

Abstract
Aim:
To investigate a novel strategy to target tumor-associated macrophages and reprogram them to an antitumor phenotype in pancreatic adenocarcinoma (PDAC).
Methods:
M2 peptides were conjugated to HA-PEG/HA-PEI polymer to form self-assembled nanoparticles with miR-125b. The efficacy of HA-PEI/PEG-M2peptide nanoparticles in pancreatic tumors from LSL-KrasG12D/+, LSL-Trp53R172H/+, Pdx1-Cre genetically engineered mice was evaluated.
Results:
In vitro M2 macrophage-specific delivery of targeted nanoformulations was demonstrated. Intraperitoneal administration of M2-targeted nanoparticles showed preferential accumulation in the pancreas of KPC-PDAC mice and an above fourfold increase in the M1-to-M2 macrophage ratio compared with transfection with scrambled miR.
Conclusion:
M2-targeted HA-PEI/PEG nanoparticles with miR-125b can transfect tumor-associated macrophages in pancreatic tissues and may have implications for PDAC immunotherapy.
Keywords: : hyaluronic acid-poly(ethylene imine) nanoparticles, intraperitoneal administration, KPC mice, microRNA transfection, pancreatic adenocarcinoma, tumor-associated macrophages
Lay abstract
In pancreatic ductal adenocarcinoma (PDAC) tumor-associated macrophages (TAM) play a major role in tumor progression. Reprogramming of TAMs from a predominant protumoral phenotype to antitumoral phenotype is a promising strategy for PDAC. CD44 targeting hyaluronic acid-poly(ethylenimine) (HA-PEI/PEG)-based nanoparticles encapsulating miR-125b and macrophage-specific delivery and accumulation in the tumor tissue of LSL-KrasG12D/+, LSL-Trp53R172H/+, Pdx1-Cre (KPC) genetically engineered mice were found. The pancreatic tumors show a switch of macrophage phenotype from protumoral to antitumoral.
Graphical abstract
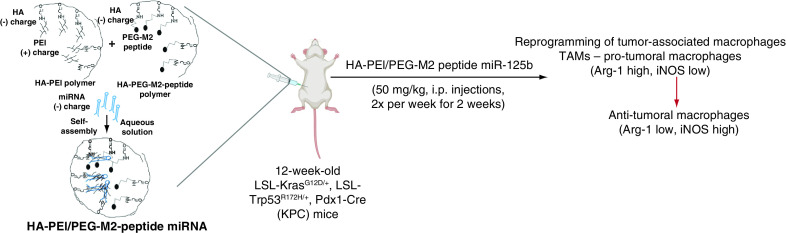
Pancreatic ductal adenocarcinoma (PDAC) is a devastating disease [1]. Surgery, which offers the only realistic hope, has a limited role, with less than 20% of patients undergoing successful resection that affects the clinical outcome. A significant hurdle in PDAC treatment stems from the presence of a complex tumor microenvironment, consisting of a stromal shell rich in fibroblasts and endothelial cells, and the tumor core, consisting of tumor cells and infiltrated tumor-associated macrophages (TAMs). Generally, macrophages are highly plastic cells, playing a crucial role in the innate and adaptive immune responses to pathogens and inflammatory stimuli. They can be activated and polarized to two distinct phenotypes: the classically activated (M1) and alternatively activated (M2) macrophages [2,3].
Among the cells associated with the tumor microenvironment, TAMs are important regulators modulating multiple steps in tumor development, including immune suppression, tumor angiogenesis, tumor growth, metastasis and chemoresistance [4]. During PDAC progression, TAMs switch to an M2-like phenotype, increasing fibroblastic morphology, upregulating mesenchymal markers and promoting proliferation and migration [5]. Not surprisingly, M2-polarized TAMs' infiltration in pancreatic cancer samples has a negative correlation with prognosis [5,6]. Therefore, repolarization of TAMs from a predominant protumor (M2) to antitumor (M1) phenotype may be a promising strategy for the treatment of aggressive tumors such as PDAC.
Previous studies have indicated that microRNAs (miRs) in the tumor microenvironment can affect TAMs' phenotype. Thus, the evaluation of molecular/cellular mechanisms underlying miRs ability to modulate TAM polarization is an area of intense investigation. In particular, miR-125b, which is expressed in macrophages at high levels compared with other immune cells, can regulate macrophage activation [7]. However, delivery of nucleic acid poses significant challenges, such as poor in vivo half-life, cell penetration incapability and instability within the cell endosomes. Among various delivery systems, nonviral vectors have been embraced for gene-delivery applications primarily due to their safety, versatility and ease of production, application and scalability [8]. Hyaluronic acid (HA) polymers are rich in hydroxyl and carboxyl functional groups that can be exploited for chemical derivatization with different modalities (Figure 1) and can specifically bind to CD44 receptors overexpressed in macrophages [9,10]. The efficacy of HA-polyethyleneimine (HA-PEI) nanoformulations encapsulating miR-125b to reprogram the TAMs to antitumor (M1) phenotype and in turn resulting in anticancer efficacy has been previously demonstrated [10–13].
Figure 1. . Synthesis of HA-PEI/PEG-M2 peptide nanoparticles.
