qian J, Nicolaou P, Chen G, Ren X, Kranias EG. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ Res. 2008;103:1270–1279. doi: 10.1161/CIRCRESAHA.108.182832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nishizawa J, Nakai A, Higashi T, Tanabe M, Nomoto S, Matsuda K, Ban T, Nagata K. Reperfusion causes significant activation of heat shock transcription factor 1 in ischemic rat heart. Circulation. 1996;94:2185–92. doi: 10.1161/01.cir.94.9.2185. [DOI] [PubMed] [Google Scholar]
- 78.Nishizawa J, Nakai A, Matsuda K, Komeda M, Ban T, Nagata K. Reactive oxygen species play an important role in the activation of heat shock factor 1 in ischemic-reperfused heart. Circulation. 1999;99:934–41. doi: 10.1161/01.cir.99.7.934. [DOI] [PubMed] [Google Scholar]
- 79.Chang J, Knowlton AA, Xu F, Wasser JS. Activation of the heat shock response: relationship to energy metabolites. A (31)P NMR study in rat hearts. Am J Physiol Heart Circ Physiol. 2001;280:H426–33. doi: 10.1152/ajpheart.2001.280.1.H426. [DOI] [PubMed] [Google Scholar]
- 80.Schmitt CA. Senescence, apoptosis and therapy-cutting the lifelines of cancer. Nat Rev Cancer. 2003;3:286–95. doi: 10.1038/nrc1044. [DOI] [PubMed] [Google Scholar]
- 81.Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–283. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Katsuumi G, Shimizu I, Yoshida Y, Minamino T. Vascular senescence in cardiovascular and metabolic diseases. Front Cardiovasc Med. 2018;5:18. doi: 10.3389/fcvm.2018.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, Alston S, Academia EC, Kilmarx S, Valdovinos A, Wang B, de Bruin A, Kennedy BK, Melov S, Zhou D, Sharpless NE, Muss H, Campisi J. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–176. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sanoff HK, Deal AM, Krishnamurthy J, Torrice C, Dillon P, Sorrentino J, Ibrahim JG, Jolly TA, Williams G, Carey LA, Drobish A, Gordon BB, Alston S, Hurria A, Kleinhans K, Rudolph KL, Sharpless NE, Muss HB. Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. J Natl Cancer. 2014;106:dju057. doi: 10.1093/jnci/dju057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, Alston S, Academia EC, Kilmarx S, Valdovinos A, Wang B, de Bruin A, Kennedy BK, Melov S, Zhou D, Sharpless NE, Muss H, Campisi J. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–176. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ruparelia N, Chai JT, Fisher EA, Choudhury RP. Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat Rev Cardiol. 2017;14:133–144. doi: 10.1038/nrcardio.2016.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hartman J, Frishman WH. Inflammation and atherosclerosis: a review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol Rev. 2014;22:147–151. doi: 10.1097/CRD.0000000000000021. [DOI] [PubMed] [Google Scholar]
- 90.Zhang S, You ZQ, Yang L, Li LL, Wu YP, Gu LQ, Xin YF. Protective effect of Shenmai injection on doxorubicin-induced cardiotoxicity via regulation of inflammatory mediators. BMC Complement Altern Med. 2019;19:317. doi: 10.1186/s12906-019-2686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang L, Chen Q, Qi H, Wang C, Wang C, Zhang J, Dong L. Doxorubicin-induced systemic inflammation is driven by upregulation of toll-like receptor tlr4 and endotoxin leakage. Cancer Res. 2016;76:6631–6642. doi: 10.1158/0008-5472.CAN-15-3034. [DOI] [PubMed] [Google Scholar]
- 92.Ridker PM, Lüscher TF. Anti-inflammatory therapies for cardiovascular disease. Eur Heart J. 2014;35:1782–1791. doi: 10.1093/eurheartj/ehu203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dehghani T, Panitch A. Endothelial cells, neutrophils and platelets: getting to the bottom of an inflammatory triangle. Open Biol. 2020;10:200161. doi: 10.1098/rsob.200161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dimasi D, Sun WY, Bonder CS. Neutrophil interactions with the vascular endothelium. Int Immunopharmacol. 2013;17:1167–75. doi: 10.1016/j.intimp.2013.05.034. [DOI] [PubMed] [Google Scholar]
- 95.Dehghani T, Panitch A. Endothelial cells, neutrophils and platelets: getting to the bottom of an inflammatory triangle. Open Biol. 2020;10:200161. doi: 10.1098/rsob.200161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Horsted F, West J, Grainge MJ. Risk of thromboembolismin patients with cancer: a systemic review and meta-analysis. PLoS Med. 2012;9:e1001275. doi: 10.1371/journal.pmed.1001275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Todorova VK, Hsu PC, Wei JY, Lopez-Candales A, Chen JZ, Su LJ, Makhoul I. Biomarkers of inflammation, hypercoagulability and endothelial injury predict early asymptomatic doxorubicin-induced cardiotoxicity in breast cancer patients. Am J Cancer Res. 2020;10:2933–2945. [PMC free article] [PubMed] [Google Scholar]
- 98.Segers VFM, Brutsaert DL, De Keulenaer GW. Cardiac remodeling: endothelial cells have more to say than just No. Front Physiol. 2018;9:382. doi: 10.3389/fphys.2018.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD. Revisiting cardiac cellular composition. Circ Res. 2016;118:400–9. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bar-Joseph H, Ben-Aharon I, Tzabari M, Tsarfaty G, Stemmer SM, Shalgi R. In vivo bioimaging as a novel strategy to detect doxorubicin-induced damage to gonadal blood vessels. PLoS One. 2011;6:e23492. doi: 10.1371/journal.pone.0023492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Khanna A, Pequeno P, Gupta S, Thavendiranathan P, Lee DS, Abdel-Qadir H, Nathan PC. Increased risk of all cardiovascular disease subtypes among childhood cancer survivors: population-based matched cohort study. Circulation. 2019;140:1041–1043. doi: 10.1161/CIRCULATIONAHA.119.041403. [DOI] [PubMed] [Google Scholar]
- 102.Luu AZ, Chowdhury B, Al-Omran M, Teoh H, Hess DA, Verma S. Role of endothelium in doxorubicin-induced cardiomyopathy. JACC Basic Transl Sci. 2018;3:861–870. doi: 10.1016/j.jacbts.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fernandez-Fernandez A, Carvajal DA, Lei T, McGoron AJ. Chemotherapy-induced changes in cardiac capillary permeability measured by fluorescent multiple indicator dilution. Ann Biomed Eng. 2014;42:2405–2415. doi: 10.1007/s10439-014-1110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wilkinson EL, Sidaway JE, Cross MJ. Cardiotoxic drugs Herceptin and doxorubicin inhibit cardiac microvascular endothelial cell barrier formation resulting in increased drug permeability. Biol Open. 2016;5:1362–1370. doi: 10.1242/bio.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.He H, Wang L, Qiao Y, Zhou Q, Li H, Chen S, Yin D, Huang Q, He M. Doxorubicin induces endotheliotoxicity and mitochondrial dysfunction via ROS/eNOS/NO pathway. Front Pharmacol. 2020;10:1531. doi: 10.3389/fphar.2019.01531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Urschel K, Cicha I. TNF-α in the cardiovascular system: from physiology to therapy. Thromb Haemost. 2015;7:9–25. [Google Scholar]
- 107.Clayton ZS, Brunt VE, Hutton DA, Casso AG, Ziemba BP, Melov S, Campisi J, Seals DR. Tumor necrosis factor alpha-mediated inflammation and remodeling of the extracellular matrix underlies aortic stiffening induced by the common chemotherapeutic agent doxorubicin. Hypertension. 2021;77:1581–1590. doi: 10.1161/HYPERTENSIONAHA.120.16759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Del Turco S, Basta G. An update on advanced glycation endproducts and atherosclerosis. Biofactors. 2012;38:266–74. doi: 10.1002/biof.1018. [DOI] [PubMed] [Google Scholar]
- 109.Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63:582–92. doi: 10.1016/j.cardiores.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 110.Chow AY, Chin C, Dahl G, Rosenthal DN. Anthracyclines cause endothelial injury in pediatric cancer patients: a pilot study. J. Clin. Oncol. 2006;24:925–8. doi: 10.1200/JCO.2005.03.5956. [DOI] [PubMed] [Google Scholar]
- 111.Gallucci G, Coccaro M, Storto G, Lapadula L, Tartarone A, Nappi A, Cammarota A, Buonerba C, Di Lorenzo G, Fusco V, Aieta M. The clinical impact of a cardiologic follow-up in breast cancer survivors: an observational study. Int J Immunopathol Pharmacol. 2010;23:1221–7. doi: 10.1177/039463201002300426. [DOI] [PubMed] [Google Scholar]
- 112.McCaffrey TA, Tziros C, Lewis J, Katz R, Siegel R, Weglicki W, Kramer J, Mak IT, Toma I, Chen L, Benas E, Lowitt A, Rao S, Witkin L, Lian Y, Lai Y, Yang Z, Fu SW. Genomic profiling reveals the potential role of TCL1A and MDR1 deficiency in chemotherapy-induced cardiotoxicity. Int J Biol Sci. 2013;9:350–60. doi: 10.7150/ijbs.6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Doroshow JH, Synold TW, Somlo G, Akman SA, Gajewski E. Oxidative DNA base modifications in peripheral blood mononuclear cells of patients treated with high-dose infusional doxorubicin. Blood. 2001;97:2839–45. doi: 10.1182/blood.v97.9.2839. [DOI] [PubMed] [Google Scholar]
- 114.Todorova VK, Beggs ML, Delongchamp RR, Dhakal I, Makhoul I, Wei JY, Klimberg VS. Transcriptome profiling of peripheral blood cells identifies potential biomarkers for doxorubicin cardiotoxicity in a rat model. PLoS One. 2012;7:e48398. doi: 10.1371/journal.pone.0048398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Todorova VK, Makhoul I, Siegel ER, Wei J, Stone A, Carter W, Beggs ML, Owen A, Klimberg VS. Biomarkers for presymptomatic doxorubicin-induced cardiotoxicity in breast cancer patients. PLoS One. 2016;11:e0160224. doi: 10.1371/journal.pone.0160224. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 116.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 117.Mehta JL, Chen J, Hermonat PL, Romeo F, Novelli G. Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1). A critical player in the development of atherosclerosis and related disorders. Cardiovasc Res. 2006;96:36–45. doi: 10.1016/j.cardiores.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 118.Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, Tcyganov E, Hashimoto A, Nefedova Y, Lin C, Partlova S, Garfall A, Vogl DT, Xu X, Knight SC, Malietzis G, Lee GH, Eruslanov E, Albelda SM, Wang X, Mehta JL, Bewtra M, Rustgi A, Hockstein N, Witt R, Masters G, Nam B, Smirnov D, Sepulveda MA, Gabrilovich DI. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. 2016;1:aaf8943. doi: 10.1126/sciimmunol.aaf8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sagar D, Gaddipati R, Ongstad EL, Bhagroo N, An LL, Wang J, Belkhodja M, Rahman S, Manna Z, Davis MA, Hasni S, Siegel R, Sanjuan M, Grimsby J, Kolbeck R, Karathanasis S, Sims GP, Gupta R. LOX-1: a potential driver of cardiovascular risk in SLE patients. PLoS One. 2020;15:e0229184. doi: 10.1371/journal.pone.0229184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bonaventura A, Montecucco F, Dallegri F, Carbone F, Lüscher TF, Camici GG, Liberale L. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc Res. 2019;115:1266–1285. doi: 10.1093/cvr/cvz084. [DOI] [PubMed] [Google Scholar]
- 121.Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. 2020;17:327–340. doi: 10.1038/s41569-019-0326-7. [DOI] [PubMed] [Google Scholar]