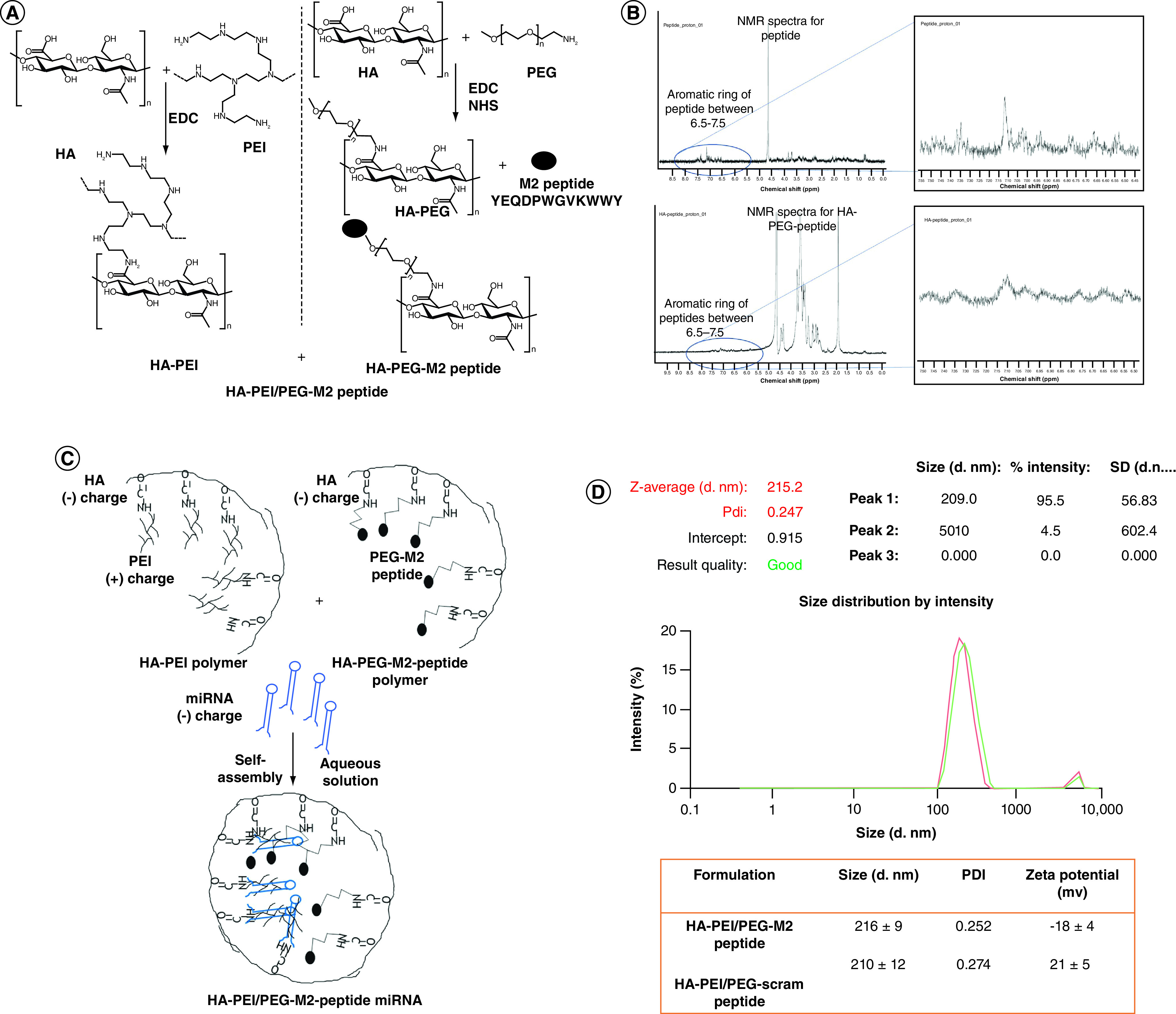
(A) Synthesis and cancerization of HA-PEI, HA-PEG and HA-PEI/HA/PEG-peptide conjugates. HA-PEI and HA-PEG-maleimide derivatives were synthesized using a simple and versatile EDC/NHS conjugation chemistry. M2 peptide was further conjugated to HA-PEG-maleimide. Both the HA polymers are used in a 1:1 ratio for encapsulation of miRNA-125b. (B) The 1H-NMR spectra of the peptide show the characteristic aromatic ring peaks between 6.5–7.5 p.p.m. The 1H-NMR spectra of the HA-PEG-peptide also shows the characteristic aromatic ring peaks between 6.5–7.5 ppm. (C) A-PEI/PEG M2 peptide miRNA nanoparticle formation (D) Hydrodynamic diameters, PDI and ζ-potentials of HA-PEI/HA-PEG-Scram peptide or HA-PEI/HA-PEG-M2 peptide nanoparticle formulations. n = 4 identically prepared samples; data are expressed as mean ± SD.
EDC: Ethyl(dimethylaminopropyl) carbodiimide; HA: Hyaluronic acid; HA-PEG: Hyaluronic acid-polyethylene glycol; HA-PEI: Hyaluronic acid-polyethyleneimine; M2: Alternatively activated macrophage; NHS: N-hydroxysuccinimide; NMR: Nuclear magnetic resonance; PDI: Polydispersity index; PEG: Poly(ethylene glycol); PEI: Polyethyleneimine; SD: Standard deviation.
For M2 macrophage-specific targeting, Cieslewicz et al. recently reported a peptide, M2pep, with the sequence YEQDPWGVKWWY, which preferentially bound to M2 macrophages upon systemic delivery [14]. The M2pep conjugated to a proapoptotic peptide showing preferential binding, rapid internalization and accumulation in murine TAMs compared with other cells [14]. In the present study, the authors used the M2pep's targeting capability and prepared HA-M2pep conjugates for incorporation in HA nanoassemblies (Figure 1). This innovative delivery system enables two levels of targeting: the HA polymer will target the CD44 receptors on the macrophage cell surface, while the M2pep peptide will allow specific targeting of TAMs in the tumor microenvironment. A hallmark of pancreatic cancer is its inability to respond to immunotherapy, with nearly all patients with PDAC failing to benefit from current immunotherapeutic drugs. Thus, there is an urgent need to explore new strategies, including those targeting TAMs, to improve the response to immunotherapy. The aim of this proof-of-concept study is to further enhance the targeting ability of HA-PEI nanoparticles (NP) to M2 macrophages in vivo.
In this study, a novel miR-based therapeutic paradigm was developed to reprogram TAMs from a protumor M2 to an antitumor M1 phenotype in the clinically relevant LSL-KrasG12D/+, LSL-Trp53R172H/+, Pdx1-Cre (KPC) genetically engineered mouse model of pancreatic cancer. It is noteworthy that KPC mice, which conditionally express endogenous mutant Kras and p53 alleles in pancreatic cells [15], develop pancreatic tumors as they age, and recapitulate pathophysiological and molecular features that resemble those observed in human PDAC [16]. This study demonstrates effective delivery, transfection and TAM repolarization with miR-125b administered with M2 peptide-conjugated HA-PEI/HA-PEG NPs in vitro and in vivo.
Materials & methods
Materials
HA 20–40 K was purchased from Lifecore Biomedical Co. (MN, USA). Branched poly(ethyleneimine) (bPEI) 10,000 Da was purchased from Polysciences Inc. (PA, USA). N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich Chemical Co. (MO, USA). Monofunctional poly(ethylene glycol)-amine (PEG2K-NH2, molecular weight 2000 Da) was purchased from Creative PEG Works (NC, USA). Dialysis membrane of 12–14 kDa molecular weight cutoff (MWCO) was obtained from Spectrum Laboratories Inc. (CA, USA). Recombinant murine IL-4 was obtained from PeproTech (NJ, USA). Cy5-NHS ester was obtained from Lumiprobe (FL, USA). Primers specific for iNOS-2, Arg-1 and β-actin were purchased from Eurofins MWG Operon (AL, USA). The M2 peptide or scrambled peptide with a spacer sequence of GGGC was synthesized at Tufts University Core Facility, MA, USA.
Cells
The J774A.1 adherent murine macrophage cell line obtained from the American Type Culture Collection (ATCC; VA, USA) was cultured in Dulbecco's modified Eagle medium (DMEM; Cellgro, VA, USA) containing 10% fetal bovine serum (FBS; HyClone, UT, USA) at 37°C and 5% CO2.
Synthesis of HA-PEI & HA-PEG-M2 conjugates
HA-PEI was synthesized as previously described [12,13]. For the synthesis of HA-PEG-M2, 50 mg of maleimide-PEG-amine was added to EDC/NHS-activated HA. Following synthesis of HA-PEG-maleimide, an M2 peptide, YEQDPWGVKWWY, or scrambled peptide, was used for conjugation with maleimide. The carboxyl group of the terminal cysteine of the peptide was reacted with the maleimide of maleimide-PEG-HA in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 7.4) at a 1:1 molar ratio while mixing under N2 at 4°C for 24 h. The peptide conjugate was then purified by dialysis (3.5 kDa). The purified polymers (HA-PEI, HA-PEG-M2 and HA-PEG-scrambled peptide) were lyophilized using a freeze dryer for 48 h (Freezone-6, Labconco Inc., MO, USA) and the conjugation was confirmed by 400 MHz 1H-NMR spectroscopy (Varian Inc., CA, USA). Cy5-NHS ester and NPs were dissolved in phosphate-buffered saline (PBS) at a 10:1 ratio for overnight reaction. The next day, they were dialyzed overnight with 12–14 kDa MWCO membrane in deionized water. The conjugate was then lyophilized for 48 h and stored at -20°C. Cy5 grafting on polymer was measured indirectly by quantifying Cy5 in dialysates using a Synergy H1 microplate reader (BioTek, VT, US).