- 122.Khomtchouk BB, Vand KA, Koehler WC, Tran DT, Middlebrook K, Sudhakaran S, Nelson CS, Gozani O, Assimes TL. HeartBioPortal: an internet-of-omics for human cardiovascular disease data. Circ Genom Precis Med. 2019;12:e002426. doi: 10.1161/CIRCGEN.118.002426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ruggeri C, Gioffré S, Achilli F, Colombo GI, D’Alessandra Y. Role of microRNAs in doxorubicin-induced cardiotoxicity: an overview of preclinical models and cancer patients. Heart Fail Rev. 2018;23:109–122. doi: 10.1007/s10741-017-9653-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Colpaert RMW, Calore M. MicroRNAs in cardiac diseases. Cells. 2019;8:737. doi: 10.3390/cells8070737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Todorova VK, Makhoul I, Wei J, Klimberg VS. Circulating miRNA profiles of doxorubicin-induced cardiotoxicity in breast cancer patients. Ann Clin Lab Sci. 2017;47:115–119. [PubMed] [Google Scholar]
- 126.Zhu S, Pan W, Song X, Liu Y, Shao X, Tang Y, Liang D, He D, Wang H, Liu W, Shi Y, Harley JB, Shen N, Qian Y. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat Med. 2012;18:1077–86. doi: 10.1038/nm.2815. [DOI] [PubMed] [Google Scholar]
- 127.Wu F, Guo NJ, Tian H, Marohn M, Gearhart S, Bayless TM, Brant SR, Kwon JH. Peripheral blood microRNAs distinguish active ulcerative colitis and Crohn’s disease. Inflamm Bowel Dis. 2011;17:241–50. doi: 10.1002/ibd.21450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Murata K, Yoshitomi H, Tanida S, Ishikawa M, Nishitani K, Ito H, Nakamura T. Plasma and synovial fluid microRNAs as potential biomarkers of rheumatoid arthritis and osteoarthritis. Arthritis Rheum. 2010;12:R86. doi: 10.1186/ar3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang H, Peng W, Ouyang X, Li W, Dai Y. Circulating microRNAs as candidate biomarkers in patients with systemic lupus erythematosus. Transl Res. 2012;160:198–206. doi: 10.1016/j.trsl.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 130.Bauer MA, Makhoul I, Hsu PC, Wei J, Su LJ, Stone A, Carter W, Todorova VK. Genome-wide DNA methylation signatures predict the early asymptomatic doxorubicin-induced cardiotoxicity in breast cancer. Proceedings of the Annual Meeting of the American Association for Cancer Research 2020; Apr 27-28 and Jun 22-24. Philadelphia (PA): AACR. Cancer Res. 2020;80(Suppl) doi: 10.3390/cancers13246291. Abstract nr LB-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pierrat OA, Paudyal A, Woodruff J, Koroleva O, Boateng SY. The exon junction complex senses energetic stress and regulates contractility and cell architecture in cardiac myocytes. Biosci Rep. 2017;37:BSR20170707. doi: 10.1042/BSR20170707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhou Z, Zheng L, Tang C, Chen Z, Zhu R, Peng X, Wu X, Zhu P. Identification of potentially relevant genes for excessive exercise-induced pathological cardiac hypertrophy in Zebrafish. Front Physiol. 2020;11:565307. doi: 10.3389/fphys.2020.565307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liao Y, Castello A, Fischer B, Leicht S, Föehr S, Frese CK, Ragan C, Kurscheid S, Pagler E, Yang H, Krijgsveld J, Hentze MW, Preiss T. The cardiomyocyte RNA-binding proteome: links to intermediary metabolism and heart disease. Cell Rep. 2016;16:1456–1469. doi: 10.1016/j.celrep.2016.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Joly S, Rhea L, Volk P, Moreland JG, Dunnwald M. Interferon regulatory factor 6 has a protective role in the host response to endotoxic shock. In: Rottenberg ME, editor. PLoS One: Public Library of Science. 2016;11:e0152385. doi: 10.1371/journal.pone.0152385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang L, Chen Q, Qi H, Wang C, Wang C, Zhang J, Dong L. Doxorubicin-induced systemic inflammation is driven by upregulation of toll-like receptor TLR4 and endotoxin leakage. Cancer Res. 2016;76:6631–6642. doi: 10.1158/0008-5472.CAN-15-3034. [DOI] [PubMed] [Google Scholar]
- 136.Xu HF, Huang TJ, Yang Q, Xu L, Lin F, Lang YH, Hu H, Peng LX, Meng DF, Xie YJ, Tan L, Qian CN, Huang BJ. Candidate tumor suppressor gene IRF6 is involved in human breast cancer pathogenesis via modulating PI3K-regulatory subunit PIK3R2 expression. Cancer Manag Res. 2019;11:5557–5572. doi: 10.2147/CMAR.S203060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Current opinion in cell biology. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 138.Gabrielson K, Bedja D, Pin S, Tsao A, Gama L, Yuan B, Muratore N. Heat shock protein 90 and ErbB2 in the cardiac response to doxorubicin injury. Cancer Res. 2007;67:1436–1441. doi: 10.1158/0008-5472.CAN-06-3721. [DOI] [PubMed] [Google Scholar]
- 139.Saha Roy S, Vadlamudi RK. Role of estrogen receptor signaling in breast cancer metastasis. Int J Breast Cancer. 2012;2012:654698. doi: 10.1155/2012/654698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Caine GJ, Stonelake PS, Lip GY, Kehoe ST. The hypercoagulable state of malignancy: pathogenesis and current debate. Neoplasia. 2002;4:465–73. doi: 10.1038/sj.neo.7900263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Visscher H, Rassekh SR, Sandor GS, Caron HN, van Dalen EC, Kremer LC, van der Pal HJ, Rogers PC, Rieder MJ, Carleton BC, Hayden MR, Ross CJ CPNDS consortium. Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline-induced cardiotoxicity in children. Pharmacogenomics. 2015;16:1065–76. doi: 10.2217/pgs.15.61. [DOI] [PubMed] [Google Scholar]
- 142.Krajinovic M, Elbared J, Drouin S, Bertout L, Rezgui A, Ansari M, Raboisson MJ, Lipshultz SE, Silverman LB, Sallan SE, Neuberg DS, Kutok JL, Laverdière C, Sinnett D, Andelfinger G. Polymorphisms of ABCC5 and NOS3 genes influence doxorubicin cardiotoxicity in survivors of childhood acute lymphoblastic leukemia. Pharmacogenomics J. 2016;16:530–535. doi: 10.1038/tpj.2015.63. [DOI] [PubMed] [Google Scholar]
- 143.Wang J, Wang Y, Zou Y, Sun K, Wang Z, Ding H, Yuan J, Wei W, Hou Q, Wang H, Liu X, Zhang H, Ji Y, Zhou X, Sharma RK, Wang D, Ahmad F, Hui R, Song L. Malignant effects of multiple rare variants in sarcomere genes on the prognosis of patients with hypertrophic cardiomyopathy. Eur J Heart Fail. 2014;16:950–7. doi: 10.1002/ejhf.144. [DOI] [PubMed] [Google Scholar]
- 144.Armenian SH, Ding Y, Mills G, Sun C, Venkataraman K, Wong FL, Neuhausen SL, Senitzer D, Wang S, Forman SJ, Bhatia S. Genetic susceptibility to anthracycline-related congestive heart failure in survivors of haematopoietic cell transplantation. Br J Haematol. 2013;163:205–13. doi: 10.1111/bjh.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Aminkeng F, Bhavsar AP, Visscher H, Rassekh SR, Li Y, Lee JW, Brunham LR, Caron HN, van Dalen EC, Kremer LC, van der Pal HJ, Amstutz U, Rieder MJ, Bernstein D, Carleton BC, Hayden MR, Ross CJ Canadian Pharmacogenomics Network for Drug Safety Consortium. A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat Genet. 2015;47:1079–84. doi: 10.1038/ng.3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chu H, Yang J, Mi S, Bhuyan SS, Li J, Zhong L, Liu S, Tao Z, Li J, Chen H. Tumor necrosis factor-alpha G-308 A polymorphism and risk of coronary heart disease and myocardial infarction: a case-control study and meta-analysis. J Cardiovasc Dis Res. 2012;3:84–92. doi: 10.4103/0975-3583.95359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tang W, Schwienbacher C, Lopez LM, Ben-Shlomo Y, Oudot-Mellakh T, Johnson AD, Samani NJ, Basu S, Gögele M, Davies G, Lowe GD, Tregouet DA, Tan A, Pankow JS, Tenesa A, Levy D, Volpato CB, Rumley A, Gow AJ, Minelli C, Yarnell JW, Porteous DJ, Starr JM, Gallacher J, Boerwinkle E, Visscher PM, Pramstaller PP, Cushman M, Emilsson V, Plump AS, Matijevic N, Morange PE, Deary IJ, Hicks AA, Folsom AR. Genetic associations for activated partial thromboplastin time and prothrombin time, their gene expression profiles, and risk of coronary artery disease. Am J Hum Genet. 2012;91:152–162. doi: 10.1016/j.ajhg.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Koch W, Hoppmann P, Michou E, Jung V, Pfeufer A, Mueller JC, Gieger C, Wichmann HE, Meitinger T, Schömig A, Kastrati A. Association of variants in the BAT1-NFKBIL1-LTA genomic region with protection against myocardial infarction in Europeans. Hum Mol Genet. 2007;16:1821–1827. doi: 10.1093/hmg/ddm130. [DOI] [PubMed] [Google Scholar]
- 149.Reiner AP, Hartiala J, Zeller T, Bis JC, Dupuis J, Fornage M, Baumert J, Kleber ME, Wild PS, Baldus S, Bielinski SJ, Fontes JD, Illig T, Keating BJ, Lange LA, Ojeda F, Müller-Nurasyid M, Munzel TF, Psaty BM, Rice K, Rotter JI, Schnabel RB, Tang WH, Thorand B, Erdmann J CARDIoGRAM Consortium. Jacobs DR Jr, Wilson JG, Koenig W, Tracy RP, Blankenberg S, März W, Gross MD, Benjamin EJ, Hazen SL, Allayee H. Genome-wide and gene-centric analyses of circulating myeloperoxidase levels in the charge and care consortia. Hum Mol Genet. 2013;22:3381–3386. doi: 10.1093/hmg/ddt189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Liu Y, Helms C, Liao W, Zaba LC, Duan S, Gardner J, Wise C, Miner A, Malloy MJ, Pullinger CR, Kane JP, Saccone S, Worthington J, Bruce I, Kwok PY, Menter A, Krueger J, Barton A, Saccone NL, Bowcock AM. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLos Genet. 2008;4:e1000041. doi: 10.1371/journal.pgen.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Castillo L, Rohatgi A, Ayers CR, Owens AW, Das SR, Khera A, McGuire DK, de Lemos JA. Associations of four circulating chemokines with multiple atherosclerosis phenotypes in a large population-based sample: results from the dallas heart study. J Interferon Cytokine Res. 2010;30:339–47. doi: 10.1089/jir.2009.0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bonaventura A, Montecucco F. CCL23 is a promising biomarker of injury in patients with ischaemic stroke. J Intern Med. 2018;283:476–478. doi: 10.1111/joim.12742. [DOI] [PubMed] [Google Scholar]
- 153.Garzorz-Stark N, Krause L, Lauffer F, Atenhan A, Thomas J, Stark SP, Franz R, Weidinger S, Balato A, Mueller NS, Theis FJ, Ring J, Schmidt-Weber CB, Biedermann T, Eyerich S, Eyerich K. A novel molecular disease classifier for psoriasis and eczema. Exp Dermatol. 2016;25:767–74. doi: 10.1111/exd.13077. [DOI] [PubMed] [Google Scholar]
- 154.Zahran A, Attia A, Mansell H, Shoker A. Contribution of diminished kidney transplant GFR to increased circulating chemokine ligand 27 level. J Inflamm. 2018;15:18. doi: 10.1186/s12950-018-0194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rassaf T, Weber C, Bernhagen J. Macrophage migration inhibitory factor in myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2014;102:321–8. doi: 10.1093/cvr/cvu071. [DOI] [PubMed] [Google Scholar]
- 156.Burger-Kentischer A, Goebel H, Seiler R, Fraedrich G, Schaefer HE, Dimmeler S, Kleemann R, Bernhagen J, Ihling C. Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation. 2002;105:1561–6. doi: 10.1161/01.cir.0000012942.49244.82. [DOI] [PubMed] [Google Scholar]