Formulation & characterization of HA-PEI/HA-PEG-M2 nanoparticles
HA-PEI/PEG and miR-125b were mixed in a 54:1 weight ratio to form the HA-PEI/HA-PEG-M2 nanosystems. Briefly, HA-PEI and HA-PEG-M2 were dissolved in PBS (pH 7.4) at 3 mg/ml in a 1:1 ratio to which miR was added at 0.5 mg/ml and stirred for 20 min at room temperature (RT). The percent encapsulation efficiency (%EE) was measured using the RiboGreen® assay kit as per the manufacturer's instructions. Dynamic light scattering (DLS) (Malvern Zetasizer Nano-S, Malvern Inc., Malvern, UK) was used to measure particle size, size distribution and Zeta-potential. The morphology of HA-PEI NPs was also assessed using transmission electron microscopy (TEM). TEM analysis was performed on an air-dried NP suspension negatively stained with uranyl acetate using a Japan Electron Optics Laboratory (JEOL) electron microscope.
Uptake of HA-PEI/HA-PEG-M2 nanoparticles by J774 macrophages
J774 murine macrophages cells were seeded on coverslips or in a 6-well plate for 2 h at 37°C with 5% CO2. Cells were then supplemented with conditioned media (IL-4, 100 ng/ml) for 6 h, followed by treatment with Cy5 labeled HA-PEI/HA-PEG-scram peptide or HA-PEI/HA-PEG-M2 peptide NPs for 2 h, 4 h, 6 h and 12 h. Cells on coverslips were then fixed with 4% paraformaldehyde (PFA) for 10 min at RT. Using a mounting media containing 4′,6-diamidino-2-phenylindole (DAPI), the coverslips were then mounted on the slide and imaged (excitation wavelength 647 nm) using a Zeiss confocal microscope (Carl Zeiss, Cambridge, UK). For fluorescence-activated cell sorting (FACS) analysis using Multilaser flow cytometry (DXP11 Analyzer), the cells in 6-well plates were fixed with 4% PFA for 10 min at RT before analysis.
In vitro polarization of J774 macrophages using miR-125b encapsulated HA-PEI/HA-PEG peptide
The ability of HA-PEI/PEG-miRNA NPs to modulate macrophage phenotype from M2 to M1 was assessed by measuring the change in the expression level of iNOS (M1 marker gene) and Arg-1 (M2 marker gene) in J774A.1 macrophages after transfection with NPs. J774 murine macrophage cells were seeded in a 6-well plate for 2 h at 37°C with 5% CO2. Cells were then supplemented with conditioned media (IL-4, 100 ng/ml) for 6 h, followed by treatment with 100 nM miR-125b encapsulated HA-PEI/HA-PEG-scram peptide or HA-PEI/HA-PEG-M2 peptide NPs. The cells were harvested at 24 h after the initial addition of the NPs for RNA isolation using a Zymo RNA Isolation kit. cDNA synthesis was performed using a Verso cDNA Synthesis kit from Thermo Fisher Scientific Inc. (MA, USA). The polarization effect was determined by quantifying the expression of iNOS and Arg-1 using qPCR (LightCycler 480, Roche, CT, USA). qPCR results were normalized to the housekeeping gene β-actin, and data were analyzed using Prism 6 software.
Polarization of macrophages using miR-125b encapsulated HA-PEI/HA-PEG-peptide in LSL-KrasG12D/+, LSL-Trp53R172H/+, Pdx1-Cre mice
The first in vivo study involved assessing the presence of HA-PEI-Cy5-M2 pep NPs in the pancreas of KPC mice. For this purpose, 12-week KPC mice were administered HA-PEI-Cy5-M2 peptide or HA-PEI Cy5-scrambled peptide (50 mg/kg) by intraperitoneal (i.p.) injection. At 2 h postadministration, all mice were euthanized, and the pancreas, lungs, liver, kidney, brain, heart and spleen were collected. All organs were imaged using the IVIS system (Perkin Elmer, MA, USA) and the intensity of the Cy5 signal was determined and compared between the groups.
The effect of i.p. administration of HA-PEI-miR-125b in repolarization of TAMs in KPC mice bearing pancreatic tumors was evaluated by assessing the phenotype of TAMs following administration of HA-PEI-miR-125b conjugated with M2 peptide. For this purpose, KPC mice bearing tumors were injected intraperitoneally (2×/week for 2 weeks) with HA-PEI-miR-125b NPs conjugated with M2 peptide or HA-PEI-scrambled miRNA NPs conjugated with M2 peptide. The expression levels of iNOS and Arg-1 (M1 and M2 markers, respectively) were evaluated in the macrophage population isolated from the pancreatic tumor tissue by qPCR, and the expression levels of Arg1 staining by immunohistochemistry (IHC). At the end of the two-week treatment, pancreas and tumors were either fixed with 4% PFA overnight at 4°C for IHC or homogenized for isolation of macrophages. The embedded pancreas/tumor tissue was sectioned (5 μm), rehydrated and stained using standard IHC protocols using F4/80 (cat. no. 70076) and Arg1 (cat. no. 93668) antibodies (Cell Signaling Technologies, MA, USA), as previously described [17,18]. Five random sections from each tissue section were quantified using ImageJ software.
TAMs were isolated by digesting pancreatic tumor tissue in DMEM media with 2 mg/ml of collagenase A (Roche) and 13 DNase I (Sigma) for 30 min at 37°C on a shaker. The reaction was halted with 5 ml of FBS (Atlanta Biologicals, Bio-Techne, MN, USA)) and cells were collected by filtering through 40 mm nylon mesh and centrifugation at 2000 r.p.m. for 5 min at 4°C. The collected cells were then resuspended in staining buffer (PBS containing 1% bovine serum albumin [BSA]). MACS magnetic selection using antimouse F4/80 microbeads (Miltenyi Biotec, CA, USA) was used for isolating F4/80+ cells. A High Pure RNA isolation kit (Roche Applied Sc, IN, USA) was used to isolate RNA from F4/80+ cells and converted cells to cDNA using capacity cDNA Reverse Transcriptase (Applied Biosystems, NY, USA). Primers against iNOS, Arg-1, CD86, CD80, CD206, CD163 and β-actin (house-keeping) genes were used for expression levels of the respective genes using qPCR (iCycler, Bio-Rad, CA, USA).
Statistical analysis
Statistical analysis was performed using Prism 7.0 software (Graph Pad Software, CA, USA). The data, obtained from at least three independent experiments, were expressed as the mean ± SD. Statistical evaluation was performed by one-factor analysis of variance (ANOVA) followed by the Tukey test for multiple comparisons. p < 0.05 was regarded as statistically significant.