- 157.Zernecke A, Bernhagen J, Weber C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation. 2008;117:1594–602. doi: 10.1161/CIRCULATIONAHA.107.729125. [DOI] [PubMed] [Google Scholar]
- 158.Park HJ, Noh JH, Eun JW, Koh YS, Seo SM, Park WS, Lee JY, Chang K, Seung KB, Kim PJ, Nam SW. Assessment and diagnostic relevance of novel serum biomarkers for early decision of ST-elevation myocardial infarction. Oncotarget. 2015;6:12970–12983. doi: 10.18632/oncotarget.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Laman JD, Schoneveld AH, Moll FL, van Meurs M, Pasterkamp G. Significance of peptidoglycan, a proinflammatory bacterial antigen in atherosclerotic arteries and its association with vulnerable plaques. Am J Cardiol. 2002;90:119–23. doi: 10.1016/s0002-9149(02)02432-3. [DOI] [PubMed] [Google Scholar]
- 160.Rohatgi A, Ayers CR, Khera A, McGuire DK, Das SR, Matulevicius S, Timaran CH, Rosero EB, de Lemos JA. The association between peptidoglycan recognition protein-1 and coronary and peripheral atherosclerosis: observations from the dallas heart study. Atherosclerosis. 2009;203:569–75. doi: 10.1016/j.atherosclerosis.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 161.Brownell NK, Khera A, de Lemos JA, Ayers CR, Rohatgi A. Association between peptidoglycan recognition protein-1 and incident atherosclerotic cardiovascular disease events: the dallas heart study. J Am Coll Cardiol. 2016;67:2310–2312. doi: 10.1016/j.jacc.2016.02.063. [DOI] [PubMed] [Google Scholar]
- 162.Klimczak-Tomaniak D, Bouwens E, Schuurman AS, Akkerhuis KM, Constantinescu A, Brugts J, Westenbrink BD, van Ramshorst J, Germans T, Pączek L, Umans V, Boersma E, Kardys I. Temporal patterns of macrophage- and neutrophil-related markers are associated with clinical outcome in heart failure patients. ESC Heart Fail. 2020;7:1190–1200. doi: 10.1002/ehf2.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Han Y, Hua S, Chen Y, Yang W, Zhao W, Huang F, Qiu Z, Yang C, Jiang J, Su X, Yang K, Jin W. Circulating PGLYRP1 levels as a potential biomarker for coronary artery disease and heart failure. J Cardiovasc Pharmacol. 2021;77:578–585. doi: 10.1097/FJC.0000000000000996. [DOI] [PubMed] [Google Scholar]
- 164.Zhou Q, Pan LL, Xue R, Ni G, Duan Y, Bai Y, Shi C, Ren Z, Wu C, Li G, Agerberth B, Sluijter JP, Sun J, Xiao J. The anti-microbial peptide LL-37/CRAMP levels are associated with acute heart failure and can attenuate cardiac dysfunction in multiple preclinical models of heart failure. Theranostics. 2020;10:6167–6181. doi: 10.7150/thno.46225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Döring Y, Drechsler M, Wantha S, Kemmerich K, Lievens D, Vijayan S, Gallo RL, Weber C, Soehnlein O. Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circ Res. 2012;110:1052–6. doi: 10.1161/CIRCRESAHA.112.265868. [DOI] [PubMed] [Google Scholar]
- 166.Edfeldt K, Agerberth B, Rottenberg ME, Gudmundsson GH, Wang XB, Mandal K, Xu Q, Yan ZQ. Involvement of the antimicrobial peptide LL-37 in human atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:1551–7. doi: 10.1161/01.ATV.0000223901.08459.57. [DOI] [PubMed] [Google Scholar]
- 167.Salamah MF, Ravishankar D, Kodji X, Moraes LA, Williams HF, Vallance TM, Albadawi DA, Vaiyapuri R, Watson K, Gibbins JM, Brain SD, Perretti M, Vaiyapuri S. The endogenous antimicrobial cathelicidin LL37 induces platelet activation and augments thrombus formation. Blood Adv. 2018;2:2973–85. doi: 10.1182/bloodadvances.2018021758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Edfeldt K, Agerberth B, Rottenberg ME, Gudmundsson GH, Wang XB, Mandal K, Xu Q, Yan ZQ. Involvement of the antimicrobial peptide LL-37 in human atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:1551–7. doi: 10.1161/01.ATV.0000223901.08459.57. [DOI] [PubMed] [Google Scholar]
- 169.Kook H, Jang DH, Kim JH, Cho JY, Joo HJ, Cho SA, Park JH, Hong SJ, Yu CW, Lim DS. Identification of plaque ruptures using a novel discriminative model comprising biomarkers in patients with acute coronary syndrome. Sci Rep. 2020;10:20228. doi: 10.1038/s41598-020-77413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Loftus IM, Naylor AR, Goodall S, Crowther M, Jones L, Bell PR, Thompson MM. Increased matrix metalloproteinase-9 activity in unstable carotid plaques. A potential role in acute plaque disruption. Stroke. 2000;31:40–47. doi: 10.1161/01.str.31.1.40. [DOI] [PubMed] [Google Scholar]
- 171.Hamed GM, Fattah MF. Clinical relevance of matrix metalloproteinase 9 in patients with acute coronary syndrome. Clin Appl Thromb Hemost. 2015;21:705–11. doi: 10.1177/1076029614567309. [DOI] [PubMed] [Google Scholar]
- 172.Hemdahl AL, Gabrielsen A, Zhu C, Eriksson P, Hedin U, Kastrup J, Thorén P, Hansson GK. Expression of neutrophil gelatinase-associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler Thromb Vasc Biol. 2006;26:136–42. doi: 10.1161/01.ATV.0000193567.88685.f4. [DOI] [PubMed] [Google Scholar]
- 173.Kai H, Ikeda H, Yasukawa H, Kai M, Seki Y, Kuwahara F, Ueno T, Sugi K, Imaizumi T. Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J Am Coll Cardiol. 1998;32:368–372. doi: 10.1016/s0735-1097(98)00250-2. [DOI] [PubMed] [Google Scholar]
- 174.Hamed GM, Fattah MF. Clinical relevance of matrix metalloproteinase 9 in patients with acute coronary syndrome. Clin Appl Thromb Hemost. 2015;21:705–711. doi: 10.1177/1076029614567309. [DOI] [PubMed] [Google Scholar]
- 175.Nurkic J, Ljuca F, Nurkic M, Jahic E, Jahic M. Biomarkers of plaque instability in acute coronary syndrome patients. Med Arh. 2010;64:103–106. [PubMed] [Google Scholar]
- 176.Pinelli A, Trivulzio S, Rossoni G, Redaelli R, Brenna S. Factors involved in sudden coagulation observed in patients with acute myocardial infarction. In Vivo. 2012;26:1021–5. [PubMed] [Google Scholar]
- 177.Alfakry H, Sinisalo J, Paju S, Nieminen MS, Valtonen V, Tervahartiala T, Pussinen PJ, Sorsa T. The association of serum neutrophil markers and acute coronary syndrome. Scand J Immunol. 2012;76:181–7. doi: 10.1111/j.1365-3083.2012.02718.x. [DOI] [PubMed] [Google Scholar]
- 178.Fertin M, Lemesle G, Turkieh A, Beseme O, Chwastyniak M, Amouyel P, Bauters C, Pinet F. Serum MMP-8: a novel indicator of left ventricular remodeling and cardiac outcome in patients after acute myocardial infarction. PLoS One. 2013;8:e71280. doi: 10.1371/journal.pone.0071280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tuomainen AM, Nyyssönen K, Laukkanen JA, Tervahartiala T, Tuomainen TP, Salonen JT, Sorsa T, Pussinen PJ. Serum matrix metalloproteinase-8 concentrations are associated with cardiovascular outcome in men. Arterioscler Thromb Vasc Biol. 2007;27:2722–8. doi: 10.1161/ATVBAHA.107.154831. [DOI] [PubMed] [Google Scholar]
- 180.Alfakry H, Sinisalo J, Paju S, Nieminen MS, Valtonen V, Tervahartiala T, Pussinen PJ, Sorsa T. The association of serum neutrophil markers and acute coronary syndrome. Scand J Immunol. 2012;76:181–92. doi: 10.1111/j.1365-3083.2012.02718.x. [DOI] [PubMed] [Google Scholar]
- 181.Pussinen PJ, Sarna S, Puolakkainen M, Öhlin H, Sorsa T, Pesonen E. The balance of serum matrix metalloproteinase-8 and its tissue inhibitor in acute coronary syndrome and its recurrence. Int J Cardiol. 2013;167:362–8. doi: 10.1016/j.ijcard.2011.12.095. [DOI] [PubMed] [Google Scholar]
- 182.Loria V, Dato I, Graziani F, Biasucci LM. Myeloperoxidase: a new biomarker of inflammation in ischemic heart disease and acute coronary syndromes. Mediators Inflamm. 2008;2008:135625. doi: 10.1155/2008/135625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, Sprecher DL, Hazen SL. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286:2136–214. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 184.Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, Munzel T, Simoons ML, Hamm CW. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 185.Maréchal P, Tridetti J, Nguyen ML, Wéra O, Jiang Z, Gustin M, Donneau AF, Oury C, Lancellotti P. Neutrophil phenotypes in coronary artery disease. J Clin Med. 2020;9:1602. doi: 10.3390/jcm9051602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ky B, Putt M, Sawaya H, French B, Januzzi JL Jr, Sebag IA, Plana JC, Cohen V, Banchs J, Carver JR, Wiegers SE, Martin RP, Picard MH, Gerszten RE, Halpern EF, Passeri J, Kuter I, Scherrer-Crosbie M. Early increases in multiple biomarkers predict subsequent cardiotoxicity in patients with breast cancer treated with doxorubicin, taxanes, and trastuzumab. J Am Coll Cardiol. 2014;63:809–16. doi: 10.1016/j.jacc.2013.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Quinn KL, Henriques M, Tabuchi A, Han B, Yang H, Cheng WE, Tole S, Yu H, Luo A, Charbonney E, Tullis E, Lazarus A, Robinson LA, Ni H, Peterson BR, Kuebler WM, Slutsky AS, Zhang H. Human neutrophil peptides mediate endothelial-monocyte interaction, foam cell formation, and platelet activation. Arterioscler Thromb Vasc Biol. 2011;31:2070–9. doi: 10.1161/ATVBAHA.111.227116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Berliner S, Rogowski O, Rotstein R, Fusman R, Shapira I, Bornstein NM, Prochorov V, Roth A, Keren G, Eldor A, Zeltser D. Activated polymorphonuclear leukocytes and monocytes in the peripheral blood of patients with ischemic heart and brain conditions correspond to the presence of multiple risk factors for atherothrombosis. Cardiology. 2000;94:19–25. doi: 10.1159/000007041. [DOI] [PubMed] [Google Scholar]
- 189.Grabie N, Hsieh DT, Buono C, Westrich JR, Allen JA, Pang H, Stavrakis G, Lichtman AH. Neutrophils sustain pathogenic CD8+ T cell responses in the heart. Am J Pathol. 2003;163:2413–2420. doi: 10.1016/S0002-9440(10)63596-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Martin FA, Murphy RP, Cummins PM. Thrombomodulin and the vascular endothelium: insights into functional, regulatory, and therapeutic aspects. Am J Physiol Heart Circ Physiol. 2013;304:H1585–97. doi: 10.1152/ajpheart.00096.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Strijbos MH, Rao C, Schmitz PI, Kraan J, Lamers CH, Sleijfer S, Terstappen LW, Gratama JW. Correlation between circulating endothelial cell counts and plasma thrombomodulin levels as markers for endothelial damage. Thromb Haemost. 2008;100:642–647. [PubMed] [Google Scholar]
- 192.Lin SM, Wang YM, Lin HC, Lee KY, Huang CD, Liu CY, Wang CH, Kuo HP. Serum thrombomodulin level relates to the clinical course of disseminated intravascular coagulation, multiorgan dysfunction syndrome, and mortality in patients with sepsis. Crit Care Med. 2008;36:683–9. doi: 10.1097/CCM.0B013E31816537D8. [DOI] [PubMed] [Google Scholar]
- 193.Wu KK. Soluble thrombomodulin and coronary heart disease. Curr Opin Lipidol. 2003;14:373–5. doi: 10.1097/00041433-200308000-00006. [DOI] [PubMed] [Google Scholar]
- 194.Salomaa V, Matei C, Aleksic N, Sansores-Garcia L, Folsom AR, Juneja H, Chambless LE, Wu KK. Soluble thrombomodulin as a predictor of incident coronary heart disease and symptomless carotid artery atherosclerosis in the atherosclerosis risk in communities (ARIC) study: a case-cohort study. Lancet. 1999;353:1729–34. doi: 10.1016/s0140-6736(98)09057-6. [DOI] [PubMed] [Google Scholar]