Results
Formulation & characterization of HA-PEI/HA-PEG-M2pep nanoparticles encapsulating miR-125b
The HA-PEG polymer was successfully conjugated to M2/scrambled peptide (Figure 1A). The 1H-NMR spectra of the peptide show the characteristic aromatic ring peaks between 6.5–7.5 p.p.m. (Figure 1B). The 1H-NMR spectra of the HA-PEG-peptide also showed the characteristic aromatic ring peaks between 6.5–7.5 p.p.m. The HA-PEI conjugation strategy and method used in this study are the same as in a previous study by the authors [12]. The 1H NMR spectrum of the HA-PEI with EDC/NHS coupling does not show the PEI peak but shows additional peaks between 2 and 3 p.p.m. due to PEI conjugation on the HA backbone. During the self-assembly process, the PEI forms the positive core that electrostatically binds miR, while the HA forms the negatively charged surface of these NPs (Figure 1C). The encapsulation of miR resulted in similar sizes with M2 (216 ± 9 nm) and scrambled peptide (210 ± 12 nm) conjugated polymer as determined by DLS (Figure 1D) and the HA-PEI NPs were imaged using a TEM (Supplementary Figure 1). The Zeta-potential on HA-PEI/HA-PEG/HA-M2pep particles was -18 ± 4 mV and -21 ± 5 mV for HA-PEI/HA-PEG/HA-scrambled peptide particles (Figure 1D). The %EE of miR-125b in HA-PEI NPs, as well as the miR-negative control, was 99%, as shown by the RiboGreen assay. Based on the start ratios of polymer:miR and the %EE, the ratio of polymer and miR-125 was maintained at 54:1 postformulation.
In vitro uptake of HA-PEI/HA-PEG-M2pep nanoparticles encapsulating miR-125b by J774 macrophages
To demonstrate that HA-PEI/HA-PEG-M2pep NPs can be taken up by tumor-associated macrophages, the NPs were labeled with Cy5 dye. J774 murine macrophages were treated with IL-4 (100 ng/ml) for 6 h prior to treatment with HA-PEI/HA-PEG/HA-M2pep particles to develop the tumor-associated M2 phenotype. After IL-4 treatment, the cells were treated with Cy5-labeled HA-PEI/HA-PEG/HA-M2pep or HA-PEI/HA-PEG/HA-scram peptide for 2, 4, 6 and 12 h. As determined by confocal microscopy (Figure 2A) and FACS analysis (Figure 2B), HA-PEI/HA-PEG/HA-M2pep NPs show a higher uptake as compared with HA-PEI/HA-PEG/HA-scram peptide NPs at 4 and 6 h. At 12 h there was a gradual decrease of fluorescence for both formulations.
Figure 2. . In vitro uptake of miR-125 encapsulated HA-PEI/PEG-M2 peptide by J774 murine macrophages.
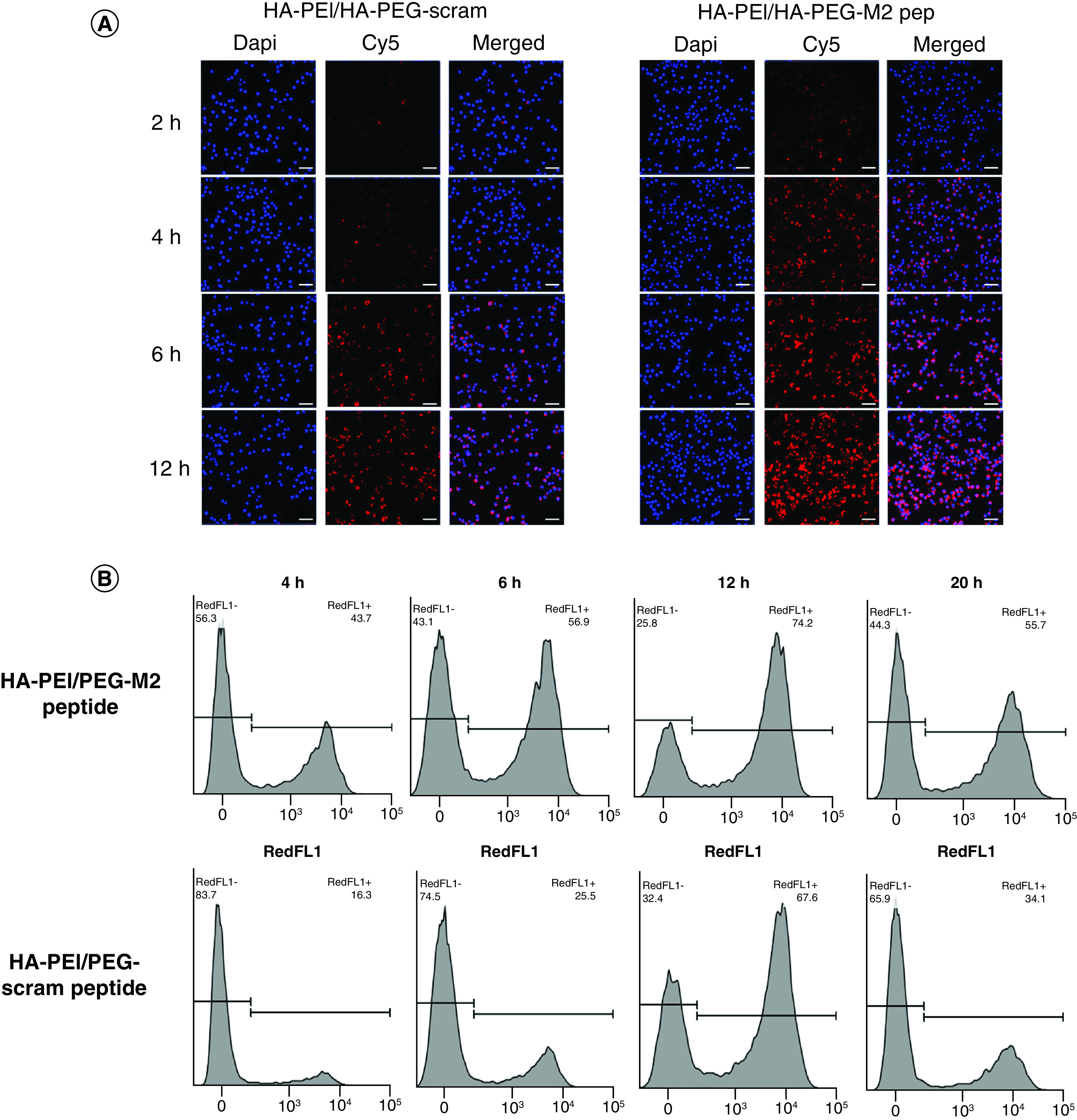
(A) J774 murine macrophage cells were treated with Interleukin-4 (100 ng/ml) for 6 h, followed by treatment with Cy5-labeled HA-PEI/HA-PEG-scram peptide or HA-PEI/HA-PEG-M2 peptide nanoparticles for 2 h, 4 h, 6 h and 12 h. Representative images of cells were stained with DAPI for confocal analysis. Scale bar indicates 50 μm. (B) J774 murine macrophage cells were treated with IL-4 (100 ng/ml) for 6 h, followed by treatment with Cy5-labeled HA-PEI/HA-PEG-scram peptide or HA-PEI/HA-PEG-M2 peptide nanoparticles for 4 h, 6 h, 12 h and 20 h. Histograms of cells after fluorescence-activated single cell sorting analysis are shown.