- 195.Tian K, Ogura S, Little PJ, Xu SW, Sawamura T. Targeting LOX-1 in atherosclerosis and vasculopathy: current knowledge and future perspectives. Ann N Y Acad Sci. 2019;1443:34–53. doi: 10.1111/nyas.13984. [DOI] [PubMed] [Google Scholar]
- 196.Hofmann A, Brunssen C, Wolk S, Reeps C, Morawietz H. Soluble LOX-1: a novel biomarker in patients with coronary artery disease, stroke, and acute aortic dissection? J Am Heart Assoc. 2020;9:e013803. doi: 10.1161/JAHA.119.013803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ehara S, Ueda M, Naruko T, Haze K, Itoh A, Otsuka M, Komatsu R, Matsuo T, Itabe H, Takano T, Tsukamoto Y, Yoshiyama M, Takeuchi K, Yoshikawa J, Becker AE. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation. 2001;103:1955–1960. doi: 10.1161/01.cir.103.15.1955. [DOI] [PubMed] [Google Scholar]
- 198.Nagase M, Hirose S, Sawamura T, Masaki T, Fujita T. Enhanced expression of endothelial oxidized low-density lipoprotein receptor (LOX-1) in hypertensive rats. Biochem Biophys Res Commun. 1997;237:496–498. doi: 10.1006/bbrc.1997.7176. [DOI] [PubMed] [Google Scholar]
- 199.Li D, Williams V, Liu L, Chen H, Sawamura T, Romeo F, Mehta JL. Expression of lectin-like oxidized low-density lipoprotein receptors during ischemia-reperfusion and its role in determination of apoptosis and left ventricular dysfunction. J Am Coll Cardiol. 2003;41:1048–55. doi: 10.1016/s0735-1097(02)02966-2. [DOI] [PubMed] [Google Scholar]
- 200.Kataoka H, Miyamoto S. Expression of lectin-like oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation. 1999;99:3110–3117. doi: 10.1161/01.cir.99.24.3110. [DOI] [PubMed] [Google Scholar]
- 201.Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, Gallant M, Martinod K, Ten Cate H, Hofstra L, Crijns HJ, Wagner DD, Kietselaer BLJH. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol. 2013;33:2032–2040. doi: 10.1161/ATVBAHA.113.301627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Döring Y, Libby P, Soehnlein O. Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights. Circ Res. 2020;126:1228–1241. doi: 10.1161/CIRCRESAHA.120.315931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shimony A, Zahger D, Gilutz H, Goldstein H, Orlov G, Merkin M, Shalev A, Ilia R, Douvdevani A. Cell free DNA detected by a novel method in acute ST-elevation myocardial infarction patients. Acute Card Care. 2010;12:109–111. doi: 10.3109/17482941.2010.513732. [DOI] [PubMed] [Google Scholar]
- 204.Mangold A, Alias S, Scherz T, Hofbauer M, Jakowitsch J, Panzenböck A, Simon D, Laimer D, Bangert C, Kammerlander A, Mascherbauer J, Winter MP, Distelmaier K, Adlbrecht C, Preissner KT, Lang IM. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res. 2015;116:1182–1192. doi: 10.1161/CIRCRESAHA.116.304944. [DOI] [PubMed] [Google Scholar]
- 205.Farkas ÁZ, Farkas VJ, Gubucz I, Szabó L, Bálint K, Tenekedjiev K, Nagy AI, Sótonyi P, Hidi L, Nagy Z, Szikora I, Merkely B, Kolev K. Neutrophil extracellular traps in thrombi retrieved during interventional treatment of ischemic arterial diseases. Thromb Res. 2019;175:46–52. doi: 10.1016/j.thromres.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 206.Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D’Angelo A, Bianchi ME, Rovere-Querini P, Manfredi AA. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12:2074–91. doi: 10.1111/jth.12710. [DOI] [PubMed] [Google Scholar]
- 207.Qi H, Yang S, Zhang L. Neutrophil extracellular traps and endothelial dysfunction in atherosclerosis and thrombosis. Front Immunol. 2017;8:928–42. doi: 10.3389/fimmu.2017.00928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–5. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Megens RT, Vijayan S, Lievens D, Döring Y, van Zandvoort MA, Grommes J, Weber C, Soehnlein O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb Haemost. 2012;107:597–598. doi: 10.1160/TH11-09-0650. [DOI] [PubMed] [Google Scholar]
- 210.Wang Y, Wang W, Wang N, Tall AR, Tabas I. Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arterioscler Thromb Vasc Biol. 2017;37:e99–e107. doi: 10.1161/ATVBAHA.117.309580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Harenberg J. Thrombin-antithrombin (TAT) complex. Lab tech in Thrombosis- a manual. Dordrecht: Springer; 1999. [Google Scholar]
- 212.Wada H, Sakuragawa N, Mori Y, Takagi M, Nakasaki T, Shimura M, Hiyoyama K, Nisikawa M, Gabazza EC, Deguchi K, Kazama M, Shiku H. Hemostatic molecular markers before the onset of disseminated intravascular coagulation. Am J Hematol. 1999;60:273–8. doi: 10.1002/(sici)1096-8652(199904)60:4<273::aid-ajh4>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 213.Asakura H, Wada H, Okamoto K, Iba T, Uchiyama T, Eguchi Y, Kawasugi K, Koga S, Mayumi T, Koike K, Gando S. Evaluation of haemostatic molecular markers for diagnosis of disseminated intravascular coagulation in patients with infections. Thrombosis and haemostasis. 2006;95:282–9. doi: 10.1160/TH05-04-0286. [DOI] [PubMed] [Google Scholar]
- 214.Valente-Acosta B, Baños-González MA, Peña-Duque MA, Martínez-Ríos MA, Quintanar-Trejo L, Aptilon-Duque G, Flores-García M, Cruz-Robles D, Cardoso-Saldaña G, de la Peña-Díaz A. Association between stable coronary artery disease and in vivo thrombin generation. Cardiol Res Pract. 2016;2016:5149825. doi: 10.1155/2016/5149825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Negreva M, Georgiev S, Prodanova K, Nikolova J. Early changes in the antithrombin and thrombin-antithrombin complex in patients with paroxysmal atrial fibrillation. Cardiol Res. 2016;7:89–94. doi: 10.14740/cr469w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bassuk SS, Rifai N, Ridker PM. High-sensitivity C-reactive protein: clinical importance. Curr Probl Cardiol. 2004;29:439–93. [PubMed] [Google Scholar]