DAPI: 4′,6-diamidino-2-phenylindole; HA-PEG: Hyaluronic acid-poly(ethylene glycol); HA-PEI: Hyaluronic acid-polyethyleneimine; M2: Alternatively activated macrophage.
In vitro repolarization of M2 macrophages using HA-PEI/HA-PEG-M2pep nanoparticles encapsulating miR-125b
Once the successful uptake of HA-PEI/HA-PEG/HA-M2pep NPs by the M2 macrophages was observed, the efficacy of HA-PEI/HA-PEG/HA-M2pep NPs loaded with miR-125b in macrophage repolarization from M2 to M1 in vitro was tested. For this purpose, the M2 phenotype was induced in the J774 macrophages by treatment with IL-4. As shown in Figure 3, IL-4 treatment significantly reduced iNOS levels and significantly increased Arg-1 levels, demonstrating M2 polarization of the macrophages. Following IL-4 administration, J774 macrophages were treated with HA-PEI/HA-PEG/HA-M2pep NPs encapsulated with miR-125b. The cells treated with HA-PEI/HA-PEG/HA-M2pep NPs and HA-PEI/HA-PEG/HA-scram pep NPs showed an increase in Arg-1 levels compared with untreated controls. However, comparing the HA-PEI/HA-PEG-M2 pep group with the HA-PEI/HA-PEG-scram pep group, HA-PEI/HA-PEG-M2 pep reduced Arg1 levels as compared with the HA-PEI/HA-PEG-scram pep treated group, when cells are pretreated with IL4 (mimicking anti-inflammatory microenvironment). A significantly higher level of iNOS was observed in cells treated with HA-PEI/HA-PEG/HA-M2pep NPs as compared with cells treated with HA-PEI/HA-PEG/HA-scram pep NPs. A significant increase in iNOS/Arg-1 levels (∼100-fold) was also observed in the HA-PEI/HA-PEG/HA-M2pep NPs treated group as compared with the HA-PEI/HA-PEG/HA-scram pep NPs (Figure 3), clearly demonstrating the enhanced ability of HA-PEI/HA-PEG/HA-M2pep NPs to repolarize macrophages from M2 to M1 phenotype.
Figure 3. . In vitro polarization of J774 macrophages using miR-125b encapsulated HA-PEI/HA-PEG-peptide.

(A) Schematic method for the in vitro macrophage repolarization study. Expression of (B) Arg-1, (C) iNOS and (D) iNOS/Arg-1 ratio in IL-4-stimulated macrophages treated with HA-PEI/HA-PEG-scram peptide or HA-PEI/HA-PEG-M2. qPCR was used for quantification of gene expression level with β-actin as the internal control. n = 4
*p < 0.05; **p < 0.01; ****p < 0.001.
HA-PEG: Hyaluronic acid-poly(ethylene glycol); HA-PEI: Hyaluronic acid-polyethyleneimine; M2: Alternatively activated macrophage; PEG: Poly(ethylene glycol); qPCR: Quantitative polymerase chain reaction.
In vivo biodistribution of HA-PEI/HA-PEG-M2pep nanoparticles
To demonstrate that HA-PEI/HA-PEG-M2pep NPs can be delivered to the tumor area in vivo, the NPs were labeled with Cy5 dye. The in vivo accumulation of HA-PEI-Cy5 NPs conjugated with M2 peptide or scrambled peptide were evaluated. KPC mice bearing pancreatic tumors were injected intraperitoneally with HA-PEI-Cy5 NPs conjugated with either M2 peptide or scrambled peptide for 2 h. Figure 4 shows that the NPs accumulated in the pancreas and liver; however, no significant difference was observed between the two formulations. No fluorescence was detected in the spleen, kidney (Figure 4), brain or lungs (not shown). These data indicate that HA-PEI NPs can reach the pancreatic tumor area.
Figure 4. . Biodistribution of HA-PEI/PEG nanoparticles in KPC mice.
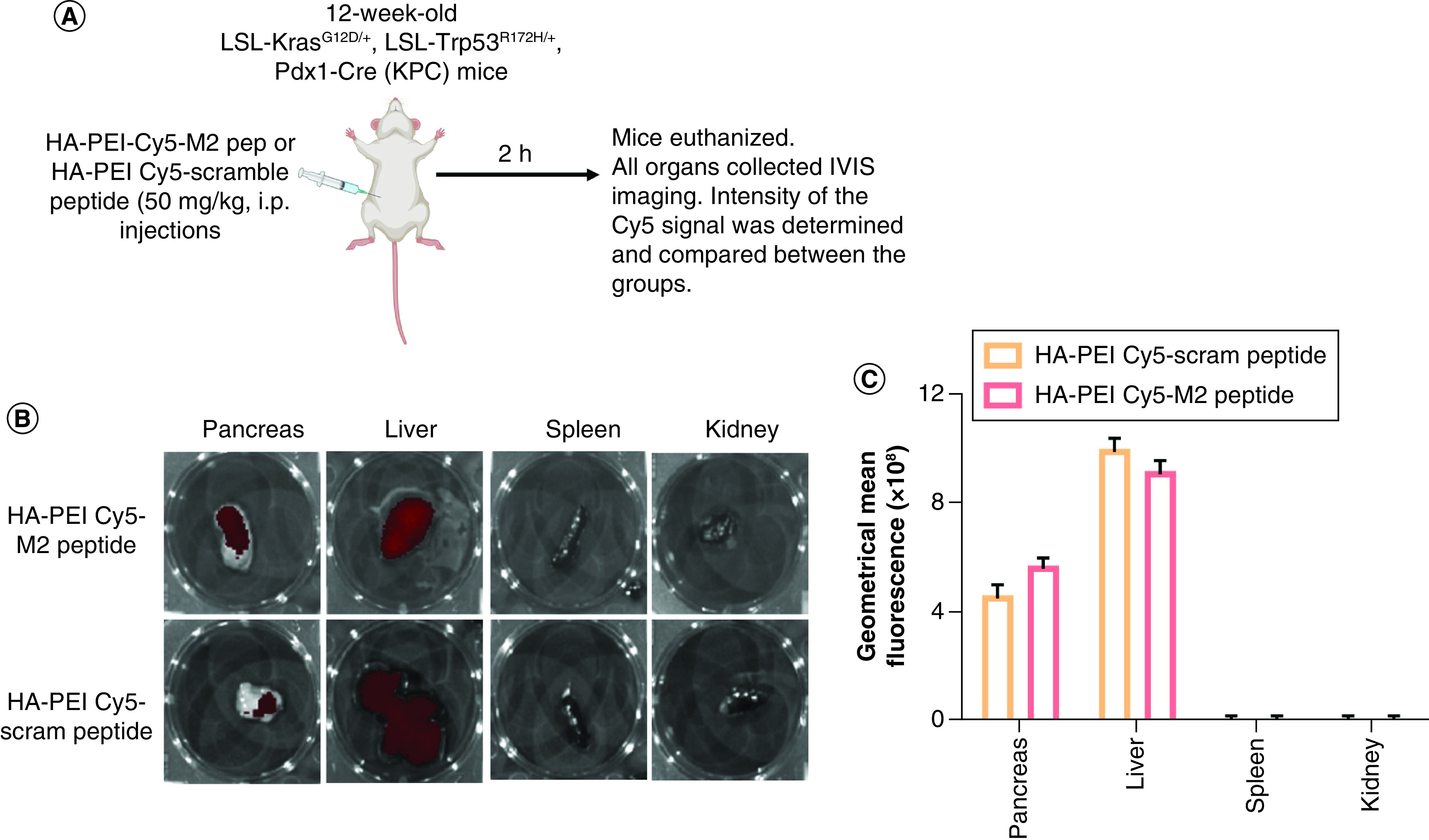
(A) Schematic method for biodistribution of HA-PEI/PEG nanoparticles in KPC mice. Presence of Cy5-labeled HA-PEI nanoparticle formulations (M2 peptide- or scrambled peptide-conjugated) in KPC pancreas. (B) The pancreas, liver, spleen and kidney tissues were isolated from 12-week-old KPC mice treated with i.p. administration of HA-PEI-Cy5 (M2 peptide- or scrambled peptide-conjugated) for 2 h. (C) The fluorescence of Cy5 was determined through imaging by the IVIS system.
HA-PEI: Hyaluronic acid-polyethyleneimine; IVIS: In Vivo Imaging Systems; KPC: KrasLSL.G12D/+; p53R172H/+; PdxCretg/+.
In vivo repolarization of M2 macrophages using HA-PEI/HA-PEG-M2pep nanoparticles encapsulating miR-125b
The effect of HA-PEI-miR-125b in the repolarization of TAMs in KPC mice bearing pancreatic tumors was investigated by assessing the phenotype of TAMs after i.p. administration of miR-125b encapsulated in HA-PEI-M2pep. For this purpose, KPC mice bearing tumors (confirmed using ultrasound), were injected (2×/week for 2 weeks) with HA-PEI-miR-125b NPs conjugated with M2 peptide or HA-PEI-scrambled miRNA NPs conjugated with M2 peptide (Figure 5A). iNOS and Arg-1 expression levels were assessed (as M1 and M2 markers, respectively) in the macrophage population isolated from the KPC pancreatic tumors by qPCR, and the Arg1 expression levels by IHC.
Figure 5. . In vivo polarization of tumor-associated macrophages using miR-125b encapsulated HA-PEI/HA-PEG-peptide in KPC mice.
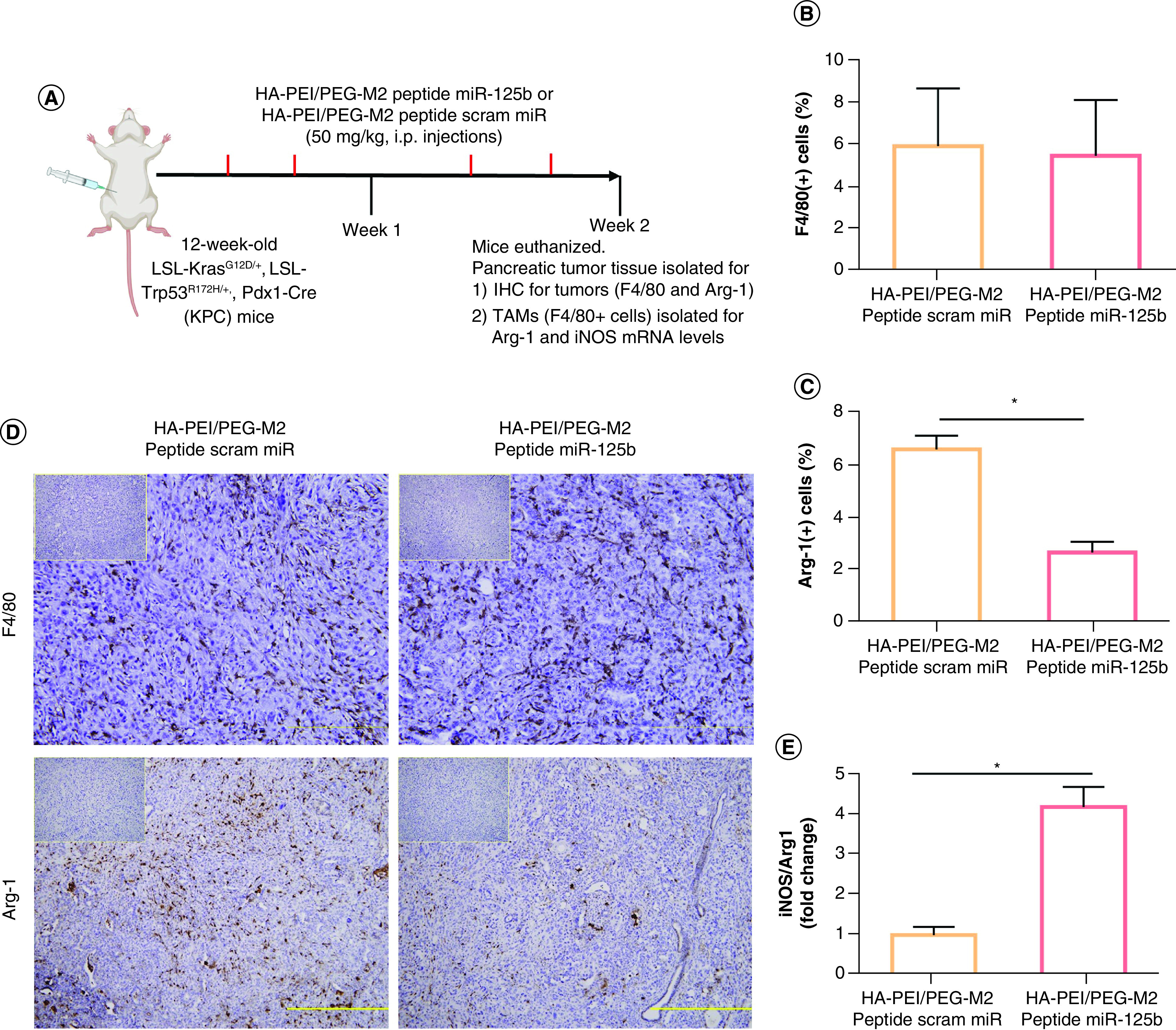
(A) Schematic method for the in vivo macrophage repolarization study in KPC mice. (B) F4/80 and (C) Arg1 immunostaining were performed on KPC tumor sections and photographs were taken at ×20 (for F4/80) and ×10 (for Arg-1) magnification. (D) The consecutive section was stained with isotype IgG as a negative staining control and is shown in the upper right corner. Results are expressed as a percent of F4/80+ or Arg1+ cells ± SD per ×20 or ×10 field, respectively. (E) The ratio of iNOS/Arg1 in KPC tumor macrophages treated with HA-PEI nanoformulations. qPCR was used for quantification of the gene expression level with β-actin as the internal control.