- 217.Hein TW, Singh U, Vasquez-Vivar J, Devaraj S, Kuo L, Jialal I. Human C-reactive protein induces endothelial dysfunction and uncoupling of eNOS in vivo. Atherosclerosis. 2009;206:61–8. doi: 10.1016/j.atherosclerosis.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Teoh H, Quan A, Lovren F, Wang G, Tirgari S, Szmitko PE, Szalai AJ, Ward ME, Verma S. Impaired endothelial function in C-reactive protein overexpressing mice. Atherosclerosis. 2008;201:318–25. doi: 10.1016/j.atherosclerosis.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 219.Voudris KV, Chanin J, Feldman DN, Charitakis K. Novel inflammatory biomarkers in coronary artery disease: potential therapeutic approaches. Curr Med Chem. 2015;22:2680–9. doi: 10.2174/0929867322666150420124427. [DOI] [PubMed] [Google Scholar]
- 220.Guo C, Zhang S, Zhang J, Liu H, Li P, Liu H, Wang Y. Correlation between the severity of coronary artery lesions and levels of estrogen, hs-CRP and MMP-9. Exp Ther Med. 2014;7:1177–1180. doi: 10.3892/etm.2014.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Devaraj S, Singh U, Jialal I. The evolving role of C-reactive protein in atherothrombosis. Clin Chem. 2009;55:229–38. doi: 10.1373/clinchem.2008.108886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Diederichsen MZ, Diederichsen SZ, Mickley H, Steffensen FH, Lambrechtsen J, Sand NPR, Christensen KL, Olsen MH, Diederichsen A, Grønhøj MH. Prognostic value of suPAR and hs-CRP on cardiovascular disease. Atherosclerosis. 2018;271:245–251. doi: 10.1016/j.atherosclerosis.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 223.Kotsis V, Antza C, Doundoulakis I, Stabouli S. Markers of early vascular ageing. Curr Pharm Des. 2017;23:3200–3204. doi: 10.2174/1381612823666170328142433. [DOI] [PubMed] [Google Scholar]
- 224.Wang X, Zhao J, Zhang Y, Xue X, Yin J, Liao L, Xu C, Hou Y, Yan S, Liu J. Kinetics of plasma von Willebrand factor in acute myocardial infarction patients: a meta-analysis. Oncotarget. 2017;8:90371–9. doi: 10.18632/oncotarget.20091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Whincup PH, Danesh J, Walker M, Lennon L, Thomson A, Appleby P, Rumley A, Lowe GD. von Willebrand factor and coronary heart disease: prospective study and meta-analysis. Eur Heart J. 2002;23:1764–70. doi: 10.1053/euhj.2001.3237. [DOI] [PubMed] [Google Scholar]
- 226.Dhanesha N, Prakash P, Doddapattar P, Khanna I, Pollpeter MJ, Nayak MK, Staber JM, Chauhan AK. Endothelial cell-derived von willebrand factor is the major determinant that mediates von willebrand factor-dependent acute ischemic stroke by promoting postischemic thrombo-inflammation. Arterioscler Thromb Vasc Biol. 2016;36:1829–37. doi: 10.1161/ATVBAHA.116.307660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Guo L, Sun G, Wang G, Ning W, Zhao K. Soluble P-selectin promotes acute myocardial infarction onset but not severity. Mol Med Rep. 2015;11:2027–33. doi: 10.3892/mmr.2014.2917. [DOI] [PubMed] [Google Scholar]
- 228.Pircher J, Engelmann B, Massberg S, Schulz C. Platelet-neutrophil crosstalk in atherothrombosis. Thromb Haemost. 2019;119:1274–1282. doi: 10.1055/s-0039-1692983. [DOI] [PubMed] [Google Scholar]
- 229.He R, Ding C, Yin P, He L, Xu Q, Wu Z, Shi Y, Su L. MiR-1a-3p mitigates isoproterenol-induced heart failure by enhancing the expression of mitochondrial ND1 and COX1. Exp Cell Res. 2019;378:87–97. doi: 10.1016/j.yexcr.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 230.Wang X, Shang Y, Dai S, Wu W, Yi F, Cheng L. MicroRNA-16-5p aggravates myocardial infarction injury by targeting the expression of Insulin receptor substrates 1 and mediating myocardial apoptosis and angiogenesis. Curr Neurovasc Res. 2020;17:11–17. doi: 10.2174/1567202617666191223142743. [DOI] [PubMed] [Google Scholar]
- 231.Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Høydal M, Autore C, Russo MA, Dorn GW 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–61. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 232.Wu T, Chen Y, Du Y, Tao J, Zhou Z, Yang Z. Serum exosomal miR-92b-5p as a potential biomarker for acute heart failure caused by dilated cardiomyopathy. Cell Physiol Biochem. 2018;46:1939. doi: 10.1159/000489383. [DOI] [PubMed] [Google Scholar]
- 233.Lv P, Zhou M, He J, Meng W, Ma X, Dong S, Meng X, Zhao X, Wang X, He F. Circulating miR-208b and miR-34a are associated with left ventricular remodeling after acute myocardial infarction. Int J Mol Sci. 2014;15:5774–5788. doi: 10.3390/ijms15045774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Liu LF, Liang Z, Lv ZR, Liu XH, Bai J, Chen J, Chen C, Wang Y. MicroRNA-15a/b are up-regulated in response to myocardial ischemia/reperfusion injury. J Geriatr Cardiol. 2012;9:28–32. doi: 10.3724/SP.J.1263.2012.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Rubiś P, Totoń-Żurańska J, Wiśniowska-Śmiałek S, Holcman K, Kołton-Wróż M, Wołkow P, Wypasek E, Natorska J, Rudnicka-Sosin L, Pawlak A, Kozanecki A, Podolec P. Relations between circulating microRNAs (miR-21, miR-26, miR-29, miR-30 and miR-133a), extracellular matrix fibrosis and serum markers of fibrosis in dilated cardiomyopathy. Int J Cardiol. 2017;231:201–206. doi: 10.1016/j.ijcard.2016.11.279. [DOI] [PubMed] [Google Scholar]