*Significant compared with the control group; p < 0.01.
HA-PEG: Hyaluronic acid-poly(ethyleneglycol); HA-PEI: Hyaluronic acid-polyethyleneimine; IHC: Immunohistochemistry; KPC: KrasLSL.G12D/+; p53R172H/+; PdxCretg/+; M2: Alternatively activated macrophage; TAM: Tumor associated macrophage.
Although the expression levels of F4/80+ macrophages in the tumor area was similar between the groups, a 60% reduction in Arg1 levels (p < 0.01) was observed in tumors from KPC mice treated with HA-PEI-miR-125b NPs conjugated with M2 peptide, compared with those treated with HA-PEI-scrambled miR (Figure 5B–D). Moreover, following i.p. administration of HA-PEI-miR-125b conjugated with M2 peptide for 2 weeks, there was a significant fourfold increase in the iNOS/Arg1 ratio levels (M1/M2 markers) in the macrophage population isolated from the pancreatic tumors, compared with mice treated with HA-PEI-scrambled miR conjugated with M2 peptide (Figure 5E). The levels of cell surface-based M1 and M2 macrophage polarization markers CD80, CD86 and CD163 as well as intracellular M2 macrophage polarization marker CD206 was evaluated (Figure 6A). The tumor-associated macrophages isolated from KPC mice treated with HA-PEI-miR-125b NPs conjugated with M2 peptide showed an increase in levels of M1 markers CD86 (∼3.5-fold increase) and CD80 (∼threefold increase) compared with the mice treated with HA-PEI-scrambled miR (Figure 6B). Additionally, the levels of both the M2 markers, CD206 and CD163, were significantly reduced in tumor-associated macrophages isolated from KPC mice treated with HA-PEI-miR-125b NPs conjugated with M2 peptide as compared with the mice treated with HA-PEI-scrambled miR (Figure 6B). Collectively, these data suggest that HA-PEI-miR-125b conjugated with M2 peptide reprograms PDAC-infiltrating TAMs from an M2-like phenotype toward an M1-like phenotype.
Figure 6. . Characterization of tumor-associated macrophages using miR-125b encapsulated HA-PEI/HA-PEG-peptide in KPC mice.

(A) Schematic method for the characterization of TAMs in KPC mice. (B) Expression levels of CD80, CD86, CD163 and CD206 in TAMs isolated from pancreatic tumor tissue of KPC mice treated with HA-PEI/PEG M2 peptide miR-125b nanoformulations or scrambled miR nanoparticles. qPCR was used for quantification of the gene expression level with β-actin as the internal control.
*Significant compared with the control group; p < 0.01.
HA-PEI: Hyaluronic acid-polyethyleneimine; PEG: Poly(ethylene glycol); KPC: KrasLSL.G12D/+; p53R172H/+; PdxCretg/+; M2: Alternatively activated macrophage; qPCR: Quantitative polymerase chain reaction; TAM: Tumor associated macrophage.
Discussion
PDAC is a deadly disease in need of novel therapeutic strategies. A significant hurdle in PDAC treatment stems from the presence of a complex tumor microenvironment, consisting of a stromal shell rich in fibroblasts and endothelial cells, and the tumor core, consisting of tumor cells and infiltrated TAMs. In this proof-of-concept study, a novel miR-based therapeutic paradigm for reprogramming TAMs from a protumor M2 to antitumor M1 phenotype was evaluated in KPC mice. Effective delivery, transfection and TAM repolarization with miR-125b administered with HA-PEI/HA-PEG-M2 peptide NPs was shown in vitro and in vivo.
Macrophages are generally highly plastic cells, playing a crucial role in the innate and adaptive immune responses to pathogens and inflammatory stimuli. They can be activated and polarized to two distinct phenotypes: the classically activated (M1) and alternatively activated (M2) macrophages. During PDAC progression, TAMs switch to M2-like phenotype increasing fibroblastic morphology, upregulating mesenchymal markers and promoting proliferation and migration. Not surprisingly, M2-polarized TAM infiltration in pancreatic cancer samples is negatively correlated with prognosis. Therefore, repolarization of TAMs from a predominant protumor (M2) to antitumor (M1) phenotype is a promising strategy for the treatment of aggressive tumors, such as PDAC. In this study, a novel strategy to specifically target M2 macrophages and reprogram them to M1-like phenotype using HA-PEI/HA-PEG-M2 peptide NPs for delivery of miR-125b was successfully designed.
Human PDAC arises from pancreatic intraepithelial neoplasias, frequently driven by activating mutations in Kras [19], followed by loss or mutation of tumor suppressors, such as p53. To confirm the findings in vivo, the KPC genetically engineered mouse model was used. This model is based on the simultaneous Cre-dependent expression of KrasG12D and a dominant-negative form of TP53 (Trp53R127H) in the pancreas [15,20]. Mice develop invasive PDAC at ∼10 weeks of age [18]. Metastases are observed in 80% of the KPC mice at the same sites observed in patients with PDAC (i.e., liver, lung and peritoneum). Moreover, pancreatic tumors show many of the immunohistochemical markers associated with human PDAC. Another crucial aspect of the KPC model is that the animals have a fully functional immune system, unlike in the immune-compromised xenograft models. In addition, KPC tumors recapitulate the dense stroma observed in human PDAC. Consequently, the KPC mouse model represents an excellent clinically relevant model to evaluate novel therapeutic strategies, including TAM repolarization [21].
Delivery of nucleic acid poses significant challenges, such as poor in vivo half-life, cell penetration incapability and instability within the cell endosomes. Among various delivery systems, nonviral vectors have been embraced for gene delivery applications primarily due to their safety, versatility and ease of production, application and scalability [9]. Actively targeting RNA nanocarriers engineered from biocompatible polymers have unique properties that can be tailored by controlling polymer composition, size, shape and surface characteristics [22]. Following systemic administration, NPs may accumulate in tumor tissues through the enhanced permeability and retention effect (EPR) [23]. Once NPs accumulate in the tumor tissue, the regulation of TAM functions may be restricted within the tumor microenvironment, reducing systemic toxicity.
The cationic polymer, PEI, was anchored onto the HA backbone, which enables self-assembly of the conjugate in the presence of negatively charged nucleic acids based on electrostatic interactions. Importantly, the chemical attachment of PEI on the HA backbone mitigates the cytotoxicity that is often associated with free PEI. Apart from using polymers for targeted delivery, several investigators have reported surface functionalization of NPs using targeting ligands, such as folic acid and epidermal growth factor peptides, to achieve tumor-specific delivery [24]. Cieslewicz et al. showed the use of M2 peptides to specifically target TAMs (M2 macrophages) [14]. We successfully conjugated this M2 peptide HA-PEG polymer and evaluated the HA-PEI/HA-PEG-M2 peptide NP system to target TAMs. This dual-targeting strategy with HA and M2 peptides led to enhanced uptake by M2 macrophages as compared with the untargeted nanoformulations, which serves as an added advantage over other HA-PEI-based nanoformulations [11,12,25]. We previously showed that i.p. administration of HA-PEI NPs brings about effective reprogramming of TAMs to an antitumoral phenotype, in turn reducing the tumor burden in an ovarian cancer mice model [13]. Based on this evidence, we used a similar dosing strategy of i.p. administration of M2 peptide targeted HA-PEI NPs in the pancreatic tumors. Using i.p. administration, we were able to increase the dosing volume as well as reduce potential toxicity. Following i.p. administration, HA-PEI/HA-PEG-M2 peptide NP encapsulating miR-125b reaches the tumor site and reprograms PDAC-infiltrating TAMs toward an M1-like phenotype, as suggested by the reduction in Arg1, CD206 and CD163 levels as well as the increase in CD80 and CD86 levels. Similarly, it has been documented that selective macrophage transfection with miR-125b using HA-PEI NPs can also effectively increase the M1-to-M2 macrophage ratio and improve antitumor efficacy in non-small cell lung cancer and ovarian cancer mouse models [12,13], highlighting that this system is not tumor restrictive.
Conclusion
The authors developed a novel miR-based therapeutic paradigm for reprogramming TAMs from a protumor M2 to antitumor M1 phenotype. Effective delivery, transfection and TAM repolarization with miR-125b administered with HA-PEI/HA-PEG-M2 peptide NPs in cells and in KPC mice was shown, strongly suggesting that i.p. administration of HA-PEI/HA-PEG-M2 peptide miR-125b nanoformulations is an effective strategy for the repolarization of TAMs. Further studies are warranted to evaluate the efficacy of this novel treatment strategy alone and in combination with current chemotherapeutic drugs as a potential anticancer therapy for PDAC.
Summary points.
In this study, the authors successfully conjugated M2 peptide to HA-PEG/HA-PEI polymers.
M2 peptide conjugated to HA-PEG polymer with HA-PEI polymer in a 1:1 ratio and can self-assemble in nanoformulations in the presence of positively charged mirRNA.
The synthesized HA-PEI/PEG M2 peptide nanoparticles (NP) can specifically target M2 macrophages in vitro and reprogram them to the antitumoral M1 phenotype.
In a LSL-KrasG12D/+, LSL-Trp53R172H/+, Pdx1-Cre (KPC) genetically engineered mice model of pancreatic ductal adenocarcinoma (PDAC), intraperitoneal administration of HA-PEI/PEG M2 peptide NPs resulted in accumulation of HA-PEI/PEG M2 peptide NPs in pancreatic tumors.
Intraperitoneal administration of HA-PEI/PEG M2 peptide NPs can reprogram M2 TAMs to antitumoral M1 phenotype in the tumor microenvironment.
Supplementary Material
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/suppl/10.2217/nnm-2021-0080
Financial & competing interests disclosure
Financial support for this work was provided by the United States National Cancer Institute of the National Institute of Health through grants R21-CA213114-01 (to MM Amiji and GG Mackenzie) and the Northeastern University-Dana Farber Cancer Center Joint Program on Cancer Drug Development. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: •• of considerable interest
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J. Clin. 70(1), 7–30 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Hao NB, Lü MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 948098 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genard G, Lucas S, Michiels C. Reprogramming of tumor-associated macrophages with anticancer therapies: radiotherapy versus chemo- and immunotherapies. Front. Immunol. 8, 828 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Squadrito ML, Etzrodt M, De Palma M, Pittet MJ. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol. 34(7), 350–359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu CY, Xu JY, Shi XYet al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Invest. 93(7), 844–854 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Mielgo A, Schmid MC. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 46(3), 131–138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaudhuri AA, So AYL, Sinha Net al. MicroRNA-125b potentiates macrophage activation. J. Immunol. 187(10), 5062–5068 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glover DJ, Lipps HJ, Jans DA. Towards safe, non-viral therapeutic gene expression in humans. Nat. Rev. Genet. 6(4), 299–310 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 6, 201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganesh S, Iyer AK, Weiler J, Morrissey DV, Amiji MM. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials 34(13), 3489–3502 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganesh S, Iyer AK, Weiler J, Morrissey DV, Amiji MM. Combination of siRNA-directed gene silencing with cisplatin reverses drug resistance in human non-small cell lung cancer. Mol. Ther. Nucleic Acids 2(7), e110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parayath NN, Parikh A, Amiji MM. Repolarization of tumor-associated macrophages in a genetically engineered nonsmall cell lung cancer model by intraperitoneal administration of hyaluronic acid-based nanoparticles encapsulating microRNA-125b. Nano Lett. 18(6), 3571–3579 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Parayath NN, Gandham SK, Leslie F, Amiji MM. Improved anti-tumor efficacy of paclitaxel in combination with MicroRNA-125b-based tumor-associated macrophage repolarization in epithelial ovarian cancer. Cancer Lett. 461, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cieslewicz M, Tang J, Yu JLet al. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc. Natl Acad. Sci. USA 110(40), 15919–15924 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This is the original study that describes the M2 peptide. We have used the M2 peptide sequence described in this study to formulate the targeted HA-PEI-/HA-PEG NPs.
- 15.Hingorani SR, Wang L, Multani ASet al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7(5), 469–483 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Gopinathan A, Morton JP, Jodrell DI, Sansom OJ. GEMMs as preclinical models for testing pancreatic cancer therapies. Dis. Model Mech. 8(10), 1185–1200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackenzie GG, Ouyang N, Xie Get al. Phospho-sulindac (OXT-328) combined with difluoromethylornithine prevents colon cancer in mice. Cancer Prev. Res. (Phila.) 4(7), 1052–1060 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mallangada NA, Vargas JM, Thomas Set al. A novel tricarbonylmethane agent (CMC2.24) reduces human pancreatic tumor growth in mice by targeting Ras. Mol. Carcinog. 57(9), 1130–1143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53(4), 549–554 (1988). [DOI] [PubMed] [Google Scholar]
- 20.Olive KP, Politi K. Translational therapeutics in genetically engineered mouse models of cancer. Cold Spring Harb. Protoc. 2014(2), 131–143 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Lee JW, Komar CA, Bengsch F, Graham K, Beatty GL. Genetically engineered mouse models of pancreatic cancer: the KPC model (LSL-Kras(G12D/+); LSL-Trp53(R172H/+); Pdx-1-Cre), its variants, and their application in immuno-oncology drug discovery. Curr. Protoc. Pharmacol. 73, 14.39.1–14.39.20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abeylath SC, Ganta S, Iyer AK, Amiji M. Combinatorial-designed multifunctional polymeric nanosystems for tumor-targeted therapeutic delivery. Acc. Chem. Res. 44(10), 1009–1017 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Nguyen CT, Tran TH, Amiji M, Lu X, Kasi RM. Redox-sensitive nanoparticles from amphiphilic cholesterol-based block copolymers for enhanced tumor intracellular release of doxorubicin. Nanomedicine 11(8), 2071–2082 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talekar M, Kendall J, Denny W, Garg S. Targeting of nanoparticles in cancer: drug delivery and diagnostics. Anti Cancer Drugs 22(10), 949–962 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Sahiner N, Sagbas S, Sahiner M, Ayyala RS. Polyethyleneimine modified poly (Hyaluronic acid) particles with controllable antimicrobial and anticancer effects. Carbohydr. Polym. 159, 29–38 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.